

Summary
Phages express anti-CRISPR (Acr) proteins to inhibit CRISPR-Cas systems that would otherwise destroy their genomes. Most acr genes are located adjacent to anti-CRISPR associated (aca) genes, which encode proteins with a helix-turn-helix DNA-binding motif. The conservation of aca genes has served as a signpost for the identification acr genes, yet the function of the proteins encoded by these genes has not been investigated. Here, we reveal that an acr associated promoter drives high levels of acr transcription immediately after phage DNA injection, and that Aca proteins subsequently repress this transcription. Without Aca activity, this strong transcription is lethal to a phage. Our results demonstrate how sufficient levels of Acr proteins accumulate early in the infection process to inhibit existing CRISPR-Cas complexes in the host cell. They also imply that the conserved role of Aca proteins is to mitigate the deleterious effects of strong constitutive transcription from acr promoters.
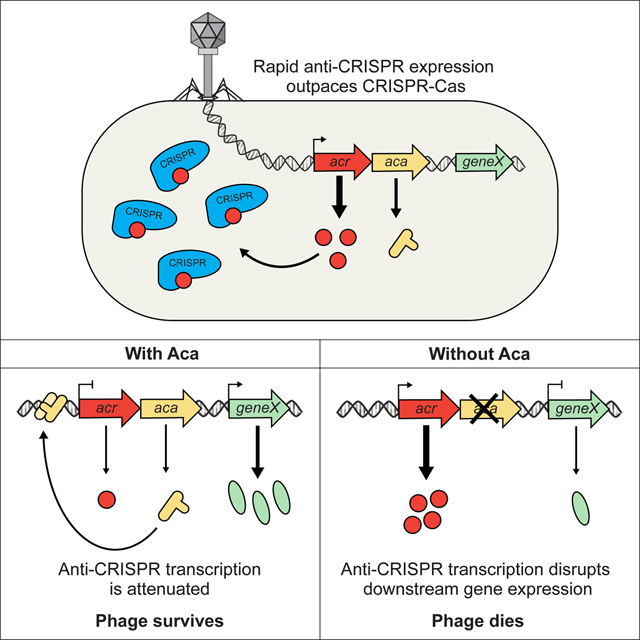
Graphical Abstract
Introduction
CRISPR-Cas systems immunize bacteria and archaea against invading genetic elements like phages by incorporating short sequences of DNA from these invaders into their chromosome (Datsenko et al., 2012; Levy et al., 2015; Yosef et al., 2012). These sequences are transcribed and processed into small RNAs known as CRISPR RNAs (crRNAs) that bind to CRISPR-associated (Cas) proteins to form ribonucleoprotein interference complexes. These complexes survey the cell, recognize foreign nucleic acids through complementarity with their crRNAs, and ultimately destroy them through the intrinsic nuclease activity of the Cas proteins (Barrangou et al., 2007; Brouns et al., 2008; Garneau et al., 2010; Marraffini and Sontheimer, 2008). CRISPR-Cas systems are diverse, comprising six distinct types, each with multiple subtypes (Makarova et al., 2015). In many bacteria, CRISPR-Cas systems are expressed in the absence of phage infection (Agari et al., 2010; Cady et al., 2011; Deltcheva et al., 2011; Juranek et al., 2012; Young et al., 2012), ensuring that they are always primed to defend against a previously encountered phage.
In response to CRISPR-Cas, phages and other mobile genetic elements endure by encoding protein inhibitors of CRISPR-Cas systems, known as anti-CRISPRs (Bondy-Denomy et al., 2013; Pawluk et al., 2016b). Anti-CRISPRs are encoded in diverse viruses and mobile elements found in the Firmicutes, Proteobacteria, and Crenarchaeota phyla. They show a tremendous amount of sequence diversity with over 40 entirely distinct anti-CRISPR protein families now identified. Among these families are inhibitors of type I-C, I-D, I-E, I-F, II-A, II-C, and V-A systems. These anti-CRISPR proteins function by preventing CRISPR-Cas systems from recognizing foreign nucleic acids or by inhibiting their nuclease activity (Bondy-Denomy et al., 2015; Chowdhury et al., 2017; Dong et al., 2017; Dong et al., 2019; Guo et al., 2017; Harrington et al., 2017; Knott et al., 2019; Pawluk et al., 2017; Wang et al., 2016).
Anti-CRISPR proteins display no common features with respect to sequence, predicted structure, or genomic location of the genes encoding them. However, anti-CRISPR genes are almost invariably found upstream of a gene encoding a protein containing a helix-turn-helix (HTH) DNA-binding domain (Figure 1). Seven different families of genes encoding these HTH-containing proteins have been designated as anti-CRISPR associated (aca). Members of aca gene families have been identified in phages, prophages, plasmids, and conjugative elements in diverse bacterial species (Bondy-Denomy et al., 2013; Marino et al., 2018; Pawluk et al., 2016a; Pawluk et al., 2016b). The ubiquity of aca genes adjacent to anti-CRISPR genes has provided a key bioinformatic tool for the identification of diverse anti-CRISPR families (Marino et al., 2018; Pawluk et al., 2016a; Pawluk et al., 2016b), and implies that they play an important role in anti-CRISPR systems. Nevertheless, their function remains unknown.
Figure 1. Anti-CRISPRs are found in diverse genomic contexts.

Schematic representation of the genome context of diverse anti-CRISPR genes. Coloured arrows represent anti-CRISPR and anti-CRISPR-associated (aca) genes as well as nearby genes encoding helix-turn-helix (HTH) motif proteins. Genes shown in the same colour represent the same family. Other genes are shown in gray and predicted functions are indicated when known. Arrows representing genes are not shown to scale. G encodes phage tail protein and I/Z encodes the protease/scaffold. Int = integrase. *indicates frameshifted/incomplete. Anti-CRISPR genes are denoted as IX#, where I represents the type of system targeted, X represents the subtype of system targeted, and # represents the protein family. See also Figure S7 and Table S2.
The goal of this work was to define the role of aca genes in anti-CRISPR biology. We investigated aca gene function using Pseudomonas aeruginosa phage JBD30 as our primary model system (Figure 1). This phage was among the first set of phages shown to use an anti-CRISPR gene for survival in the presence of CRISPR-Cas (Bondy-Denomy et al., 2013). The anti-CRISPR operon of JBD30 and other closely related phages is located between operons encoding phage structural proteins. In JBD30, a single anti-CRISPR (acr) gene, acrIF1, is followed directly by an aca gene, known as aca1. Aca1 is conserved (>50% identity) among diverse anti-CRISPR encoding phages and prophages in Pseudomonas species (Pawluk et al., 2016b). Since Aca1 possesses a HTH DNA-binding motif, we reasoned that it might be involved in regulating anti-CRISPR gene transcription. To address this, we have investigated the transcript levels of the acrIF1 gene of JBD30 throughout its infection cycle. We found that anti-CRISPR transcription occurs at high level early in the phage infection process, and that Aca1 represses this transcription. Remarkably, we found that the repressor activity of Aca1 is essential for phage survival irrespective of CRISPR-Cas. We also showed that other Aca protein families act as repressors of anti-CRISPR transcription. This crucial function of Aca likely explains its ubiquity in anti-CRISPR operons.
Results
The acrIF1 gene is robustly transcribed from its own promoter at the onset of phage infection
To investigate the potential role of aca1 in regulating acr transcription, we first evaluated the dynamics of anti-CRISPR gene expression over the course of phage infection in P. aeruginosa strain PA14, which encodes a type I-F CRISPR-Cas system. A lysate of phage JBD30 was mixed with PA14 and samples were removed at successive time points after phage addition. By extracting RNA from these samples, which span the lytic cycle of JBD30 (~70 minutes, Figure S1), and quantifying the level of anti-CRISPR acrIF1 transcripts using quantitative reverse transcriptase PCR (RT-qPCR), we found that acrIF1 transcription was easily detectable within 6 minutes after infection (Figure 2A). There was more than a 100-fold increase in the level of anti-CRISPR transcripts within the first 20 minutes of infection. Remarkably, acrIF1 transcription initiated earlier and at a considerably higher level than that of the transposase gene (A), which is expected to be one of the first genes transcribed in JBD30. JBD30 is very similar to the well characterized E. coli phage Mu in its genome organization and composition, placing it in the Mu-like phage family. The transposase is expressed early in infection as it is required for the first step in the life cycle of Mu-like phages, which involves transposition of the phage genome into that of the host (Marrs and Howe, 1990). Also, by comparison with phage Mu, we expected transcription of gene G, a gene required for phage morphogenesis located directly upstream of the acrIF1 gene, to increase late in the infection process. In accord with this expectation, G transcript did not accumulate to appreciable levels until 50 minutes after phage addition. Overall, these data show that acrIF1 transcription is very high early in the infection process, presumably allowing the anti-CRISPR protein to accumulate sufficiently to inhibit the pre-formed CRISPR-Cas complexes present in the cell.
Figure 2. acrIF1 expression is driven by a promoter region that includes binding sites for Aca1 (A).

Relative transcription levels of phage genes were measured by RT-qPCR at the indicated times after infection of PA14 by phage JBD30. Transcriptional levels are shown of the anti-CRISPR gene (acrIF1), an early expressed gene (A, transposase), and a late expressed gene (G, a tail component) during one round of phage infection at a multiplicity of infection (MOI) of 5. Levels were normalized to the geometric mean of the transcript levels of the housekeeping genes: clpX and rpoD. Data are represented as mean ± SEM from three independent experiments. (B) Multiple nucleotide sequence alignment of anti-CRISPR phages from the stop codon of the Mu G homologue (G stop) to the start codon of the anti-CRISPR genes (acr start). Bioinformatically predicted promoter elements (BPROM; Solovyev and Salamov, 2011) −10 and −35 are shown. Inverted repeats are indicated by red boxes. A common sequence in both repeats is underlined. Positions sharing greater than 85% identity are coloured according to nucleotide. (C) The putative acr promoter region from phage JBD30 was cloned upstream of a promoterless lacZ expression vector (lacZ+acrIF1 upstream) and β-galactosidase activity was measured in P. aeruginosa strain PA14. The mean ± SEM of three independent assays is shown. (D) Tenfold dilutions of wild-type (JBD30), anti-CRISPR gene frameshift mutant (JBD30acrfs) and acr promoter mutant (JBD30ΔPacr) phage lysates were applied to lawns of PA14 and PA14ΔCRISPR. (E) EMSAs were performed using a dsDNA fragment with the sequence shown, which encompasses the acr promoter region. The IR1 and IR2 mutants contained the triple and quadruple base substitutions indicated under the DNA sequence. Representative non-denaturing polyacrylamide gels stained with SYBR gold are shown. Purified Aca1 was added to the DNA at concentrations of 10 nM, 50 nM, 100 nM and 250 nM. The dash sign (−) indicates no added protein. (F) The acr promoter region from JBD30 either wild-type (WT), or bearing IR1 and/or IR2 mutations was cloned upstream of a promoterless lacZ gene. β-galactosidase activity was measured in PA14 (−Aca1) or in a JBD30 lysogen (+Aca1). The mean ± SEM β-galactosidase activity relative to the wild-type promoter is shown (n ≥ 3). (G) Tenfold dilutions of phage JBD30 lysates carrying the indicated inverted repeat mutations were applied to lawns of PA14 or PA14ΔCRISPR. Representative images of three replicates are shown. See also Figures S1and S2.
The distinct transcription profile of the acrIF1 gene implied that it possessed its own promoter. A DNA sequence alignment of the region upstream of diverse acr genes from phages related to JBD30 revealed a conserved predicted promoter (Figure 2B). This region from phage JBD30 was cloned upstream of a promoterless lacZ reporter gene carried on a plasmid. The presence of the putative acrIF1 promoter increased β-galactosidase activity by approximately 15-fold when compared to the control lacking a promoter, demonstrating that this DNA sequence can direct robust transcription in P. aeruginosa (Figure 2C). To confirm that this promoter was responsible for anti-CRISPR gene expression during phage infection, we created a JBD30 mutant phage (JBD30ΔPacr) lacking this region. In a plaquing assay, the JBD30ΔPacr mutant phage replicated robustly on PA14 lacking a functional CRISPR-Cas system (PA14ΔCRISPR), but in the presence of CRISPR-Cas phage replication was equivalent to that of a JBD30 mutant bearing a frameshift mutation in acrIF1 (acrfs) (Figure 2D). These data imply that the identified promoter drives acrIF1 transcription during infection.
Aca1 acts on the acr promoter
Aca1 proteins are bioinformatically predicted to contain a HTH DNA-binding motif (Figure S2A). HTH-containing proteins are generally dimeric and bind to inverted repeat sequences (Luscombe et al., 2000). We identified two such sites with very similar sequences which we refer to as IR1 and IR2. IR1 lies upstream of the −35 region of the acrIF1 promoter and IR2 lies between the −35 and −10 regions (Figure 2B). To determine whether Aca1 could bind to the acr promoter region, purified Aca1 was mixed with a 110 bp dsDNA fragment containing the acr promoter and an electrophoretic mobility shift assay (EMSA) was performed. Incubation of the promoter-containing fragment with Aca1 resulted in a concentration-dependent shift in the mobility of the fragment, which was not observed with a non-specific DNA sequence (Figure S2B). At higher Aca1 concentrations, a second shifted band was observed, consistent with the presence of two Aca1 binding sites within this fragment. The dissociation constant (Kd) of this interaction was approximately 50 nM (Figure S2C). This Kd value is 10 to 100-fold weaker than other well-known HTH-containing proteins (Gilbert and Muller-Hill, 1967; Kamionka et al., 2004; Liu and Matthews, 1993; Nelson and Sauer, 1985). A 53 bp fragment encompassing only IR1 and IR2 of the acr promoter also bound to Aca1 and displayed two shifted bands by EMSA (Figure 2E). To determine if the two shifted bands represented Aca1 binding at both IR1 and IR2 sites, point mutations were introduced into each inverted repeat to abolish their symmetry. Fragments bearing mutations in either IR1 or IR2 still bound to Aca1, but only a single shifted band was observed, while no shift was observed with a fragment bearing mutations in both sites. These results demonstrated that Aca1 binds the acrIF1 promoter at both the IR1 and IR2 sites.
Given the binding of Aca1 to the acrIF1 promoter and the position of IR1 relative to the core promoter elements, we speculated that Aca1 might contribute to the strong transcription of the anti-CRISPR gene early in infection. To determine if Aca1 binding to the acrIF1 promoter modulates its transcriptional activity, we measured the activity of this promoter in the presence of Aca1 using the lacZ reporter assay described above. Contrary to our expectation, the presence of Aca1 led to a five-fold reduction in β-galactosidase reporter activity (Figure 2F). The repressive activity of Aca1 depended on the presence of an intact IR2 site, suggesting that this site is active in vivo. By contrast, the IR1 site was not required for repression despite being bound by Aca1 in vitro. The in vivo function of the Aca1 binding sites in the acrIF1 promoter were assessed by crossing the inverted repeat mutations into phage JBD30 through in vivo recombination (Bondy-Denomy et al., 2013). Despite the marked effect of the IR1 and IR2 mutations on Aca1 DNA binding in vitro, introduction of these mutations into the phage genome caused no significant decrease in the viability of the mutant phages on either PA14 or PA14ΔCRISPR (Figure 2G).
Aca1 repressor activity is required for phage viability
To further investigate the role of the Aca1 DNA-binding activity, we introduced amino acid substitutions within the putative HTH region of Aca1 that were expected to reduce DNA-binding (Figure S2D). Substitutions with Ala at Arg33 or Arg34 and an Arg33/Arg34 double mutant each partially reduced the DNA-binding activity of Aca1 in vitro, while substituting Arg44, which is predicted to be in the major groove recognition helix, completely abolished Aca1 DNA-binding (Figure 3A). The DNA-binding activity of these mutants was also measured using the lacZ reporter assay. Consistent with the in vitro data, the R44A mutant displayed very little repressor activity on the acrIF1 promoter (Figure 3B). The activity of the R33A/R34A double mutant was intermediate between the R44A mutant and the R33A and R34A single mutants, corroborating the in vitro changes in DNA-binding activity observed for these mutants.
Figure 3. Uncontrolled expression from the anti-CRISPR promoter is detrimental to phage viability.

(A) Representative EMSA with indicated Aca1 mutants using the 110 bp upstream region from phage JBD30 as a substrate. Purified protein was added at concentrations of 10 nM, 50 nM, 100 nM, 250 nM and 500 nM. The dash sign (−) indicates no added protein. Non-denaturing acrylamide gels stained with SYBR gold are shown. (B) The acr promoter region from phage JBD30 was cloned upstream of a promoterless lacZ gene. β-galactosidase activity was measured in a wild-type JBD30 lysogen (WT Aca1), JBD30 Aca1 mutant lysogens as indicated, and wild-type PA14 with no prophage (−). The mean ± SEM from three independent experiments relative to the wild-type Aca1 JBD30 lysogen is shown. (C) Lysates of phage JBD30 (WT or Aca1 R44A mutant) were spotted in 10-fold serial dilutions on lawns of PA14, PA14ΔCRISPR, or on PA14ΔCRISPR bearing a plasmid that expresses Aca1. These phages are targeted by the CRISPR-Cas system in the absence of anti-CRISPR activity. Representative images from three replicates are shown. (D) Lysates of wild-type JBD30, JBD30acaR44A mutant phage and suppressor JBD30acaR44A phage were spotted in 10-fold serial dilutions on bacterial lawns of PA14 or on PA14ΔCRISPR. The sequence of the suppressor phage demonstrating the loss of the −35 element from the acr promoter when compared to the sequence of the parent phage is shown below. (E) The transcript levels of the acrIF1 and aca1 genes in phages bearing acr promoter operator mutants and aca mutants were determined by RT-qPCR. Expression levels were normalized to the geometric mean of the transcript levels of two bacterial housekeeping genes: clpX and rpoD. The mean ± SEM is shown (n = 3). Assays were performed in PA14ΔCRISPR lysogens of the indicated phages. See also Figure S2 and S3.
The Aca1 DNA-binding mutants were subsequently crossed into phage JBD30. Unexpectedly, we were able to isolate phages carrying the mutations affecting Arg33 and Arg34, but not the mutation affecting Arg44. The R44A mutant phage could only be obtained by plating on cells expressing wild-type Aca1 from a plasmid, suggesting that the Aca1 DNA-binding activity is essential for phage viability. Using high titre lysates of JBD30aca1R44A produced in the presence of Aca1, we discovered that this phage was unable to replicate (titre reduced >106-fold) on PA14 or PA14ΔCRISPR (Figure 3C). By contrast, the mutant phages encoding Aca1 substitutions at the Arg33 or Arg34 positions formed plaques at levels approaching that of the wild-type phage (Figure S3A). These data demonstrate that intermediate reductions of Aca1 DNA-binding activity have little effect on phage viability, but that a complete loss of Aca1 DNA-binding activity is lethal.
Although the JBD30aca1R44A phage replicated very poorly on the PA14ΔCRISPR strain, plating high concentrations of this phage did lead to the appearance of revertant plaques at a low frequency (<1×10−6). Sequencing the anti-CRISPR regions of several of these revertants revealed that they still carried the aca1R44A mutation. Most also displayed a 25 bp deletion encompassing the −35 region of the acrIF1 promoter (Figure 3D). These suppressors were able to plate to the same level as wild-type JBD30 on the PA14ΔCRISPR strain, but showed a marked reduction in titre on PA14. This is what we would expect to see if the acr promoter were impaired as demonstrated in Figure 2D. This result implies that the inviability of the aca1R44A mutant phage likely arises from the high transcription level at the acr promoter. Hence, the deletion of a critical portion of this promoter is able to restore viability.
To verify the transcriptional effects of mutations in the JBD30 acr promoter and aca1 gene, we performed RT-qPCR on strains lysogenized with mutant phages (i.e. the phage genomes were integrated into the PA14ΔCRISPR genome to form a prophage). In the lysogenic state, acr expression must persist to prevent the host CRISPR-Cas system from targeting the prophage, which would be lethal. Performing assays in the lysogenic state also allowed us to assess transcription levels at a steady state as opposed to the dynamic situation existing during phage infection. Both the acrIF1 and aca1 genes were transcribed from the JBD30 prophage (Figure 3E). The transcription of both genes was more than 20-fold lower in the phage mutant lacking the acr promoter, confirming the key role of this promoter in expression of both of these genes. By contrast, the JBD30aca1R44A mutant displayed vastly increased levels of acrIF1 and aca1 transcription (100-fold and 20-fold increases, respectively). Prophages expressing Aca1 mutants that bound DNA at somewhat reduced levels in vitro (i.e. substitutions at Arg33 and Arg34, Figure 3A) also displayed increased transcription of the acrIF1 and aca1 genes, but not nearly to the same degree as the JBD30aca1R44A mutant. Mutations in IR2 that that caused loss of repression in the lacZ reporter assay (Figure 2F) also resulted in increased acrIF1 and aca transcription. However, this increase was similar to that of the JBD30acaR33A/R34A mutant, which was 15-fold lower than the JBD30acaR44A mutant. The reduced transcription level of the IR2 and IR1+IR2 operator mutants as compared to the transcription level of the aca1R44A mutant may be due to the base substitutions in the operators causing a reduction in the promoter strength. We investigated this issue by combining the IR1+IR2 operator mutant with aca1R44A within the phage. This mutant phage was fully viable on the PA14ΔCRISPR strain (Figure S3B), consistent with the mutations in the Aca1 operator sites also reducing promoter strength. This result also explains why the IR1+IR2 operator mutant phage is viable even though Aca1 cannot repress this promoter.
Taken together, our data imply that the uniquely high transcription level from the acr promoter resulting from the aca1R44A mutant causes the inviability of the JBD30acaR44A phage while phages bearing other aca1 or acr promoter mutations retained their replicative ability. It is notable that examination of plaque sizes resulting from infection by wild-type and JBD30 phages bearing other aca1 mutations showed that the Arg33 and Arg34 substitutions measurably decreased phage replication (Figure S3C). Thus, the more modest increases in acr promoter activity seen for these mutants still influenced phage viability.
Although our results using the aca1R44A mutant suggest that the key role of Aca1 is to repress acr transcription, it is possible that the lethal effect of this mutant could have been the result of a toxic gain-of-function. To rule out this scenario, we plated phages on cells expressing the Aca1 R44A mutant from the acrIF1 promoter on a high copy number plasmid. This high expression of the Aca1 R44A mutant prior to phage infection had no effect on phage viability (Figure S3D). Plasmid-based overexpression of AcrIF1 in the same manner also had no effect on phage viability, implying that the non-viability of JBD30acaR44A is not due to lethal overaccumulation of Aca1 R44A or AcrIF1.
acr promoter activity is strong during early infection independent of Aca1
To directly address the role of Aca1 early in the phage infection process, we infected cells with wild-type JBD30 or the JBD30aca1R44A mutant, and measured transcript accumulation using RT-qPCR. Very early in infection, acrIF1 transcripts accumulated to high levels in both wild-type and mutant phage (Figure 4A). At the 30 and 40 minute time points, acrIF1 transcripts accumulated to levels 85 to 140- fold higher, respectively, in the JBD30aca1R44A mutant as compared to wild-type, highlighting the repressor activity of Aca1. The transcription of the transposase gene varied relatively little between the wild-type and mutant phage (Figure 4B). It should be noted that transcription of both the acrIF1 and transposase genes was observed earlier in these experiments than in those shown in Figure 2A due to the use of a higher multiplicity of infection to improve the limit of detection in this assay. These results clearly demonstrate that Aca1 is not required for the early activation of acr transcription. Consequently, the importance of Aca1 must be derived from its ability to repress the acr promoter.
Figure 4. Loss of Aca1 repressor activity affects the transcription of the gene immediately downstream of the anti-CRISPR locus.

The transcription of the indicated phage genes from wild-type JBD30 and JBD30aca1R44A during one-round of infection was determined by RT-qPCR. The genes assayed were: acrIFI (A); transposase, an early expressed gene (B); I/Z, the scaffold gene, which lies immediately downstream of aca1 (C); and G, a late gene lying directly upstream of the acr gene (D). Expression levels were normalized to the geometric mean of the transcript levels of two bacterial housekeeping genes: clpX and rpoD. The mean ± SEM of three independent experiments is shown. Assays were performed in PA14ΔCRISPR at a MOI of 8.
Loss of Aca1 repressor activity alters the transcription of downstream genes
In light of the results above, we postulated that the loss of viability observed for the JBD30aca1R44A mutant was brought about by uncontrolled transcription from the strong acr promoter. With the expectation that this inappropriate acr transcription might perturb the transcription of downstream genes, we measured the transcript levels of the phage protease/scaffold (I/Z) gene, which lies immediately downstream of the anti-CRISPR locus. Surprisingly, I/Z gene transcript levels in the JBD30acaR44A mutant phage were dramatically decreased relative to wild-type phage, reaching nearly a 100-fold difference at the later timepoints (Figure 4C). By contrast, the G gene, which lies immediately upstream of the anti-CRISPR locus, displayed less than 10-fold differences in transcript levels between the wild-type and mutant phages (Figure 4D).
Based on genomic comparison with phage Mu, the I/Z gene is situated at the beginning of an operon that contains genes required for capsid morphogenesis (Hertveldt and Lavigne, 2008). The observed decrease in I/Z transcript level likely extends to other essential genes within this operon. Thus, the JBD30acaR44A mutant phage would lack sufficient levels of these morphogenetic proteins required for particle formation. This explains the observed loss of phage viability regardless of the CRISPR-Cas status of the host. Defects in virion morphogenesis could also lead to the small plaque phenotype observed in the partially incapacitated Aca1 mutants (Figure S3C). In further experiments, we determined that the JBD30aca1R44A phage forms lysogens with the same frequency as the wild-type phage (Figure S4). Since lysogen formation does not require particle formation, this finding is consistent with the hypothesis that Aca1 debilitation causes a defect in phage morphogenesis in the case of JBD30.
To determine whether the readthrough transcription from the strong acr promoter contributes to the loss of viability of the JBD30aca1R44A phage, we inserted a single copy of the rrnB T1 transcriptional terminator (Orosz et al., 1991) immediately downstream of the aca1 stop codon. Insertion of this terminator resulted in a greater than 1000-fold increase in the plaquing efficiency of the JBD30aca1R44A mutant phage (Figure S5), implying that readthrough transcription is indeed contributing to the loss of phage viability in the absence of Aca1 repressor activity. Addition of this terminator did not reduce viability of the wild-type phage. The incomplete recovery of the viability of the JBD30aca1R44A mutant phage suggests that other factors affecting viability may also be at play, or that the terminator used was not strong enough to stop readthrough transcription from the acr promoter.
Aca1 can act as an “anti-anti-CRISPR”
Since Aca1 is a repressor of the acr promoter, we postulated that excessive Aca1 expression might inhibit the replication of phages requiring anti-CRISPR activity for viability in the presence of CRISPR-Cas. To test this hypothesis, we plated phage JBD30 on PA14 cells in which Aca1 was expressed from a plasmid. We found that phage replication was inhibited by more than 100-fold in the presence of plasmid-expressed Aca1 as compared to cells carrying an empty vector (Figure 5A). This loss of phage replication was CRISPR-Cas dependent, as plasmid-expressed Aca1 had no effect on phage replication in the PA14ΔCRISPR strain, indicating that the impairment of phage replication results from a decrease in anti-CRISPR expression. Importantly, phages bearing mutations in IR2, which is the binding site required for Aca1-mediated repression of the acr promoter, were able to replicate in the presence of excess Aca1. On the other hand, a phage mutated in the IR1 site, which binds Aca1 but does not mediate repression, replicated more poorly than wild-type in the presence of Aca1. This confirms that binding of IR2 by Aca1 is required for repression of acr transcription in vivo, and indicates that likely IR1 titrates Aca1 away from IR2 and thereby lessens the repressive effect of Aca1. The DNA-binding activity of Aca1 is necessary to reduce JBD30 replication, as the overexpression of the Aca1 R44A mutant has no impact on JBD30 replication in the presence of CRISPR-Cas (Figure 5B).
Figure 5. Overexpression of Aca1 inhibits phage-borne anti-CRISPRs.

(A) Tenfold dilutions of lysates of JBD30 carrying the indicated mutations in the Aca1 binding sites (IR1 and IR2) were applied to lawns of PA14 or PA14ΔCRISPR expressing wild-type Aca1 from a plasmid. A representative image of at least three replicates is shown. (B) Tenfold dilutions of phage JBD30 lysate carrying the indicated inverted repeat mutation were applied to lawns PA14 or PA14ΔCRISPR expressing the R44A Aca1 mutant from a plasmid. A representative image from three biological replicates is shown.
Overall, the inhibitory effect of Aca1 on acr-dependent phage replication further bolsters our conclusion that Aca1 is a repressor of the acr promoter. This observation also raises the intriguing possibility that expression of Aca1 could be co-opted by bacteria as an “anti-anti-CRISPR” mechanism for protection against phages or other mobile genetic elements carrying anti-CRISPR genes.
Members of other Aca families are also repressors of anti-CRISPR promoters
Genes encoding active anti-CRISPR proteins have been found in association with genes encoding HTH motif-containing proteins that are completely distinct in sequence from Aca1. For example, aca2 has been found in association with five different families of anti-CRISPR genes in diverse species of Proteobacteria (Pawluk et al., 2016a; Pawluk et al., 2016b). Genes encoding homologues of Aca3, another distinctive HTH-containing protein, have been identified in association with three different type II-C anti-CRISPR genes (Pawluk et al., 2016a). To investigate the generality of Aca function, we determined whether representative members of Aca2 and Aca3 (Figures S6A and S6B) families also function as repressors of anti-CRISPR transcription.
By aligning the intergenic regions found immediately upstream of anti-CRISPR genes associated with aca2, we detected a conserved inverted repeat sequence that could act as a binding site for Aca2 proteins (Figure S6C). The same alignment approach also revealed an inverted repeat sequence that could act as a binding site for Aca3 (Figure S6D). To investigate the functions of Aca2 and Aca3, the acr/aca regions from Pectobacterium phage ZF40 and from N. meningitidis strain 2842STDY5881035 were investigated as representatives of the Aca2 and Aca3 families, respectively. The putati6ve promoter regions of the anti-CRISPR genes in these two operons (Figures S6C and S6D) were cloned upstream of a promoterless lacZ reporter gene carried on a plasmid. When assayed in E. coli, both regions mediated robust transcription of the reporter as detected by measuring β-galactosidase activity in cell extracts (Figure 6). Co-expression of Aca2ZF40 and Aca3Nme with their putative cognate promoters resulted in 100-fold and 20-fold reductions in β-galactosidase activity, respectively. Co-expression of aca2 with the aca3 operon promoter construct or vice versa did not exhibit any repression, demonstrating that the repressor activities of Aca2 and Aca3 are specific to their associated promoters. Overall these data show that, similar to Aca1, both Aca2 and Aca3 are repressors of anti-CRISPR transcription.
Figure 6. Members of other Aca families are repressors of putative anti-CRISPR promoters.

Promoter regions of acrIF1 from Pseudomonas phage JBD30, and putative promoter regions of acrIF8 from Pectobacterium phage ZF40, and acrIIC3 from a N. meningitidis prophage were cloned upstream of a promoterless lacZ gene. β-galactosidase activity was measured in the absence and presence of the indicated plasmid expressed Aca proteins in E. coli. The cognate Aca for each promoter is underlined. The mean ± SEM from three biological replicates is shown. See also Figure S6.
Anti-CRISPR protein is not packaged into phage particles
While we have demonstrated that anti-CRISPR are robustly expressed at the onset of phage infection to protect phage DNA from CRISPR-Cas mediated degradation, additional protection would be provided if anti-CRISPR proteins were injected along with the DNA into the cell. Packaging of phage-encoded inhibitors of bacterial defence systems has been documented. For example, E. coli phages T4 and P1 both incorporate protein inhibitors of restriction endonucleases into their capsids and deliver them along with their genomes to protect against host defences (Bair et al., 2007; Iida et al., 1987; Piya et al., 2017). Since these anti-restriction proteins are present at greater than 40 copies per phage particle, we expected to detect anti-CRISPR protein in the phage particles if they were packaged. To assay for the presence of anti-CRISPR protein in particles of the AcrIF1-encoding phage JBD30, we performed mass spectrometry on purified phage particles. While we were able to detect AcrIF1 in partially purified phage samples, this protein was not detected after further purification by buffer exchange, implying that AcrIF1is not incorporated into phage particles (Figures 7A and 7B). By contrast, all expected phage viral particle proteins were detected (Table S1).
Figure 7. Anti-CRISPR AcrIF1 is not incorporated into the JBD30 virion.

(A) Analysis of absolute protein abundance of phage proteins before and after buffer exchange. Phage particle proteins are highlighted in blue. AcrIFI is shown in red. (B) Estimated concentration of AcrIF1 in phage particles before and after additional purification by buffer exchange. (C) Schematic representation of the genomic context of AcrIF1 from phage JBD30. The anti-CRISPR region (outlined red) was inserted into a transposon, which was used to randomly introduce the anti-CRISPR region into phage JBD44. F and G encode phage head and tail morphogenesis proteins, respectively. I/Z encodes the protease/scaffold and T encodes the major head protein. (D) Tenfold dilutions of lysates of phage JBD44 and phage JBD44 carrying the JBD30 anti-CRISPR locus (JDB44::acr) were applied on lawns of PA14 and PA14 CRISPR expressing a crRNA targeting phage JBD44 (JBD44 crRNA) from a plasmid. A representative image of at least three biological replicates is shown. See also Table S1.
For anti-CRISPR proteins to be packaged into phage particles, recognition between a virion protein and the anti-CRISPR would likely be required. This requirement has been described in other bacteriophages. For example, protein packaged into T4 particles are incorporated by a specific interaction with a core capsid scaffolding protein (Hendrix and Duda, 1998; Hong and Black, 1993), and restriction enzyme inhibitors of phage P1 are incorporated through a specific assembly pathway (Piya et al., 2017). Thus, an anti-CRISPR from one phage would not be expected to function within the context of a completely different phage. To investigate this issue, we incorporated the anti-CRISPR region of phage JBD30 (Figure 7C) into random locations in the genome of the unrelated D3-like P. aeruginosa phage JBD44 using transposon mutagenesis. Even though the virion proteins of JBD44 are completely unrelated to those of JBD30, the acrIF1 region inserted into JBD44 was still able to confer resistance against the type I-F CRISPR-Cas system of PA14. The plaque-forming ability of wild-type JBD44 was robustly inhibited when targeted by the PA14 CRISPR-Cas system, while JBD44 phages carrying the anti-CRISPR region (JBD44::acr) were protected from CRISPR-Cas mediated inhibition (Figure 7D). Additionally, the level of plaquing by JBD44::acr phages was the same regardless of the presence or absence of a CRISPR-Cas system, suggesting that these phages exhibit full anti-CRISPR activity. These results demonstrate that AcrIF1 retains full functionality in the genomic context of an entirely different phage, implying that interaction between the anti-CRISPR and other phage components (including the virion proteins) is not required. This experiment also demonstrates the transferability of acr operons, and their ability to function in completely different genomic contexts.
Discussion
Given the increasing numbers of anti-CRISPR families are being discovered, we expect that they exert a strong and widespread influence on CRISPR-Cas function, and have been important drivers of CRISPR-Cas evolution. The ubiquity of the association between Aca proteins and anti-CRISPRs implies that Aca proteins play a crucial role in anti-CRISPR systems; thus, an investigation into their function was timely. Here we have shown that Aca1 is a repressor of anti-CRISPR transcription and is critical for phage particle production. Since we found no evidence that AcrIF1 is incorporated into phage particles and injected into host cells along with phage DNA, our results imply that phage survival in the face of pre-formed CRISPR-Cas complexes in the host cell is dependent upon rapid high-level transcription of the anti-CRISPR gene from a powerful promoter. However, the placement of such strong constitutive promoters within the context of a gene-dense, intricately regulated phage genome is likely to result in the dysregulation of critical genes and a decrease in fitness. The inclusion of repressors within anti-CRISPR operons to attenuate transcription once sufficient anti-CRISPR protein has accumulated solves this problem. We surmise that the widespread presence of aca genes within anti-CRISPR operons has been vital for the spread of these operons by horizontal gene transfer, allowing them to incorporate at diverse positions within phage genomes without a resulting decrease in phage viability. In addition, our work shows that Aca proteins have the potential to broadly inhibit anti-CRISPR expression, effectively acting as anti-anti-CRISPRs, which could have applications in CRISPR-based antibacterial technologies (Greene, 2018; Pursey et al., 2018).
One question with respect to anti-CRISPR operon function is how rapid high-level expression of anti-CRISPR proteins can be achieved when a repressor of the operon is produced simultaneously. Since Aca proteins are not present when phage DNA is first injected, initial transcription of anti-CRISPR operons is not impeded. In most anti-CRISPR operons the acr genes precede the aca gene and are thus translated first, allowing anti-CRISPR proteins to accumulate earlier. In addition, the aca ribosome binding sites in the aca1, aca2, and aca3 controlled operons tested in this work are predicted to be considerably weaker than those of associated acr genes, which would result in slower accumulation of the Aca proteins (Espah Borujeni et al., 2014; Salis et al., 2009; Seo et al., 2013). The presence of two binding sites for Aca1 in the acr promoter, only one of which mediates repression, may also serve to delay the repressive activity of Aca1. Evidence for this is seen in Figure 5A, where the replication of a phage lacking IR1 is inhibited to a greater extent than wild-type by plasmid-based expression of Aca1, presumably because Aca1 is normally titrated away from the IR2 site by binding to IR1. Other mechanisms to fine-tune the balance of anti-CRISPR and Aca protein levels, such as differential protein and/or mRNA stability, may also play a role in some cases. Another notable feature of Aca1 is its weaker DNA-binding affinity as compared to other well-known HTH repressor, such as the lambda, lac, and tet repressors, presumably making Aca proteins weaker repressors. This is likely a functional requirement of Aca proteins as some level of anti-CRISPR transcription is necessary for the survival of prophages, which would be targets of host CRISPR-Cas systems in the absence of anti-CRISPR activity.
In the case of phage JBD30, the absence of Aca1 function led to a dramatic increase in acr operon transcription and abrogation of phage particle production. This loss of viability appeared, at least in part, to result from a large decrease in the transcription of essential genes downstream of the acr operon (Figure 4C). We believe that this decrease is caused by readthrough transcription from the acr promoter, a conclusion supported by the rescue of phage viability upon insertion of a transcriptional terminator downstream of the aca1R44A gene. In an interesting parallel, strong transcriptional readthrough from early promoters in E. coli phage T7 is lethal if it is not attenuated by a phage protein that inhibits E. coli RNA polymerase (Savalia et al., 2010). Although it seems counterintuitive that strong upstream transcription would attenuate downstream transcription, this type of transcriptional interference has been shown to result from RNA polymerase pausing over downstream promoters, dislodgement of transcription factors, or DNA supercoiling effects from high rates of transcription (Hao et al., 2017; Liu and Wang, 1987; Palmer et al., 2009; Palmer et al., 2011; Scheirer and Higgins, 2001).
Another question is why would anti-CRISPR operons include a repressor to decrease transcription rather than a strong downstream transcriptional terminator to prevent readthrough transcription. Depending on where an acr operon has been inserted, transcription may need to proceed through the anti-CRISPR region for essential genes to be transcribed. Thus, inserting a terminator could be detrimental. Additionally, maintaining a very high level of transcription would unnecessarily sap host resources. Consistent with this idea, highly expressed genes have generally been found to have a lower likelihood of horizontal transfer because of their greater potential to disrupt recipient physiology (Park and Zhang, 2012; Sorek et al., 2007). Our observation that terminator insertion did not completely rescue the aca1R44A mutant also suggests that unrestrained transcription from the acr promoter may reduce viability through other mechanisms. In general, we expect that strong anti-CRISPR-associated promoters would reduce evolutionary fitness when placed in most locations within mobile DNA elements if these promoters were unregulated. Reductions in fitness could be due to various mechanisms of gene misregulation depending upon the specific insertional position of the acr operon. The great diversity of acr operon insertion points in mobile elements is emphasized by the distinct locations observed for nine different acr operons controlled by Aca2 (Figure S7A). It can be seen that the acr promoter/aca operator regions and Aca2 coding regions of these operons are remarkably conserved at the nucleotide level, while the Acr encoding regions in between are highly diverse (Figure S7B), highlighting the importance of Aca regulation of the acr promoter. It was recently shown that anti-CRISPR-expressing phages like JBD30 cooperate to inhibit CRISPR-Cas systems. Initial phage infections may not result in successful phage replication, but anti-CRISPR protein accumulating from infections aborted by CRISPR-Cas activity leads to “immunosuppression” that aids in subsequent phage infections (Borges et al., 2018; Landsberger et al., 2018). Through demonstrating that anti-CRISPR genes are expressed quickly after infection, we provide an explanation for how anti-CRISPR protein accumulates even when phage genomes are ultimately destroyed by the CRISPR-Cas system. In the anti-CRISPR-expressing archaeal virus, SIRV2, the acrID1 gene was also transcribed at high levels early in infection, supporting the generalizability of this mechanism of anti-CRISPR action (Quax et al., 2013).
STAR Methods
Contact for Reagent and Resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alan R. Davidson (alan.davidson@utoronto.ca).
Experimental Model and Subject Details
Microbes
Pseudomonas aeruginosa strains (UCBPP-PA14 and UCBPP-PA14 CRISPR mutant derivatives) and Escherichia coli strains (DH5α, SM10λpir, BL21(DE3)) were cultured at 37°C in lysogeny broth (LB) or on LB agar supplemented with antibiotics at the following concentrations when appropriate: ampicillin, 100 μg ML−1 for E. coli; carbenicillin, 300 μg mL−1 for P. aeruginosa; gentamicin, 30 μg mL−1 for E. coli and 50 μg mL−1 for P. aeruginosa.
Phages
Pseudomonas aeruginosa phages JBD44, JBD30 and JBD30 derivatives, DMS3 and DMS3 derivatives were propagated on PA14ΔCRISPR and stored in SM buffer (100 mM NaCl, 8 mM Mg2SO4, 50 mM Tris-HCl pH 7.5, 0.01% w/v gelatin) over chloroform at 4°C.
Method Details
Phage plaque and spotting assays
For spotting assays, 150 μL of overnight culture was added to 4 mL of molten top agar (0.7%) supplemented with 10 mM MgSO4 and poured over prewarmed LB agar plates containing 10 mM MgSO4 and antibiotic as needed. After solidification of the top agar lawn, 10-fold serial dilutions of phage lysate were spotted on the surface. The plates were incubated upright overnight at 30°C.
For plaque assays, 150 μL of overnight culture was mixed with an appropriate amount of phage and incubated at 37°C for 10 minutes. The bacteria/phage mixture was added to 4 mL of molten top agar (0.7%) supplemented with 10 mM MgSO4 and poured over prewarmed LB agar plates containing 10 mM MgSO4 and antibiotic as needed. The plates were incubated upright overnight at 30°C. Plaques were counted and expressed as the number of plaque forming units (PFU) mL−1. Plaque sizes were analyzed using ImageJ (Schneider et al., 2012). Images of plaque assays were converted to 8-bit (greyscale). The image threshold was then adjusted to isolate plaques from the image background. The area of each plaque was measured in pixels squared. Image sizes were calibrated using the diameter of the petri dish in the image.
Phage infection time course
Overnight cultures of PA14 or PA14ΔCRISPR were subcultured 1:100 into LB and grown with shaking at 37°C to an OD600 of 0.4. After removing 1 mL of culture for an uninfected control, phage JBD30 was added at a multiplicity of infection (MOI) of 5 or 8. Samples were removed after 0, 2, 4, 6, 8, 10, 20, 30, 40, 50, 60, and 70 minutes. Cells were pelleted and flash frozen. One round of infection was stopped at 70 minutes post phage addition. To help synchronize the infection, cells were pelleted 10 minutes post phage addition and resuspended in fresh pre-warmed LB.
Lysogens were subcultured 1:100 from overnight cultures and grown for 5 hours prior to RNA extraction.
RNA extraction and RT-qPCR
Cell pellets were resuspended in 800 μL LB and mixed with 100 μL lysis buffer (40 mM sodium acetate, 1% SDS, 16 mM EDTA) and 700 μL acid phenol:chloroform pre-heated at 65°C. The mixture was incubated at 65°C for 5 minutes with regular vortexing and centrifuged at 12,000 × g for 10 minutes at 4°C. The aqueous layer was collected, extracted with chloroform, and precipitated with ethanol. Total RNA was resuspended in water and subsequently treated with DNase (TURBO DNA-free kit, Ambion) according to the manufacturer’s instructions. cDNA was synthesized using SuperScript IV VILO master mix (Invitrogen) and quantified using PowerUp SYBR green master mix (Applied Biosystems) with primers listed in Table S. For the purpose of quantification, standards were generated by PCR. Data were analyzed using BioRad CFX manager 3.1 software.
aca gene and promoter region cloning
aca1 and its associated promoter region were PCR amplified from lysates of phage JBD30 using the primers listed in Table S4. aca1 was cloned as a NcoI/HindIII restriction fragment into pHERD30T (for anti-CRISPR activity assays in P. aeruginosa) or into BseR1/HindIII cut p15TV-L (for protein expression and purification in E. coli with an N-terminal His6 tag). The promoter region was cloned as a NcoI/HindIII restriction fragment into the promoterless β-galactosidase reporter shuttle vector pQF50 (Farinha and Kropinski, 1990).
The anti-CRISPR locus from Pectobacterium phage ZF40 (NC_019522.1: 19220–19999) and the anti-CRISPR upstream region and Aca3 coding sequence from Neisseria meningitidis strain 2842STDY5881035 (NZ_FERW01000005.1: 56624–56978; NZ_FERW01000005.1: 55654–55893) were synthesized as gBlocks (Integrated DNA Technologies). aca2 and aca3 were PCR amplified from their respective gBlocks using primers list in Table S4. Each fragment was gel purified and cloned into pCM-Str using isothermal assembly (Gibson et al., 2009). The anti-CRISPR upstream regions from ZF40 and N. meningitidis were amplified by PCR and cloned as a NcoI/HindIII restriction fragment into pQF50. All plasmids were verified by sequencing.
β-galactosidase reporter assays
β-galactosidase reporter plasmids were transformed into DH5α and PA14. Overnight cultures of transformed cells were subcultured 1:100 and grown for ~3 hours with shaking (OD600 = 0.4–0.7). β-galactosidase activity was then quantified using a method derived from Zhang and Bremer, 1995. Briefly, 20 μL of culture was mixed with 80 μL of permeabilization solution (0.8 mg mL−1 CTAB, 0.4 mg mL−1 sodium deoxycholate, 100 mM Na2HPO4, 20 mM KCl, 2 mM MgSO4, 5.4 μL mL−1 β- mercaptoethanol) and incubated at 30°C for 30 minutes. 600 μl of substrate solution (60 mM Na2HPO4, 40 mM NaH2PO4, 1 mg mL−1 o-nitrophenyl-β-galactosidase) was added and the reaction was allowed to proceed at 30°C for 30 minutes to 1.5 hours. The reaction was stopped with the addition of 700 μL of 1 M Na2CO3, A420 and A550 were measured, and Miller Units were calculated.
Aca1 amino acid substitutions
Key residues of the Aca1 HTH domain were identified using HHPRED and modeled onto the helix-turn-helix domain of PlcR (PDB: 3U3W) using PyMol to generate a reference homology model of Aca1. Alanine substitutions at key Aca1 residues were introduced by site-directed mutagenesis with Phusion polymerase (Thermo Scientific) in either pHERD30T (for P. aeruginosa activity assays) or p15TV-L (for protein expression and purification in E. coli).
Purification of AcrIF1 and Aca1 proteins
Overnight cultures of E. coli BL21(DE3) carrying the appropriate AcrIF1 or Aca1 expression plasmid were subcultured 1:100 and grown with shaking at 37°C to an OD600 of 0.5. Protein expression was induced with 1 mM IPTG for 4 hours at 37°C. Cells were lysed by sonication in binding buffer (20 mM Tris-HCl pH 7.5, 250 mM NaCl, 5 mM imidazole). Clarified lysates were batch bound to Ni-NTA agarose resin (Qiagen) at 4°C for 1 hour, passed through a column at room temperature, and washed extensively with binding buffer containing 30 mM imidazole. Bound protein was eluted with binding buffer containing 250 mM imidazole and dialyzed overnight at 4°C in buffer containing 10 mM Tris-HCl pH 7.5 and 150 mM NaCl. All Aca1 mutant purified at levels similar to wild-type. Proteins were purified to greater than 95% homogeneity as assessed by Coomassie-stained SDS-PAGE.
Electrophoretic mobility shift assay
Varying concentrations of purified Aca1 or Aca1 mutants were mixed with 20 ng of target DNA (gel purified PCR product or annealed oligo) in binding buffer (10 mM HEPES pH 7.5, 1 mM MgCl2, 20 mM KCl, 1 mM TCEP, 6% v/v glycerol; final reaction volume 20 μL) and incubated on ice for 20 minutes. The DNA-protein complexes were separated by gel electrophoresis at 100 V on a 6% native 0.5X TBE polyacrylamide gel. Gels were stained at room temperature with Sybr gold (Invitrogen) and visualized according to the supplier’s instructions. Bands were quantified using Image Lab 6.0 software (BioRad). The percent DNA bound was plotted as a function of Aca1 concentration in Prism 7.0 (GraphPad).
Annealed oligos were generated by mixing complementary oligonucleotides in a 1:1 molar ratio in annealing buffer (10 mM Tris, pH 7.5, 50 mM NaCl, 1 mM EDTA), heating at 95°C for 5 minutes, and cooling slowly to room temperature.
Operator and Aca1 mutant phage construction
Point mutations were introduced into each inverted repeat of the acr promoter on a recombination cassette (JBD30 genes 34 to 38; Bondy-Denomy et al., 2013) by site-directed mutagenesis using primers listed in Table S4. Alanine substitutes of key Aca1 residues were introduced into the wild-type JBD30 recombination cassette by site-directed mutagenesis. Mutant phages were then generated using in vivo recombination as previously described by Bondy-Denomy et al., 2013. All mutations were verified by sequencing.
Anti-CRISPR promoter deletion in phage JBD30
A recombination cassette consisting of genes 34 to 38 of phage JBD30 (anti-CRISPR locus with large flanking regions) in plasmid pHERD20T was previously generated (Bondy-Denomy et al., 2013). This plasmid was linearized by PCR using primers that excluded the acr promoter, and then re-circularized using In-fusion HD technology (Clontech) to generate a recombination cassette with an acr promoter deletion. Using this cassette, mutant phages were generated as previously described (Bondy-Denomy et al., 2013).
Terminator insertion into phage JBD30
The rrnB T1 terminator from plasmid pMQ70 (GenBank: DQ642035.1; Shanks et al., 2006) was PCR amplified using primers listed in Table S4 and inserted into the JBD30 gene 34 to 38 recombination cassette in pHERD20T by isothermal assembly (Gibson et al., 2009). Terminator insertion phages were generated by in vivo recombination as previously described (Bondy-Denomy et al., 2013)
Lysogen construction
P. aeruginosa lysogens were generated by either streaking out cells to single colonies from the center of a phage-induced zone of clearing or by plating cells infected with phage and isolating single colonies. The presence of a prophage was confirmed by resistance to superinfection from the phage used to generate the lysogen.
Bioinformatics
Protein sequence similarity searches were performed with PSI-BLAST (Altschul et al., 1997). Protein sequence alignments were performed with MAFFT (Katoh et al., 2002), and nucleotide sequence alignments were performed with ClustalO (Sievers et al., 2011). HHPred was used to predict the location of HTH motifs (Soding et al., 2005).
Mass spectrometry of the JBD30 virion
Phage particles were purified from lysates by two sequential rounds of cesium chloride density gradient ultracentrifugation (Sambrook and Russell, 2006). Following ultracentrifugation, further purification was achieved by buffer exchange into gelatin-free SM using a 100 kDa MWCO centrifugal filter unit (MilliporeSigma). For mass spectrometry experiments, Ni-NTA affinity purified AcrIF1 was further purified by size exclusion chromatography as previously described (Bondy-Denomy et al., 2015).
For mass spectrometry, phage particles were sonicated in SM buffer without gelatin on ice. All samples (phage particles or purified AcrIF1 protein) were reduced in 10 mM dithiothreitol for 30 minutes at room temperature in the dark, then alkylated with 10 mM of iodoacetamide for 30 minutes at room temperature in the dark. The total protein concentration of the phage particles was determined by Bradford assay. Samples were digested with trypsin (Promega) at a 1:100 enzyme:substrate ratio at 37 °C overnight. Following digestion, samples were acidified with addition of 10% trifluoroacetic acid and desalted on MiniSpin C18 tips (The Nest Group). The eluates were then dried by SpeedVac, and resuspended in 4% formic acid, 2% acetonitrile in water for MS analysis. For AcrIF1, a standard curve with protein concentrations of 0.05, 0.1, 0.2, 0.5, 0.75, 1, 2, and 5 fmol μL−1 was prepared. Samples were then analyzed by liquid chromatography tandem-mass spectrometry on a Q-Exactive Plus (Thermo Fisher) mass spectrometer equipped with an EASY-nLC 1200 system (Thermo Fisher). Peptides were loaded on a 75 μm ID column packed with 25 cm of Reprosil C18 1.9 μm, 120 Å particles (Dr. Maisch GmbH). The compositions of mobile phase A and B were 0.1% formic acid in water and 0.1% formic acid in 80% acetonitrile, respectively. Digested peptide mixtures were separating from 1 to 56 minutes with 4% to 22% B over a 75 minute gradient at a flow rate of 300 nL minute−1.
Purified phage particle peptides were analyzed via a discovery based acquisition method (data-dependent acquisition, DDA) to estimate their absolute abundance using the iBAQ calculation (Schwanhausser et al., 2011) in the MaxQuant program (Cox et al., 2014; Cox and Mann, 2008). DDA spectra were searched against AcrlF1 sequence and JBD30 protein databases.
The phage particles as well as purified AcrIF1 were also analyzed via a parallel reaction monitoring (PRM) targeted proteomic approach of only the AcrIF1 protein. Quantitation of AcrlF1 was based on the fragmentation of the precursor peptide VENVVNGK ion 429.7376++ m/z to the product ions [y3] 318.1772+, [y4] 417.2456+, and [y7] 759.3995+ m/z found to be the most analytically robust. All three transitions ion peak area intensities were integrated and summed for quantitation. All PRM data was analyzed with Skyline (MacLean et al., 2010).
PRM targets for AcrIF1
| Analyte | Isolation m/z | Transition m/z |
|---|---|---|
| VENVVNGK | 429.737617 | 429.737617++ to [y7] 759.399543+, 429.737617++ to [y4] 417.245609+, 429.737617++ to [y3] 318.177195+ |
| IENAMNEISR | 588.787512 | 588.787512++ to [y8] 934.441091+, 588.787512++ to [y7] 820.398163+, 588.787512++ to [y6] 749.361049+, 588.787512++ to [y5] 618.320565+ |
anti-CRISPR locus introduction into JBD44
The anti-CRISPR locus of phage JBD30 was PCR amplified and cloned as a SalI restriction fragment into the transposon of pBTK30 (Goodman et al., 2004). This construct was transformed into E. coli SM10λpir. Conjugation was then used to move the transposon into a JBD44 lysogen of PA14. Following conjugation, lysogens were grown to log phase (OD600 = 0.5) and prophages were induced with mitomycin C (3 μg mL−1). Lysates were plated on lawns of PA14 expressing a crRNA targeting phage JBD44 from pHERD30T to isolate phages carrying and expressing the anti-CRISPR locus.
Aca3 misannotation
A nucleotide alignment of several anti-CRISPR loci from Neisseria meningitidis revealed that many aca3 homologs had one to two in-frame start codons (ATG) upstream of their annotated start that would result in a N-terminal extension of 8 to 10 amino acid residues. aca3 was cloned with and without this N-terminal extension. Aca3 repressor activity was best with the inclusion of the N-terminal extension (sequence shown below with new residues in bold). Thus, this version was used in all experiments presented here. All other Aca protein sequences are as annotated.
Aca3: MKMRRIWRAGMIDNPELGYTPANLKAIRQKYGLTQKQVADITGATLSTAQKWE AAMSLKTHSDMPHTRWL LLLEYVRNL
Quantification and statistical analysis
All experiments were performed with at least three biological replicates (n ≥ 3). Statistical parameters are reported in the Figure Legends.
Data and code availability
This study did not generate/analyze any datasets/codes.
Supplementary Material
Supplementary Figure 1. Infection time course of phage JBD30, Related to Figures 2A and 4
Tenfold dilutions of phage samples collected through the course of phage infection in wild-type PA14 were applied to lawns of wild-type PA14. Phages and cells were mixed at time = 0. The “input” is the phage titer immediately after phage addition. This number decreases by 20 minutes as all the added phages have adsorbed to cells, but new particles have not yet been produced. During the first 50 minutes after infection, few phages are produced while the full phage burst appears to have occurred by 70 minutes.
Supplementary Figure 2. Bioinformatic and functional analysis of Aca1, Related to Figures 2 and 3
(A) Multiple sequence alignment of Aca1 homologs from the indicated phages and bacteria. The position of the predicted HTH motif is outlined in a black box. Arrows indicate R33, R34, and R44 that were subject alanine substitution. (B) Representative EMSAs with Aca1 using the 110 bp anti-CRISPR upstream region from phage JBD30 as a substrate. Purified protein was added at concentrations of 10 nM, 50 nM, 100 nM, 250 nM and 500 nM. The dash sign (−) indicates that no protein was added. Non-denaturing acrylamide gels stained with SYBR gold are shown. (C) Quantification of DNA bound by Aca1 in EMSAs. Error bars represent the standard deviation of the mean of three replicates. (D) To indicated their position relative to a DNA substrate, residues R33, R34, and R44 (highlighted in red) of JBD30 Aca1 were modeled onto the HTH DNA binding domain of the virulence regulator PlcR in complex with DNA (PDB: 3U3W) from Bacillus thuringiensis (Grenha et al., 2013).
Supplementary Figure 3. Effect of Aca1 mutations and overexpression on phage viability, Related to Figure 3
(A) Tenfold dilutions of lysates of the JBD30 phage carrying the indicated Aca1 mutation were applied to lawns of PA14 and PA14ΔCRISPR. A representative image from three biological replicates is shown. (B) Tenfold dilutions of lysates of JBD30 carrying the indicated mutations in Aca1 binding sites and/or Aca1 were applied to lawns PA14ΔCRISPR overexpressing wild-type Aca1 from a plasmid. (C) The plaque sizes (area) of the Aca1 partial DNA binding mutants in phage JBD30 were quantified on the PA14ΔCRISPR strain. The average size is shown relative to that of wild-type JB30 phage. Averages were calculated from three independent plaque assays, where >100 plaques were measured. Error bars represent the standard error of the mean. Representative plaque images are shown. (D) Tenfold dilutions of wild-type (JBD30), anti-CRISPR gene frameshift mutant (JBD30acrfs) phage, and acr promoter deletion phage (JBD30ΔPacr) lysates were applied to lawns PA14ΔCRISPR overexpressing AcrIF1 and R44A Aca1 from their native promoter on a plasmid.
Supplementary Figure 4. Insertion of a transcriptional terminator downstream of the anti-CRISPR operon partially rescues JBD30acaR44A viability, Related to Figure 4
Lysates of phage JBD30 (WT or Aca1 R44A mutant) with or without an inserted rrnB T1 terminator downstream of aca1 were spotted in 10-fold serial dilutions on bacterial lawns of wild-type P. aeruginosa PA14, a CRISPR deletion version of PA14 (PA14ΔCRISPR), or on the deletion strain expressing Aca1 from a plasmid. Representative images from three biological replicates incubated for 42 hours are shown.
Supplementary Figure 5. Phage JBD30 lysogen formation is unaffected by the R44A Aca1 substitution, Related to Figure 4
The PA14ΔCRISPR strain was infected with wild-type JBD30 (WT Aca1) or JBD30aca1R44A (R44A Aca1) at the same multiplicity of infection and plated to obtain single colonies. Lysogens were identified by cross-streaking the colonies over top of a line of phage lysate. The mean ± SEM percentage of lysogens formed in three independent infection assays where 100 colonies were screened relative to the wild-type phage is shown.
Supplementary Figure 6. Multiple sequence alignment of other Aca proteins and their respective anti-CRISPR upstream regions, Related to Figure 6
(A) Multiple sequence alignment of Aca2 proteins from diverse Proteobacteria. The predicted helix-turn-helix motif is outlined in a black box. (B) Multiple sequence alignment of Aca3 proteins from different strains of Neisseria meningitidis. The predicted helix-turn-helix motif is outlined in a black box. (C) Multiple nucleotide sequence alignment of the region immediately upstream of the anti-CRISPR genes found in association with aca2 in panel A. A putative Aca2 binding site is outlined in a black box. Positions with ≥ 60% identity are coloured (D) Multiple nucleotide sequence alignment of the region immediately upstream of the anti-CRISPR genes found in association with aca3 in panel B. A putative binding site for Aca3 is outlined in a black box. Nme, Neisseria meningitidis; numbers indicate strain. Positions with >60% identity are coloured. See also Table S3.
Supplementary Figure 7. aca2 genes found in diverse genomic contexts share conserved sequences, Related to Figures 1 and S6
(A)Schematic representation of the genome context of aca2 associated anti-CRISPR operons. Coloured arrows represent anti-CRISPR and anti-CRISPR-associated (aca) genes. Genes shown in gray represent the genomic context. Predicted functions are indicated when known. Arrows representing genes are not shown to scale. TF = transcription factor. Anti-CRISPR genes are denoted as IX#, where I represents the type of system targeted, X represents the subtype of system targeted, and # represents the protein family. (B) Multiple nucleotide sequence alignment of the region immediately upstream of the anti-CRISPR genes found in association with aca2 to the stop codon of aca2. A putative aca2 binding site as well as the acr and aca2 start codon are outlined in black boxes. Positions sharing ≥ 60% identity are coloured. See also Table S3.
Anti-CRISPR genes are transcribed to high levels quickly after phage infection
Aca proteins are ubiquitous DNA-binding proteins encoded in anti-CRISPR operons
Aca proteins repress anti-CRISPR transcription
Aca function obviates deleterious effects of high anti-CRISPR transcription
Anti-CRISPR associated (Aca) proteins function as repressors of anti-CRISPR transcription.
Acknowledgments
The authors would like to thank Karen Maxwell, Emma Brownlie, Cayla Burk, and Veronique Taylor for useful comments on the manuscript. The authors would also like to thank Susan Kelso for help with the graphical abstract. This work was supported by NIH grants to N.J.K. (P50GM082250 and U01MH115747) and J. B.-D. (DP5-OD021344 and R01GM127489), and by Canadian Institutes of Health Research grants to A.R.D. (MOP-130482 and FDN-15427).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors have filed for a patent related to this work.
References
- Agari Y, Sakamoto K, Tamakoshi M, Oshima T, Kuramitsu S, and Shinkai A (2010). Transcription profile of Thermus thermophilus CRISPR systems after phage infection. J Mol Biol 395, 270–281. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bair CL, Rifat D, and Black LW (2007). Exclusion of glucosyl-hydroxymethylcytosine DNA containing bacteriophages is overcome by the injected protein inhibitor IPI*. J Mol Biol 366, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, and Davidson AR (2015). Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, and Davidson AR (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Zhang JY, Rollins MF, Osuna BA, Wiedenheft B, and Bondy-Denomy J (2018). Bacteriophage Cooperation Suppresses CRISPR-Cas3 and Cas9 Immunity. Cell 174, 917–925 e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady KC, White AS, Hammond JH, Abendroth MD, Karthikeyan RS, Lalitha P, Zegans ME, and O’Toole GA (2011). Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 157, 430–437. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. (2017). Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell 169, 47–57 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, and Semenova E (2012). Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun 3, 945. [DOI] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, and Charpentier E (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Guo M, Wang S, Zhu Y, Wang S, Xiong Z, Yang J, Xu Z, and Huang Z (2017). Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–439. [DOI] [PubMed] [Google Scholar]
- Dong L, Guan X, Li N, Zhang F, Zhu Y, Ren K, Yu L, Zhou F, Han Z, Gao N, et al. (2019). An anti-CRISPR protein disables type V Cas12a by acetylation. Nat Struct Mol Biol 26, 308–314. [DOI] [PubMed] [Google Scholar]
- Espah Borujeni A, Channarasappa AS, and Salis HM (2014). Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res 42, 2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha MA, and Kropinski AM (1990). Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J Bacteriol 172, 3496–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, and Moineau S (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Gilbert W, and Muller-Hill B (1967). The lac operator is DNA. Proc Natl Acad Sci U S A 58, 2415–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, and Lory S (2004). A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell 7, 745–754. [DOI] [PubMed] [Google Scholar]
- Greene AC (2018). CRISPR-Based Antibacterials: Transforming Bacterial Defense into Offense. Trends Biotechnol 36, 127–130. [DOI] [PubMed] [Google Scholar]
- Grenha R, Slamti L, Nicaise M, Refes Y, Lereclus D, and Nessler S (2013). Structural basis for the activation mechanism of the PlcR virulence regulator by the quorum-sensing signal peptide PapR. Proc Natl Acad Sci U S A 110, 1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T, Earl LA, et al. (2017). Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell 171, 414–426 e412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N, Palmer AC, Dodd IB, and Shearwin KE (2017). Directing traffic on DNA-How transcription factors relieve or induce transcriptional interference. Transcription 8, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu JJ, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. (2017). A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell 170, 1224–1233 e1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW, and Duda RL (1998). Bacteriophage HK97 head assembly: a protein ballet. Adv Virus Res 50, 235–288. [DOI] [PubMed] [Google Scholar]
- Hertveldt K, and Lavigne R (2008). Bacteriophages of Pseudomonas In Pseudomonas (Wiley-VCH Verlag GmbH & Co. KGaA; ), pp. 255–291. [Google Scholar]
- Hong YR, and Black LW (1993). Protein folding studies in vivo with a bacteriophage T4 expression-packaging-processing vector that delivers encapsidated fusion proteins into bacteria. Virology 194, 481–490. [DOI] [PubMed] [Google Scholar]
- Iida S, Streiff MB, Bickle TA, and Arber W (1987). Two DNA antirestriction systems of bacteriophage P1, darA, and darB: characterization of darA- phages. Virology 157, 156–166. [DOI] [PubMed] [Google Scholar]
- Juranek S, Eban T, Altuvia Y, Brown M, Morozov P, Tuschl T, and Margalit H (2012). A genome-wide view of the expression and processing patterns of Thermus thermophilus HB8 CRISPR RNAs. RNA 18, 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamionka A, Bogdanska-Urbaniak J, Scholz O, and Hillen W (2004). Two mutations in the tetracycline repressor change the inducer anhydrotetracycline to a corepressor. Nucleic Acids Res 32, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, and Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Thornton BW, Lobba MJ, Liu JJ, Al-Shayeb B, Watters KE, and Doudna JA (2019). Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat Struct Mol Biol 26, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberger M, Gandon S, Meaden S, Rollie C, Chevallereau A, Chabas H, Buckling A, Westra ER, and van Houte S (2018). Anti-CRISPR Phages Cooperate to Overcome CRISPR-Cas Immunity. Cell 174, 908–916 e912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne R, Ceyssens PJ, and Robben J (2009). Phage proteomics: applications of mass spectrometry. Methods Mol Biol 502, 239–251. [DOI] [PubMed] [Google Scholar]
- Levy A, Goren MG, Yosef I, Auster O, Manor M, Amitai G, Edgar R, Qimron U, and Sorek R (2015). CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LF, and Wang JC (1987). Supercoiling of the DNA template during transcription. Proc Natl Acad Sci U S A 84, 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, and Matthews KS (1993). Dependence of trp repressor-operator affinity, stoichiometry, and apparent cooperativity on DNA sequence and size. J Biol Chem 268, 23239–23249. [PubMed] [Google Scholar]
- Luscombe NM, Austin SE, Berman HM, and Thornton JM (2000). An overview of the structures of protein-DNA complexes. Genome Biol 1, REVIEWS001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, Rauch BJ, Walton RT, Berry JD, Joung JK, Kleinstiver BP, et al. (2018). Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 362, 240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrs CF, and Howe MM (1990). Kinetics and regulation of transcription of bacteriophage Mu. Virology 174, 192–203. [DOI] [PubMed] [Google Scholar]
- Nelson HC, and Sauer RT (1985). Lambda repressor mutations that increase the affinity and specificity of operator binding. Cell 42, 549–558. [DOI] [PubMed] [Google Scholar]
- Orosz A, Boros I, and Venetianer P (1991). Analysis of the complex transcription termination region of the Escherichia coli rrnB gene. Eur J Biochem 201, 653–659. [DOI] [PubMed] [Google Scholar]
- Palmer AC, Ahlgren-Berg A, Egan JB, Dodd IB, and Shearwin KE (2009). Potent transcriptional interference by pausing of RNA polymerases over a downstream promoter. Mol Cell 34, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AC, Egan JB, and Shearwin KE (2011). Transcriptional interference by RNA polymerase pausing and dislodgement of transcription factors. Transcription 2, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, and Zhang J (2012). High expression hampers horizontal gene transfer. Genome Biol Evol 4, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, et al. (2016a). Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 167, 1829–1838 e1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Shah M, Mejdani M, Calmettes C, Moraes TF, Davidson AR, and Maxwell KL (2017). Disabling a Type I-E CRISPR-Cas Nuclease with a Bacteriophage-Encoded Anti-CRISPR Protein. mBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Staals RH, Taylor C, Watson BN, Saha S, Fineran PC, Maxwell KL, and Davidson AR (2016b). Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol 1, 16085. [DOI] [PubMed] [Google Scholar]
- Piya D, Vara L, Russell WK, Young R, and Gill JJ (2017). The multicomponent antirestriction system of phage P1 is linked to capsid morphogenesis. Mol Microbiol 105, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursey E, Sunderhauf D, Gaze WH, Westra ER, and van Houte S (2018). CRISPR-Cas antimicrobials: Challenges and future prospects. PLoS Pathog 14, e1006990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax TE, Voet M, Sismeiro O, Dillies MA, Jagla B, Coppee JY, Sezonov G, Forterre P, van der Oost J, Lavigne R, et al. (2013). Massive activation of archaeal defense genes during viral infection. J Virol 87, 8419–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis HM, Mirsky EA, and Voigt CA (2009). Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, and Russell DW (2006). Purification of Bacteriophage lambda Particles by Isopycnic Centrifugation through CsCl Gradients. CSH Protoc 2006. [DOI] [PubMed] [Google Scholar]
- Savalia D, Robins W, Nechaev S, Molineux I, and Severinov K (2010). The role of the T7 Gp2 inhibitor of host RNA polymerase in phage development. J Mol Biol 402, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheirer KE, and Higgins NP (2001). Transcription induces a supercoil domain barrier in bacteriophage Mu. Biochimie 83, 155–159. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, and Selbach M (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Seo SW, Yang JS, Kim I, Yang J, Min BE, Kim S, and Jung GY (2013). Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab Eng 15, 67–74. [DOI] [PubMed] [Google Scholar]
- Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, and O’Toole GA (2006). Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environ Microbiol 72, 5027–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Biegert A, and Lupas AN (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33, W244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V, and Salamov A (2011). Automatic Annotation of Microbial Genomes and Metagenomic Sequences In Metagenomics and its applications in agriculture, biomedicine, and environmental studies, Li RW, ed. (New York: Nova Science; ), pp. 61–78. [Google Scholar]
- Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, and Rubin EM (2007). Genome-wide experimental determination of barriers to horizontal gene transfer. Science 318, 1449–1452. [DOI] [PubMed] [Google Scholar]
- Wang X, Yao D, Xu JG, Li AR, Xu J, Fu P, Zhou Y, and Zhu Y (2016). Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat Struct Mol Biol 23, 868–870. [DOI] [PubMed] [Google Scholar]
- Yosef I, Goren MG, and Qimron U (2012). Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res 40, 5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Dill BD, Pan C, Hettich RL, Banfield JF, Shah M, Fremaux C, Horvath P, Barrangou R, and Verberkmoes NC (2012). Phage-induced expression of CRISPR-associated proteins is revealed by shotgun proteomics in Streptococcus thermophilus. PloS one 7, e38077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Bremer H (1995). Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J Biol Chem 270, 11181–11189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Infection time course of phage JBD30, Related to Figures 2A and 4
Tenfold dilutions of phage samples collected through the course of phage infection in wild-type PA14 were applied to lawns of wild-type PA14. Phages and cells were mixed at time = 0. The “input” is the phage titer immediately after phage addition. This number decreases by 20 minutes as all the added phages have adsorbed to cells, but new particles have not yet been produced. During the first 50 minutes after infection, few phages are produced while the full phage burst appears to have occurred by 70 minutes.
Supplementary Figure 2. Bioinformatic and functional analysis of Aca1, Related to Figures 2 and 3
(A) Multiple sequence alignment of Aca1 homologs from the indicated phages and bacteria. The position of the predicted HTH motif is outlined in a black box. Arrows indicate R33, R34, and R44 that were subject alanine substitution. (B) Representative EMSAs with Aca1 using the 110 bp anti-CRISPR upstream region from phage JBD30 as a substrate. Purified protein was added at concentrations of 10 nM, 50 nM, 100 nM, 250 nM and 500 nM. The dash sign (−) indicates that no protein was added. Non-denaturing acrylamide gels stained with SYBR gold are shown. (C) Quantification of DNA bound by Aca1 in EMSAs. Error bars represent the standard deviation of the mean of three replicates. (D) To indicated their position relative to a DNA substrate, residues R33, R34, and R44 (highlighted in red) of JBD30 Aca1 were modeled onto the HTH DNA binding domain of the virulence regulator PlcR in complex with DNA (PDB: 3U3W) from Bacillus thuringiensis (Grenha et al., 2013).
Supplementary Figure 3. Effect of Aca1 mutations and overexpression on phage viability, Related to Figure 3
(A) Tenfold dilutions of lysates of the JBD30 phage carrying the indicated Aca1 mutation were applied to lawns of PA14 and PA14ΔCRISPR. A representative image from three biological replicates is shown. (B) Tenfold dilutions of lysates of JBD30 carrying the indicated mutations in Aca1 binding sites and/or Aca1 were applied to lawns PA14ΔCRISPR overexpressing wild-type Aca1 from a plasmid. (C) The plaque sizes (area) of the Aca1 partial DNA binding mutants in phage JBD30 were quantified on the PA14ΔCRISPR strain. The average size is shown relative to that of wild-type JB30 phage. Averages were calculated from three independent plaque assays, where >100 plaques were measured. Error bars represent the standard error of the mean. Representative plaque images are shown. (D) Tenfold dilutions of wild-type (JBD30), anti-CRISPR gene frameshift mutant (JBD30acrfs) phage, and acr promoter deletion phage (JBD30ΔPacr) lysates were applied to lawns PA14ΔCRISPR overexpressing AcrIF1 and R44A Aca1 from their native promoter on a plasmid.
Supplementary Figure 4. Insertion of a transcriptional terminator downstream of the anti-CRISPR operon partially rescues JBD30acaR44A viability, Related to Figure 4
Lysates of phage JBD30 (WT or Aca1 R44A mutant) with or without an inserted rrnB T1 terminator downstream of aca1 were spotted in 10-fold serial dilutions on bacterial lawns of wild-type P. aeruginosa PA14, a CRISPR deletion version of PA14 (PA14ΔCRISPR), or on the deletion strain expressing Aca1 from a plasmid. Representative images from three biological replicates incubated for 42 hours are shown.
Supplementary Figure 5. Phage JBD30 lysogen formation is unaffected by the R44A Aca1 substitution, Related to Figure 4
The PA14ΔCRISPR strain was infected with wild-type JBD30 (WT Aca1) or JBD30aca1R44A (R44A Aca1) at the same multiplicity of infection and plated to obtain single colonies. Lysogens were identified by cross-streaking the colonies over top of a line of phage lysate. The mean ± SEM percentage of lysogens formed in three independent infection assays where 100 colonies were screened relative to the wild-type phage is shown.
Supplementary Figure 6. Multiple sequence alignment of other Aca proteins and their respective anti-CRISPR upstream regions, Related to Figure 6
(A) Multiple sequence alignment of Aca2 proteins from diverse Proteobacteria. The predicted helix-turn-helix motif is outlined in a black box. (B) Multiple sequence alignment of Aca3 proteins from different strains of Neisseria meningitidis. The predicted helix-turn-helix motif is outlined in a black box. (C) Multiple nucleotide sequence alignment of the region immediately upstream of the anti-CRISPR genes found in association with aca2 in panel A. A putative Aca2 binding site is outlined in a black box. Positions with ≥ 60% identity are coloured (D) Multiple nucleotide sequence alignment of the region immediately upstream of the anti-CRISPR genes found in association with aca3 in panel B. A putative binding site for Aca3 is outlined in a black box. Nme, Neisseria meningitidis; numbers indicate strain. Positions with >60% identity are coloured. See also Table S3.
Supplementary Figure 7. aca2 genes found in diverse genomic contexts share conserved sequences, Related to Figures 1 and S6
(A)Schematic representation of the genome context of aca2 associated anti-CRISPR operons. Coloured arrows represent anti-CRISPR and anti-CRISPR-associated (aca) genes. Genes shown in gray represent the genomic context. Predicted functions are indicated when known. Arrows representing genes are not shown to scale. TF = transcription factor. Anti-CRISPR genes are denoted as IX#, where I represents the type of system targeted, X represents the subtype of system targeted, and # represents the protein family. (B) Multiple nucleotide sequence alignment of the region immediately upstream of the anti-CRISPR genes found in association with aca2 to the stop codon of aca2. A putative aca2 binding site as well as the acr and aca2 start codon are outlined in black boxes. Positions sharing ≥ 60% identity are coloured. See also Table S3.
Data Availability Statement
This study did not generate/analyze any datasets/codes.