

Abstract
Objective.
Genome-wide association studies (GWASs) for epithelial ovarian cancer (EOC) have focused largely on populations of European ancestry. We aimed to identify common germline variants associated with EOC risk in Asian women.
Methods.
Genotyping was performed as part of the OncoArray project. Samples with >60% Asian ancestry were included in the analysis. Genotyping was performed on 533,631 SNPs in 3238 Asian subjects diagnosed with invasive or borderline EOC and 4083 unaffected controls. After imputation, genotypes were available for 11,595,112 SNPs to identify associations.
Results.
At chromosome 6p25.2, SNP rs7748275 was associated with risk of serous EOC (odds ratio [OR] = 1.34, P = 8.7 × 10−9) and high-grade serous EOC (HGSOC) (OR = 1.34, P = 4.3 × 10−9). SNP rs6902488 at 6p25.2 (r2 = 0.97 with rs7748275) lies in an active enhancer and is predicted to impact binding of STAT3, P300 and ELF1. We identified additional risk loci with low Bayesian false discovery probability (BFDP) scores, indicating they are likely to be true risk associations (BFDP <10%). At chromosome 20q11.22, rs74272064 was associated with HGSOC risk (OR = 1.27, P = 9.0 × 10−8). Overall EOC risk was associated with rs10260419 at chromosome 7p21.3 (OR = 1.33, P = 1.2 × 10−7) and rs74917072 at chromosome 2q37.3 (OR = 1.25, P = 4.7 × 10−7). At 2q37.3, expression quantitative trait locus analysis in 404 HGSOC tissues identified ESPNL as a putative candidate susceptibility gene (P = 1.2 × 10−7).
Conclusion.
While some risk loci were shared between East Asian and European populations, others were population-specific, indicating that the landscape of EOC risk in Asian women has both shared and unique features compared to women of European ancestry.
Keywords: Genome-wide association study, Asian ancestry, Epithelial ovarian cancer, Enhancer, eQTL, Gene regulation
1. Introduction
Epithelial ovarian cancers (EOC) are a diverse group of tumors occuring predominantly among postmenopausal women. Epidemiological and lifestyle risk factors include a family history of ovarian and breast cancer [1], nulliparity [2], and no oral contraceptive use [3]. Germline mutations in highly penetrant genes, including BRCA1 [4] and BRCA2 [5], are associated with a >15% lifetime risk of ovarian cancer [6]; but these mutations are present in only 10–15% of EOC cases. In the general population, common genetic variants identified using genome-wide association studies (GWASs) have been found to confer more modest disease risks with odds ratios generally ≤1.5. To date, GWASs have identified >30 regions of the genome harboring common variants associated with EOC risk (reviewed in Jones et al. [7]), with all but one of these studies reporting risk variants identified in women of European ancestry [8–19]. There is one report of a GWAS performed in 1057 Asian cases and 1799 controls (Han Chinese), with replication in 492 EOC cases and 1004 controls. This study identified two genome-wide significant risk regions at 9q22.33 and 10p11.21; however, neither region appears significantly associated with EOC risk in European populations [9].
EOC incidence varies by race and/or ethnicity; women in Asian, North African and Middle Eastern countries tend to have lower rates of EOC than women in northern European and Baltic countries [20,21]. Women from Asia migrating to the United States and Europe retain their comparatively reduced rates of EOC [22]. In addition, Asian women, particularly those from China, Korea, Vietnam and the Philippines, tend to be significantly younger at diagnosis, and are more likely to be diagnosed with early stage (stage I) disease. Consequently, they have better 5-year survival rates compared to EOC cases in white women of European ancestry [23,24]. The distribution of histological subtypes also differs between Asian and European women. Asian women are more likely than European women to develop clear cell or mucinous EOCs and less likely to develop serous EOC [23,25]. Much of the variation in EOC incidence rates by race or ethnicity may be attributable to differences in reproductive, lifestyle, and environmental risk factors; but variations in the underlying genetics are also likely to contribute to the observed differences.
The international Genetic Associations and Mechanisms in Oncology (GAME-ON) project designed a custom Illumina genotyping array (the ‘OncoArray’), containing over 530,000 variants to identify genetic risk factors across multiple tumor types, and in different racial or ethnic groups [26]. The OncoArray includes risk-associated SNPs identified from GWAS meta-analyses of breast, colorectal, lung, ovarian and prostate cancers; risk regions associated with numerous other phenotypes (both cancer and non-cancer); and a genome-wide SNP ‘backbone’ to enable agnostic GWASs to be performed for each phenotype [26]. The OncoArray has been used to genotype >500,000 subjects, including ~53,000 individuals from EOC case-control studies, of which ~7000 subjects are of East Asian ancestry. Here, we report genetic association analyses of EOC case-control subjects of East Asian ancestry genotyped on the OncoArray.
2. Methods
2.1. Study samples
All subjects included in this analysis were of Asian descent (see below) and provided written informed consent. Data and blood samples were collected under protocols approved by the Institutional Review Boards or Ethics Committee at each institution. All constituent studies and host institutions are listed in Supplementary Table 1. The OCAC OncoArray data set comprised 63 genotyping project/case-controls sets (Supplementary Table 1). Some studies (e.g. SEARCH) contributed samples to more than one genotyping project and some case-control sets are a combination of multiple individual studies. Post-QC sample numbers, by histology, for subjects of Asian ancestry are shown in Table 1.
Table 1:
Number of participants by analysis stratum. Cases and controls by study stratum. US represents a collection of all US studies with fewer than 50 Asian subjects; EUR represents all European studies with fewer than 50 Asian subjects.
| Analysis stratum |
Controls | HGSOC | LGSOC | EnOC | CCOC | MOC | Other invasive |
All invasive cases |
Serous borderline |
Mucinous borderline |
All borderline |
Total samples |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUS | 18 | 41 | 2 | 12 | 18 | 3 | 2 | 78 | 4 | 8 | 12 | 108 |
| CAN | 49 | 57 | 0 | 15 | 20 | 7 | 8 | 107 | 5 | 8 | 13 | 169 |
| CHI | 2032 | 878 | 12 | 193 | 50 | 92 | 310 | 1535 | 33 | 30 | 63 | 3630 |
| DOV | 45 | 31 | 0 | 8 | 11 | 1 | 4 | 55 | 6 | 7 | 13 | 113 |
| EUR | 32 | 12 | 1 | 4 | 7 | 4 | 3 | 31 | 3 | 0 | 3 | 66 |
| HAW | 408 | 103 | 1 | 45 | 31 | 26 | 13 | 219 | 25 | 23 | 48 | 675 |
| JPN | 230 | 72 | 2 | 10 | 35 | 13 | 4 | 136 | 5 | 8 | 13 | 379 |
| KRA | 682 | 194 | 9 | 25 | 28 | 12 | 16 | 284 | 1 | 15 | 16 | 982 |
| LAX | 92 | 53 | 2 | 15 | 22 | 15 | 3 | 110 | 25 | 11 | 36 | 238 |
| MAS | 157 | 62 | 0 | 36 | 23 | 14 | 11 | 146 | 2 | 2 | 4 | 307 |
| MEC | 28 | 10 | 0 | 2 | 2 | 1 | 4 | 19 | 0 | 0 | 0 | 47 |
| STA | 60 | 24 | 3 | 9 | 10 | 8 | 7 | 61 | 10 | 4 | 14 | 135 |
| SWH | 135 | 45 | 0 | 17 | 10 | 13 | 45 | 130 | 0 | 0 | 0 | 265 |
| US | 115 | 33 | 4 | 13 | 11 | 3 | 6 | 70 | 14 | 8 | 22 | 207 |
| Total | 4083 | 1615 | 36 | 404 | 278 | 212 | 436 | 2981 | 133 | 124 | 257 | 7321 |
2.2. Genotype data and quality control (QC)
Genotyping was performed at five locations: University of Cambridge, Center for Inherited Disease Research (CIDR), National Cancer Institute (NCI), Genome Quebec and Mayo Clinic. OncoArray sample QC was similar to that carried out for the other projects (as described in Pharoah et al., 2013 [11,16,17,27]). Samples were excluded if (1) genotyping call rate was <95%, (2) heterozygosity was excessively low or high, (3) if they were not female or had ambiguous sex, or (4) were duplicates (cryptic or intended). Duplicates and close relatives were identified using in-house software that calculates a concordance matrix for all individuals. Samples with concordance >0.86 were flagged as duplicates and samples with concordance between 0.74 and 0.86 were flagged as relatives. The comparison was performed among all the OncoArray samples, and all the previously genotyped samples. Concordance statistics were used to identify cryptic duplicates and expected duplicates whose genotypes did not match, and we attempted to resolve these with the study investigators. If a discrepancy could not be resolved both samples were excluded. For confirmed cryptic duplicates and relatives, we retained one sample in the analysis. For case-control pairs we excluded the control, while for case-case and control-control pairs we excluded the sample with the lower call rate.
SNP QC was carried out according to the OncoArray QC Guidelines [26]. Only SNPs that passed QC for all consortia were used for imputation. We excluded SNPs with a call rate < 95%, SNPs deviating from Hardy-Weinberg equilibrium (P < 10−7 in controls and P < 10−12 in cases) and SNPs with concordance <98% among 5280 duplicate pairs. For the imputation, we additionally excluded SNPs with a MAF < 1% and a call rate < 98% and SNPs that could not be linked to the 1000 genomes reference or differed significantly in frequency from the 1000 genomes (European frequency) and a further 1128 SNPs where the cluster plot was judged to be inadequate. Of the 533,631 SNPs which were manufactured on the array, 494,813 SNPs passed the initial QC and 470,825 SNPs were used for imputation. Samples with overall heterozygosity <5% or > 40% were excluded.
2.3. Ancestry analysis and imputation
Intercontinental ancestry was calculated using the software package FastPop (http://sourceforge.net/projects/fastpop/) [28]. Consistent with other OncoArray studies, only the samples with >60% Asian ancestry, were included in the analyses reported here. Principal component analysis for the OncoArray data was carried out using data from 33,661 uncorrelated SNPs (pair-wise r2 <0.1) with minor allele frequency >0.05 using an in-house program (available at http://ccge.medschl.cam.ac.uk/software/pccalc/). Principal components analysis for the other genotype data sets was carried out as previously described [16].
We performed imputation separately for each genotyping project data set. We imputed genotypes into the reference panel from the 1000 Genomes Project (v3 October 2014) [29]. We initially used a two-step procedure, which involved pre-phasing in the first step and imputation of the phased data in the second, to improve computation efficiency. We carried out pre-phasing using Shape-IT [30] and the subsequent imputation using 1MPUTE2 [31]. We then performed more accurate imputation for any region with a SNP with P < 10−6. The boundaries of these regions were set +/− 500 kb from the most significant SNP in the region. The single-step imputation used 1MPUTE2 without pre-phasing with some of the default parameters modified. These included an increase of the MCMC iterations to 90 (out of which the first 15 were used as burn-in), an increase of the buffer region to 500 kb and increasing to 100 the number of haplotypes used as templates when phasing observed genotypes.
2.4. Association analyses
We excluded SNPs from the association analysis if their imputation accuracy was r2 < 0.3 or their minor allele frequency (MAF) was <0.01. In total, genotypes for 11,595,112 million variants were available for analysis. We evaluated the association between genotype and disease using the imputed genotype dosage (log-additive genetic models) in a logistic regression model. We carried out initial genome-wide analyses separately for OncoArray, COGS and the five GWAS datasets and pooled the results using a fixed effects meta-analysis. The analyses were adjusted for study and for population substructure by including the eigenvectors of project-specific principal components as covariates in the model (nine for OncoArray, five for COGS, two for UK GWAS, and two for the US, BWH and POL GWAS, and a single PC for the MAY GWAS). The number of eigenvectors chosen was based on the point of inflection of a scree plot. After one-step imputation of the genotypes in the regions of interest we used genotype dosages in a single logistic regression model with adjustment for each genotyping project/study combination and nineteen principal components. Principal components were set to zero for samples not included in a given project. We used custom written software for the analysis.
To assess the magnitude of confounding caused by cryptic population substructure, we calculated inflation in the test statistics (λ) by dividing the median of the test statistic by 0.455 (the median for the χ2 distribution on 1 degree of freedom). The inflation was converted to an equivalent inflation for a study with 1000 cases and 1000 controls (λ1000) by adjusting by effective study size:
where n is the number of cases and m is the number of controls in each study stratum, k. There was a small inflation of the test statistics for the all invasive analysis (λ = 1.057, λ1000 = 1.015).
EOC is a heterogeneous phenotype with five major histotypes for invasive disease – high-grade serous ovarian cancer (HGSOC), low-grade serous ovarian cancer (LGSOC), mucinous ovarian cancer (MOC), endometrioid ovarian cancer (EnOC) and clear cell ovarian cancer (CCOC) – and two histotypes of borderline disease – serous and mucinous. The pattern of association across the different histotypes varies for the known OC risk loci [8]. We therefore carried out the association analysis on the following nine histotypes: all invasive disease; HGSOC; LGSOC; all invasive serous; serous borderline; LGSOC and borderline serous combined; EnOC; CCOC; and mucinous invasive/mucinous borderline combined. QQ plots are shown in Supplementary Fig. 1.
2.5. Identifying candidate causal SNPs in each susceptibility region
To identify a set of variants most likely to contain the true underlying causal association – the candidate causal variants – we excluded SNPs with causality odds of <1:100 by comparing the likelihood of each SNP from the association analysis with the likelihood of the most strongly associated SNP.
2.6. Expression quantitative trait locus (eQTL) analyses
Expression QTL analyses were conducted using three data sets. We used an in-house generated RNA-sequencing data set of 105 primary normal ovarian surface epithelial cell (OSEC) and 60 fallopian tube secretory epithelial cell (FTSEC) cultures (Lawrenson et al., submitted). All samples were sequenced following a ribo-depletion library preparation method, and all specimens were genotyped on the OncoArray. We also performed tumor-specific eQTL analyses using TCGA HGSOC data set [32]. The sample size for women of Asian ancestry was too small in each data set to perform an eQTL analysis restricted specifically to Asians and so we decided to leverage the entire (cosmopolitan ancestry) data sets for eQTL analysis. For the TCGA analysis, we focused on all genes and samples (n = 404) that had matched gene expression (measured on the Agilent 1 M microarray), CpG methylation (measured on the Illumina Infinium HumanMethylation27 BeadChip), copy number alteration (called using the Affymetrix SNP 6.0 array), and germline genotype (called using the Affymetrix SNP 6.0 array) data available. Genotypes were imputed into the 1000 Genomes October 2014 (Phase 3, version 5) European reference panel for all three data sets [29]. Expression QTL analyses were performed using linear regression as implemented in the R package Matrix eQTL [33]. Prior to eQTL analyses the effects of tumor copy number and methylation on gene expression were regressed out as previously described [34]. We focused on cis-acting eQTL relationships between candidate causal risk SNPs and all genes up to 1 Mb on either side of these SNPs. Two-sided P-values are reported.
2.7. Functional annotation of variants
We used shell scripts with bedtools (http://bedtools.readthedocs.org/en/latest/) to generate overlap data between all variants in each associated region including likely causal SNPs and bed file versions of all the biofeature data used. The overlap data thus obtained were converted to matrix form by means of python scripts and then sorted in Microsoft Excel.
To test for locus-specific tissue enrichment of variants, H3K27 acetylation peaks were collated from public sources or from in-house data (all listed in Supplementary Table 2). Overlaps were counted for the all SNPs against which genotypes were imputed in 1000 genomes for each H3K27ac dataset. The fraction of causal SNPs with overlaps was then tested for significance against this background for each cell type in the H3K27ac datasets using the hypergeometric distribution. Finally, p values were adjusted for multiple comparisons using Bonferroni’s method.
2.8. Data availability statement
The summary results for all imputed SNPs reported in this paper are available at: https://doi.org/10.17863/CAM.25845
3. Results
3.1. Association analyses
Of the samples genotyped on the OncoArray, 21,879 EOC cases and 29,224 controls from OCAC met our quality control (QC) criteria (see Methods). Intercontinental ancestry was determined using FastPop [28] to select samples with >60% Asian ancestry for inclusion in association analyses (Fig. 1a). This analysis inferred Asian ancestry in 2981 women with invasive EOC (1615 high-grade serous, 36 low-grade serous, 404 endometrioid, 278 clear cell and 212 mucinous EOC; 436 ‘other’), 257 women with borderline EOC (133 serous and 124 mucinous), and 4083 controls. Most samples were collected through population-based studies conducted in eastern Asia (Fig. 1b), with around 25% of specimens collected in studies conducted in the US, Europe and Australia. The number of participants by analysis stratum is listed in Table 1. Data from the 1000 Genomes Project reference panel [29] were used to impute genotypes for 11,403,952 common variants (minor allele frequency, MAF > 1%). Histology was ascertained for 2802 (86.5%) Asian EOC cases in total. The Asian populations had higher frequencies of the clear cell (9.9%) and mucinous (12.0%) histological subtypes (‘histotypes’) and lower frequency of the high-grade serous histotype (57.6%) compared to the non-Asian populations (7.0%, 7.2% and 66.4%, respectively) (Fig. 1c).
Fig. 1.
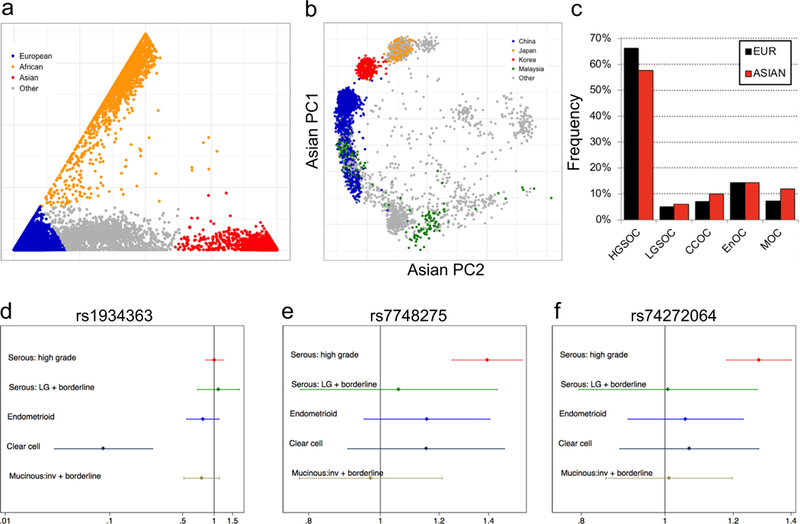
Novel loci associated with EOC risk in Asian women. (a-b) Principal component analysis (PCA) of 21,879 EOC cases and 29,224 controls (a) identified a total of 7321 subjects with Asian ancestry which were stratified by country-of-origin (b); (c) Clear cell and mucinous EOC is more common in Asian women than women of European ancestry. (d-f) Forest plots showing the three genome-wide significant risk regions for EOC identified in women of Asian ancestry.
Association analyses were performed for all East Asian subjects considering histotypes combined, all invasive cases, all borderline cases, and for each histotype separately (Table 1). There was little evidence of inflation of the test statistic (λ1000 for all invasive analysis = 1.015). We identified three risk associations at two different loci reaching genome-wide significance (effect allele frequency, EAF ≥0.05, P < 5 × 10−8) (Table 2 and Fig. 1d-f). There was no evidence of heterogeneity by study for these associations. At chromosome 6p25.2, rs7748275 was associated with risk of serous EOC (OR = 1.4, P = 8.7 × 10−9). The Bayesian false-discovery probability (BFDP [35]) for this association was 9% assuming a maximum likely OR of 1.2 and a prior of 1:10,000, or 1% assuming a prior of 1:1000). This SNP was also associated with risk of high-grade serous ovarian cancer (HGSOC) specifically (OR = 14, P = 4.3 × 10−9, BFDP = 24% or 3% assuming a prior of 1:10,000 or 1:1000, respectively).
Table 2:
New genome-wide histotype-specific EOC risk associations in Asian women.
| Histotype | SNP rsID | Chr | Position | Risk Allele | RAF | r2a | OR | LCL | UCL | P-valueb | P-hetc | BFDP (%) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1:10,000 | 1:1000 | ||||||||||||
| CCOC | rs1934363 | 10 | 28,773,478 | C | 0.94 | 0.90 | 11 | 5.3 | 24 | 3.0 × 10−10 | 0.98 | 100 | 100 |
| Serous | rs7748275 | 6 | 3,581,089 | A | 0.16 | 0.97 | 1.4 | 1.2 | 1.5 | 8.7 × 10−9 | 0.19 | 9 | 1 |
| HGSOC | rs7748275 | 6 | 3,581,089 | A | 0.16 | 0.97 | 1.4 | 1.2 | 1.5 | 4.3 × 10−8 | 0.19 | 24 | 3 |
Average imputation r2 across the six datasets;
From analysis of imputed genotyped derived from one-step imputation (see methods);
test for heterogeneity of effect between study strata in OCAC; chr, chromosome; RAF, risk allele frequency; LCL, lower 95% confidence limit; UCL, upper 95% confidence limit; HGSOC, high grade ovarian cancer; CCOC, clear cell ovarian cancer; BFDP, Bayes false discovery probability, assuming a prior of 1:1000 or 1:10,000 and maximum likely true odds ratio of 1.2. Position is genome build 37.
At chromosome 10p12.1, SNP rs1934363 was associated with risk of clear cell ovarian cancer (CCOC) (odds ratio (OR) = 10.9, P = 3.0 × 10−10); however, the Bayesian false-discovery probability (BFDP [35]) for this association was close to 100% (Table 2) strongly suggesting that it is likely to be a false positive result. SNPs rs1934363 and rs7748275 were not significantly associated with EOC risk in Europeans genotyped on the OncoArray (Supplementary Table 3).
We also identified one-hundred and twenty-six loci that showed marginal evidence of risk associations (P-values 1 × 10−5 to 5 × 10−8) (Supplementary Table 4). Twenty-eight of these loci are more likely than not to be true associations based on a BFDP score < 50% for the index SNP (Supplementary Table 5). This included a locus at chromosome 20q11.22, where SNP rs74272064 (kgp7556451) was associated with both HGSOC (OR = 1.27, P = 9.0 × 10−8, BFDP = 3% assuming a prior of 1:1000), and serous EOC (P = 1.5 × 10−7); and a locus at chromosome 7p21.3, where SNP rs10260419 was associated with overall EOC risk (OR = 1.33, P = 1.2 × 10−7, BFDP = 5% assuming a prior of 1:1000); and a locus at chromosome 2q37.3, where SNP rs74917072 was associated with overall risk of invasive EOC (OR = 1.25, P = 4.7 × 10−7, BFDP = 7% assuming a prior of 1:1000).
Finally, we compared EOC risk associations in European and East Asian populations [8]. Of the 28 East Asian risk loci with a BFDP score < 50%, only one showed evidence of an association in European women (rs10260419, P = 3.4 × 10−4 for HGSOC risk) (Supplementary Table 5). However, of the 30 loci previously identified in European women, associations in this East Asian population were in the same direction for 22, 17 of which have a one-sided P-value <0.2. While the P-values for these loci were modest in the Asian population, the BFDPs indicate they are all likely to be true associations (BFDP range 0.5–21%, Supplementary Table 6). The most significant association was for rs10069690 at chromosome 5p15, which lies within an intron of TERT, identified in European subjects (OncoArray East Asian study OR = 1.21, P = 3.18 × 10−4) [18]. To explore the global genetic architecture in the two populations we calculated a polygenic risk score using published European hits (excluding mucinous associations) and found that European risk scores do predict cancer in East Asians (HGSOC OR = 1.76 per unit increase in polygenic risk score, P = 8.6 × 10−6), confirming that European risk loci contribute to EOC risk in the East Asian population. Finally, we compared our results to the previous Han Chinese ovarian cancer GWAS [9]. Neither of the risk regions identified in this report (at 9q22.33 and 10p11.21) were associated with EOC risk in our study (Supplementary Table 7). Conversely, none of the 126 regions identified in our study showed evidence of association in the prior GWAS (Supplementary Table 4).
3.2. Functional annotation of risk variants
We performed functional annotation for the genome-wide significant 6p25.2 risk region and the 28 loci with a BFDP score < 50% (Supplementary Tables 2 & 8). These loci were associated with overall EOC, serous EOC or HGSOC-specific risk. For each region we used imputation-based fine mapping to identify SNP sets that represent the most likely candidate causal risk variants (SNPs with likelihood odds >1:100 of being the causal variant at each locus; see Methods and Supplementary Table 8). In total we identified 1283 candidate causal risk SNPs across the 29 regions. These variants were intersected with catalogues of regulatory biofeature data generated using chromatin immunoprecipitation sequencing (ChIP-seq) of primary EOC tissues and normal EOC precursor cells (fallopian tube secretory epithelial cells (FTSECs) and ovarian surface epithelial cells (OSECs)). The biofeature annotated in the most cell types was active chromatin, indicated by H3K27ac ChIP-seq signal. Other marks included H3K4me1, CCCTC-binding factor (CTCF), associated with insulators, PAX8, a transcription factor overexpressed in many EOCs [36,37], and regions of open chromatin catalogued using Formaldehyde-Assisted Isolation of Regulatory Elements sequencing (FAIRE-seq) [38]. The full list of EOC-relevant epigenetic data sets used is provided in Supplementary Table 2.
Twenty-nine percent of SNPs (368/1283) overlapped one or more biofeatures present in relevant cell types (Supplementary Table 8). SNPs rs6058070 and rs6087592 at chromosome 20q11.22 (linked with r2 = 0.97) coincided with 44 and 43 unique biofeature data sets, respectively. Rs6058070 and rs6087592 show expression quantitative trait locus (eQTL) associations for the MAP1LC3A (microtubule-associated protein 1 light chain 3 alpha) gene in GTEx tissues, but not in EOC related samples (OSEC, FTSEC or HGSOC). At the 6p25.2 locus, out of seven candidate causal SNPs, only one, rs6902488, coincided with biofeatures in EOC-relevant cells, specifically marks of active chromatin (H3K27ac and H3K4me1) present in two independent immortalized OSEC lines [38,39]. SNP rs6902488 lies within a putative enhancer close to the center of a 265 kb intergenic region between SLC22A23 and PXDC1. We used MotifbreakR [40] to predict the function of rs6902488 and found this SNP alters the binding of transcription factors (TFs) STAT3, p300 and ELF1. Stronger binding was associated with the risk-conferring (T) allele for all three TFs (Supplementary Table 9, Fig. 2).
Fig. 2.
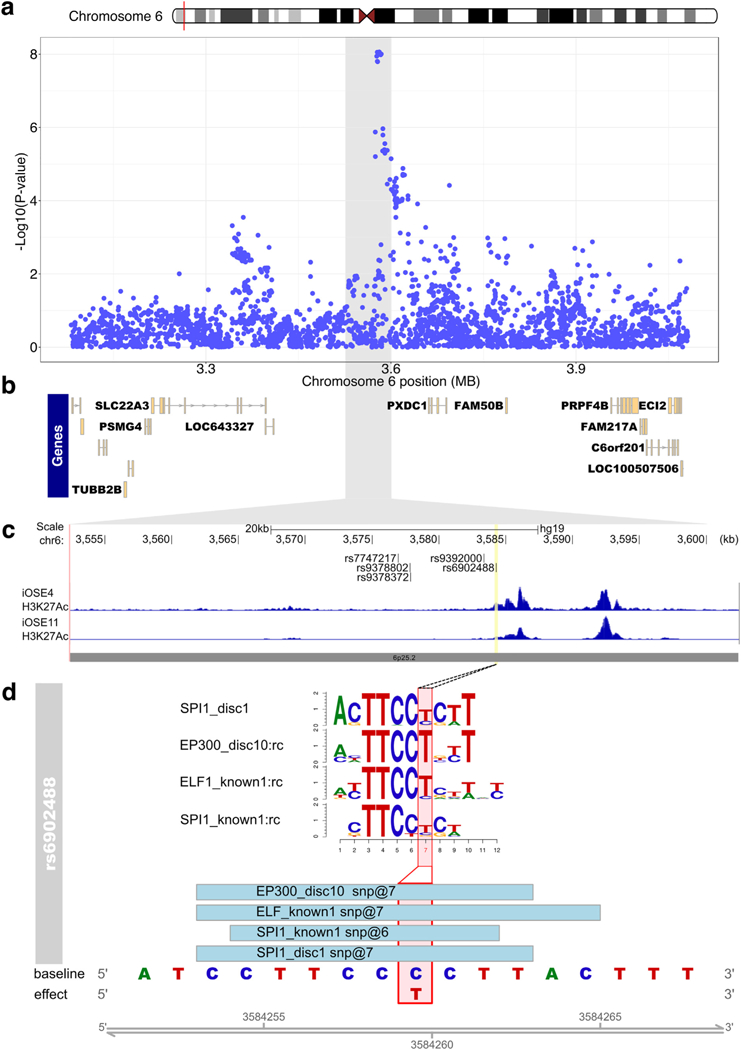
Functional annotation of risk variants at the 6p25.2 risk locus. (a) Regional association plot for serous cancers, centered on rs7748275, with genes in the regions indicated in panel (b). The grey highlighted region corresponds indicates the interval on chromosome 6 in panel a that is shown in panel (c). (c) ChIP-seq in OC-relevant cell types. The locations of the top 6 candidate causal alleles are indicated. (d) SNP rs6902488 significantly alters binding sites for 4 transcription factors. Position weight matrices are shown for each, the height of each letter indicates the importance of that base in the motif.
We also evaluated locus-specific enrichment of tissue specific H3K27ac signals with candidate causal risk SNPs based on the hypothesis that multiple SNPs may be working together to mediate risk (Fig. 3). Variants at chromosome 20q11.21 were enriched in PAX8 binding sites and regions of active chromatin in EOC cell lines and FTSECs; the 9q22.1 risk locus showed strong enrichment of SNPs within active chromatin in primary tumors; and six of the seven risk SNPs at 3p23 were located in CTCF peaks suggesting they may play a role in gene repression. Risk SNPs at the 4p15.33, 21q22.1, 6q26 and 20p11.32 loci also showed enrichment in active and or/poised regulatory elements detected in ovarian cancer relevant cell types.
Fig. 3.
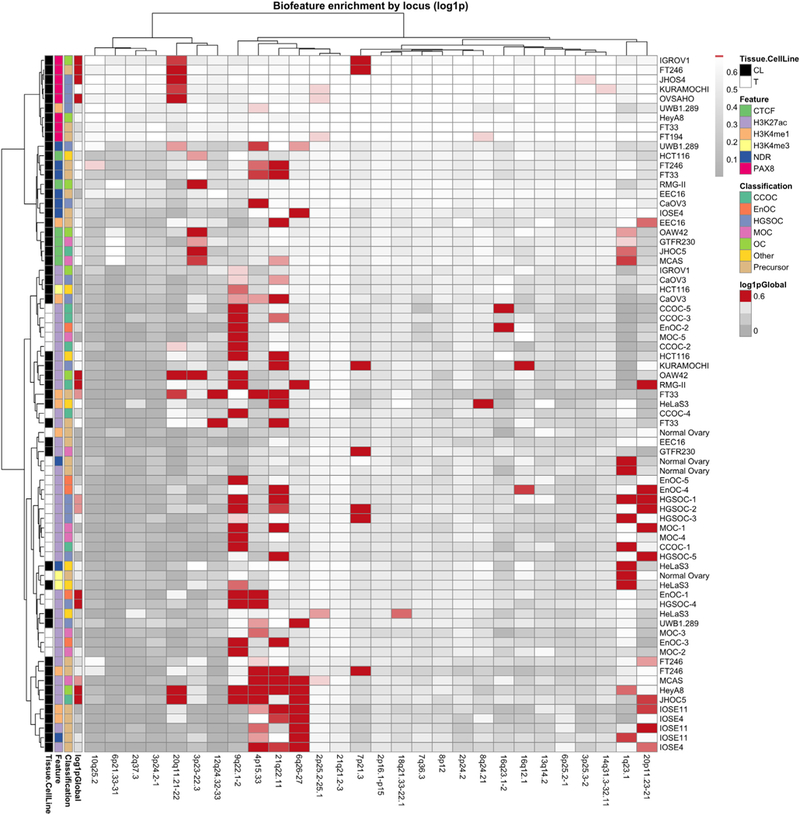
Risk loci can be stratified by biofeature enrichment patterns. All loci with BFDP <0.5 were intersected with 73 biofeature data sets for EOC-relevant cell types. Red color indicates a statistically significant enrichment of SNPs within a certain biofeature (rows) for a particular locus (columns). Global enrichment for all loci collectively is indicated on the far left of the heatmap. Clustering was performed to aggregate loci that exhibit similar patterns of enrichment, using the Ward method. CL, cell line; NDR, nucleosome depleted region; T, tumor.
3.3. Expression quantitative trait locus (eQTL) analyses to identify target genes at risk loci
We performed eQTL analysis using three whole transcriptomic data sets: (1) 404 primary HGSOCs from The Cancer Genome Atlas (TCGA) [32]; (2) 105 primary OSEC samples; and (3) 60 primary FTSEC samples. We regressed out the effects of copy number and methylation on gene expression in tumors [34]. For all three data sets we evaluated associations between the 1283 candidate causal risk SNPs spanning the 29 loci and gene expression for all genes within a 1 Mb window spanning the index SNPs at each locus. We applied a threshold of P < 3.9 × 10−5 for eQTL associations based on correction for testing 1283 SNPs. Where multiple SNPs at the same locus were associated with expression of the same gene due to linkage disequilibrium, we report the strongest associations.
We identified a significant eQTL signal for SNP rs34307842 associated with ESPNL expression at the 2q37.3 locus in primary HGSOCs (PeQTL = 1.2 × 10−7, Fig. 4, Supplementary Table 10). The GT allele of rs34307842 was associated with increased ESPNL expression and increased HGSOC risk. We examined the epigenomic landscape at this locus and identified a putatively functional variant, rs12620528, which is in linkage disequilibrium with an eQTL SNP for ESPNL (rs10929255, r2 = 0.86 in Chinese and Japanese populations) (Fig. 5). This SNP lies within an intron of UBE2F, around 130 kb centromeric to ESPNL. SNP rs12620528 coincides with active chromatin detected in primary ovarian tumors and significantly alters binding motifs for TEAD factors, NFKB and SPI1 (Fig. 5c,d).
Fig. 4.
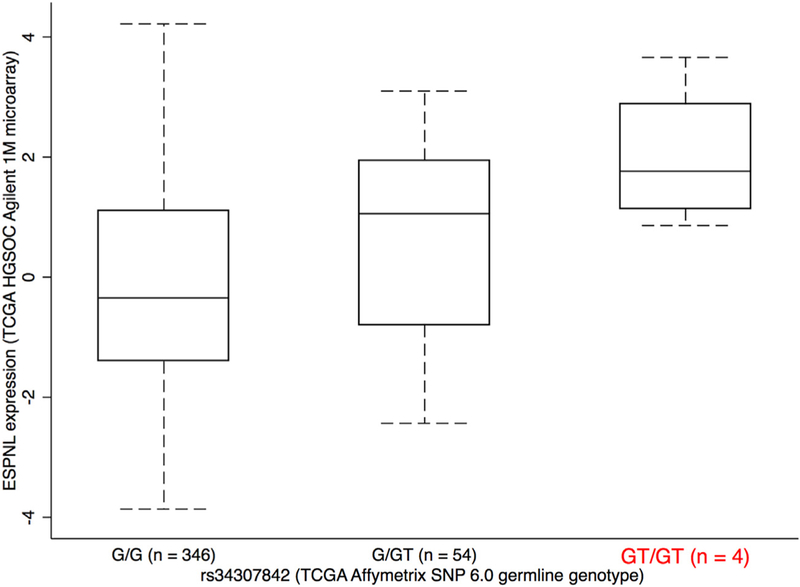
ESPNL expression is associated with rs34307842 genotype. Increased expression is associated with the GT genotype, which is associated with increased risk. Other variants that are also eQTLs for this gene are listed in Supplementary Table 10.
Fig. 5.
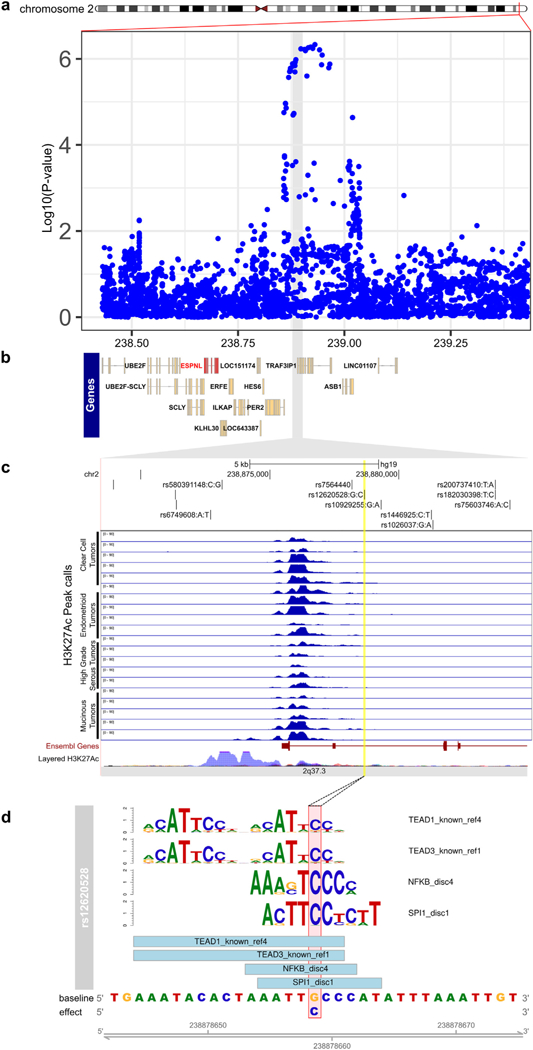
Functional analysis of the 2q37 risk locus. (a) Regional association plot for all invasive cancers, with genes in the regions indicated in panel (b). (c) ChIP-seq in OC-relevant cell types. The locations of the top 6 candidate causal alleles are indicated. (d) SNP rs6902488 significantly alters binding sites for 4 transcription factors. Position weight matrices are shown for each, the height of each letter indicates the importance of that base in the motif.
Significant eQTLs (PeQTL < 3.9 × 10−5) were also identified for 9 genes at 3 additional sub-genome-wide significant risk regions in primary FTSECs and for 41 genes spanning 6 sub-genome-wide significant risk regions in OSECs (Supplementary Tables 11 & 12). Notably, at 6q21.32 the index risk SNP rs72492309 (Prisk = 2.2 × 10−6, BFDP = 26%) was associated with expression of the HLA-DRB1, HLA-DRB6, and HLA-DQB1 genes in both FTSECs and OSECs (PeQTL < 3.9 × 10−5). Other significant associations were either cell-type specific or were not replicated between OSECs and FTSECs (Supplementary Tables 11 and 12).
4. Discussion
This study reports the identification of a novel genome-wide significant locus at chromosome 6p25.2 associated with risk of serous and high-grade serous ovarian cancer in East Asian women. We also identified several candidate EOC risk regions that did not reach statistical thresholds of genome-wide significance but where the Bayesian false discovery probabilities (BFDPs) provided additional evidence that the risk associations may be real. There are several likely reasons why we did not identify additional EOC risk regions at genome-wide levels of significance (P < 5 × 10−8). Firstly, this study included 2981 and 257 subjects diagnosed with invasive and borderline EOC, respectively, and so was not strongly powered to identify risk alleles with this level of statistical stringency. Disease heterogeneity will also have affected our ability to identify risk loci; studies in European subjects have shown that common variant risk regions differ for different ovarian cancer histotypes [8,15,19]. Our power to detect associations for specific histotypes in East Asian women was extremely limited given that this study included few cases of rare histologies.
This study would also have been limited by the content of the genotyping array (OncoArray) – the GWAS backbone was primarily designed for European populations and the custom content was largely based on meta-analyses performed in European subjects. Several studies for other phenotypes have shown that the spectrum of disease risk can vary substantially across different populations. Because data from large-scale genotyping in East Asian populations were not included in the design of the OncoArray, there will be bias against risk alleles that are common in these populations but rare in Europeans. The current study supports other findings that show both similarities and differences in risk variants between East Asian and European populations. None of the most significant EOC risk regions identified in this study were associated with risk in European populations; although of the 30 confirmed EOC risk loci so far reported in Europeans, around two-thirds showed evidence of association in our East Asian study. This includes variants at 8q24, which are associated with the risk of multiple cancer types, including ovarian, breast, prostate and colorectal [17,41]. Variants at 8q24 have been previously implicated in candidate SNP studies for ovarian cancer risk in Asian women [42]; and were nominally significant in both this study (BFDP = 12%) and the previous Han Chinese GWAS (Supplementary Table 5). This study did not replicate findings from a three-staged GWAS analysis of a homogenous population of around 2500 Han Chinese women with ovarian cancer, and ~4000 controls, even though we included women of Han Chinese other Chinese ancestry in our analyses. It may be that the previously reported loci did not replicate as they are only associated with EOC risk in Han Chinese and not across other Asian ethnic groups [9].
Similar to most other GWASs, the majority of risk associated SNPs identified in this study, including all candidate causal SNPs at the 6p25.2 locus, were non-coding, suggesting their functional impact is mediated through non-coding elements that regulate gene expression. We leveraged epigenomic data profiled in primary ovarian tumors and cell types representing the precursors of EOC (OSECs and FTSECs) to identify putative functional targets of risk SNPs. At 6p25.2 a risk SNP rs6902488 is located in a region of active chromatin in OSECs, with the alternative allele at this SNP predicted to strongly enhance the binding of three proteins with a known role in EOC biology. STAT3 is a member of the STAT family of transcription factors and has been posited as a therapeutic target for EOC, particularly in chemoresistant cells [43]. P300 is a transcriptional co-activator known to bind to enhancers in many cell types and works in concert with other transcription factors to orchestrate downstream transcriptional changes. Finally, ELF1 is an ETS domain transcription factor and closely related to ELF4, which has previously been implicated as a transforming factor in EOC [44]. Further functional experiments will be required to evaluate which of these factors play a role in mediating risk at this locus.
It is unlikely that our epigenomic analyses have captured all possible functional noncoding elements. For example, data representing disease specific transcription factors (e.g. WT1) are not available for the major EOC histotypes and their precursor tissues. This may explain why so few of the candidate causal risk variants we identified at 6p25.2 and other risk loci intersect putative functional biofeatures. Future investigations will require more comprehensive analysis of the non-coding architecture of disease relevant tissues to identify the likely causal risk SNPs and target gene(s).
We identified 28 genomic regions where the Bayesian false discovery probability (BFDP) score provided additional evidence that these loci confer susceptibility to EOC even though the risk associations for these regions failed to reach genome-wide significance. Of particular interest is a locus at chromosome 2q37.3 which is associated with overall EOC risk. A BFDP calculation indicated this is likely to be a true association (for the index SNP rs74917072, P = 4.65 × 10−7 and BFDP = 7%). We identified candidate causal risk SNPs at this locus with significant eQTL associations (top P = 1.2 × 10−7) for the Espin-Like (ESPNL) gene using expression data from primary HGSOCs. ESPNL is a little studied gene with homology to Espin (ESPN). Both ESPNL and ESPN are involved in actin bundling in hair cell stereocilia in the inner ear (40) and there is some evidence that both are expressed in the normal fallopian tube in the Human Protein Atlas (proteinatlas.org). ESPNL has not previously been implicated in cancer, but we speculate that it may play a role in EOC cell motility in response to chemical stimuli.
We also explored eQTLs in novel RNA-seq data generated in OSECs and FTSECs. In both cell types we identified significant eQTL associations after Bonferroni-correction between candidate causal risk alleles and Major Histocompatibility Complex, Class II, DR Beta 1 (HLA-DRB1), DR Beta 6 Pseudogene (HLA-DRB6), and DQ Beta 1 (HLA-DQB1) gene expression at a sub-genome-wide significant risk locus at 6q21.32. The index EOC risk SNP (rs72492309) at this locus is 154 kb away from rs2647012, a genome-wide significant index SNP associated with follicular lymphoma risk in Europeans that has previously been shown to have cis-regulatory effects on HLA-DQB1, HLA-DRB1, and HLA-DRB6 expression in lymphoblastoid cell lines [45]. SNP rs72492309 is also 91 kb from rs9272143, a SNP known to be associated with both cervical cancer risk (P = 2.8 × 10−17) and HLA-DRB1 expression (P = 4 × 10−7) [46]. SNPs at this locus are associated at genome-wide significance with inflammatory bowel disease [47], blood pressure [48], autism [49], and Alzheimer’s disease [50]. Class II HLA genes are predominantly expressed by antigen presenting cells in the immune system, but can be expressed by other cell types, albeit at lower levels. HLA proteins are localized to the surface of cells where they present cellular peptides to the immune system to enable identification and clearance of invading pathogens. Clustered somatic mutations in HLA genes in certain cancers point to a role for HLA deregulation during tumorigenesis, and may contribute to immune escape during neoplastic transformation. Mild immune deregulation could feasibly affect cancer susceptibility by modulating tumor suppressive immune pathways.
In summary, this study reports novel EOC susceptibility regions identified in East Asian populations, and a risk spectrum for common variants that includes some population-specific regions. Much larger genetic association studies, based on genetic variance catalogued in East Asian populations are warranted to characterize the full spectrum of risk variation in EOC cases in these populations. Such studies are also needed to define risks associated with different EOC histotypes that may partly explain variations in the clinical presentation of disease in Asian compared to non-Asian populations.
Supplementary Material
HIGHLIGHTS.
This study analyzed genotyping data from >7,000 individuals of Asian descent to find risk loci for epithelial ovarian cancer
We identified two novel genome-wide significant loci, plus evidence of association at an additional 28 regions
eQTL analyses in 404 TCGA tumors highlight ESPNL as a novel susceptibility gene for ovarian cancer
OCAC Acknowledgements
We thank study participants, doctors, nurses, clinical and scientific collaborators, health care providers and health information sources who have contributed to the constituent studies.
We also thank other OCAC members who provided 50 samples or fewer to the current analysis: James Brenton (CAM), Ralf Butzow (HOC), Florian Heitz (HSK, ICN), Graham Giles (MCC), Michelle Hildebrandt (MDA), Line Bjorge (NOR), Laus Hogdall (PVD), Susana Banerjee (RMH), Kristen Moysich (RPC), Peter Fasching (BAV), Digna Velez Edwards (BVU), Joellen Schildkraut (DKE and NCO), Jenny Chang-Claude (GER), Drakoulis Yannoukakos (GRC), Thilo Doerk (HMO), Francesmary Modugno (HOP), Bart Kiemeney (NTH), Diether Lambrechts (BEL), Javier Benitez (CNI), Renee Fortner (EPC), Meir Stampfer (NHS), Tanja Pejovic (ORE), Usha Menon (UKO), Penny Webb (OPL), Taymaa May (UHN), Daniel Cramer (NEC), Anna Defazio (WMH), Dale P. Sandler (SIS), Harvey A. Risch (TOR), Darya Prokofieva (HUO), Beth Y. Karlan (LAX), Douglas Levine (MSK), David Huntsman (VAN), Hoda Anton-Culver (UCI), V. Wendy Setiawan (MEC), Nicolas Wentzensen (PLC).
The Australian Ovarian Cancer Study (AOCS) Management Group (D. Bowtell, A. deFazio, D. Gertig, A. Green, P. Webb); ACS Investigators (A. Green, P. Parsons, N. Hayward, P. Webb, D. Whiteman) and collaborators (see http://www.aocstudy.org) (ACS). Gilian Peuteman, Thomas Van Brussel, Annick Van den Broeck and Joke De Roover for technical assistance (BEL); The dataset(s) used for the analyses described for BioVU were obtained from Vanderbilt University Medical Center’s BioVU which is supported by institutional funding, the 1S10RR025141–01 instrumentation award, and by the Vanderbilt CTSA grant UL1TR000445 from NCATS/NIH. (BVU); CRUK; the University of Cambridge; NIHR Cambridge Biomedical Research Centre (CAM); the Australian Cancer Database (MCC); The Total Cancer Care™ Protocol and the Collaborative Data Services and Tissue Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292), Merck Pharmaceuticals and the state of Florida (MOF). The following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, and WY (NHS). Caroline Baynes and Don Conroy (SEA); I. Jacobs, M. Widschwendter, E. Wozniak, A. Ryan, J. Ford and N. Balogun (UKO); Gynecological Oncology Biobank at Westmead, a member of the Australasian Biospecimen Network-Oncology group (WMH).
We also wish to pay tribute to the contribution of Professor Brian Henderson to the GAME-ON consortium.
Funding
The OCAC OncoArray genotyping project was funded through grants from the U.S. National Institutes of Health (NIH) (CA1X01HG007491–01, U19-CA148112, R01-CA149429 and R01-CA058598); Canadian Institutes of Health Research (MOP-86727) and the Ovarian Cancer Research Fund (OCRF).
Genotyping of the iCOGS array was funded by the European Union [HEALTH-F2–2009-223175], Cancer Research UK [C1287/A10710, C1287/A10118, C12292/A11174], NIH grants (R01 CA114343, R01 CA149429, R01 CA176016, R01 CA128978, R01 CA116167, R01 CA176785 and R01-CA058598), the GAME-ON initiative (1U19 CA148537,1U19 CA148065 and 1U19 CA148112), an NCI Specialized Program of Research Excellence (SPORE) in Breast Cancer (P50 CA116201), the Canadian Institutes of Health Research (CIHR) (MOP-86727), the CIHR Team in Familial Risks of Breast Cancer, the Ministère de [‘Économie, Innovation et Exportation du Québec (#PSR-SIIRI-701), Komen Foundation for the Cure, the Breast Cancer Research Foundation (BCRF), the OCRF and National Health and Medical Research Council of Australia grant #1017028.
Funding for the US GWAS and Mayo GWAS was provided by NIH R01 CA114343 and U19-CA148112. The UK GWAS genotyping and data analysis were supported by Cancer Research UK (C490/A8339) and the Wellcome Trust (076113). The expression Quantitative Trait Locus analyses results published here are in part also based upon data generated by The Cancer Genome Atlas (TCGA) Research Network. This work was also supported by the National Natural Science Foundation of China (81320108022, 81502877), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) in China (IRT_14R40), Tianjin Science and Technology Committee Foundation (16JCYBJC26600), National Human Genetic Resources Sharing Service Platform (2005DKA21300), The National Key Research and Development program of China: The Net construction of human genetic resource Bio-bank in North China (2016YFC1201703).
The OCAC database management is supported by a grant from the OCRF thanks to donations by the family and friends of Kathryn Sladek Smith (PPD/RPCI.07).
Funding for individual studies are provided by ULTR000445 (BVU); R01-CA112523, R01-CA087538, (DOV); R01-CA058598, N01-CN-55424, N01-PC-67001 (HAW); K07-CA080668, R01-CA095023, P50-CA159981, NIH/National Center for Research Resources/General Clinical Research Center grant MO1-RR000056 (HOP); National Center for Advancing Translational Sciences (NCATS) UL1TR000124 (LAX); R01-CA122443, P30-CA15083, P50-CA136393 (MAC/MAY); R01-CA054281, R01-CA164973, R01-CA063464 (MEC); R01-CA114343 (MOF); R01-CA076016 (NCO); R01-CA054419, P50-CA105009 (NEC); R01-CA067262, UM1 CA176726, UM1 CA186107, P01 CA087969, R01 CA049449 (NHS); R01CA160669 (OVA); P50 CA159981, R01CA126841 (RPC); National Institute of Environmental Health Sciences (NIEHS) (Z01 ES044005) (SIS); U01-CA071966, R01-CA016056, U01-CA069417 (STA); R01 CA063678, R01 CA063682 (TOR); R01-CA058860 (UCI); P01CA17054, P30CA014089, R01CA061132, N01PC67010, R03CA113148, R03CA115195, N01CN025403 (USC).
A study acronym JPN was supported in part by Grants-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology, consisting of Priority Areas of Cancer (No. 17015018), Innovative Areas (No. 221S0001), JSPS KAKENHI Grant (No. 16H06277 and 26253041) and Grant-in-Aid for the Third Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare. The authors thank study participants and research staff of the Shanghai Women’s Health Study (SWHS) for their contributions and commitment to this project. The SWHS is supported in part by National Cancer Institute grants (U.S.A) UM1 CA182910.
U.S. Department of Defense (DoD) Congressionally Directed Medical Research Program (CDMRP): DAMD17–01-1–0729 (AUS); DAMD17–02-1–0669 (HOP); W81XWH-07–0449 (MDA); DAMD17–02-1–0666 (NCO); W81XWH-10–1-02802 (NEC).
American Cancer Society (ACS) Early Detection Professorship (SIOP-06–258-01-COUN) (LAX); ACS (CRTG-00–196-01-CCE); Ovarian Cancer Research Fund (OCRF) (DKE); Roswell Park Cancer Institute Alliance Foundation (RPC); Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; Fraternal Order of Eagles (MAC/MAY); OHSU Foundation (ORE); Intramural Research Program of the National Cancer Institute (SIS); Lon V Smith Foundation grant LVS-39420 (UCI); California Cancer Research Program (0001389 V-20170, 2110200) (USC). KL is funded by a K99/R00 Pathway to Independence Award from the NCI (R00CA184415). Additional support came from Sao Paulo Research Foundation (FAPESP) grants: 2015/07925–5, 2017/08211–1 (MASF, FS, HN) and institutional grants from Henry Ford Hospital (HN).
Breast Cancer Now, Institute of Cancer Research (ICR) and NHS funding to the National Institutes of Health Research (N1HR) Biomedical Research Centre (BGS); Cambridge N1HR Biomedical Research Centre and Cancer Research UK (CRUK) Cambridge Cancer Centre (CAM and SEA); CRUK C490/A10119, C490/A10124 (SEA); The Eve Appeal (The Oak Foundation) and N1HR University College London Hospitals Biomedical Research Centre (UKO); CRUK, Royal Marsden Hospital (RMH).
National Health & Medical Research Council (NHMRC) of Australia 199600 and 400281 (AUS, AOCS), 209057, 251533, 396414 and 504715 (MCC), RBH, APP1025142 (OPL) and 310670 and 628903 (WMH); Cancer Councils of New South Wales, Victoria, Queensland, South Australia and Tasmania, and Cancer Foundation of Western Australia under Multi-State Application Numbers 191, 211 and 182 (AUS/AOCS); Cancer Council Victoria (MCC); Brisbane Women’s Club (OPL); Cancer Institute NSW 11/TRC/1–06 and 12/R1G/1–17 (WMH). GCT & PW are supported by Fellowships from NHMRC. KAP is an Australian National Breast Cancer Foundation Practitioner Fellow.
Canadian Institutes of Health Research grant (MOP-86727) (OVA); Princess Margaret Cancer Centre Foundation-Bridge for the Cure (UHN); BC Cancer Foundation, VGH & UBC Hospital Foundation (VAN).
ELAN Funds of the University of Erlangen-Nuremberg (BAV); Nationaal Kankerplan (BEL); Instituto de Salud Carlos 111 (PI 12/01319); Ministerio de Economía y Competitividad (SAF2012–35779) (CN1); German Federal Ministry of Education and Research, Programme of Clinical Biomedical Research (01 GB 9401) and the German Cancer Research Center (DKFZ) (GER); European Union (European Social Fund - ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) - Research Funding Program of the General Secretariat for Research & Technology: SYN11_10_19 NBCA (GRC); Helsinki University Research Fund (HOC); Rudolf-Bartling Foundation (HMO and HUO); Radboud University Medical Centre (NTH); Herlev Hospitals Forskningsråd, Direktør Jacob Madsens og Hustru Olga Madsens fond, Arvid Nilssons fond, Gangsted fonden, Herlev Hospitals Forskningsråd and Danish Cancer Society (PVD).
The coordination of EPIC (EPC) is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer (IARC). The national cohorts are supported by Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); ERC-2009-AdG232997 and Nordforsk, Nordic Centre of Excellence Programme on Food, Nutrition and Health (Norway); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Swedish Cancer Society, Swedish Research Council and County Councils of Skåne and Västerbotten (Sweden); CRUK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (MRC) (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom).
Footnotes
All authors read and approved the final manuscript.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ygyno.2019.02.023.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- [1].Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- [2].Adami HO, Hsieh CC, Lambe M, Trichopoulos D, Leon D, Persson I, et al. Parity, age at first childbirth, and risk of ovarian cancer. Lancet 1994;344:1250–4. [DOI] [PubMed] [Google Scholar]
- [3].Collaborative Group on Epidemiological Studies of Ovarian CancerV. Beral, Doll R, Hermon C, Peto R, Reeves G, Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls, Lancet 371 (2008) 303–314, 10.1016/S0140-6736(08)60167-1. [DOI] [PubMed] [Google Scholar]
- [4].Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- [5].Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995;378:789–92. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- [6].Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The breast cancer linkage consortium. Am. J. Hum. Genet. 1998;62:676–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jones MR, Kamara D, Karlan BY, Pharoah PDP, Gayther SA, Genetic epidemiology of ovarian cancer and prospects for polygenic risk prediction, Gynecol. Oncol. 147 (2017) 705–713, 10.1016/j.ygyno.2017.10.001. [DOI] [PubMed] [Google Scholar]
- [8].Phelan CM, Kuchenbaecker KB, Tyrer JP, Kar SP, Lawrenson K, Winham SJ, et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017;49:680–91. doi: 10.1038/ng.3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen K, Ma H, Li L, Zang R, Wang C, Song F, et al. Genome-wide association study identifies new susceptibility loci for epithelial ovarian cancer in Han Chinese women. Nat. Commun. 2014;5:4682. doi: 10.1038/ncomms5682. [DOI] [PubMed] [Google Scholar]
- [10].Permuth-Wey J, Lawrenson K, Shen HC, Velkova A, Tyrer JP, Chen Z, et al. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat. Commun. 2013;4:1627. doi: 10.1038/ncomms2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 2015; 47:164–71. doi: 10.1038/ng.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bolton KL, Tyrer J, Song H, Ramus SJ, Notaridou M, Jones C, et al. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat. Genet. 2010;42:880–4. doi: 10.1038/ng.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kar SP, Beesley J, Amin Al Olama A, Michailidou K, Tyrer J, Kote-Jarai Zs, et al. Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov 2016;6:1052–67. doi: 10.1158/2159-8290.CD-15-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lawrenson K, Iversen ES, Tyrer J, Weber RP, Concannon P, Hazelett DJ, et al. Common variants at the CHEK2 gene locus and risk of epithelial ovarian cancer. Carcinogenesis 2015;36:1341–53. doi: 10.1093/carcin/bgv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kelemen LE, Lawrenson K, Tyrer J, Li Q, Lee JM, Seo J-H, et al. Genome-wide significant risk associations for mucinous ovarian carcinoma. Nat. Genet. 2015;47:888–97. doi: 10.1038/ng.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pharoah PDP, Tsai Y-Y, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat. Genet. 2013;45:362–70, 370e1. doi: 10.1038/ng.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goode EL, Chenevix-Trench G, Song H, Ramus SJ, Notaridou M, Lawrenson K, et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet. 2010;42:874–9. doi: 10.1038/ng.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 2013;45:371–84, 384e1. doi: 10.1038/ng.2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen H, Fridley BL, Song H, Lawrenson K, Cunningham JM, Ramus SJ, et al. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat. Commun. 2013;4:1628. doi: 10.1038/ncomms2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wu AH, Pearce CL, Tseng C-C, Pike MC, African Americans and Hispanics remain at lower risk of ovarian cancer than non-Hispanic whites after considering nongenetic risk factors and oophorectomy rates, Cancer Epidemiol. Biomark. Prev. 24 (2015) 1094–1100, 10.1158/1055-9965.EPI-15-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shirley MH, Barnes I, Sayeed S, Finlayson A, Ali R, Incidence of breast and gynaecological cancers by ethnic group in England, 2001–2007: a descriptive study, BMC Cancer 14 (2014) 979, 10.1186/1471-2407-14-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Herrinton LJ, Stanford JL, Schwartz SM, Weiss NS, Ovarian cancer incidence among Asian migrants to the United States and their descendants, J. Natl. Cancer Inst. 86 (1994) 1336–1339. [DOI] [PubMed] [Google Scholar]
- [23].Fuh KC, Shin JY, Kapp DS, Brooks RA, Ueda S, Urban RR, et al. Survival differences of Asian and Caucasian epithelial ovarian cancer patients in the United States. Gynecol. Oncol. 2015;136:491–7. doi: 10.1016/j.ygyno.2014.10.009. [DOI] [PubMed] [Google Scholar]
- [24].Goodman MT, Howe HL, Descriptive epidemiology of ovarian cancer in the United States, 1992–1997, Cancer 97 (2003) 2615–2630, 10.1002/cncr.11339. [DOI] [PubMed] [Google Scholar]
- [25].Tung K-H, Goodman MT, Wu AH, McDuffie K, Wilkens LR, Kolonel LN, et al. Reproductive factors and epithelial ovarian cancer risk by histologic type: a multiethnic case-control study. Am. J. Epidemiol. 2003;158:629–38. [DOI] [PubMed] [Google Scholar]
- [26].Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, et al. The oncoarray consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev. 2017;26:126–35. doi: 10.1158/1055-9965.EPI-16-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Song H, Ramus SJ, Tyrer J, Bolton KL, Gentry-Maharaj A, Wozniak E, et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat. Genet. 2009;41:996–1000. doi: 10.1038/ng.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li Y, Byun J, Cai G, Xiao X, Han Y, Cornelis O, et al. FastPop: a rapid principal component derived method to infer intercontinental ancestry using genetic data. BMC Bioinformatics 2016;17:122. doi: 10.1186/s12859-016-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Delaneau O, Coulonges C, Zagury J-F, Shape-IT: new rapid and accurate algorithm for haplotype inference, BMC Bioinformatics 9 (2008) 540, 10.1186/1471-2105-9-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].van Leeuwen EM, Kanterakis A, Deelen P, Kattenberg MV, Genome of the Netherlands Consortium, Slagboom PE, et al. , Population-specific genotype imputations using minimac or IMPUTE2, Nat. Protoc. 10 (2015) 1285–1296, 10.1038/nprot.2015.077. [DOI] [PubMed] [Google Scholar]
- [32].Cancer Genome Atlas Research Network, Integrated genomic analyses of ovarian carcinoma, Nature 474 (2011) 609–615, 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shabalin AA, Matrix eQTL: ultra fast eQTL analysis via large matrix operations, Bioinformatics 28 (2012) 1353–1358, 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li Q, Seo J-H, Stranger B, McKenna A, Pe’er I, Laframboise T, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013;152:633–41. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wakefield J, A Bayesian measure of the probability of false discovery in genetic epidemiology studies, Am. J. Hum. Genet. 81 (2007) 208–227, 10.1086/519024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Adler EK, Corona RI, Lee JM, Rodriguez-Malave N, Mhawech-Fauceglia P, Sowter H, et al. The PAX8 cistrome in epithelial ovarian cancer. Oncotarget 2017;8:108316–32. doi: 10.18632/oncotarget.22718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Elias KM, Emori MM, Westerling T, Long H, Budina-Kolomets A, Li F, et al. Epigenetic remodeling regulates transcriptional changes between ovarian cancer and benign precursors. JCI Insight 2016;1. doi: 10.1172/jci.insight.87988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Coetzee SG, Shen HC, Hazelett DJ, Lawrenson K, Kuchenbaecker K, Tyrer J, et al. Cell-type-specific enrichment of risk-associated regulatory elements at ovarian cancer susceptibility loci. Hum. Mol. Genet. 2015;24:3595–607. doi: 10.1093/hmg/ddv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lawrenson K, Sproul D, Grun B, Notaridou M, Benjamin E, Jacobs IJ, et al. Modelling genetic and clinical heterogeneity in epithelial ovarian cancers. Carcinogenesis 2011;32:1540–9. doi: 10.1093/carcin/bgr140. [DOI] [PubMed] [Google Scholar]
- [40].Coetzee SG, Coetzee GA, Hazelett DJ, motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites, Bioinformatics 31 (2015) 3847–3849, 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ghoussaini M, Song H, Koessler T, Al Olama AA, Kote-Jarai Z, Driver KE, et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J. Natl. Cancer Inst. 2008;100:962–6. doi: 10.1093/jnci/djn190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Han J, Zhou J, Yuan H, Zhu L, Ma H, Hang D, et al. Genetic variants within the cancer susceptibility region 8q24 and ovarian cancer risk in Han Chinese women. Oncotarget 2017;8:36462–8. doi: 10.18632/oncotarget.16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Duan Z, Foster R, Bell DA, Mahoney J, Wolak K, Vaidya A, et al. Signal transducers and activators of transcription 3 pathway activation in drug-resistant ovarian cancer. Clin. Cancer Res. 2006;12:5055–63. doi: 10.1158/1078-0432.CCR-06-0861. [DOI] [PubMed] [Google Scholar]
- [44].Ando M, Kawazu M, Ueno T, Koinuma D, Ando K, Koya J, et al. Mutational landscape and antiproliferative functions of ELF transcription factors in human cancer. Cancer Res. 2016;76:1814–24. doi: 10.1158/0008-5472.CAN-14-3816. [DOI] [PubMed] [Google Scholar]
- [45].Smedby KE, Foo JN, Skibola CF, Darabi H, Conde L, Hjalgrim H, et al. GWAS of follicular lymphoma reveals allelic heterogeneity at 6p21.32 and suggests shared genetic susceptibility with diffuse large B-cell lymphoma. PLoS Genet. 2011;7:e1001378. doi: 10.1371/journal.pgen.1001378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen D, Gyllensten U, A cis-eQTL of HLA-DRB1 and a frameshift mutation of MICA contribute to the pattern of association of HLA alleles with cervical cancer, Cancer Med 3 (2014) 445–452, 10.1002/cam4.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Goyette P, Boucher G, Mallon D, Ellinghaus E, Jostins L, Huang H, et al. High-density mapping of the MHC identifies a shared role for HLA-DRB1*01:03 in inflammatory bowel diseases and heterozygous advantage in ulcerative colitis. Nat. Genet. 2015; 47:172–9. doi: 10.1038/ng.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Warren HR, Evangelou E, Cabrera CP, Gao H, Ren M, Mifsud B, et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat. Genet. 2017;49:403–15. doi: 10.1038/ng.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium, Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia, Mol Autism 8 (2017) 21, 10.1186/s13229-017-0137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014;10:e1004606. doi: 10.1371/journal.pgen.1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary results for all imputed SNPs reported in this paper are available at: https://doi.org/10.17863/CAM.25845