

Abstract
Assembly of Kaposi’s sarcoma-associated herpesvirus (KSHV) begins at a bacteriophage-like portal complex that nucleates formation of an icosahedral capsid with capsid-associated tegument complexes (CATCs) and facilitates translocation of an ∼150-kb dsDNA genome, followed by acquisition of a pleomorphic tegument and envelope. Because of deviation from icosahedral symmetry, KSHV portal and tegument structures have largely been obscured in previous studies. Using symmetry-relaxed cryo-EM, we determined the in situ structure of the KSHV portal and its interactions with surrounding capsid proteins, CATCs, and the terminal end of KSHV’s dsDNA genome. Our atomic models of the portal and capsid/CATC, together with visualization of CATCs’ variable occupancy and alternate orientation of CATC-interacting vertex triplexes, suggest a mechanism whereby the portal orchestrates procapsid formation and asymmetric long-range determination of CATC attachment during DNA packaging prior to pleomorphic tegumentation/envelopment. Structure-based mutageneses confirm that a triplex deep binding groove for CATCs is a hotspot that holds promise for antiviral development.
Keywords: cryo-EM, microscopy, Kaposi's sarcoma-associated herpesvirus, herpesvirus, gammaherpesvirus, virology, capsid, portal, tegument, genome
Graphical Abstract
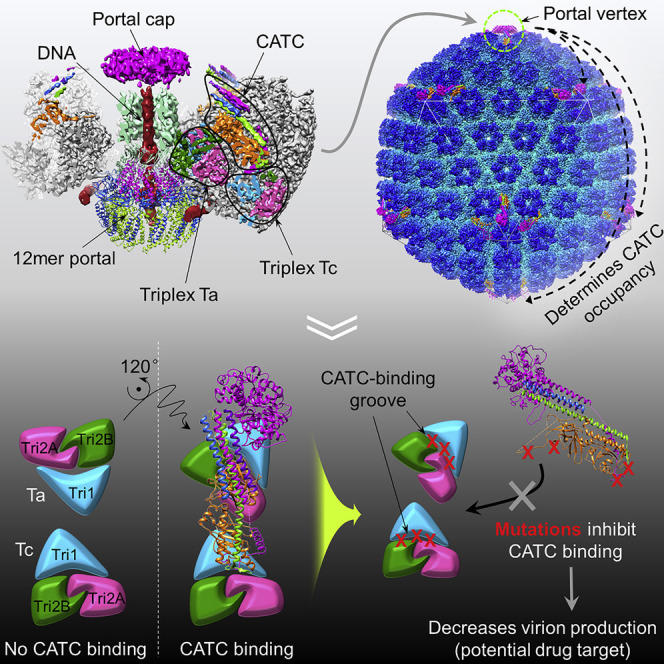
Highlights
-
•
Genome-packing portal and capsid-associated tegument complexes (CATCs) resolved
-
•
Partial and asymmetric CATC occupancy introduces structural variability
-
•
CATC binding introduces 120° counterclockwise rotation of triplex Ta on the capsid
-
•
Structure-based mutageneses reveal binding hotspot for antiviral development
Resolution of KSHV’s asymmetric icosahedral structure is achieved via symmetry-relaxed cryo-EM using sequential localized classification. Interactions between the DNA-translocating portal protein, associated capsid and tegument proteins, and the viral genome reveal surprising variability in tegument protein occupancy and orientation plasticity.
Introduction
First discovered in 1994 associated with tumor lesions in AIDS patients in Los Angeles (Chang et al., 1994), Kaposi’s sarcoma-associated herpesvirus (KSHV) has since been shown to cause endemic cancers in sub-Saharan Africa, the greater Mediterranean, and the Xinjiang region of China (Ganem, 2010, Giffin and Damania, 2014). KSHV is a member of the herpesvirus subfamily Gammaherpesvirinae, which also includes Epstein-Barr virus (EBV), the first identified human oncovirus. Like all herpesviruses, assembly of an infectious KSHV virion starts at a bacteriophage-like portal complex that putatively nucleates the formation of a T = 16 icosahedral capsid, which, at maturation, is composed of major capsid protein (MCP), small capsid protein (SCP), αβ2 heterotrimers of the Tri1 monomer and Tri2 dimer and is decorated by capsid-associated tegument complexes (CATCs) (Cardone et al., 2012). Upon establishment of an initial procapsid, the portal facilitates the translocation of KSHV’s ∼150-kb genome. This key process involves recruitment of an ATP-driven terminase (Yang et al., 2007, Heming et al., 2017) to the unique portal vertex to recognize, package, and cleave concatemeric viral double-stranded (ds)DNA, which, in conjunction with CATC’s critical supporting roles (Heming et al., 2017), give rise to viable genome-containing nucleocapsids (Adelman et al., 2001, Beard et al., 2002). Unlike the comparatively high occupancies of capsid-associated tegument proteins in alphaherpesviruses (Dai and Zhou, 2018, Wang et al., 2018) and betaherpesviruses (Liu et al., 2018, Yu et al., 2017), KSHV CATC binding sites are markedly partially and/or more flexibly occupied, leading to poorly resolved CATC structures in prior icosahedral reconstructions of KSHV (Dai et al., 2014, Dai et al., 2018). The CATC nonetheless plays a critical role in the release of a pleomorphic virion because the recruitment of outer tegument proteins and a glycoprotein-sporting envelope leading to virion egress depend on interactions of various viral proteins with constituents of the CATC (Owen et al., 2015, Sathish et al., 2012).
In the absence of an in situ KSHV portal and CATC structures, how a single portal protein (pORF43) orchestrates the rise of a robust capsid and how order is maintained in the subsequently complex and variable processes of tegumentation and envelopment remains unknown. An invaluable body of pioneering work has been accomplished so far on the prototypical herpesvirus herpes simplex virus type 1 (HSV-1) portal, including microscopy studies confirming its localization at a capsid vertex (Cardone et al., 2007), its dodecameric stoichiometry (Rochat et al., 2011), and, more recently, the impressive identification and reconstruction of the 5-fold vertex region surrounding the portal (McElwee et al., 2018). Here we present the first atomic structures of a gammaherpesvirus portal vertex in KSHV, which allowed us to use structure-guided mutageneses to identify a viable drug target. Simultaneously, our method of symmetry relaxation and sequential localized classification enabled us to dissect a heretofore puzzling schema of variable CATC occupancy at capsid vertices, providing insights into portal-seeded capsid assembly and how structural plasticity arises from a well-defined and highly ordered capsid.
Results
Resolving Symmetry Mismatches in the KSHV Capsid
To determine high-resolution structures of KSHV’s unique portal vertex, we imaged frozen-hydrated KSHV virions, obtaining 44,328 virion particle images. Prior KSHV capsid reconstructions, although informative, have been calculated with icosahedral symmetry applied, obscuring non-icosahedrally related structures (Dai et al., 2018, Trus et al., 2001, Wu et al., 2000). Here we developed a workflow (Figures S1 and S2 ) to determine structures of the non-icosahedrally arranged components. This sub-particle data processing procedure, employing sequential localized classification with symmetry relaxation, allows stepwise reconstruction of selected regions of a viral particle. Briefly, two rounds of sub-particle classification were performed to relax icosahedral and local symmetries. In the first round, 12 “sub-particles” of capsid vertices were extracted from each virion image using coordinates calculated from an initial icosahedral reconstruction of the virion particle. We then performed 3D classification with 5-fold symmetry applied to sort out the unique portal vertex sub-particle from the 11 penton vertex sub-particles for each virion image. Subsequent refinement yielded a 4.3-Å resolution reconstruction of the portal vertex with 5-fold (C5) symmetry (first visualized in HSV-1; McElwee et al., 2018), revealing high-resolution structures of the capsid components surrounding the portal (Table 1 ; Figures S1, S2A, and S2D). A second round of sub-particle classification further relaxed C5 symmetry at the portal vertex, and subsequent refinement with C12 symmetry resulted in a 4.7-Å reconstruction, revealing high-resolution features of the dodecameric portal (Table 1; Figures S1, S2A, and S2E). Using a specific orientation determined in our C12 classification, we then calculated an asymmetric (C1) reconstruction of the entire dsDNA-containing capsid at 7.6 Å (Table 1; Figures 1 A, S1, S2B, and S2G; Video S1) and a C1 reconstruction of the portal vertex at 5.2 Å (Table 1; Figures 1B, S1, S2A, and S2F). Our C1 structures reveal concentrically packed layers of DNA with an inter-duplex distance of ∼25 Å, quasi-5-fold-organized capsid and tegument densities, a quasi-12-fold-symmetric portal dodecamer, and asymmetric terminal DNA within a DNA translocation channel capped by a distinctive density visible at lower thresholds (Figures 1A and 1B). This stepped implementation of symmetry expansion and sub-particle classification thus proved effective in teasing apart the multiple symmetry mismatches present in herpesvirus capsids.
Figure S1.

Sequential Localized Classification and Sub-particle Reconstruction, Related to Table 1 and STAR Methods
Flowchart illustrates the application of sequential localized classification and reconstruction to resolve symmetry-mismatched structures of portal and penton vertices.
Figure S2.

Resolution Assessment of Reconstructions, Related to Table 1 and STAR Methods
(A-C) Gold-standard FSC curves of all cryoEM reconstructions. Based on the 0.143 criterion, the resolutions of our C1 portal vertex, C5 portal vertex, and C12 portal reconstructions are 5.2-Å, 4.3-Å, and 4.7-Å, respectively. The resolution of our C1 capsid reconstruction is 7.6-Å, and the resolution of our penton vertex reconstructions with and without CATC are 3.8-Å and 3.7-Å, respectively. (D-I) Density maps colored by local resolution estimated from ResMap (Kucukelbir et al., 2014). Note that despite an FSC estimated resolution of 4.7-Å, the vast majority of our C12 map reached a resolution of 4.0-Å or better, thus enabling atomic model building.
Table 1.
Cryoelectron Microscopy (Cryo-EM) and Modeling Results
| Cryo-EM Data Collection, Refinement, and Validation Statistics | |||||||
|---|---|---|---|---|---|---|---|
| C1 Virion Capsid (EMDB: EMD-20430) | C1 Portal Vertex (EMDB: EMD-20431) | C5 Portal Vertex (EMDB: EMD-20432, PDB: 6PPB) | C12 Portal (EMDB: EMD-20437, PDB: 6PPI) | C1 Penton Vertex Register, CATC-Absent (EMDB: EMD-20433, PDB: 6PPD) | C1 Penton Vertex Register, CATC-Binding (EMDB: EMD-20436, PDB: 6PPH) | ||
| Data Collection and Processing | |||||||
| Magnification | 14,000 | 14,000 | 14,000 | 14,000 | 14,000 | 14,000 | |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 | 300 | |
| Electron exposure (e−/Å2) | 25 | 25 | 25 | 25 | 25 | 25 | |
| Defocus range (μm) | −1 to −3 | −1 to −3 | −1 to −3 | −1 to −3 | −1 to −3 | −1 to −3 | |
| Pixel size (Å) | 2.06 | 1.03 | 1.03 | 1.03 | 1.03 | 1.03 | |
| Symmetry imposed | C1 | C1 | C5 | C12 | C1 | C1 | |
| Initial particle images (no.) | 44,328 | 44,328 | 44,328 | 44,328 | 44,328 | 44,328 | |
| Final particle images (no.) | 39,073 | 39,073 | 39,773 | 39,073 | 1,521,505 (sub-particles) | 928,740 (sub-particles) | |
| Map resolution (Å) | 7.6 | 5.2 | 4.3 | 4.7 | 3.7 | 3.8 | |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | |
| Estimated resolution range (Å) | 7.0-8.5 | 4.0-6.0 | 3.5-5.5 | 3.5-5.5 | 3.5-4.8 | 3.5-4.8 | |
| Map sharpening B factor (Å2) | 0.0 | 180.5 | 177.4 | 200.0 | 177.4 | 180.4 | |
| Model refinement | |||||||
| Model-to-map fit, mask CC | N/A | N/A | 0.787 | 0.798 | 0.765 | 0.769 | |
| Model Composition | |||||||
| Non-hydrogen atoms | 62,671 | 39,504 | 68,769 | 73,502 | |||
| Protein residues | 7,953 | 4,956 | 8,728 | 9,326 | |||
| Ligands | N/A | N/A | N/A | N/A | |||
| Mean Isotropic B Factor (Å2) | |||||||
| Protein | 50.49 | 204.94 | 40.81 | 33.40 | |||
| Ligand | N/A | N/A | N/A | N/A | |||
| Root-Mean-Square Deviations | |||||||
| Bond lengths (Å) | 0.005 | 0.003 | 0.008 | 0.007 | |||
| Bond angles (°) | 0.923 | 0.680 | 1.043 | 1.014 | |||
| Validation | |||||||
| MolProbity score | 1.55 | 1.54 | 1.63 | 1.60 | |||
| Clashscore | 4.23 | 4.21 | 4.78 | 4.79 | |||
| Rotamer outliers (%) | 0.31% | 0.00% | 0.64% | 0.49% | |||
| Ramachandran Statistics | |||||||
| Outliers (%) | 0.11% | 0.00% | 0.20% | 0.17% | |||
| Allowed (%) | 4.92% | 4.89% | 5.42% | 4.85% | |||
| Favored (%) | 94.97% | 95.11% | 94.38% | 94.97% | |||
The table shows cryo-EM data collection, reconstruction, and model validation results and values. See also Figures S1 and S2.
Figure 1.

In Situ Structure of the KSHV Portal and Its Interactions with Packaged DNA
(A) Clipped view of KSHV C1 capsid reconstruction, showing packaged dsDNA within the capsid and unique portal vertex.
(B) Enlarged view of the portal vertex region, showing terminal DNA held in the portal’s DNA translocation channel and DNA encircling the portal’s lower periphery.
(C) pORF43 model, shown as rainbow-colored ribbons (blue, N terminus → red, C terminus). Inset depictions are ribbon-and-stick in C12 mesh density.
(D) Two opposing pORF43 portal subunits, colored by domains assigned according to dsDNA phage portal structures (Lebedev et al., 2007, Lokareddy et al., 2017, Sun et al., 2015), shown superimposed with Gaussian-filtered C1 portal vertex density.
(E) Clip (top) and β-hairpin (bottom) slices define two narrow constrictions within the DNA translocation channel.
(F) Electrostatic surface potential rendering of the portal’s DNA translocation channel.
(G) C12 density shaded by the corresponding pORF43 domain (Gaussian-filtered C1 density shown for lower-resolution clip turret). Mint green cylinders represent helix-like structures observed to extend from the clip in C1 reconstructions as in (B) but disordered in the C12 density. Three consecutive pORF43 subunits illustrate the daisy-chained 2+1 β sheet augmentation facilitated by the modeled clip’s three β strands (insets). Unmodeled turret densities extend from the middle ring of β strands (dotted black circles).
Overview of the C1 reconstruction of DNA-containing capsid, including the unique portal vertex.
Structure of the DNA-Translocating Portal
Using our C12 portal reconstruction, we atomically modeled KSHV’s 605-amino acid (aa) portal protein pORF43 (Table 1; Figure 1C; Video S2). Despite a lack of sequence homology, our pORF43 model showed striking similarities with structures of phage portal proteins in domain organization and topology (Lebedev et al., 2007, Lokareddy et al., 2017, Sun et al., 2015). We thus named the five domains of our pORF43 model in a fashion analogous to phage portal proteins: wing (aa 9–45 and 126–245), wall (aa 46–125 and 496–553), stem (aa 246–272 and 454–477), clip (aa 273–453 [aa 281–412 unmodeled]), and β-hairpin (aa 478–495) (Figure 1D). The N-terminal eight residues (aa 1–8) and C-terminal 52 residues (aa 554–605) were disordered and also not modeled. Also similar to phages, 12 copies of pORF43 arrange in a vaguely flying saucer-like ring (Lebedev et al., 2007, Lokareddy et al., 2017, Sun et al., 2015). In agreement with previous tomographic (Cardone et al., 2007, Chang et al., 2007) and intermediate-resolution (McElwee et al., 2018, Rochat et al., 2011) visualizations of herpesvirus portals, we observe pORF43 portal docking in the capsid through interactions between the portal’s wing domains and the surrounding MCP floor and, additionally, between the base of the portal clip and the Tri1 subunit of periportal triplexes (Figure 1B). Clip, stem, β-hairpin, and wall domains largely form the interior of the portal channel (i.e., DNA translocation channel) through which DNA is threaded into the capsid during genome packaging (Figures 1D and 1E).
Depicts the C12 reconstruction and atomic model of pORF43, highlighting domains and notable structural features.
Of the channel-lining domains, β-hairpin and clip domains form the most constricted regions of the channel at ∼28 Å and ∼32 Å in diameter, respectively (Figures 1D and 1E). Notably, both regions contain characteristic β sheets and interact with DNA in our mature virion state. Arranged radially, 12 β-hairpins comprise an apertured disk perpendicular to the portal channel axis (Figure 1E). Interestingly, residues that line the channel are markedly positively charged above this aperture, likely facilitating interactions with negatively charged DNA (Figure 1F). Twelve sets of three-stranded β sheets roughly parallel to the channel axis form the base of the clip region (Figure 1G). Each β sheet is the result of a 2+1 augmentation motif, where two parallel clip β strands from each pORF43 subunit are augmented in anti-parallel fashion by a single clip β strand from its clockwise neighbor (when viewing toward the capsid interior) (Figure 1G, insets); this results in a “daisy-chained” ring structure, conceivably ideal for propagating and coordinating conformation changes among subunits in the dodecameric complex during dsDNA translocation.
In contrast to phage portals, a turret-like density arises from our KSHV clip, extending distally toward the portal-capping density (Figures 1B and 1G). Although we were unable to model this clip turret, we identified what appeared to be helical structures comprising the turret walls (Figure 1G), consistent with pORF43 secondary structure and disorder predictions, which show strong helix propensity in this region (aa 281–412) (Figure S3 A). During DNA packaging, the distal end of this turret is the putative docking site of terminase (Yang et al., 2007, Heming et al., 2017). The lower resolution of the clip turret is thus likely a result of inherent plasticity in the structure to accommodate interactions with various partners during different stages of viral assembly (e.g., terminase during active DNA packaging and the portal cap after packaging).
Figure S3.

Bioinformatics Predictions and Analyses, Related to Figures 1 and 4
(A) Secondary structure and disorder prediction for pORF43 obtained from Phyre2 (Kelley et al., 2015), annotated by pORF43 atomic model domains for reference as per key. (B) Structure-based pairwise alignment of HSV-1 pUL17 and KSHV pORF32, performed using MatchMaker in Chimera (Pettersen et al., 2004).
A Portal-Effected Global Distribution of CATC
Our C1 portal vertex reconstruction revealed tegument densities sitting atop the periportal triplexes Ta and Tc (Figures 1B and S2F). The morphology of these densities—a helix bundle supported by a triplex-bridging base—resemble that of CATCs identified around penton vertices in previous icosahedral reconstructions of KSHV (Dai et al., 2014) and of neurotropic HSV-1 (Dai and Zhou, 2018) and HSV-2 (Wang et al., 2018). However, peripenton CATC densities in the KSHV icosahedral reconstruction were distinctively weaker than surrounding capsid proteins and only discernible when low-pass-filtered to ∼6-Å, suggesting low CATC occupancy and/or flexibility (Dai et al., 2014); in contrast, the HSV-1 icosahedral reconstruction indicated full CATC occupancy (Dai and Zhou, 2018). Interestingly, our C1 reconstruction of the KSHV portal vertex reveals the presence of five CATC densities with a strength comparable with that of underlying capsid elements, indicating full occupancy of the five CATC registers surrounding the portal vertex. This preferred association of CATC with the portal vertex over the remaining 11 (penton) vertices suggests an important role of CATC at the portal vertex, to be discussed later.
Our C1 reconstruction of the genome-containing capsid also indicates a curious binding pattern of CATCs to penton vertices in a manner dictated by the portal vertex. When displayed at a density threshold appropriate for capsid proteins, our C1 capsid reconstruction shows two adjacent (i.e., ortho) CATCs bound to each portal-proximal penton vertex, one CATC to each portal-distal penton vertex, and no visible CATCs at the portal-opposite penton vertex (Figure 2 A). Incrementally decreasing the density display threshold reveals additional CATCs of progressively weaker density (indicating progressively lower occupancies at those registers) manifesting at penton vertices in a “portal-outward” manner (Figures 2B and S4 ). Importantly, within each penton vertex, CATCs appear to selectively bind registers on the portal side of a hypothetical “equatorial” bisecting the penton vertex, with a clear preference for the most portal-side register (Figure S4). Adherence to this “portal-side equatorial rule” is exemplified by the observation that portal-proximal penton vertices (with two registers portal-side of the equatorial) max out at two CATC copies as the density display threshold is decreased, whereas portal-distal penton vertices (with three portal-side registers) max out at three CATC copies. Portal-opposite vertices can bind the CATC at any register (i.e., no preferred register), up to five CATCs in total, because every register is portal-side (Figures 2B and S4).
Figure 2.

A Portal-Effected Distribution of CATC
(A) Global view of C1 capsid reconstruction showing consensus-patterned (i.e., averaged) occupancy of CATCs at penton vertices.
(B) Same as in (A) but with only CATC densities (Gaussian-filtered [2σ], pORF19 heads removed for clarity) displayed at three progressively lower thresholds. The left panel is at a threshold where capsid proteins are optimally displayed. Densities in the right panel are numbered from strongest to weakest.
See also Figure S4.
Figure S4.

Density Strength as an Indicator of CATC Binding Occupancy, Related to Figures 2 and 3
Chart displays capsid vertices at progressively lower density display thresholds. CATCs appear to bind only portal-side registers of each penton vertex. Red arrows indicate the direction of the portal vertex with respect to each penton vertex location. Dashed red lines indicate each respective penton vertex’s equatorial (where applicable in relation to the portal-side equatorial rule).
Given that CATC occupancy varies among vertices in a non-random fashion, the existence of CATCs of varying densities indicates that CATC occupancy varies even among capsids and is thus not fully determined in our C1 reconstruction (i.e., the averaging of capsids with differing occupancies/binding patterns inherently obscures information on specific occupancy). Thus, to accurately assess the specific occupancy of penton vertex CATCs and to understand the structural basis of a CATC’s discriminatory association with portal and penton vertices, we relaxed 5-fold symmetry for penton vertex sub-particles and performed 3D focused classification of their CATC-binding registers (Figure S1). Using a mask encompassing the region surrounding one CATC, four resulting classes were obtained. Although three classes clearly lacked CATC densities, one class contained a CATC density of a quality comparable with surrounding capsid protein densities (Figure S1). 37.9% of masked sub-particles (i.e., 37.9% of penton vertex registers) were assigned to this CATC-binding class, slightly higher than the ∼30% occupancy estimated in our previous KSHV icosahedral study (Dai et al., 2014).
We further distinguished between possible CATC binding occupancies and permutations at the five registers of a single penton vertex using geometry-based sub-particle classification. In all, eight possible permutations of zero to five CATCs can bind a penton vertex, all of which were observed and reconstructed in our analyses (Figure 3 A). In agreement with trends observed in our C1 capsid reconstruction, penton vertices with two adjacent CATCs bound were most abundant (ortho-CATC-binding, at 38.6% of penton vertex sub-particles), followed by penton vertices with a single CATC bound (one-CATC-binding, at 21.9% of sub-particles). Five-CATC-binding penton vertices were rarest, as expected, at 0.2% of sub-particles because, barring deviations to the C1 capsid-observed consensus binding pattern, these should be limited to the portal-opposite vertices of highly bound capsids. Finally, we tallied the total number of penton CATCs in each capsid from our classified penton vertex sub-particles. The resulting histogram follows a sharp, slightly left-skewed Gaussian distribution peaking at approximately 23 penton CATCs per capsid (Figure 3B). Intriguingly, additional CATC binding falls off sharply after capsids have bound 30 penton CATCs, which happens to be the theoretical maximum of a capsid with “full” penton vertex CATC binding in compliance with the portal-side equatorial rule. In all, these results—the full occupancy of portal CATC registers, the portal vertex-referenced directional binding of CATC within penton vertices, and the portal-dictated maximum of allowed binding registers—strongly suggest that the nucleating portal effects long-range (allosteric) structural influence on the penton vertices of each capsid.
Figure 3.

Geometry-Based Sub-particle Classification of Penton Vertices
(A) Penton vertex sub-particles were classified into eight classes of varying CATC stoichiometries and permutations and reconstructed individually. The resulting eight reconstructions are shown, with the capsid colored radially and the CATC in gold.
(B) Histogram describing population-wide global penton CATC occupancies.
See also Figure S4.
CATC Structures at Portal and Penton Vertices
Further 3D refinement of classes obtained from our masked classification of penton vertex sub-particles yielded CATC-binding and CATC-absent reconstructions of penton vertex registers at 3.8 Å and 3.7 Å, respectively (Table 1; Figures S1, S2C, S2H, and S2I). From the CATC binding reconstruction, we identified and modeled the constituents of CATC, which include two copies of pORF19, two copies of pORF64, and one copy of pORF32 (Table 1; Figures 4 A, 4B, and S5 A); this is in one-to-one correspondence with pUL25, pUL36, and pUL17, respectively, of HSV-1 CATC (Dai and Zhou, 2018).
Figure 4.

CATC Structures at Portal and Penton Vertices
(A) Sharpened CATC-binding penton vertex register density used to model CATC elements.
(B) Ribbon models of the CATC helix bundle in a coiled-coil arrangement of pORF64 and pORF19, two copies each.
(C and D) C1 reconstructions of the portal vertex (C) and penton vertex with one CATC occupancy (D), colored according to the key. Insets depict the colored C1 density within the Gaussian-filtered C1 map (gray), showing lower-resolution features and interactions. Connections between CATC helix bundles and the portal cap putatively formed by pORF19 head domains are visible in the filtered map (C, inset) and circled in black. One head domain of pORF19 is clearly visible in (D). Putative head domains of the second pORF19 copy in each CATC are circled in green (C and D, insets).
(E) Homology model of the pORF19 head domain docked into the C1 penton vertex density reveals a finger-like motif (∼aa 457–468) interacting with penton MCP.
(F) Superposition of the HSV-1 pUL25 head domain (PDB: 2F5U) with the docked pORF19 head domain homology model demonstrates conservation of the head domain structure between HSV-1 and KSHV.
(G and H) Ribbon model of pORF32 colored according to the domains in HSV-1 pUL17 (H).
(I and J) Schematics depicting Ta and Tc triplex orientations in the absence (I) and presence (J) of CATC at vertices. Insets (capsid interior view) show that the Ta Tri1 N-anchor retains a conserved orientation despite CATC-binding Ta’s 120° counterclockwise apical rotation.
See also Figures S3 and S5 and Video S3.
Figure S5.

High- and Low-Resolution Density Features of the CATC, Related to Figure 4
(A) Densities of B-factor–sharpened and unsharpened maps of penton CATC. Density of the loop between the N-terminal helix regions of magenta “upper” pORF19 is only visible in the unsharpened map. Boxed regions correspond to colored inset boxes illustrating residue features (ribbon-and-stick) in density (mesh). (B) C1 reconstruction of penton vertex with one CATC bound, colored as per key. Dotted black circle denotes density putatively assigned to the second pORF19 head domain (density shown at a lower threshold than that of surrounding features). Inset displays the C1 reconstruction in (A) fitted within the same reconstruction Gaussian-filtered at 3.0σ to showcase connectivity between the weaker (green) putative pORF19 head domain and CATC helix bundle. (C) CATC density shown with both pORF19 head domains (at lower thresholds). Note that a helical density feature can be observed in the putative (green) pORF19 head domain density.
Comparing reconstructions of the portal vertex and a CATC-binding penton vertex reveals nearly identical structures of their associated CATCs (Figures 4C and 4D). At both vertices, pORF32 bridges the triplexes Ta and Tc and supports a four-membered helix bundle composed of the N-terminal segments of two pORF19 subunits (aa 62–104) and the very C-terminal segments of two pORF64 subunits (aa 2,596–2,635) (Figures 4B–4D). Obvious differences aside—e.g., a portal in the portal vertex, an SCP-decorated penton in the penton vertex, or CATC occupancies—the two vertices also differ in the presence of a disk-like portal cap at the distal ends of the portal vertex’s CATC helix bundles (Figure 4C, magenta) versus a globular density cocked to the right of the penton vertex’s CATC helix bundle (Figure 4D, magenta). Because this globular density’s size and proximity to CATC (Figure 4E) are reminiscent of the flexibly tethered head domain of pUL25 in HSV-1 (a homolog of KSHV pORF19), we interpret this density as the head domain of one of the pORF19 copies in the CATC. Indeed, the crystal structure of an HSV-1 pUL25 head domain (PDB: 2F5U) (Bowman et al., 2006) satisfactorily fits into the globular density (Figure 4F), lending support to its assignment as pORF19. We then proceeded to construct a homology model of the pORF19 head domain (aa 127–546) using PDB: 2F5U as a template (Figures 4E and 4F). From the homology model, we estimate that aa 457–468 map to a finger-like density that inserts between an adjacent penton MCP and SCP, serving as the sole interacting residues between CATC and the penton protrusion (Figure 4E).
Of note, because the globular density accommodates only one copy of pORF19/pUL25 head domain (whereas the corresponding region in HSV-1 CATC accommodates two), we previously asserted—based on a 6-Å icosahedral reconstruction—that the KSHV CATC had a different stoichiometry than the HSV-1 CATC (Dai et al., 2014). But, as noted in a subsequent study demonstrating differences in globular head domain arrangement between KSHV and neurotropic alphaherpesviruses (Dai et al., 2014, Liu et al., 2017), and as our present sub-particle reconstructions reveal, the KSHV CATC does, in fact, bear the same stoichiometry/architecture as the HSV-1 CATC. These findings indicate that the second head domain of pORF19 is present but perhaps flexibly tethered elsewhere. Indeed, a second globular density gradually appears to the left of the penton vertex’s CATC helix bundle at lower resolutions/density thresholds (Figures 4D, green circle, S5B, and S5C).
In the KSHV portal vertex, secondary structures in the portal cap beyond the CATC helix bundles are not resolved as in the penton vertex’s globular density. Nonetheless, several lines of evidence support interpretation of the portal cap as pORF19. First, CATC helix bundles clearly connect with the portal cap at lower resolutions/density thresholds (Figure 4C, black circles). Second, the portal cap density is about the size of five pORF19 head domains and further exhibits five weaker globular densities attached at its periphery (Figure 4C, green circle), analogous to the globular density assigned to the second pORF19 head domain at the penton vertex (Figures 4D, green circle, S5B, and S5C). Third, functional data indicate that HSV-1 pUL25 (pORF19’s homolog), but not HSV-1 pUL36 (pORF64’s homolog), is critical for viral genome encapsidation, especially near the termination of genome packaging (McNab et al., 1998, Ogasawara et al., 2001). Moreover, pUL25 plays an important role in docking the incoming capsid at host cell nuclear pores and gating the release of the viral genome (Huffman et al., 2017, Pasdeloup et al., 2009). It is therefore plausible that pORF19 could be intimately associated with the portal channel, and the portal cap is certainly poised in a prime position to execute pORF19’s roles in regulating the retention and release of the viral genome.
CATC Binding Related to Triplex Orientation
Despite similar architecture, KSHV and HSV-1 CATCs bind their underlying triplexes in somewhat different fashions. In both viruses, the pORF32-equivalent CATC subunit (pUL17 in HSV-1) functions as a structural framework facilitating capsid association of the other four subunits that make up the CATC helix bundle. Side-by-side comparison of pORF32 and pUL17 models reveals similar core structures of separate N- and C-terminal β sheet-rich domains positioned beneath a central α helix–rich arch (Figures 4G and 4H, orange). However, several prominent features visible in pUL17 are missing in pORF32, accounting for pORF32’s 249 fewer residues: a “hump,” characterized by four short helices at the top of the arch that constrain and orient CATC’s helix bundle in HSV-1; an extended helix; and a short helix bundle at the vertex-proximal end of pUL17 (Dai and Zhou, 2018). Notably, pUL17’s short helix bundle makes direct contact with the underlying triplex Ta, almost solely mediating CATC-Ta binding in HSV-1. In pORF32, nearly the entirety of this region is disordered and/or invisible in both portal and penton vertex reconstructions save for a 13-aa “anchoring loop” (AL; aa 220–232) (Figure 4G, yellow). Although pORF32’s 93 unmodeled residues in this region may potentially account for the critical Ta-interacting helix bundle, a structure-based sequence alignment of pORF32 with pUL17 suggests that the Ta-binding short helix bundle in pUL17 is missing, even in the pORF32 sequence (Figure S3B).
The importance of pORF32’s AL for Ta binding becomes apparent in light of an alternate morphology of CATC-decorated KSHV triplexes not observed at the CATC-decorated penton vertices of HSV-1. A comparison of our models of KSHV triplexes with and without the CATC (Table 1; Figures 4I and 4J) reveals that all Ta triplexes underlying CATCs experience an ∼120° counterclockwise axial rotation relative to canonical undecorated triplex Ta (cf. Figures 4I and 4J). Our models suggest that this rotation may be due to the steric specificity of pORF32’s Ta-binding AL. Specifically, a 120° counterclockwise rotation of Ta exposes a deep groove running between the molecular boundaries of Tri1 and the Tri2A/B dimer that facilitates docking of pORF32’s AL and, thus, CATC. Strikingly, despite CATC-decorated Ta’s large degree of apical (i.e., main body) rotation, our structures show that Tri1’s N-anchor—the characteristic N terminus of Tri1 subunits that penetrates the capsid floor—maintains a conserved orientation in the capsid interior at penton vertices regardless of Ta’s apical orientation (and, by extension, regardless of CATC decoration) (Figures 4I and 4J, respective insets). (Periportal Ta triplexes, which are all CATC-decorated, exhibit 120° counterclockwise apical rotation, but their Tri1 N-anchors are disordered and appear to adopt a unique configuration.)
In the context of capsid assembly, our observations allow several structure-based inferences. First, the presence of a uniform Tri1 N-anchor orientation (barring periportal triplexes) supports the notion that initial triplex attachment to the procapsid, putatively through the Tri1 N-anchor, precedes the stage of CATC binding (reviewed in Heming et al., 2017). Second, whatever the means by which the portal effects triplex orientation and, thus, CATC binding (or perhaps vice versa) presumably occurs at a stage of procapsid maturation before triplexes are fully “stapled” to the MCP floor by main-body interactions (Zhou et al., 2014). This is predicated upon the necessity for triplex Ta to have sufficient rotational freedom to permit adoption of a CATC-bound orientation. Third, for reasons one and two, CATC binding occupancy is highly unlikely to be predetermined but, rather, determined with the simultaneous maturation of the procapsid, although this is subject to influence by the portal, as demonstrated previously.
Targeting CATC-Triplex Interaction Hotspots
At the vertex-proximal end of CATC, interactions with triplex Ta occur exclusively through pORF32. An N-terminal helix (NH; aa 2–14) and two strands from pORF32’s N-terminal β-barrel domain directly contact the apical surfaces of Ta’s Tri2A/B dimer (Figures 5 A–5C; Video S3). Additionally, the aforementioned AL binds in Ta’s deep hydrophobic cleft between Tri1 and the Tri2A/B dimer (Figures 5B–5D; Video S3). AL binding involves three pORF32 hydrophobic residues—Val222, Leu224, and Phe226 (Figure 5D)—as well as β sheet-like hydrogen bonding between the AL and an adjacent Ta Tri1 strand (Figure 5E).
Figure 5.

Key CATC-Triplex Interactions Defined by Structure-Guided Mutageneses
(A) Ribbon models of triplex-complexed CATC colored according to the key.
(B and C) Enlarged views illustrate how pORF32′s anchoring loop (AL) and N-terminal helix (NH) interact with Ta (displayed as surface).
(D) The AL (ribbon-and-stick superimposed with mesh density) binds a hydrophobic groove between the Tri1 and Tri2 dimer (cyan, hydrophilic → maroon, hydrophobic).
(E) The AL forms β sheet-like hydrogen bonds (dotted lines) with an adjacent Tri1 strand (some side chains are hidden for clarity).
(F and G) Consecutive enlarged views showing CATC-Tc interaction via upper pORF19, which interacts with Tc Tri1 via a helical motif (G). Dotted lines represent hydrogen bonds. Lower pORF19 facilitates two sets of β sheets (starred in F and H) at the CATC’s vertex-distal end.
(H and I) The flexible N-terminal residues of upper and lower pORF19 could not be modeled, although the disordered density from the two pORF19 copies can be traced (H, dashed lines) and observed (I, gray density).
(J–M) Structure-guided mutageneses of pORF32 (J and K) and pORF19 (L and M), confirming the importance of their respective triplex-binding segments in infectious virion production. Overexpressing pORF32 mutants with deletions of N-terminal helix (ΔNH) and/or anchoring loop (ΔAL) in KSHV lytic-replicating cells reduced virion production normalized to the control (vector) (J). Likewise, overexpressing pORF19 with truncated N-terminal 17 or 28 residues reduced virion production (L). Expression of mutant pORF32 (K) and pORF19 (M) was verified by western blotting with an anti-FLAG antibody. Expression of cellular GAPDH was examined as an internal control.
(N–P) Mutageneses of triplex protein residues at the CATC-binding groove. Measures of viral titer (N), genome replication (O), and RNA transcription (P) are shown. The dashed line in (N) indicates the detection limit. Data are mean ± SEM (n = 3 biologically independent samples).
Depicts interactions between the CATC and underlying Ta and Tc triplexes through pORF32′s “NH” and “AL” regions and pORF19’s N-terminal residues.
At the vertex-distal end of CATC, pORF32 sits atop, but does not directly bind to, triplex Tc. Instead, CATC-Tc interactions rely on the N-terminal tails of CATC’s two pORF19 subunits, which extend distally from the helix bundle and descend beneath pORF32 to contact Tc (Figures 5A and 5F). Contact with Tc occurs mainly via a short helical motif (aa 20–24) from the N-terminal tail of the “upper” (magenta) pORF19 subunit. An adjacent helix belonging to Tc Tri1 (aa 264–275) orthogonal to the pORF19 helical motif fits within the motif’s helical groove (Figure 5F), with Tri1’s Thr269 and Arg268 and pORF19’s Arg22 contributing hydrogen bonds to this intermolecular interaction (Figure 5G). In contrast to upper pORF19, “lower” (green) pORF19 exhibits no apparent direct contacts with Tc in our visible structure. Nonetheless, lower pORF19 plays an important role in lashing CATC’s constituents as a collective unit. Chiefly, lower pORF19’s N-terminal tail contributes two β strands (aa 23–25 and 31–33) to form two sets of β sheet interactions—the first with one β strand from pORF32 (aa 319–321) and upper pORF19 (aa 30–32) each (Figure 5F, starred) and the second with a β strand from pORF32 (aa 311–313) (Figure 5H, starred). These two sets of β sheets fasten the vertex-distal end of CATC in a quasi β-barrel.
The first 17 and 21 residues of upper and lower pORF19’s N-terminal tails, respectively, are flexible and, thus, unmodeled (Figure 5H). However, we speculate that some of these unmodeled residues may also bind Tc, augmenting previously described CATC-Tc interactions at upper pORF19’s short helical motif. Particularly, strong unassigned densities roughly three aa in length occupy the Tri1-Tri2A/B surface groove of Tc (Figure 5I), analogous to pORF32’s AL occupying triplex Ta’s groove (Figures 5B–5D). Given that the N-terminal tail of upper pORF19 appears to extend away from Tc, and given the proximity of lower pORF19’s Nth-most residue to Tc’s groove, the unassigned densities likely belong to the unmodeled N-terminal residues of lower pORF19. If interactions here are also similar to AL binding at Ta (i.e., in part involving β sheet-like hydrogen bonds between adjacent backbones), then pORF19 binding at this site may not necessarily be sequence-specific, thus accounting for the unresolved side-chain densities because of averaging.
To validate our structural interpretation of the roles of the pORF32 NH and AL in CATC-Ta binding, we constructed three pORF32 mutants with deletions in either the NH or AL or both, named 32ΔNH, 32ΔAL, and 32ΔNH-ΔAL. We posited that these “loss-of-Ta-interaction” pORF32 mutants might serve as dominant negatives if NH and/or AL are critical for CATC-Ta binding. Essentially, pORF32 mutants expressed in KSHV-replicating cells should compete against wild-type (WT) pORF32 (32WT) for incorporation into CATC so that mutant-incorporated CATC would be deficient in capsid association, inhibiting KSHV virion formation. Indeed, expression of 32ΔNH and 32ΔAL reduced virion production to 30.7% and 27.8% of that of the control, respectively, whereas expression of 32ΔNH-ΔAL inhibited levels to 12.1% (Figures 5J and 5K). Importantly, viral DNA replication and viral RNA transcription were not significantly affected (Figures S6 A and S6C). These results confirm that the pORF32 NH and AL are both important for KSHV virion production, supporting our interpretation that these aa stretches mediate CATC-Ta binding.
Figure S6.

Overexpressing pORF32/pORF19 Mutants Does Not Significantly Affect KSHV DNA Replication or Gene Expression, Related to Figure 5
(A-B) Viral genome replication in cells overexpressing wild-type or mutant forms of pORF32 (A) or pORF19 (B). 293T-KSHV cells were transfected with corresponding expression plasmids or empty vector as control, and then induced with NaB and TPA to facilitate KSHV lytic replication. Total DNA was extracted from cells, and genome replication was determined by qPCR. (C-D) Viral RNA transcription in cells transfected with wild-type or mutant forms of pORF32 (C) or pORF19 (D). Total RNA was extracted from the same cells as (A-B). Viral RNA transcripts were quantified by RT-qPCR and presented as x-fold changes over RNA level of empty vector transfected cells. Data is mean ± SEM (n = 3 biologically independent samples).
Similarly, to test our hypothesis that pORF19’s N-terminal tail is important for CATC capsid association and, hence, function, we generated two pORF19 mutants: 19ΔN28, which deletes the first N-terminal 28 residues of pORF19 (i.e., all pORF19 residues in contact with triplex Tc), and 19ΔN17, which deletes pORF19’s first 17 residues (i.e., retains all pORF19 residues visible in our structure but deletes the disordered, unmodeled N-terminal tails). Compared with WT pORF19 (19WT), 19ΔN28 expression reduced virion production to 9.7% of that of the control without significantly affecting viral DNA replication or RNA transcription (Figures 5L, 5M, S6B, and S6D), indicating that pORF19 N-terminal interactions with Tc (Figures 5F–5I) are important for viral efficacy. Remarkably, expression of 19ΔN17 also inhibited virion production to 30.6% of that of the control (Figures 5L, 5M, S6B, and S6D), suggesting that pORF19’s first 17 residues of pORF19 are required for optimal pORF19-Tc binding. These findings are therefore consistent with our speculation that lower pORF19’s unmodeled N-terminal residues (aa 1–21) bind Tc’s hydrophobic groove, enhancing CATC-Tc binding.
Finally, we mutated residues in the hydrophobic triplex groove, creating six mutants: Tri1-I280R, Tri1-L278R/I280R/L283E, Tri2-A216R, Tri2-L217R, Tri2-A216R/L217R, and Tri2-V244R (Figure 5I). These mutants affect binding grooves indiscriminately at both Ta and Tc triplexes (and, therefore, CATC binding at both its vertex-proximal and vertex-distal ends). All mutants yielded decreased virion production (Figure 5N), whereas neither viral DNA replication nor RNA transcription were significantly affected (Figures 5O and 5P). Impressively, virion production of mutant Tri2-A216R/L217R was under the detection limit (Figure 5N, UD).
Discussion
Prospect of an Antiviral KSHV Inhibitor
The visualization of pORF32 and pORF19 peptide fragments binding in the respective deep grooves of triplex Ta and Tc (Figures 5A–5I) and our demonstration of their importance in viral replication (Figures 5J–5P) open exciting prospects for future KSHV inhibitor design and drug development. Currently, most available drugs countering herpesvirus infections are nucleic acid analogs that target viral DNA synthesis. Unfortunately, these often elicit negative side effects, including frequent induction of drug-resistant mutant viruses and varying kinds of toxicity (Gilbert et al., 2002, Reusser, 1996, Walling et al., 2003). That the DNA-packaging portal vertex is critically involved in the production of virus progeny and that the five portal vertex CATC registers exhibit full CATC occupancy—which suggests that disrupting a single portal-associated CATC might abolish the assembly of infectious virion—presents the possibility of a novel antiviral target without homology with cellular proteins. Therefore, a CATC-specific inhibitor would be very potent, and the deep groove on triplexes can serve as a structurally informed target for focused antiviral development.
CATC Binding Implications for Capsid Assembly and Diversity
The portal is widely believed to serve as the nucleating nexus of herpesvirus procapsid formation (Motwani et al., 2017, Newcomb et al., 2005)—although capsid-like particles have been observed in capsid protein-expressing insect cells and cell-free systems in the absence of portal protein (Newcomb et al., 1994, Perkins et al., 2008, Tatman et al., 1994)—by facilitating initial interactions with scaffold-tethered MCP subunits and portal-adjacent triplexes (Deng et al., 2008, Zhou et al., 1998). A metastable spherical procapsid then forms as follows. Tethering by the scaffolding protein brings together additional MCP molecules. The scaffolding core ensures proper curvature and size of the capsid in what is known as the “rope” mechanism (Deng et al., 2008), and heterotrimeric triplexes, with the N-terminal anchor of Tri1 (pORF62) traversing the capsid shell, “plug” every 3-fold hole between MCP capsomers. Our data showing that CATC-triplex Ta association correlates with a 120° apical rotation of Ta (Figure 4I and 4J)—but not the 3-fold-related 240°, eliminating the possibility of this being a stochastic event—indicate that the determination of CATC binding (and, likely, binding of CATC itself or at least a subunit of CATC; Thurlow et al., 2006) occurs not long after procapsid formation, when the triplex’s apical orientation has yet to be fixed by procapsid maturation. Procapsid maturation is concomitant with DNA packaging; head-full is likely sensed through capsid elements and relayed by the portal through the turret-like structure to the externally located terminase for cleavage of the concatemeric genome (Yang et al., 2007), and the spring-loaded dsDNA genome is corked inside the capsid by the portal cap, putatively formed by five portal-adjacent CATCs (Figure 1B).
That capsid structures are rigidly symmetric reflects the capsid’s well-defined and conserved role in packaging and protecting the viral genome, in marked contrast to both form and function of the largely pleomorphic tegument layer. Accordingly, all three sub-families of herpesviruses have highly conserved capsid proteins and structures but only partially conserved tegument and envelope proteins because these more often reflect specific host cell adaptations. Among tegument proteins, CATC is unique in possessing a structural role and, thus, being relatively conserved, but even so, a surprising finding here is that, unlike the full occupancy of CATC at capsid vertices in neurotropic HSV-1 and HSV-2 (Dai et al., 2014, Dai and Zhou, 2018, Wang et al., 2018), CATC occupancy in KSHV at penton vertices is only partial and varies even among KSHV capsids (Figures 2 and 3). That KSHV CATC occupancy appears to be dictated at least in part by the portal not only underscores the portal’s aforementioned nucleation role but further spotlights its allosteric effect in defining an emergent first level of variability that, importantly, demarcates a departure from symmetry in KSHV metastructure. A second constructive level of variability is facilitated by pORF64, which accounts for two of the five subunits in CATC and is the largest tegument protein, with 2,635 residues folded into multiple domains joined by presumably flexible linkers (the vast majority of pORF64 is thus invisible in our structure). Some of these domains recruit other tegument proteins and bind the endodomains of envelope glycoproteins (Rozen et al., 2008, Sathish et al., 2012), introducing pleomorphic variability. Last, the majority of tegument proteins, including pORF64, have been shown to be capable of being packaged into non-infectious virus-like vesicles in the absence of capsids (Gong et al., 2017), suggesting an alternate pathway of tegument incorporation into virions independent of the capsid/CATC, implicating yet another level of assembly-driven variability.
In light of the tegument’s primary role in manipulating host cells to facilitate virus replication, the multiple layers of structural variability at play in the tegument provide unique opportunities in the context of herpesvirus adaptability. In much the same way that more genetically fluid RNA viruses such as influenza viruses (Harris et al., 2006, Vahey and Fletcher, 2019), filoviruses (Bharat et al., 2011), and coronaviruses (Goldsmith et al., 2004) benefit from heterogeneous compositions to rapidly respond to selective pressures, structural pleomorphism may provide a crucial avenue of diversity in more genetically constrained dsDNA viruses (because of lower mutation rates) like KSHV, resulting in an increase in evolutionary bandwidth. In essence, facing a relative lack of intraspecies genetic diversity (the tradeoff is more viable progeny), adaptability regarding the ability to manipulate hosts is, instead, implemented at a structural level. Our findings regarding KSHV CATC occupancy offer a pertinent example: given that HSV pUL36 (KSHV pORF64’s homolog) is involved in axonic transport of alphaherpesvirus capsids (Dai and Zhou, 2018, Luxton et al., 2005), the absence of full CATC occupancy in non-neurotropic KSHV perhaps reflects its lack of need for long-range neuronal transport. The emergent transition from rigidly symmetric structures to less-structured compartments described here thus sheds light on a delicate balance of conservation and adaptation that delineates the genetic arms race between herpesvirus and host.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-FLAG M2 antibody | Sigma-Aldrich | Cat#F3165; RRID: AB_259529 |
| Monoclonal anti-GAPDH antibody | Abcam | Cat#AB181602; RRID: AB_2630358 |
| Bacterial and Virus Strains | ||
| Human herpesvirus 8 strain JSC-1 clone BAC16 (KSHV-BAC16) | Gift from Dr. Jae U. Jung at the University of Southern California (USC) | Genebank: Q994935.1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium butyrate | Sigma-Aldrich | Cat#303410; CAS 156-54-7 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#D9891; CAS 24390-14-5 |
| 12-O-Tetradecanoylphorbol-13-acetate (TPA) | Sigma-Aldrich | Cat#P8139; CAS: 16561-29-8 |
| hygromycin B | Invivogen | Cat#ant-hg-5; CAS: 31282-04-9 |
| Critical Commercial Assays | ||
| Purelink RNA Mini Kit | ThermoFisher Scientific | Cat#12183018A |
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | Cat#18080093 |
| SuperSignal West Femto Maxmium Sensitivity Substrate | ThermoFisher Scientific | Cat#34095 |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat#E5520S |
| Deposited Data | ||
| Structure of C1 virion capsid | This manuscript | EMD-20430 |
| Structure of C1 portal vertex | This manuscript | EMD-20431 |
| Structure of C5 portal vertex | This manuscript | EMD-20432, PDB-6PPB |
| Structure of C12 portal | This manuscript | EMD-20437, PDB-6PPI |
| Structure of C1 penton vertex register, CATC-absent | This manuscript | EMD-20433, PDB-6PPD |
| Structure of C1 penton vertex register, CATC-binding | This manuscript | EMD-20436, PDB-6PPH |
| Experimental Models: Cell Lines | ||
| Human 293T cells | ATCC | CRL-3216 |
| 293T-KSHV | This manuscript | N/A |
| iSLK-KSHV-BAC16 | Jae U. Jung at USC | N/A |
| Oligonucleotides | ||
| Primers for cloning: | Integrated DNA Technologies | N/A |
|
ORF32: 5′-AC CGT CAG ATC CGC TAG AGA TCT GCC ACC ATG GAT GCG CAT GCT ATC AAC-3′ and 5′-CC GCG TCG ACG GTA CCA GAT CTA CTT GTC GTC ATC GTC TTT GTA GTC GCC ATA GCG GCC TCG AAT G-3′ | ||
|
ORF19: 5′-C CGT CAG ATC CGC TAG AGA TCT GCC ACC ATG GAC TAC AAA GAC GAT GAC GAC AAG GGT GGC GGA GGT TCT ATG CTG ACAT CAG AAA GGT C-3′ and 5′-CC GCG TCG ACG GTA CCA GAT CTA AAC GAC CGC GAG GAC CAC-3′ | ||
| Primers for qPCR: | Integrated DNA Technologies |
N/A |
|
GAPDH: 5′-TGC ACC ACC AAC TGC TTA GC-3′ and 5′- GGC ATG GAC TGT GGT CAT GAG-3′ | ||
|
RTA: 5′-CAC AAA AAT GGC GC AAG ATG A-3′ and 5′- TGG TAG AGT TGG GCC TTC AGT T-3′ | ||
|
ORF59: 5′-TTG GCA CTC CAA CGA AAT ATT AGA A-3′ and 5′-CGG GAA CCT TTT GCG AAG A-3′ | ||
|
K8.1: 5′-GTA ACC GTG TGC CAT TTT CTG-3′ and 5′-TCC CAG CAA TAA ACC CAC AG-3′ | ||
| Recombinant DNA | ||
| RedTrackCMV | James Bamburg Lab, unpublished | Addgene Plasmid #50957; RRID: Addgene_50957 |
| Expression plasmids of pORF32 and its mutants | This manuscript | N/A |
| Expression plasmids of pORF19 and its mutants | This manuscript | N/A |
| Software and Algorithms | ||
| Relion2.1 | Scheres, 2012 | https://www3.mrc-lmb.cam.ac.uk/relion/ |
| Sub-particle reconstruction scripts | This manuscript | https://github.com/procyontao/Herpesportal |
| UCSF Chimera 1.11.2 | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Coot 0.8.6.1 | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/; |
| Phenix 1.13 | Adams et al., 2010 | https://www.phenix-online.org/; |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Z. Hong Zhou (Hong.Zhou@UCLA.edu).
Experimental Model and Subject Details
Viruses and cell lines
KSHV virions were isolated from iSLK-KSHV-BAC16 cells, received as a gift from Dr. Jae U. Jung of the University of Southern California. Dr. Jung’s group previously established the iSLK-KSHV-BAC16 cell line, which supports robust KSHV lytic replication and the production of KSHV virions as previously described (Brulois et al., 2012, Dai et al., 2014, Dai et al., 2018). iSLK-KSHV-BAC16 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin streptomycin, 1 μg/mL puromycin, 250 μg/mL G418, and 1,200 μg/mL hygromycin B. To induce KSHV lytic replication and thus KSHV virion production, cells were treated with 1 μg/mL doxycycline plus 1 mM sodium butyrate (NaB) for three days, after which tissue culture supernatant was collected. KSHV virions were pelleted by ultracentrifugation at 80,000 g for 1 hour, then resuspended in phosphate buffered saline (PBS) and further purified in 15%–50% (w/v) sucrose density gradient sedimented at 100,000 g for 1 hour. In addition to cryoEM studies, these KSHV virions were also used to infect human 293T cells at a MOI of 3. 293T cells infected with wild-type KSHV virions (termed 293T-KSHV) or transfected with triplex mutant KSHV BACs were selected and maintained in DMEM supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 100 μg/mL hygromycin B (Dai et al., 2018). These 293T cells harboring KSHV wild-type or triplex protein mutants were used for all functional analyses as described below in METHOD DETAILS.
Method Details
CryoEM and icosahedral reconstruction
Aliquots of 2.5 μL purified virion sample were applied onto Quantifoil R2/1 Cu grids, manually blotted with filter paper and plunge-frozen in liquid ethane. Super-resolution movies of purified wild-type KSHV intact virions were recorded on a Gatan K2 direct electron detection camera in counting mode with a pixel size of 1.03-Å/pixel at the specimen scale. The 26 frames in each movie were subjected to drift correction using motioncorr (Li et al., 2013) and averaged to produce one micrograph. Defocus for each micrograph was determined by CTFFind3 (Mindell and Grigorieff, 2003), and a total of 44,328 viral particles were picked manually. Because the size of boxed particles (1,440 × 1,440 pixels) was so large that the cumulative dataset required an unrealistic amount of computer memory for computation, boxed particles were normalized and binned four times prior to implementing standard icosahedral reconstruction procedures in Relion (Scheres, 2012). Using a Gaussian ball as the initial reference, auto-refinement for icosahedral reconstruction was performed imposing I3 symmetry, generating an icosahedral map with one of the twelve 5-fold axes aligned on the z axis.
Sub-particle extraction from icosahedron vertices and local focus value calculation
As illustrated in Figure S1, we extracted twelve sub-particles corresponding to the twelve capsid vertices for each KSHV virion based on the icosahedral orientation described above. To do so, we first expanded the icosahedral symmetry of the particles using relion_particle_symmetry_expand, generating 60 icosahedrally-related orientations for each particle. Each orientation has three Euler angles denoted as parameters within the Relion star files: rot (_rlnAngleRot), tilt (_rlnAngleTilt), and psi (_rlnAnglePsi). We then defined the orientation for each of the twelve vertices from the 60 icosahedrally-related orientations as follows. First, we noted that because our icosahedral reconstruction was performed using I3 symmetry, there are 5 redundant orientations relative to each vertex that differ only in their rot angles (i.e., the in-plane rotation angle about the z axis). For this reason, we classified the 60 orientations into twelve groups each, with five orientations per group that differ only in their rot angles. We then randomly chose one of the five orientations in each group as the orientation of that vertex, thereby defining the orientations for all twelve vertices of each capsid.
Next, we determined the location of each vertex sub-particle on the viral particle image. The two-dimensional Cartesian positions (x, y) of each sub-particle on their respective particle image were calculated using the following formula:
| (1) |
where d is the distance from the center of the reconstructed capsid to the vertex in pixels and C is the center of the 2D projection image (in our case, the projection center is at [720, 720], so C = 720 pixels). Because icosahedral reconstruction was performed with four times-binned particles, Ox and Oy are four times the offset distance (_rlnOriginX and _rlnOriginY in Relion) of each particle image relative to the projection center of the icosahedral reconstruction. Finally, sub-particles (384 × 384 pixels) containing only vertices, henceforth termed “vertex sub-particles,” were extracted from original unbinned particle images based on their calculated positions using relion_preprocess without further normalization.
Our sub-particle reconstruction method also enabled a more accurate determination of the defocus for each sub-particle, thus alleviating the well-documented depth-of-focus problem (DeRosier, 2000, Zhang and Zhou, 2011). The defocus value of each vertex sub-particle was calculated based on its location with the following formula:
| (2) |
where is the original defocus and is the new defocus for each vertex.
Classification and refinement of vertex sub-particles with 5-fold symmetry
To identify the unique portal vertex from among the 12 icosahedral vertices for each virus, we classified all vertex sub-particles with 5-fold symmetry (Figure S1). No rotational orientation search was allowed during classification (using the –skip_rotate parameter in Relion), though the center for each vertex sub-particle was refined with a ± 3 pixel offset search. The initial reference for classification was a 30-Å reconstruction of the vertex sub-particles using relion_reconstruct. Although the portal vertex lacks true 5-fold symmetry, this classification with 5-fold symmetry imposed successfully distinguished between penton and portal vertices. Through 50 iterations of 3D classification, four classes were ultimately generated, with one class in particular exhibiting markedly different structures (i.e., a blurry central channel with a rod-like density) (Figure S1). Additionally, this class contained 7.9%, or approximately 1/12th of the vertex sub-particles, consistent with the expectation that one out of twelve capsid vertices in each particle is a portal vertex. We thus considered this class the portal vertex class. In rare instances, two or more vertices from a capsid were classified into the portal vertex class, likely due to the low quality of these individual particles and/or errors in classification. These redundant sub-particles were removed as follows: if two or more vertices from the same virus particle were assigned to the portal vertex class, only the vertex sub-particle with the highest _rlnMaxValueProbDistribution score was retained. Upon removing all redundant particles, 39,773 vertex sub-particles remained and were deemed sub-particles of the portal vertex, henceforth referred to as “portal vertex sub-particles.” 3D auto-refinement with 5-fold symmetry imposed was then performed on these portal vertex sub-particles with only a local search for orientation determination. Using relion_postprocess, the final resolution of this C5 reconstruction was calculated with two independently refined maps from halves of the dataset with gold-standard FSC at the 0.143 criterion (Rosenthal and Henderson, 2003), and determined to be 4.3-Å (Figures S2A and S2D). This reconstruction of the portal vertex contains a well-resolved 5-fold–arranged capsid and tegument, but a smeared portal dodecamer density due to symmetry mismatch.
Reconstructing the portal dodecamer with 12-fold symmetry
From the portal vertex sub-particles, we further extracted sub-particles containing only the portal dodecamer in order to reconstruct the 12-fold symmetric portal. The positions of portal dodecamers on portal vertex sub-particles were determined using the above formula (1). The Euler angles (rot, tilt, and psi), Ox, and Oy are the orientation parameters of the portal vertex sub-particles; d is the distance from the center of the dodecamer to the center of the portal vertex sub-particle reconstruction (–100 pixels); and C is the center of the 2D projection image of the portal vertex sub-particle (192 pixels). The sub-particles of portal dodecamers (192x192 pixels), henceforth referred to as “dodecamer sub-particles” were then extracted with relion_preprocess using these parameters.
To reconstruct the portal dodecamer, we expanded the 5-fold symmetry of the dodecamer sub-particles using relion_particle_symmetry_expand, generating five unique orientations for each dodecamer sub-particle. We then applied 3D classification with 12-fold symmetry imposed and without rotational orientation search, which, after 100 iterations, yielded five classes of similar structures with a rotational difference of approximately 72° between classes. Ideally, each of the five expanded orientations of a dodecamer sub-particle should be assigned to exactly one of the five classes, such that each class should contain 20% of the symmetry-expanded sub-particles. After removing redundant particles as previously described—only particles with the highest _rlnMaxValueProbDistribution score was retained—the five classes contained 39,073, 37,753, 36,797, 37,270, 38,031 particles, respectively. As the five reconstructed classes were of the same quality upon visual inspection, we chose the class with the most abundant particles for 3D refinement with 12-fold symmetry imposed and limited to local orientation search. As with the previous reconstruction, the resolution of this C12 portal dodecamer reconstruction was calculated with relion_postprocess using gold-standard FSC at the 0.143 criterion (Rosenthal and Henderson, 2003), and determined to be ∼4.7-Å. However, both a visual assessment of the portal’s density and local resolution estimate derived from ResMap (Kucukelbir et al., 2014) indicate the majority of the portal itself has a resolution of ∼4.0-Å (Figures S2A and S2E), thereby permitting ab initio modeling. The lower resolution estimate obtained from FSC calculation likely results from the unresolved, flexible regions of the portal and/or the surrounding DNA and protein densities that deviate from 12-fold symmetry, which are present in the map and therefore factored into the estimation.
Asymmetric reconstruction of the portal vertex and virion
Since each portal orientation determined from the previous round of portal dodecamer classification was selected from one of the five expanded orientations of each portal vertex sub-particle, these orientations can be used for 3D refinement of the portal vertex and whole virion without symmetry. The asymmetric auto-refinement for both portal vertex sub-particles and virion particles was thus performed with a local search for orientations determined from the classification of the portal dodecamer. Due to the large computational requirement for refinement of the whole virion, we performed this refinement using two times-binned particles. The resolution of the portal vertex and whole virion C1 reconstructions was determined by relion_postprocess to be 5.2-Å and 7.6-Å, respectively (Figures S2A, S2B, S2F, and S2G), according to gold-standard FSC at the 0.143 criterion (Rosenthal and Henderson, 2003).
Focused classification of symmetry-relaxed penton vertex sub-particles
Our classification of vertex sub-particles identified not only the portal vertex class, but also generated three classes of penton vertex (Figure S1). The sub-particles of these three classes were combined and deemed “penton vertex sub-particles.”
Due to the 5-fold symmetry surrounding capsid vertices, CATC can bind to any of five registers at the penton vertex. To determine the structures of both CATC-bound and CATC-absent registers, we expanded the 5-fold symmetry of penton vertex sub-particles by relion_particle_symmetry_expand, producing 2,450,245 symmetry-expanded sub-particles of penton vertex. To create a mask for focused classification of CATC, one CATC-containing region with its corresponding triplex Ta was manually traced using volume_tracer in Chimera (Pettersen et al., 2004), after which a mask encompassing the traced region was created by relion_mask_create in Relion. Focused classification of the masked region was then performed on symmetry-expanded penton vertex sub-particles with neither angular nor offset search (using the –skip_align parameter in Relion). Since classification was performed on such a small area relative to the whole reconstruction, we specified a tau factor of 10 during classification (Scheres, 2016). After 90 iterations, four classes were generated, among which only one class had apparent CATC density corresponding to 37.9% of symmetry-expanded masked regions, consistent with the occupancy calculated from a previous study (Dai et al., 2014). We therefore considered this a CATC-binding class. The other three classes, though of slightly differing map quality, clearly lacked CATC density while sharing the same triplex Ta orientation. We therefore regarded these three classes as CATC-absent classes.
Refinement of the CATC-binding and CATC-absent structures without symmetry
We next performed separate 3D auto-refinements without symmetry on both penton vertex sub-particles in the CATC-binding class and in the CATC-absent classes. Orientations for sub-particles were determined by local search around each sub-particle’s predetermined orientation from the mask refinement. A 30-Å reconstruction of penton vertex obtained through relion_reconstruct was used as the initial reference for refinements. The final resolutions of the CATC-binding and CATC-absent reconstructions determined by relion_postprocess were 3.8-Å and 3.7-Å, respectively, according to gold-standard FSC at the 0.143 criterion (Rosenthal and Henderson, 2003; Figure S2C). Of note, because focused classification was performed with respect to the masked area, only structural features within the masked area (which encompasses one CATC and an associated triplex Ta) of the refined structure are genuine, while the other registers of CATC and triplex Ta are of mixed occupancy and/or conformations (Figure S1) (these will be fully separated in the following procedure). Local resolution estimation by ResMap (Kucukelbir et al., 2014) indicates the masked area of both CATC-binding and CATC-absent reconstructions reached a resolution of ∼3.5-Å (Figures S2H and S2I).
Geometry-based classification and reconstruction of penton vertices
To distinguish all possible CATC-binding occupancies and binding permutations at the five registers of a penton vertex, we performed occupancy and geometry-based classification. To reiterate, capsid vertices can theoretically be bound by any number of CATCs from zero up to five. Moreover, in cases of two CATCs bound to a penton vertex, the two CATCs can either occupy registers adjacent to each other or registers with a gap (i.e., an empty register) in between. We named these two binding patterns “ortho-CATC–binding” and “meta-CATC–binding,” respectively. In instances of three CATCs bound to a penton vertex, the two vacant registers can either be adjacent or have a gap (i.e., an occupied register) between them, analogous to the case of two vertex-bound CATCs. We thus named these two permutations “ortho-CATC–absent” and “meta-CATC–absent,” respectively. Altogether, eight classes of penton vertices are possible. These are: zero-, one-, four-, and five-CATC–binding vertices; ortho- and meta-CATC–binding vertices (two CATCs bound); and ortho- and meta-CATC–absent vertices (three CATCs bound).
We then classified all penton vertex sub-particles into the eight classes as follows. We first noted that focused classification with expanded symmetry enabled us to determine whether the masked area of a penton vertex sub-particle (for each register) fell into a CATC-binding or CATC-absent class. We then counted the number of orientations (out of five symmetry-expanded orientations per sub-particle) assigned to the CATC-binding class for each penton vertex sub-particle. This counted number of orientations, ranging from zero to five, reflects the number of CATCs bound to that particular vertex in that viral particle. If two CATCs were determined to bind that vertex, the angle between the two CATC-binding orientations was calculated. If the calculated angle was ∼72°, this particular penton vertex was ortho-CATC–binding; otherwise, it was deemed a meta-CATC–binding vertex. An analogous calculation for CATC-absent registers was also performed when three CATCs were found to bind a vertex. We thus classified each penton vertex sub-particle into one of the eight possible classes.
Classified penton vertex sub-particles were then used for subsequent auto-refinement. Given that five expanded orientations for each vertex were already each assigned to either a CATC-binding or CATC-absent class (due to masked classification of the CATC-binding region), orientations for vertex sub-particles were determined as follows. The orientations for zero- and five-CATC–binding vertex sub-particles were randomly selected from their five orientations in the CATC-absent and CATC-binding classes, respectively. Orientations of one-CATC–binding and four-CATC–binding vertices were chosen as their single orientation in the CATC-binding and CATC-absent classes, respectively. Where each vertex bound two CATCs, orientations were chosen as one of the two orientations in the CATC-binding class, such that the second (i.e., non-chosen) orientation was a 72° and 144° counterclockwise rotation from the chosen orientation, for ortho- and meta-CATC-binding vertices, respectively. Similarly, the orientations for ortho- and meta-CATC–absent classes were chosen such that the non-chosen CATC-absent orientation was a 72° and 144° counterclockwise rotation from the assigned orientation, respectively. For all auto-refinements, sub-particle orientations were determined by local angular and offset searches about these selected orientations, and initial references were 30-Å reconstructions of the sub-particles generated by relion_reconstruct. 5-fold symmetry was imposed during refinements of zero- and five-CATC–binding vertices, while refinements of all other binding occupancies were carried out without symmetry. The resolutions of the resulting eight reconstructions determined by relion_postprocess according to gold-standard FSC at the 0.143 criterion were 5.8-Å, 4.7-Å, 6.9-Å, and 9.0-Å for zero-, one-, four-, and five-CATC–binding vertices, respectively; 4.1-Å and 5.3-Å for ortho- and meta-CATC–binding vertices, respectively; and 4.7-Å and 5.0-Å for ortho- and meta-CATC–absent vertices, respectively.
As the orientation information of all CATC-binding and CATC-absent registers and vertex sub-particles were conserved in their respective header files in our workflow, we were able to trace each register and vertex back to their original particle image. This enabled us to conduct a survey of the global number of CATCs in each capsid and generate a histogram tallying this data (Figure 3B).
Atomic and homology modeling
Atomic models were built ab initio following established protocol for model building from cryoEM density maps as described in great detail recently (Yu et al., 2018). Briefly, we assigned amino acid residues by correlating predicted secondary structures from Phyre2 (Kelley et al., 2015) and visible side-chain features in the mainchain density. Initial Cα backbone traces were generated using the baton_build utility in COOT (Emsley et al., 2010), and full-atom registered mainchains were manually refined using regularize_zone to improve both geometry and fit in our cryoEM density. Subsequent iterations of real space refinement in Phenix (Adams et al., 2010) and manual refinement in COOT were then applied to optimize the atomic models.
To build the homology model of pORF19 globular head domain (aa 127-546), we first generated an initial model utilizing an extension of Modeler ( Webb and Sali, 2017 ) in Chimera (Pettersen et al., 2004), using the HSV-1 pUL25 head domain X-ray crystal structure (PDB: 2F5U) as a template. The generated model was then rigidly fitted into the density map of a CATC-binding penton vertex, manually adjusted in COOT, then refined with Phenix real space refinement to improve fit and geometry.
Construction of pORF32/pORF19 plasmids
To generate a C-terminal FLAG-tagged expression plasmid of pORF32 (32WT), we PCR-amplified its coding region using primers 5′-AC CGT CAG ATC CGC TAG AGA TCT GCC ACC ATG GAT GCG CAT GCT ATC AAC-3′ and 5′-CC GCG TCG ACG GTA CCA GAT CTA CTT GTC GTC ATC GTC TTT GTA GTC GCC ATA GCG GCC TCG AAT G-3′, and further cloned the PCR product into RedTrackCMV vector using an NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs). The pORF19 coding region was PCR-amplified using primers 5′-C CGT CAG ATC CGC TAG AGA TCT GCC ACC ATG GAC TAC AAA GAC GAT GAC GAC AAG GGT GGC GGA GGT TCT ATG CTG ACAT CAG AAA GGT C-3′ and 5′-CC GCG TCG ACG GTA CCA GAT CTA AAC GAC CGC GAG GAC CAC-3′, and also cloned into RedTrackCMV vector to generate an N-terminal FLAG-tagged expression plasmid (19WT). pORF32 mutants and pORF19 mutants were generated by PCR-based deletion mutagenesis from 32WT and 19WT, respectively. Sequences of the PCR-amplified fragments and their correct insertion in the plasmid were verified by sequencing.
Titration of infectious KSHV virions
The concentration of infectious KSHV virions was determined as previously described (Gong et al., 2016). Briefly, 293T-KSHV cells were transfected with tegument protein expression plasmids or the empty vector as control. At 16 h post-transfection, cells were treated with 0.5 mM NaB plus 25 ng/mL 12-O-Tetradecanoylphorbol-13-acetate (TPA) to induce KSHV lytic replication. Three days later, supernatants were collected, centrifuged at 10,000 g for 10 min at 4°C to remove cellular debris, serially diluted in DMEM with 10% FBS, and then used to infect 293T cells in 96-well plates by spinoculation (3,000 g for 1 h at 30°C). Two days post-infection, GFP-positive cell clusters containing two or more cells were counted under a fluorescence microscope to determine the titer of KSHV virion. Infectious units (IU) are expressed as the number of GFP-positive cell clusters in each well at a specific dilution of the viral stock.
Measuring viral DNA replication and RNA transcription by real-time PCR
Total DNA was isolated from 293T-KSHV cells induced with NaB plus TPA, and viral genome copy numbers were determined by real-time PCR using primers for the essential viral gene ORF59. Total RNA was extracted from cells with Purelink RNA Mini Kit (Thermo Fisher Scientific), treated with DNase I, then reverse-transcribed using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) and random hexamers. Real-time PCR was then performed with the following primers to detect the corresponding DNA or RNA transcripts. Host house-keeping gene GAPDH: 5′-TGC ACC ACC AAC TGC TTA GC-3′ and 5′-GGC ATG GAC TGT GGT CAT GAG-3′; KSHV immediate early gene RTA: 5′-CAC AAA AAT GGC GC AAG ATG A-3′ and 5′-TGG TAG AGT TGG GCC TTC AGT T-3′; KSHV early gene ORF59: 5′-TTG GCA CTC CAA CGA AAT ATT AGA A-3′ and 5′-CGG GAA CCT TTT GCG AAG A-3′; KSHV late gene K8.1: 5′-GTA ACC GTG TGC CAT TTT CTG-3′ and 5′-TCC CAG CAA TAA ACC CAC AG-3′.
Western blotting and antibodies
Cells were lysed in 1X western blotting loading buffer, resolved by SDS-PAGE gel electrophoresis, and transferred onto PVDF membrane. Proteins were detected with antibodies against FLAG-epitope (Sigma-Aldrich) or GAPDH (Abcam). HRP-conjugated secondary antibodies (Cell Signaling Technology) were used for detection with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Construction of Tri1 and Tri2 KSHV mutants
KSHV-BAC16 plasmid was modified according to a previously described method (Brulois et al., 2012, Gong et al., 2016). Briefly, DNA fragments of KSHV ORF62 (Tri1) and ORF26 (Tri2) with defined mutations were used to replace the wild-type sequence in KSHV-BAC16 plasmid by homologous recombination. Restriction patterns of mutated KSHV BAC plasmids were verified by comparison to that of wild-type KSHV-BAC16 plasmid to ensure overall genome integrity. Fragments containing mutations were PCR-amplified from BAC plasmids and sequenced to confirm that all mutations were correct. Mutant BAC plasmids were transfected into 293T cells individually, followed by selection with 100 μg/mL hygromycin B for one month to generate cell lines latently infected by a specific KSHV mutant virus. As described above, KSHV lytic replication was induced by treatment of cells with 0.5 mM NaB plus 25 ng/mL TPA. Three days later, supernatants were collected for determining titers of infectious KSHV virions, while cells were harvested for measuring viral DNA replication and RNA transcription as described above.
Quantification and Statistical Analysis
Statistical comparisons between groups were made using Student’s t test calculated in Microsoft Excel. Data and error bars displayed for measured relative virion production, viral titer, genome replication levels, and RNA transcription levels (shown in Figures 5J, 5L, 5N–5P, and S6A–S6D) are presented as mean ± SEM (n = 3 biologically independent samples).
Data and Code Availability
Six cryoEM maps generated during this study have been deposited in the Electron Microscopy Data Bank (EMDB) and are available under accession numbers EMD-20430 (C1 virion capsid reconstruction), EMD-20431 (C1 portal vertex reconstruction), EMD-20432 (C5 portal vertex reconstruction), EMD-20437 (C12 portal reconstruction), EMD-20433 (C1 penton vertex register, CATC-absent reconstruction), and EMD-20436 (C1 penton vertex register, CATC-binding reconstruction). Atomic models corresponding to EMD-20432, EMD-20437, EMD-20433, and EMD-20436 have been deposited in the Protein Data Bank (PDB) and are available under accession numbers PDB-6PPB, PDB-6PPI, PDB-6PPD, and PDB-6PPH, respectively. Sub-particle reconstruction scripts used in our workflow have been deposited on GitHub and can be accessed here: https://github.com/procyontao/Herpesportal.
Acknowledgments
We thank the Bioinformatics Center of the University of Science and Technology of China, School of Life Science, for providing supercomputing resources for this project. This research has been supported in part by grants from the National Key R&D Program of China (2017YFA0505300 and 2016YFA0400900 and NIH (GM071940, DE025567, DE028583, DE027901, and AI094386 to Z.H.Z and CA177322, CA091791, and DE023591 to R.S.). We acknowledge the use of resources at the Electron Imaging Center for Nanomachines supported by UCLA and by instrumentation grants from the NIH (1S10RR23057 and 1U24GM116792) and NSF (DBI-1338135 and DMR-1548924).
Author Contributions
Z.H.Z. and R.S. conceived the project and supervised the research. D.G. prepared samples and carried out mutagenesis and functional studies. X.D. and Y.-T.L. determined the structures. J.J. and X.D. built atomic models. Z.H.Z., X.D., J.J., Y.-T.L., D.G., and R.S. analyzed the results and wrote the paper. Z.H.Z., R.S., and G.-Q.B. managed resources and supervised the research. All authors reviewed and approved the paper.
Declaration of Interests
The authors declare no competing interests.
Published: August 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.07.035.
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman K., Salmon B., Baines J.D. Herpes simplex virus DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl. Acad. Sci. USA. 2001;98:3086–3091. doi: 10.1073/pnas.061555698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard P.M., Taus N.S., Baines J.D. DNA cleavage and packaging proteins encoded by genes U(L)28, U(L)15, and U(L)33 of herpes simplex virus type 1 form a complex in infected cells. J. Virol. 2002;76:4785–4791. doi: 10.1128/JVI.76.10.4785-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat T.A., Riches J.D., Kolesnikova L., Welsch S., Krähling V., Davey N., Parsy M.L., Becker S., Briggs J.A. Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLoS Biol. 2011;9:e1001196. doi: 10.1371/journal.pbio.1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman B.R., Welschhans R.L., Jayaram H., Stow N.D., Preston V.G., Quiocho F.A. Structural characterization of the UL25 DNA-packaging protein from herpes simplex virus type 1. J. Virol. 2006;80:2309–2317. doi: 10.1128/JVI.80.5.2309-2317.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brulois K.F., Chang H., Lee A.S., Ensser A., Wong L.Y., Toth Z., Lee S.H., Lee H.R., Myoung J., Ganem D. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 2012;86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G., Winkler D.C., Trus B.L., Cheng N., Heuser J.E., Newcomb W.W., Brown J.C., Steven A.C. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology. 2007;361:426–434. doi: 10.1016/j.virol.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G., Heymann J.B., Cheng N., Trus B.L., Steven A.C. Procapsid assembly, maturation, nuclear exit: dynamic steps in the production of infectious herpesvirions. Adv. Exp. Med. Biol. 2012;726:423–439. doi: 10.1007/978-1-4614-0980-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y., Cesarman E., Pessin M.S., Lee F., Culpepper J., Knowles D.M., Moore P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chang J.T., Schmid M.F., Rixon F.J., Chiu W. Electron cryotomography reveals the portal in the herpesvirus capsid. J. Virol. 2007;81:2065–2068. doi: 10.1128/JVI.02053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X., Zhou Z.H. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science. 2018;360 doi: 10.1126/science.aao7298. eaao7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X., Gong D., Wu T.T., Sun R., Zhou Z.H. Organization of capsid-associated tegument components in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014;88:12694–12702. doi: 10.1128/JVI.01509-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X., Gong D., Lim H., Jih J., Wu T.T., Sun R., Zhou Z.H. Structure and mutagenesis reveal essential capsid protein interactions for KSHV replication. Nature. 2018;553:521–525. doi: 10.1038/nature25438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B., O’Connor C.M., Kedes D.H., Zhou Z.H. Cryo-electron tomography of Kaposi’s sarcoma-associated herpesvirus capsids reveals dynamic scaffolding structures essential to capsid assembly and maturation. J. Struct. Biol. 2008;161:419–427. doi: 10.1016/j.jsb.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosier D.J. Correction of high-resolution data for curvature of the Ewald sphere. Ultramicroscopy. 2000;81:83–98. doi: 10.1016/s0304-3991(99)00120-5. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 2010;120:939–949. doi: 10.1172/JCI40567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffin L., Damania B. KSHV: pathways to tumorigenesis and persistent infection. Adv. Virus Res. 2014;88:111–159. doi: 10.1016/B978-0-12-800098-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C., Bestman-Smith J., Boivin G. Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms. Drug Resist. Updat. 2002;5:88–114. doi: 10.1016/s1368-7646(02)00021-3. [DOI] [PubMed] [Google Scholar]
- Goldsmith C.S., Tatti K.M., Ksiazek T.G., Rollin P.E., Comer J.A., Lee W.W., Rota P.A., Bankamp B., Bellini W.J., Zaki S.R. Ultrastructural characterization of SARS coronavirus. Emerg. Infect. Dis. 2004;10:320–326. doi: 10.3201/eid1002.030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong D., Kim Y.H., Xiao Y., Du Y., Xie Y., Lee K.K., Feng J., Farhat N., Zhao D., Shu S. A Herpesvirus Protein Selectively Inhibits Cellular mRNA Nuclear Export. Cell Host Microbe. 2016;20:642–653. doi: 10.1016/j.chom.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong D., Dai X., Xiao Y., Du Y., Chapa T.J., Johnson J.R., Li X., Krogan N.J., Deng H., Wu T.T., Sun R. Virus-Like Vesicles of Kaposi’s Sarcoma-Associated Herpesvirus Activate Lytic Replication by Triggering Differentiation Signaling. J. Virol. 2017;91:e00362-17. doi: 10.1128/JVI.00362-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A., Cardone G., Winkler D.C., Heymann J.B., Brecher M., White J.M., Steven A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA. 2006;103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heming J.D., Conway J.F., Homa F.L. Herpesvirus Capsid Assembly and DNA Packaging. Adv. Anat. Embryol. Cell Biol. 2017;223:119–142. doi: 10.1007/978-3-319-53168-7_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman J.B., Daniel G.R., Falck-Pedersen E., Huet A., Smith G.A., Conway J.F., Homa F.L. The C Terminus of the Herpes Simplex Virus UL25 Protein Is Required for Release of Viral Genomes from Capsids Bound to Nuclear Pores. J. Virol. 2017;91 doi: 10.1128/JVI.00641-17. e00641-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A., Sigworth F.J., Tagare H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev A.A., Krause M.H., Isidro A.L., Vagin A.A., Orlova E.V., Turner J., Dodson E.J., Tavares P., Antson A.A. Structural framework for DNA translocation via the viral portal protein. EMBO J. 2007;26:1984–1994. doi: 10.1038/sj.emboj.7601643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Mooney P., Zheng S., Booth C.R., Braunfeld M.B., Gubbens S., Agard D.A., Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.T., Jiang J., Bohannon K.P., Dai X., Gant Luxton G.W., Hui W.H., Bi G.Q., Smith G.A., Zhou Z.H. A pUL25 dimer interfaces the pseudorabies virus capsid and tegument. J. Gen. Virol. 2017;98:2837–2849. doi: 10.1099/jgv.0.000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Dai H., Jih J., Chan K., Trang P., Yu X., Balogun R., Mei Y., Liu F., Zhou Z.H. Different capsid-binding patterns of the β-herpesvirus-specific tegument protein pp150 (pM32/pUL32) in murine and human cytomegaloviruses. bioRxiv. 2018 doi: 10.1101/420604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokareddy R.K., Sankhala R.S., Roy A., Afonine P.V., Motwani T., Teschke C.M., Parent K.N., Cingolani G. Portal protein functions akin to a DNA-sensor that couples genome-packaging to icosahedral capsid maturation. Nat. Commun. 2017;8:14310. doi: 10.1038/ncomms14310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxton G.W.G., Haverlock S., Coller K.E., Antinone S.E., Pincetic A., Smith G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA. 2005;102:5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee M., Vijayakrishnan S., Rixon F., Bhella D. Structure of the herpes simplex virus portal-vertex. PLoS Biol. 2018;16:e2006191. doi: 10.1371/journal.pbio.2006191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab A.R., Desai P., Person S., Roof L.L., Thomsen D.R., Newcomb W.W., Brown J.C., Homa F.L. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 1998;72:1060–1070. doi: 10.1128/jvi.72.2.1060-1070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell J.A., Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Motwani T., Lokareddy R.K., Dunbar C.A., Cortines J.R., Jarrold M.F., Cingolani G., Teschke C.M. A viral scaffolding protein triggers portal ring oligomerization and incorporation during procapsid assembly. Sci. Adv. 2017;3:e1700423. doi: 10.1126/sciadv.1700423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W., Homa F.L., Thomsen D.R., Ye Z., Brown J.C. Cell-free assembly of the herpes simplex virus capsid. J. Virol. 1994;68:6059–6063. doi: 10.1128/jvi.68.9.6059-6063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W., Homa F.L., Brown J.C. Involvement of the portal at an early step in herpes simplex virus capsid assembly. J. Virol. 2005;79:10540–10546. doi: 10.1128/JVI.79.16.10540-10546.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara M., Suzutani T., Yoshida I., Azuma M. Role of the UL25 gene product in packaging DNA into the herpes simplex virus capsid: location of UL25 product in the capsid and demonstration that it binds DNA. J. Virol. 2001;75:1427–1436. doi: 10.1128/JVI.75.3.1427-1436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D.J., Crump C.M., Graham S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses. 2015;7:5084–5114. doi: 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdeloup D., Blondel D., Isidro A.L., Rixon F.J. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 2009;83:6610–6623. doi: 10.1128/JVI.02655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins E.M., Anacker D., Davis A., Sankar V., Ambinder R.F., Desai P. Small capsid protein pORF65 is essential for assembly of Kaposi’s sarcoma-associated herpesvirus capsids. J. Virol. 2008;82:7201–7211. doi: 10.1128/JVI.00423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Reusser P. Herpesvirus resistance to antiviral drugs: a review of the mechanisms, clinical importance and therapeutic options. J. Hosp. Infect. 1996;33:235–248. doi: 10.1016/s0195-6701(96)90010-9. [DOI] [PubMed] [Google Scholar]
- Rochat R.H., Liu X., Murata K., Nagayama K., Rixon F.J., Chiu W. Seeing the portal in herpes simplex virus type 1 B capsids. J. Virol. 2011;85:1871–1874. doi: 10.1128/JVI.01663-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal P.B., Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Rozen R., Sathish N., Li Y., Yuan Y. Virion-wide protein interactions of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008;82:4742–4750. doi: 10.1128/JVI.02745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathish N., Wang X., Yuan Y. Tegument Proteins of Kaposi’s Sarcoma-Associated Herpesvirus and Related Gamma-Herpesviruses. Front. Microbiol. 2012;3:98. doi: 10.3389/fmicb.2012.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H.W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H. Processing of Structurally Heterogeneous Cryo-EM Data in RELION. Methods Enzymol. 2016;579:125–157. doi: 10.1016/bs.mie.2016.04.012. [DOI] [PubMed] [Google Scholar]
- Sun L., Zhang X., Gao S., Rao P.A., Padilla-Sanchez V., Chen Z., Sun S., Xiang Y., Subramaniam S., Rao V.B., Rossmann M.G. Cryo-EM structure of the bacteriophage T4 portal protein assembly at near-atomic resolution. Nat. Commun. 2015;6:7548. doi: 10.1038/ncomms8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatman J.D., Preston V.G., Nicholson P., Elliott R.M., Rixon F.J. Assembly of herpes simplex virus type 1 capsids using a panel of recombinant baculoviruses. J. Gen. Virol. 1994;75:1101–1113. doi: 10.1099/0022-1317-75-5-1101. [DOI] [PubMed] [Google Scholar]
- Thurlow J.K., Murphy M., Stow N.D., Preston V.G. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J. Virol. 2006;80:2118–2126. doi: 10.1128/JVI.80.5.2118-2126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trus B.L., Heymann J.B., Nealon K., Cheng N., Newcomb W.W., Brown J.C., Kedes D.H., Steven A.C. Capsid structure of Kaposi’s sarcoma-associated herpesvirus, a gammaherpesvirus, compared to those of an alphaherpesvirus, herpes simplex virus type 1, and a betaherpesvirus, cytomegalovirus. J. Virol. 2001;75:2879–2890. doi: 10.1128/JVI.75.6.2879-2890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahey M.D., Fletcher D.A. Low-Fidelity Assembly of Influenza A Virus Promotes Escape from Host Cells. Cell. 2019;176:281–294.e19. doi: 10.1016/j.cell.2018.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walling D.M., Flaitz C.M., Nichols C.M. Epstein-Barr virus replication in oral hairy leukoplakia: response, persistence, and resistance to treatment with valacyclovir. J. Infect. Dis. 2003;188:883–890. doi: 10.1086/378072. [DOI] [PubMed] [Google Scholar]
- Wang J., Yuan S., Zhu D., Tang H., Wang N., Chen W., Gao Q., Li Y., Wang J., Liu H. Structure of the herpes simplex virus type 2 C-capsid with capsid-vertex-specific component. Nat. Commun. 2018;9:3668. doi: 10.1038/s41467-018-06078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B., Sali A. Protein Structure Modeling with MODELLER. Methods Mol. Biol. 2017;1654:39–54. doi: 10.1007/978-1-4939-7231-9_4. [DOI] [PubMed] [Google Scholar]
- Wu L., Lo P., Yu X., Stoops J.K., Forghani B., Zhou Z.H. Three-dimensional structure of the human herpesvirus 8 capsid. J. Virol. 2000;74:9646–9654. doi: 10.1128/jvi.74.20.9646-9654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Homa F., Baines J.D. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 2007;81:6419–6433. doi: 10.1128/JVI.00047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Jih J., Jiang J., Zhou Z.H. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150. Science. 2017;356:1350. doi: 10.1126/science.aam6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu I., Nguyen L., Avaylon J., Wang K., Lai M., Zhou Z.H. Building atomic models based on near atomic resolution cryoEM maps with existing tools. J. Struct. Biol. 2018;204:313–318. doi: 10.1016/j.jsb.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zhou Z.H. Limiting factors in atomic resolution cryo electron microscopy: no simple tricks. J. Struct. Biol. 2011;175:253–263. doi: 10.1016/j.jsb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.H., Macnab S.J., Jakana J., Scott L.R., Chiu W., Rixon F.J. Identification of the sites of interaction between the scaffold and outer shell in herpes simplex virus-1 capsids by difference electron imaging. Proc. Natl. Acad. Sci. USA. 1998;95:2778–2783. doi: 10.1073/pnas.95.6.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.H., Hui W.H., Shah S., Jih J., O’Connor C.M., Sherman M.B., Kedes D.H., Schein S. Four levels of hierarchical organization, including noncovalent chainmail, brace the mature tumor herpesvirus capsid against pressurization. Structure. 2014;22:1385–1398. doi: 10.1016/j.str.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overview of the C1 reconstruction of DNA-containing capsid, including the unique portal vertex.
Depicts the C12 reconstruction and atomic model of pORF43, highlighting domains and notable structural features.
Depicts interactions between the CATC and underlying Ta and Tc triplexes through pORF32′s “NH” and “AL” regions and pORF19’s N-terminal residues.
Data Availability Statement
Six cryoEM maps generated during this study have been deposited in the Electron Microscopy Data Bank (EMDB) and are available under accession numbers EMD-20430 (C1 virion capsid reconstruction), EMD-20431 (C1 portal vertex reconstruction), EMD-20432 (C5 portal vertex reconstruction), EMD-20437 (C12 portal reconstruction), EMD-20433 (C1 penton vertex register, CATC-absent reconstruction), and EMD-20436 (C1 penton vertex register, CATC-binding reconstruction). Atomic models corresponding to EMD-20432, EMD-20437, EMD-20433, and EMD-20436 have been deposited in the Protein Data Bank (PDB) and are available under accession numbers PDB-6PPB, PDB-6PPI, PDB-6PPD, and PDB-6PPH, respectively. Sub-particle reconstruction scripts used in our workflow have been deposited on GitHub and can be accessed here: https://github.com/procyontao/Herpesportal.