

Abstract
Eukaryotic RNAs are heavily processed including co- and post-transcriptional formation of various 5′ caps. In snRNAs and snoRNAs the canonical 7mG cap is hypermethylated at the N2-position while in higher eukaryotes and viruses 2′-O-methylation of the first transcribed nucleotide yields the cap1 structure. The function and potential dynamics of several RNA-cap modifications has not been fully elucidated, necessitating preparative access to these caps. However, introduction of these modifications during chemical solid-phase synthesis is challenging and enzymatic production of defined short and uniform RNAs also faces difficulties. In this work, we combined chemical synthesis of RNA with site-specific enzymatic methylation using the methyltransferases hTGS1, GlaTgs2 and CMTR1. We show that RNAs with di-and trimethylated caps as well as RNAs with caps methylated at the 2′-O-position of the first transcribed nucleotide can be conveniently prepared. These highly modified RNAs with a defined and uniform sequence are hard to access by in vitro transcription or chemical synthesis alone.
Keywords: AdoMet, SAM, cap, snRNA, RNA
Graphical Abstract
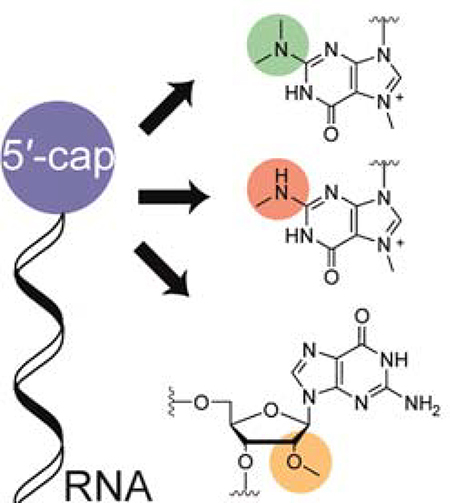
Enzymatic production of various RNA 5′-caps: For the production of capped RNAs we combined chemical and enzymatic synthesis with site-specific methyltransferases to obtain highly cap-modified RNAs. We hereby offer a facile and convenient route for the large scale production of these specific transcripts, ultimately facilitating research on RNA function and dynamics.
Introduction
The RNA 5′ cap is a hallmark of transcripts made by the eukaryotic RNA-polymerase II and consists of an N7-methyl guanosine linked to the first transcribed nucleotide via a unique 5′−5′ triphosphate bridge. It enhances stability by protecting against degradation by exoribonucleases and provides a platform for numerous RNA-protein interactions underlying processes such as splicing, export, translation initiation and degradation.[1–8] Furthermore, capped RNAs serve as a marker for the innate immune system to distinguish endogenous from pathogenic RNAs.[9]
The RNA cap can be subject to additional methylations at various distinct sites. In small nuclear RNAs (snRNAs) or small nucleolar RNAs (snoRNAs) the N2-position of the terminal guanosine is dimethylated resulting in formation of a 2,2,7-trimethylguanosine (TMG or m3G) cap.[10] The human trimethylguanosinesynthase 1 (hTgs1) converts N7-methyl G caps into hypermethylated TMG-caps in the cytoplasm, and only TMG-capped snRNAs are reimported into the nucleus where they are assembled into spliceosomes.[11–13] In Giardia lamblia, a homologous TGS (GlaTgs2) transfers only a single methyl group onto the N2-position, however its endogenous function is still unknown.[14,15]
Additional cap modifications in eukaryotic RNAs comprise methylations at the 2′-O-position of the first and/or second transcribed nucleotide, yielding the cap1 or cap2 structure, respectively.[16,17] In trypanosomes, methylation of up to four nucleotides has been observed in the so-called splice-leader RNA (cap4).[18,19] Formation of cap1 and cap2 structures in humans are catalyzed by CMTR1 and CMTR2, respectively.[20,21] In contrast to previous results, recent findings suggest that the 2′-O-methylation is constitutive, however its precise function remains to be elucidated.[22] While 2′-O-methylation was shown to have an enhancing effect on ribosome binding and translation, Belanger et al. reported that knockdown of CMTR1 did not impair overall cell viability in vivo.[20]
As recently discovered, further modifications can include methylation at the N6-position if the first transcribed nucleotide is an adenosine in mRNAs and snRNAs. This modification occurs only on 2′-O-methylated RNAs, yielding 6mAm, and was theorized to happen co-transcriptionally upon cap1 formation and to influence mRNA stability.[22,23] In snRNAs, recent findings suggest that the methylation at the N6-position of the first 2′-O-methyl adenosine is dynamic, indicating a regulatory function of 6mAm in splicing or other snRNA-dependent mRNA processing reactions.[24]
While all modifications mentioned above are observed on a phenomenological level and many of the enzymes installing them are known, their biogenesis, dynamics and endogenous functions are not fully elucidated. Difficulties associated with the large scale production of these cap-modified RNAs limit the availability and hence research regarding their endogenous functions.
In these two last decades, the synthesis of 5′-capped RNAs has been achieved following different routes based on enzymatic, chemical or chemo-enzymatic methods.[25] Each strategy presents advantages and limitations, and in particular the production of 7mGppp-RNAs of defined sequences and in great amounts is a bottleneck for their use as tools for mechanistic and structural studies of RNA world. Therefore, some years ago we developed a method using the combination of chemical synthesis of short Gppp-RNAs followed by enzymatic N7-methylation of guanine in the cap structure.[26] Thus great quantities of 5′−7mGppp-RNAs of various lengths (from 2 to 21 nt), of any sequence and carrying different cap structures (cap, cap0, cap1) were obtained with high purity. It should be noted that the incorporation of the 2′-O-methyl nucleosides is possible via solid-phase synthesis as corresponding phosphoramidites are readily available, yielding easy access to cap1 RNAs (5′−7mGpppNm-RNAs). In contrast, the final site-specific N2-mono- and di-methylation of cap RNAs proved difficult by chemical methylation agents which are not exclusively specific for the G in the cap and would also methylate guanosines within the RNA sequence. Nevertheless, using diverse synthetic strategies several groups succeeded in the chemical synthesis of 5′-terminal TMG-capped RNAs. First, Sekine et al. described the condensation of the m3G 5′-monophosphate with an appropriately protected tri-ribonucleotide 5′-diphosphate prepared in solution and later on solid-support.[27,28] Several years later, Stromberg et al. proposed a modified synthesis of a m3G 5′-diphosphate which was used in reaction with 5′-monophosphate RNAs.[29] Another capping method was the attachment of m3G cap to RNAs via click chemistry.[30,31] The corresponding conjugates were obtained by coupling an azido-functionalized m3G-cap with the activated triple bond linker on the RNA in solution. However, it is noteworthy that the synthesis of the m3G-capping reagent from G requires a tedious series of reactions involving at least four steps. In our work aiming at TMG-capped RNAs and following the same strategy as for the synthesis of Gppp-RNAs,[26] another way would be to react N2-dimethyl GDP with the chemically synthesized short RNAs instead of using the standard commercially available GDP. However, the lack of convenient availability of 2m,2mGDP hampers use of this capping reagent therefore another strategy combining chemical and enzymatic reactions was preferred.
On the other hand, the enzymatic preparation of specific cap-modified RNAs via in vitro run-off transcription starting from a cap-analog is common for long 7mGpppG-RNAs. mRNAs with >1000 nt length can be readily made, whereas solid-phase synthesis is limited to a length of approximately 200 nucleotides. However, for more specialized caps and RNAs where the first transcribed nucleotide is not a G, enzymatic production is often not possible, due to lack of the respective cap analog or limited transcription yields (T7 RNA polymerase prefers to start with a G). The fact that T7 polymerase can add additional nucleotides at the 3′ end yielding non-uniform products hampers application for production of short RNAs.
In this study, we utilized enzymatic strategies for the production of higher cap-structures, starting from either chemically or enzymatically produced RNAs bearing the N7-methyl-G cap (Scheme 1). For the preparation of 2,7-dimethyl- or 2,2,7-trimethylguanosine capped RNAs in preparative scale from chemically synthesized RNAs, the enzymes GlaTgs2 or hTgs1 were used, respectively. For generation of the cap1 structure, the enzyme CMTR1 was employed, acting on either chemically or enzymatically prepared RNAs. Here, exploiting the human 2′-O-MTase CMTR1 for the post-transcriptional cap1 production represents an interesting and more cost-effective alternative to commercial cap analogs, which are often associated with low yields. While the co-substrate specificities of hTgs1 and GlaTgs2 were investigated in previous studies, we were curious to whether CMTR1 is able to transfer non-natural moieties from AdoMet-analogs.[32]
Scheme 1.

Schematic representation of 5′-cap methylation by the means of different methyltransferases. RNA equipped with a cap0 structure, prepared either chemically or enzymatically, can be further methylated by cap-modifying methyltransferases. The N2-methyltransferases GlaTgs2 and hTgs1convert the N7-methyl guanosine into the di-or tri-methyl mRNA caps 2m,7mGppp or 2m,2m,7mGppp, respectively. The first transcribed nucleotide can be methylated at its 2′-position by the human 2′-O-methyltransferase CMTR1.
Results and Discussion
Here we used the same route for the synthesis of 5′−7mGppp-RNAs 1–4 (Table 1) as previously described.[26] Chemical synthesis of the corresponding RNA sequences was performed on solid-support with the commercially available 2′-O-pivaloyloxymethyl (PivOM) phosphoramidites based on a method developed by our group.[33] The 5′-terminal adenosine of RNA sequences was either an unmodified A (1,2), methylated at the 2′-OH (Am) (3), or methylated at the 2′-OH and N6 position (6mAm) (4). The commercial 2′-O-Me adenosine phosphoramidite was used to introduce Am at the 5′-end of RNA (3). For the synthesis of 6mAm-RNA (4), the preparation of a 6mAm phosphoramidite building block was performed by a selective one-step N6-methylation of the 2′-O-Me A phosphoramidite.[34] After RNA assembly completion, the 5′-OH of each RNA bound to solid support was converted in 5′-H-phosphonate which was subsequently oxidized in 5′-phosphoromidazolidate ready for coupling with guanosine diphosphate (GDP) to give the solid-supported 5′-Gppp-RNAs. All these reactions were performed on solid-phase enabling the removal of excess reagents and making the synthesis more convenient. The capping reaction with the commercial GDP was derived from a reaction on solid-support with pyrophosphate to afford 5′-triphosphate DNA or RNA with good yields and fair purity.[35] Then, a last deprotection step with a simple aqueous ammonia treatment for 3 h at room temperature is required to release 5′-Gppp-RNAs from the support. Finally, after purification by anion-exchange HPLC (Figures S1–S4), the 5′-Gppp-RNAs were submitted to purified recombinant human (guanine N7)-methyl transferase (N7-hMTase) in the presence of S-adenosyl-l-methionine (SAM or AdoMet) to yield 7mGppp-RNAs 1–4 as pure compounds after a simple filtration through cartridges (Figure 1).[26]
Table 1.
Data for 7mGppp-RNAs 1–4 and RNAs with hypermethylated caps 5–7.
| N° | RNA 5′-sequences-3′ | Length | Calc (m/z) [a] | Found[a] |
|---|---|---|---|---|
| 1 | 7mGpppAUAU | 4-mer | 1726.97 | 1726.97 |
| 2 | 7mGpppAGUUGUUAGUCUACGUGG | 18-mer | 6268.63 | 6268.90 |
| 3 | 7mGpppAmGUUGUUAGUCUACGUGGA | 19-mer | 6611.86 | 6612.97 |
| 4 | 7mGppp6mAmCACUUGCUUUUGACACAACU | 21-mer | 7124.21 | 7123.15 |
| 5 | 2m,7mGpppAUAU | 4-mer | 1741.00 | 1741.26 |
| 6 | 2m,2m,7mGpppAUAU | 4-mer | 1755.02 | 1755.22 |
| 7 | 2m,2m,7mGppp6mAmCACUUGCUUUUGACACAACU | 21-mer | 7152.26 | 7152.32 |
| 8 | 7mGpppGAUC | 4-mer | 1741.99 | 1741.70 |
MALDI-TOF characterization in negative mode [M+-2H]−
Figure 1.

Analyses of synthesized 7mGpppRNAs 1-4: RP-HPLC profiles and corresponding MALDI-TOF mass spectra (insets). Sequences are indicated above the respective RP-HPLC chromatograms.
Enzymatic mono N2-methylation with GlaTgs and double N2-methylation with hTgs1
First attempts of N2-methylation were performed with 7mGpppAUAU 1 as a model to assay the conditions of the reaction for both methyltransferases GlaTgs and hTgs1. 7mGppp-RNA 1 (150 μM) was incubated with 500 μM of SAM, 5 μM of GlaTgs or 5 μM of hTgs1 in reaction buffer (50 mM Tris-HCl, 5 mM DTT, 50 mM NaCl, pH 8).[36] The enzymatic methylations were monitored at 260 nm by reversed-phase HPLC (RP-HPLC).
Using the described enzymatic system, we typically observed quantitative N2-methylation and double N2-methylation efficiencies after an incubation time of 2 h at 37 °C with GlaTgs or hTgS1, respectively (Figure 2). In the RP-HPLC chromatograms, the order of elution for the three different methylated species was in agreement with the increasing number of methyl groups in the guanosine of the cap. The retention time of 2m,7mGpppAUAU 5 (Rt=7.33 min) was evidently higher than the N7-methylated substrate 1 (Rt=6.88 min) but lower than the trimethylated 2m,2m,7mGpppAUAU 6 (Rt=7.66 min). Moreover, the success of the N2-methylations was unambiguously ascertained by MALDI-TOF characterization of 5 and 6 (Table 1).
Figure 2.

A) Reaction scheme of the mono- and dimethylation of the N2-position by GlaTgs2 and hTgs1. B) RP-HPLC analysis of methylation reactions of 7mGpppAUAU after 2 h incubation (black traces) with GlaTgs and C) after 2.5 h incubation with hTgs1 in comparison with the substrate 7mGpppAUAU (red traces). MALDI-TOF mass spectra of 2m,7mGpppAUAU 5 and 2m,2m,7mGpppAUAU 6 are shown in insets. D) RP-HPLC analysis of N2-methylation reactions of 7mGpppA-(N)17-RNA 2 (18-mer) after 6 h incubation with hTgs1 at 2.5 μM (black trace) and 5 μM (blue trace) in comparison with the N7-methylated substrate 2 (red trace).
Enzymatic double N2-methylation of 7mGpppA-(N)17 2 with hTgs1
Next, we tested the double N2-methylation with hTgs1 at two concentrations (2.5 μM and 5 μM) with a longer RNA sequence (2). After 6 h incubation with hTgs1, the double N2-methylation was incomplete at both enzyme concentrations indicating that the reaction was much slower than with a short RNA sequence. It is noteworthy that while the retention times of the RNA 18-mers A-(N)17 bearing three differently methylated caps with 7mG, 2m,7mG and 2m,2m,7mG were very similar in the chromatogram, they could be distinguished but not isolated separately (Figure 2D). The enzyme concentration seems to play an important role in the reaction efficacy since at 5 μM, the final trimethylated 2m,2m,7mGpppA-(N)17 was formed in larger proportions than at 2.5 μM. Nevertheless, in order to preserve enzyme, the assays were subsequently performed with 2.5 μM hTgs1.
Enzymatic double N2-methylation of 7mGpppAm(N)17 3 with hTgs1
To check if a 2′-OMe nucleotide at the 5′-end of an RNA sequence might hamper N2-methylation of G in the cap with hTgs1, the reaction was assayed on 7mGpppAm(N)17 3, testing two different conditions for optimization purposes. First, the substrate 3 was directly used in the reaction mixture containing the remaining SAM and SAH from the previous N7-methylation reaction. Even after 15 h incubation with 2.5 μM hTgs1, the double N2-methylation was not complete, nevertheless the mono- and the double N2-methylated cap RNA could be observed in the HPLC chromatogram showing that a 2′-OMe nucleotide at the 5′-end is not detrimental to enzymatic methylation (Figure S5A).
In the second test, the SAM and SAH from the N7-methylation were removed through a NAP-cartridge by gel exclusion chromatography before the reaction with 2.5 μM hTgs1. After 6 h incubation, even though the double N2-methylation was not complete, the rate of the reaction was faster than in the first test where some SAH was present (Figure S5B). The removal of SAH released from the N7-methylation reaction seems crucial for the success of N2-methylation to completion, in line with reports about SAH being an inhibitor of MTases.[14]
Enzymatic double N2-methylation of 7mGppp6mAm(N)20 4 with hTgs1
We applied the optimized conditions for N2-methylation with hTgs1 to the 7mG-capped RNA 4, which is of special interest due to its double methylated adenosine 6mAm as the first 5′-nucleotide of the RNA sequence. This modified ribonucleotide is found in particular at the transcription-start nucleotide of Sm-class spliceosomal snRNAs. The dimethylated m2-snRNAs are major targets of the RNA demethylase FTO which removes the methyl group from N6 adenosine to give m1-snRNAs.[24]
To determine whether FTO demethylates snRNAs with a mono-methylated cap, 7mGppp6mAm or a tri-methylated cap 2m,2m,7mGppp6mAm we prepared RNA 7 from 4 (Table 1).[24] The double N2-methylation was performed on 4 (150 μM) with hTgs1 (2.5 μM) in using SAM at 1 mM instead of 0.5 mM concentration as previously used. This greater excess of SAM (SAM/SAH ratio > 0.5) should avoid inhibition of the reaction by the released SAH. Indeed, after 7.5 h incubation with hTgs1, the trimethylated cap 2m,2m,7mGppp6mAm(N)20 7 corresponds to the major peak in the HPLC chromatogram and the MALDI-TOF mass analysis ascertained its characterization (Figure 3). This compound 7 was purified by RP-HPLC and 48 nmoles of pure material were isolated and used in experiments investigating FTO demethylation dynamics of snRNAs.[24]
Figure 3.

A) N2-double methylation by hTgs1 of a 7mG-capped RNA (4) with 6mAm as the first nucleotide yielding 7. B) RP-HPLC analysis of double N2-methylation reaction of 7mGppp 21-mer 4 after 7.5 h incubation with hTgs1 at 2.5 μM (black trace) in comparison with the N7-methylated substrate 4 (red trace). C) MALDI-TOF mass spectrum (right panel) of the trimethylated capped 21-mers 2m,2m,7mGppp6mAmCACUUGCUUUUGACACAACU 7.
Enzymatic 2′-O-methylation of RNA by CMTR1
Next, we were interested in installing a 2′-O-methyl group at the 5΄-terminal nucleotide of an RNA sequence, using the corresponding human MTase CMTR1 (Figure 4A). CMTR1 was identified in 2010 by the group of Pelletier and was crystallized by the group of Bujnicki in 2014.[20,37] To this point, formation of the cap1 structure using CMTR1 has only been performed in analytical scale using either radioactive [3H-methyl]-SAM or 32P labeled RNA as a means to characterize the enzyme.[20,37] Here, we use the truncated construct CMTR1160–549, as the full-length protein was reported to be insoluble.[37]
Figure 4.

A) Schematic representation of 2′-O modification by CMTR1 using AdoMet and AdoMet analogs SeAdoYn and AdoEnYn. B) Probing the catalytic efficiency of CMTR1160–549 by incubating the RNA substrate with varying ratios of enzyme. A short model RNA 7mGpppGAUC 8 (1 nmol) was incubated with varying amounts of CMTR160–549, a ten-fold excess of AdoMet and MTAN. Upon reaction, RNA was digested using snake venom phosphodiesterase and alkaline phosphatase and analyzed by RP-HPLC. C) Time course experiment of the in vitro 2′-O-methylation of 7mGpppGAUC by CMTR1160–549. Samples were taken after 0, 5, 10, 15, 30, 60, 90, 120 and 240 minutes and analyzed as described above. D) In vitro methylation assay of synthetic 7mGpppGAUC RNA 8 with CMTR1 using AdoMet, SeAdoYn or AdoEnYn as co-substrates. UV-chromatograms (260 nm, black traces) and the extracted ion-counts (EIC, blue traces) of the methylated (top panel), propargylated (middle panel) and hexenynylated (bottom panel) products are shown. E) In vitro translation experiment of Renilla luciferase RNA capped with the ARCA (3′-O-Me-7mGpppG) cap upon CMTR160–549 2′-O-methylation.
For the CMTR1 methylation assay, a short synthetic model RNA 7mGpppGAUC 8 was prepared by the above mentioned methods. The RNA substrate was incubated with CMTR1160–549, a ten-fold excess of SAM and MTAN for the degradation of S-adenosyl-L-homocysteine. After the reaction, RNA was digested using snake venom phosphodiesterase (SVP) and subsequently dephosphorylated by alkaline phosphatase (AP). As SVP also cleaves the triphosphate bridge of the 5′-cap, single nucleosides could then be analyzed via RP-HPLC or LC-MS. After SVP degradation, formation of a new peak eluting after adenosine was observed in HPLC chromatograms, which was correlated to a decrease in signal intensity of guanosine. Identity of this new peak as 2′-O-methyl guanosine was confirmed via LC-MS (Figure 4D). The extracted ion counts for 2′-OMe-guanosine revealed two peaks. By MS2 fragmentation the first peak was identified as the N7-methyl guanosine which has the same mass as 2′-OMe-G (Figure S6). Encouraged by these results, we investigated whether CMTR1160–549 can also be used to generate the cap1 structure on longer RNAs. To this end, a 24 nt long RNA was produced via in vitro transcription and capped with the Vaccinia capping enzyme. We found, CMTR1160–549 to be active on this longer model RNA too (Figure S7).
Probing CMTR1 efficiency
We were interested to use CMTR1160–549 (Figure S8) to enzymatically convert chemically or enzymatically prepared RNAs with a cap0 into cap1-containing RNAs. To improve the conversion, we optimized the reaction conditions of the biotransformation. To this end, the enzyme to RNA ratio necessary for quantitative conversion was determined (Figure 4B). We found that over 90 minutes at 37 °C, 20 mol% enzyme are sufficient to provide roughly 80% of cap1 RNA. A corresponding time-course experiment with 20 mol% of CMTR1160–549 revealed no further product formation after 90 minutes (Figure 4C).
Next, we investigated the co-substrate specificity of CMTR1160–549. While GlaTgs shows slight promiscuity, the engineered variant GlaTgs2-V34A is known to transfer various alkene and alkyne chains from corresponding SAM (or AdoMet) analogs in near quantitative fashion and even bulkier benzylic moieties – albeit to a lower extent. At the same time, we found hTgs1 to not accept any AdoMet analogs in previous studies.[32] To this end, CMTR1160–549 was incubated with the AdoMet analogs SeAdoYn and AdoEnYn (Figure S9), the synthetic 7mGpppGAUC 8 equipped with a cap0 and MTAN. Reaction progress was monitored via HPLC. While SAM as co-substrate led to formation of a new peak, no new product was detectable when either SeAdoYn or AdoEnYn were used (expected products, Figure S9). This was further confirmed by LC-MS, where the respective products could not be detected (Figure 4D).
Assessing biological activity of CMTR1-treated RNA
To test, whether 2′-O-methylated and capped RNA was still translated, we prepared a >1000 nt mRNA coding for Renilla luciferase. This mRNA was enzymatically produced with an ARCA (3′-O-Me-7mGpppG) cap via in vitro run-off transcription. This RNA was then subjected to CMTR1160–549 mediated cap1 formation, precipitated upon reaction and subjected to in vitro translation. We found that the modified mRNA was still translated (Figure 4E).
In conclusion, we have shown that chemically or enzymatically prepared RNAs equipped with the canonical N7-G cap can be readily converted into m3G-capped, m2G-capped or 2′-O-methylated RNAs, using the MTases hTgs1, GlaTgs2 or CMTR1, respectively. Alternatively, cap1 methyltransferases from the Vaccinia virus can be used either during or after the enzymatic capping reaction. Furthermore, a novel trinucleotide cap analog, equipped with a cap1 structure was recently introduced (CleanCap). However, the corresponding MTases and the trinucleotide cap analog are expensive. Direct production of RNA equipped with the cap1 structure by common in vitro run-off transcription from respective dinucleotide cap-analogs, however, has not been reported to the best of our knowledge.
Our chemo-enzymatic strategy presents a convenient and cost-effective alternative over the use of cap-analogs, which are tedious to synthesize if not commercially available. Mono or di-methylation of the N2 position was shown on N7Gppp-RNAs prepared by chemical synthesis and is independent of sequence or length of the substrate. This allowed access to interesting structural motifs such as hypermethylated cap RNAs with a 6mAm nucleoside in the first position, facilitating research in the methylation and demethylation dynamics of 6mAm in snRNAs.[24] Regarding CMTR1, we found that near-quantitative 2′-O-methylation of the 5′-terminal nucleotide is achieved on RNA substrates of different lengths, prepared either chemically or enzymatically.
Supplementary Material
Acknowledgements
We thank Janusz Bujnicki for the plasmid coding for CMTR1 and Ann-Marie Lawrence-Dörner for excellent technical assistance. We thank Dr. Wolfgang Dörner and Stephanie Wulff for MS measurements. S.R.J. was supported by NIH grant R01CA186702. A.R. thanks the DFG for financial support (RE2796/2-1 and RE2796/6-1).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Experimental Section
Experimental details are given in the Supporting Information.
References
- [1].Edery I, Sonenberg N, Proc. Natl. Acad. Sci 1985, 82, 7590–7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Köhler A, Hurt E, Nat. Rev. Mol. Cell Biol 2007, 8, 761–773. [DOI] [PubMed] [Google Scholar]
- [3].Sonenberg N, Hinnebusch AG, Cell 2009, 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Müller-McNicoll M, Neugebauer KM, Nat. Rev. Genet 2013, 14, 275–287. [DOI] [PubMed] [Google Scholar]
- [5].Shimotohno K, Kodama Y, Hashimoto J, Miura KI, Proc. Natl. Acad. Sci 1977, 74, 2734–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Konarska MM, Padgett RA, Sharp PA, Cell 1984, 38, 731–736. [DOI] [PubMed] [Google Scholar]
- [7].Furuichi Y, LaFiandra A, Shatkin AJ, Nature 1977, 266, 235–239. [DOI] [PubMed] [Google Scholar]
- [8].Wang Z, Jiao X, Carr-Schmid A, Kiledjian M, Proc. Natl. Acad. Sci 2002, 99, 12663–12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pichlmair A, Reis e Sousa C, Immunity 2007, 27, 370–383. [DOI] [PubMed] [Google Scholar]
- [10].Hausmann S, Zheng S, Costanzo M, Brost RL, Garcin D, Boone C, Shuman S, Schwer B, J. Biol. Chem 2008, 283, 31706–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pellizzoni L, EMBO Rep. 2007, 8, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Patel SB, Bellini M, Nucleic Acids Res 2008, 36, 6482–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Matera AG, Wang Z, Nat. Rev. Mol. Cell Biol 2014, 15, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hausmann S, Altura MA, Witmer M, Singer SM, Elmendorf HG, Shuman S, J. Biol. Chem 2005, 280, 12077–86. [DOI] [PubMed] [Google Scholar]
- [15].Hausmann S, Shuman S, J. Biol. Chem 2005, 280, 32101–32106. [DOI] [PubMed] [Google Scholar]
- [16].Wei C-M, Gershowitz A, Moss B, Cell 1975, 4, 379–386. [DOI] [PubMed] [Google Scholar]
- [17].Furuichi Y, Morgan M, Shatkin AJ, Jelinek W, Salditt-Georgieff M, Darnell JE, Proc. Natl. Acad. Sci 1975, 72, 1904–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ullu E, Tschudi C, Proc. Natl. Acad. Sci 1991, 88, 10074–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zamudio JR, Mittra B, Campbell DA, Sturm NR, Mol. Microbiol 2009, 72, 1100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bélanger F, Stepinski J, Darzynkiewicz E, Pelletier J, J. Biol. Chem 2010, 285, 33037–33044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Werner M, Purta E, Kaminska KH, Cymerman IA, Campbell DA, Mittra B, Zamudio JR, Sturm NR, Jaworski J, Bujnicki JM, Nucleic Acids Res. 2011, 39, 4756–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Akichika S, Hirano S, Shichino Y, Suzuki T, Nishimasu H, Ishitani R, Sugita A, Hirose Y, Iwasaki S, Nureki O, et al. , Science (80-. ). 2018, 0080, eaav0080. [DOI] [PubMed] [Google Scholar]
- [23].Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur J-J, Chen Q, et al. , Nature 2017, 541, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mauer J, Sindelar M, Despic V, Guez T, Hawley B, Vasseur J-J, Rentmeister A, Gross SS, Pellizzoni L, Debart F, et al. , Nat Chem Biol 2019, accepted for publication, DOI: 10.1038/s41589-019-0231-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Muttach F, Muthmann N, Rentmeister A, Beilstein J Org. Chem 2017, 13, 2819–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Thillier Y, Decroly E, Morvan F, Canard B, Vasseur J-J, Debart F, RNA 2012, 18, 856–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sekine M, Kadokura M, Satoh T, Seio K, Wada T, Fischer U, Sumpter V, Lührmann R, J. Org. Chem 1996, 61, 4412–4422. [DOI] [PubMed] [Google Scholar]
- [28].Kadokura M, Wada T, Seio K, Moriguchi T, Huber J, Lührmann R, Sekine M, Tetrahedron Lett. 2001, 42, 8853–8856. [Google Scholar]
- [29].Moreno PMD, Wenska M, Lundin KE, Wrange Ö, Strömberg R, Smith CIE, Nucleic Acids Res. 2009, 37, 1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Honcharenko M, Romanowska J, Alvira M, Jezowska M, Kjellgren M, Edvard Smith CI, Strömberg R, RSC Adv. 2012, 2, 12949. [Google Scholar]
- [31].Wojtczak BA, Warminski M, Kowalska J, Lukaszewicz M, Honcharenko M, Smith CIE, Strömberg R, Darzynkiewicz E, Jemielity J, RSC Adv. 2016, 6, 8317–8328. [Google Scholar]
- [32].Schulz D, Holstein JM, Rentmeister A, Angew. Chemie Int. Ed 2013, 52, 7874–7878. [DOI] [PubMed] [Google Scholar]
- [33].Lavergne T, Bertrand J-R, Vasseur J-J, Debart F, Chem. - A Eur. J 2008, 14, 9135–9138. [DOI] [PubMed] [Google Scholar]
- [34].Kruse S, Zhong S, Bodi Z, Button J, Alcocer MJC, Hayes CJ, Fray R, Sci. Rep 2011, 1, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zlatev I, Lavergne T, Debart F, Vasseur J-J, Manoharan M, Morvan F, Org. Lett 2010, 12, 2190–2193. [DOI] [PubMed] [Google Scholar]
- [36].Benarroch D, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Shuman S, RNA 2010, 16, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Smietanski M, Werner M, Purta E, Kaminska KH, Stepinski J, Darzynkiewicz E, Nowotny M, Bujnicki JM, Nat. Commun 2014, 5, 3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.