

The increasing prevalence of multidrug-resistant Helicobacter pylori strains has created an urgent need for alternative therapeutic regimens that complement the current antibiotic treatment strategies for H. pylori eradication; however, this is greatly hampered due to a lack of “druggable” targets. Although the CFAs are present in H. pylori cytoplasmic membranes at high levels, their physiological role has not been established. In this report, deletion of the CFA synthase CfaS was shown to attenuate acid and drug resistance, immune escape, and gastric colonization of H. pylori. These findings were validated by inhibition of the CfaS activity with the tool compound dioctylamine. These studies identify this enzyme as an attractive target for further drug discovery efforts against H. pylori.
KEYWORDS: cyclopropane fatty acid, Helicobacter pylori, acid resistance, drug target, mouse gastric colonization
ABSTRACT
Cyclopropane fatty acids (CFAs) are synthetized by the addition of a methylene group from S-adenosyl-l-methionine across the carbon-carbon double bonds of unsaturated fatty acid chains of membrane phospholipids. This fatty acid cyclopropanation, catalyzed by the CFA synthase (CfaS) enzyme, occurs in many bacteria, including the human pathogen Helicobacter pylori. Although the cyclopropane modification was reported to play a key role in the adaptation in response to environmental stress, its role in H. pylori remains unknown. In this study, we showed that H. pylori HP0416 encodes a functional CfaS. The enzyme was demonstrated to be required for acid resistance, antibiotic resistance, intracellular survival and mouse gastric colonization, and cell membrane integrity. Moreover, the tool compound dioctylamine, which acts as a substrate mimic, directly inhibits the CfaS function of H. pylori, resulting into sensitivity to acid stress, increased antibiotic susceptibility, and attenuated abilities to avoid macrophage killing and to colonize mouse stomachs. These results validate CfaS as a promising antibiotic target and provide new potentials for this recognized target in future anti-H. pylori drug discovery efforts.
IMPORTANCE The increasing prevalence of multidrug-resistant Helicobacter pylori strains has created an urgent need for alternative therapeutic regimens that complement the current antibiotic treatment strategies for H. pylori eradication; however, this is greatly hampered due to a lack of “druggable” targets. Although the CFAs are present in H. pylori cytoplasmic membranes at high levels, their physiological role has not been established. In this report, deletion of the CFA synthase CfaS was shown to attenuate acid and drug resistance, immune escape, and gastric colonization of H. pylori. These findings were validated by inhibition of the CfaS activity with the tool compound dioctylamine. These studies identify this enzyme as an attractive target for further drug discovery efforts against H. pylori.
INTRODUCTION
Helicobacter pylori infects an estimated 50% of the global population and is an important cause of peptic ulcer disease and gastric cancer (1, 2). It is recommended that treatment to eradicate the organism be initiated following firm diagnosis (3). In particular, recent studies have demonstrated that H. pylori eradication could reduce the incidence of gastric cancer (4, 5). Initial treatment is triple therapy, consisting of a proton pump inhibitor and two broad-spectrum antibiotics, or quadruple therapy, in which bismuth is added to the triple therapy (6). However, the complicated regimens of multiple pills combined with the nonselectivity of the antibiotics can lead to poor patient compliance and various side effects, compromising the effectiveness of these treatments (7). Moreover, increased antibiotic usage worldwide has led to antibiotic resistance in H. pylori, resulting in falling success rates for H. pylori eradication (8). Novel targeted anti-H. pylori treatments and antibiotics would be beneficial, as they would reduce the effects on the microbiota and potentially diminish resistance. To develop new treatments, it is necessary to understand the molecular strategies utilized by H. pylori in surviving the harsh conditions in humans and to identify novel and attractive drug targets.
Since H. pylori infects and survives the extremely acidic (pH < 3.0) environment of the human stomach, it has to evolve mechanisms for acid resistance. Besides highly active urease, which catalyzes the hydrolysis of urea into ammonia and scavenges protons to mitigate acidity (9), H. pylori seems to process other defense systems and enzymes to support acid resistance. The bacterial cell envelope may play an important role during these processes, as it protects the cell from environmental stress, maintains cell shape and rigidity, acts as a molecular sieve, and provides a platform for communication with the environment (10, 11). H. pylori membrane phospholipid has a unique fatty acid composition, in that the cyclopropane fatty acid (CFA) cis-11,12-methyleneoctadecanoic acid (C19:0 cyc) constitutes a large proportion (approximately 30%) of total fatty acids (12–14). It is generally thought currently that this energetically expensive CFA modification (CFA synthesis requires 3 ATP molecules per cyclopropane ring formed) of membrane lipids is an important strategy employed by bacteria to quickly adapt to a drastic perturbation of the environment. Indeed, CFAs produced by some bacteria have been shown to provide protection against various forms of environmental stress, including low oxygen tension, hydrogen peroxide, desiccation, and high ionic strength and/or osmolarity (15–17). However, there is debate regarding their roles in the resistance of bacteria to a lethal pH. For instance, the CFA content has been demonstrated to be a major factor in the acid resistance of Escherichia coli (18), whereas the cyclopropanation of membrane lipids is not essential for survival under acidic conditions for Lactococcus lactis subsp. cremoris and Pseudomonas putida (19, 20). Nevertheless, the exact physiological impact of CFAs on H. pylori remains unclear.
CFAs are synthesized by modification of the acyl chains of membrane phospholipids through methylenation of unsaturated fatty acyl (UFA) chains (16). This conversion is catalyzed by the CFA synthase enzyme (CfaS), which uses S-adenosyl-l-methionine (SAM) as the methylene donor (21). The E. coli CfaS was the first soluble cyclopropane-ring-synthesizing enzyme of known amino acid sequence (22). The enzymes of this family are homologous in both primary sequence and tertiary structure, and cfaS genes have been identified in the genomes of many bacteria, including Lactococcus lactis (19), Salmonella enterica (23), Brucella abortus (24), and Sinorhizobium meliloti (25). Moreover, pathogenic Mycobacterium species carry several CfaSs and methyltransferases, which are responsible for synthesis of the cyclopropane rings and methyl branches, respectively, of mycolic acids (26, 27). These enzymes have been strongly implicated in pathogenesis and identified as attractive targets for new antituberculosis drugs (28, 29). To date, however, there appear to have been no related studies in H. pylori. The locus tag hp0416 was annotated as the cfaS gene in H. pylori, but no genetic or metabolite analyses have been conducted to support functional assignment.
In this study, we address the suitability of hp0416-encoded CfaS as a potential target for anti-H. pylori agents. We demonstrate that CfaS can be targeted by a tool compound and that this inhibition impairs acid and antibiotic resistance, intracellular survival, mouse gastric colonization, and cell wall structure in H. pylori.
RESULTS
The hp0416-encoded CfaS is dispensable for H. pylori growth.
The genome of H. pylori strain 26695 contains an open reading frame (ORF) (hp0416) that is predicted to encode a 45.4-kDa protein of 389 amino acids with a SAM-dependent methyltransferase domain (amino acids 164 to 271) that is conserved in all CfaSs; it shows 31 to 37% identity to E. coli CfaS and M. tuberculosis methyltransferases that modify mycolic acids (see Fig. S1 in the supplemental material). Note that HP0416 has identical orthologues in all sequenced strains of H. pylori. To analyze the activity of the protein encoded by hp0416, we constructed an hp0416 knockout mutant by allelic exchange and complemented the mutant with an improved pIR203C04 complementation system (Fig. S2) (30). The CfaS activity of HP0416 was demonstrated by fatty acid composition analysis of phospholipids from wild-type strain, Δhp0416 strain, and complementation strain cells. As shown in Fig. 1A, the two major fatty acids in strain 26695 were tetradecanoic acid (C14:0) and C19:0 cyc, in agreement with previous studies (12, 31). In contrast, C19:0 cyc was not detected in the mutant cells and was replaced by the precursor, cis-vaccenic acid (C18:1) (Fig. 1B). Note that relative levels of two saturated fatty acids, namely, palmitic acid (C16:0) and stearic acid (C18:0), were also significantly increased in the mutant cells (Table S2). Complementation restored the ability to generate C19:0 cyc (Fig. 1D), suggesting that HP0416 functions as a CfaS responsible for fatty acid cyclopropanation in H. pylori.
FIG 1.

GC-MS chromatograms of fatty acid methyl esters from total phospholipids of the H. pylori strains (strain 26695 background). The fatty acids were derivatized to their methyl esters and then analyzed by GC-MS. The C19:0 cyc composition represents the mean of results from three independent experiments. WT, wild-type; WT+Dioc (50 μM), strain 26695 cells grown with 50 μM dioctylamine.
We then determined whether CfaS affects H. pylori growth. The growth curve for each H. pylori strain was determined in brain heart infusion (BHI) broth by measuring bacterial density. Compared with the parent strain 26695, the ΔcfaS mutant showed a moderate growth impairment, with a longer lag time at neutral pH, while the complemented strain showed a growth phenotype similar to that of the wild-type strain (Fig. 2A). In addition, the wild-type strain 26695 and the complemented strain grew exponentially until 40 h, with the same generation times of 4.0 h, whereas the ΔcfaS mutant grew exponentially until 55 h, with a generation time of 6.7 h (Table S3). Comparable growth reduction was observed for ΔcfaS mutants constructed in two other H. pylori backgrounds, i.e., the mouse-adapted strain NSH57 and clinical isolate strain HP159 (data not shown). These data indicated that H. pylori growth was partially but not completely inhibited by the loss of fatty acid cyclopropanation. Note that C19:0 cyc supplementation did not restore normal growth to the ΔcfaS mutant (Fig. S3). This result is confirmed by the finding that no C19:0 cyc was detected in the mutant cells when they were cultured in the presence of C19:0 cyc (data not shown), indicating that C19:0 cyc was not easily incorporated into H. pylori cells.
FIG 2.

Loss of fatty acid cyclopropanation or dioctylamine inhibition of H. pylori growth. (A) Growth curves for H. pylori strain 26695, strain 26695 ΔcfaS, strain 26695 ΔcfaS::cfaS, strain 26695 supplemented with 10 μM dioctylamine (Dioc), and strain 26695 supplemented with 50 μM dioctylamine at 37°C in BHI broth containing FCS. (B) Dioctylamine resistance. H. pylori strain 26695 overexpressing CfaS (HpS49) was more resistant to dioctylamine than strain 26695 with vector alone (HpS48). The data are means, with standard error bars, from three independent determinations, analyzed by Student's t test.
Dioctylamine inhibits fatty acid cyclopropanation in H. pylori by targeting CfaS.
To investigate the phenotypic consequences caused by loss of the cyclopropanation modification, we sought a chemical inhibitor of H. pylori CfaS. Dioctylamine was recently identified as an inhibitor of E. coli CfaS (32) and M. tuberculosis mycolic acid methyltransferases (28). Since these CfaSs share some amino acid identity, dioctylamine might also inhibit H. pylori CfaS by acting as a substrate mimic. We first used a recently developed CfaS colorimetric assay to examine the inhibition of dioctylamine, and we determined the 50% inhibitory concentration (IC50) of dioctylamine for H. pylori CfaS to be 63.81 μM (Fig. S4). Next, we determined whether the dioctylamine treatment affected the enzymatic activity of H. pylori CfaS in vivo. The fatty acid profiles showed that the CFAs represented 28% of the total phospholipid fatty acids in strain 26695 but this composition sharply decreased to 4% for the strain treated with 50 μM dioctylamine (Table S2). We also observed that strain 26695 in broth supplemented with 50 μM dioctylamine exhibited a retarded growth phenotype, which is similar to that of the ΔcfaS mutant (Fig. 2A). The generation time for strain 26695 supplemented with 50 μM dioctylamine was 6.6 h, close to that for the ΔcfaS mutant. Thus, the inhibition of growth by dioctylamine could be the direct result of extremely low levels of the cyclopropanation modification. To confirm the result, we overexpressed this enzyme in a high-copy-number plasmid and tested for resistance to dioctylamine. The strain overexpressing CfaS (HpS49) grew exponentially with similar generation times (4.7 ± 0.5 versus 4.3 ± 0.8) when supplemented with or without 50 μM dioctylamine (Fig. 2B; also see Table S3 in the supplemental material), indicating partial resistance. As expected, the control strain (HpS48) supplemented with dioctylamine had a longer lag time and generation time than those without dioctylamine. These data indicate that dioctylamine inhibits the lipid modifications and thus the growth of H. pylori by targeting CfaS. Note that dioctylamine may have other targets, since the ΔcfaS mutant retains some sensitivity to dioctylamine (data not shown).
Fatty acid cyclopropanation is required for acid resistance.
Fatty acid cyclopropanation in E. coli has been shown to play a role in survival under acidic conditions (18). We then characterized the ΔcfaS mutant for its sensitivity to low-pH conditions. Wild-type H. pylori strain 26695 or ΔcfaS mutant cells were treated by suspension in buffer at different pH values (5.0 or 3.0), and the survival rates were subsequently determined. As shown in Fig. 3A, treatment at pH 5.0 for 1 h did not have a significant effect on survival of the wild-type cells, whereas the same treatment killed approximately 99.9% of the ΔcfaS mutant cells. Furthermore, after acid (pH 3.0) treatment, the survival ability of the ΔcfaS mutant was approximately 2 or 4 orders of magnitude lower than that of the parent strain within 30 min or 1 h of shock, respectively (Fig. 3B and C). Thus, the ΔcfaS mutant was more sensitive to acid stress than the wild-type strain. In addition, we used a clinical isolate, the HP159 strain, to confirm whether the fatty acid cyclopropanation is required for survival under acidic conditions. Similar findings were obtained with the HP159 background, with its ΔcfaS mutant consistently displaying much less acid sensitivity than the wild-type strain after challenge at pH 5.0 and pH 3.0 (Fig. 3). Note that the complementation of CFA function restored the acid sensitivity to the wild-type level. We also observed that the wild-type cells treated with 50 μM dioctylamine, which sharply decreased the CFA content to 14% of the level in untreated cells, had acid sensitivity similar to that of the ΔcfaS mutants on both the strain 26695 and strain HP159 backgrounds (Fig. 3), suggesting that inhibition of fatty acid cyclopropanation attenuates the acid resistance of H. pylori.
FIG 3.
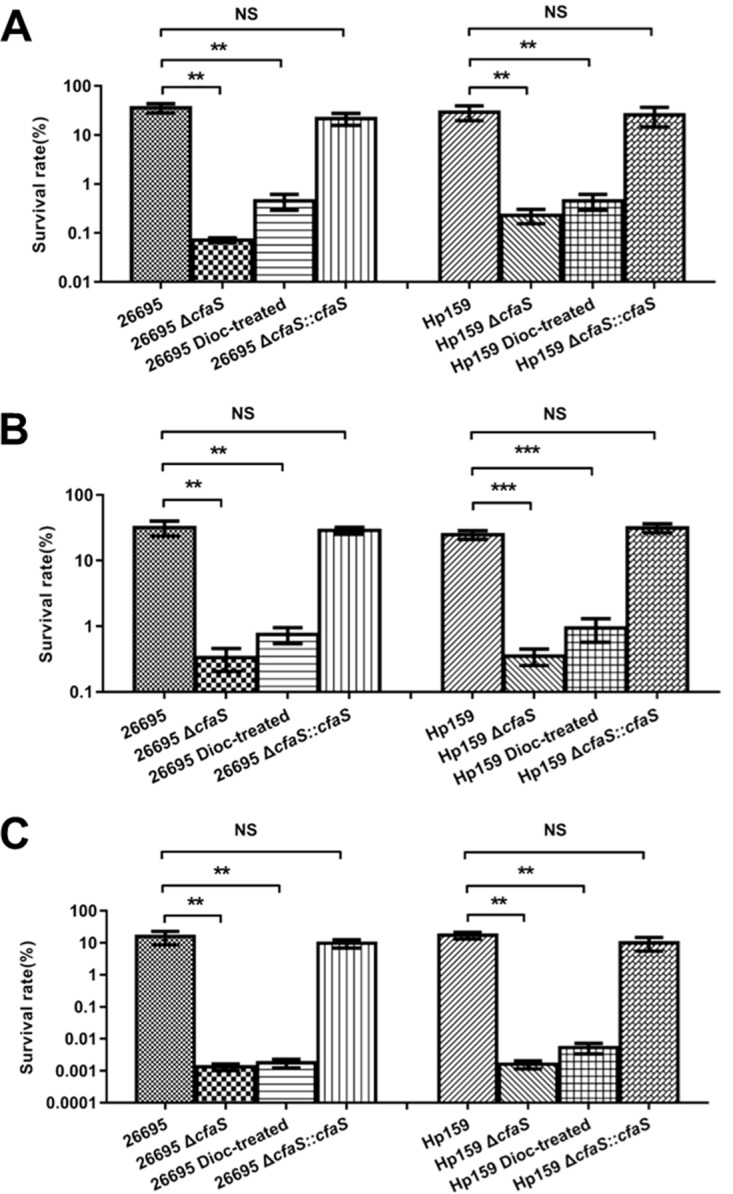
Acid sensitivity of H. pylori strains. All strains were grown to an OD600 of 0.3 and subjected to acid shock at pH 5.0 for 1 h (A), at pH 3.0 for 0.5 h (B), or at pH 3.0 for 1 h (C). The values are the percent cell survival after treatment, relative to the survival at pH 7.0. The data are means, with standard error bars, from three independent determinations, analyzed by Student's t test. **, P < 0.01; ***, P < 0.001; NS, no significant difference (P > 0.05); 26695 Dioc-treated and HP159 Dioc-treated denote cells grown with 50 μM dioctylamine before acid shock.
Fatty acid cyclopropanation plays an essential role in gastric colonization in a mouse model of infection and survival in murine macrophages.
The ecological importance of CfaS, which was found to contribute to acid resistance in vitro (see above), was subsequently tested in vivo via a mouse colonization assay. C57BL/6 mice were orogastrically infected with the H. pylori wild-type NSH57 strain (a mouse-adapted derivative of the G27 strain), its ΔcfaS mutant, or the complemented strain. Three weeks after infection, the colonization loads of the mice were assessed by quantitative cultures of stomach homogenates. Bacterial enumeration showed that the ΔcfaS mutant was completely deficient in its ability to colonize mice, while the geometric mean of colonization by the parental wild-type strain reached 3.5 × 105 CFU/g stomach (Fig. 4A). The colonization ability of the complemented strain was restored to a level close to that of the wild-type strain. We also tested the ΔcfaS mutant in a 1:1 competition experiment with wild-type bacteria. The ΔcfaS mutant represented around 0.5% of the wild-type population in the stomach after 3 weeks of colonization (Fig. 4B). The high degree of impairment of the ΔcfaS mutant when competing with the wild-type strain demonstrates that H. pylori CfaS mediates resistance against gastric acid secretion during stomach colonization. To further validate CfaS as a promising target for the development of anti-H. pylori therapeutics, we tested dioctylamine efficacy in mice infected with the H. pylori strain NSH57. Dioctylamine (35 mg/kg) was administered by oral gavage once daily for 3 consecutive days after infection. After 2 days of treatment, there was a 2-log-unit reduction in bacterial burden in the dioctylamine-treated mice, compared with phosphate-buffered saline (PBS)-treated control mice (Fig. S5), demonstrating that dioctylamine inhibits bacterial proliferation during the infection of mice. These data together confirmed that targeting CfaS is a valid and potentially promising therapeutic strategy.
FIG 4.

Mouse stomach colonization levels of H. pylori strains. (A) Mice were inoculated with PBS (negative control) or H. pylori strains four times (2-day intervals); a dose of 4 × 108 viable cells in 0.4 ml PBS per mouse was administered by gavage each time. After 3 weeks, the stomachs were harvested, homogenized, diluted, and plated, and the number of CFU was determined 3 to 5 days after harvest. Each point represents the CFU count from one mouse stomach, and the solid horizontal lines represent the geometric means of the colonization numbers for each group. Error bars represent the standard deviations derived from 8 mice (strain NSH57 infected), 10 mice (strain NSH57 ΔcfaS infected), or 8 mice (strain NSH57 ΔcfaS::cfaS infected) per group, as analyzed by Student's t test. **, P < 0.01; NS, no significant difference (P > 0.05). The detection limit of the assay is 100 CFU/g stomach (shown with a dashed line). (B) Mice were inoculated with strains NSH57 and NSH57 ΔcfaS together four times (2-day intervals); a dose of 2 × 108 viable cells of each stain in a total of 0.4 ml PBS per animal was administered each time. After 3 weeks of infection, colonization of H. pylori in mouse stomachs was examined and the competitive index (CFU of mutant bacteria/CFU of wild-type bacteria) was determined. The horizontal lines represent the geometric means of the colonization proportion. Error bars represent the standard deviations derived from 6 mice.
H. pylori has the ability to evade elimination by host innate immune responses, including macrophages, thus establishing a persistent gastric infection. We then performed macrophage killing assays, using the murine macrophage cell line RAW 264.7, to determine whether CfaS plays such a role. Similar numbers (5 × 108 CFU/ml) of the H. pylori wild-type strain, the ΔcfaS mutant strain, or the complemented strain were inoculated into the macrophages. After extracellular killing by gentamicin and incubation within macrophages, the numbers of surviving cells of the strains were compared. Two hours after incubation, a slight difference was noticed between the wild-type strain and the mutant strain (Fig. 5), but that difference was statistically significant only at the 90% level (P < 0.1). However, the average number of recovered ΔcfaS mutant cells decreased significantly more rapidly than did the number of wild-type cells after that point. Twenty-four hours after incubation, about 0.2% viable cells of the wild-type strain 26695 survived, while the survival rate of the ΔcfaS mutant was 100-fold lower than that of the wild-type strain (Fig. 5A). Note that the complemented strain exhibited a survival phenotype similar to that of wild-type strain. We also observed that dioctylamine treatment sharply decreased the intracellular survival of the wild-type cells, whose recovery abilities remained close to those of the ΔcfaS mutants on both the strain 26695 and strain HP159 backgrounds (Fig. 5). These results indicate a role for CfaS in the survival of H. pylori within macrophages.
FIG 5.

Survival of H. pylori strains in macrophage RAW 264.7 cells, determined with the gentamicin killing assay. Similar numbers of the strains were inoculated into the macrophages. After extracellular killing by gentamicin and incubation within macrophages, samples were removed at the times indicated and the numbers of surviving CFU per milliliter of H. pylori cells were determined by serial dilution and plate counting. The survival rates are the percent cell survival after incubation within macrophages, relative to the survival at time zero. The data are means, with standard error bars, from three independent determinations, analyzed by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The strains, in strain 26695 (A) and strain HP159 (B) backgrounds, are indicated; 26695 Dioc-treated and HP159 Dioc-treated denote the cells grown with 50 μM dioctylamine before inoculation into the macrophages.
Fatty acid cyclopropanation mediates resistance to antibiotics.
This dioctylamine treatment was shown previously to facilitate the uptake of some antibiotics via the mycobacterial membrane, thus increasing the susceptibility to them in M. tuberculosis (28). To test whether the cyclopropanation contributes to antibiotic resistance in H. pylori, we conducted antibiotic sensitivity assays using MIC tests. As shown in Fig. 6, CFA deletion resulted in 4- to 16-fold reductions in resistance to all four tested antibiotics (ampicillin, metronidazole, levofloxacin, and clarithromycin), on both the strain 26695 and strain HP159 backgrounds. We also found that the complementation of CFA function restored the antibiotic sensitivity to the wild-type level. The wild-type strains treated with dioctylamine had antibiotic sensitivity similar to that of the ΔcfaS mutants (Fig. 6). These results suggested that CfaS activity plays a regulatory role in the resistance to antibiotics. We then measured the uptake of the hydrophobic fluorescent probe N-phenyl-1-naphthylamine (NPN) to determine the outer membrane permeability of H. pylori in response to dioctylamine treatment. We found a significant increase in the fluorescence signal of NPN penetration into dioctylamine-treated cells, an effect that was most evident after 10 min but was sustained after 20 min (Fig. 7A). As expected, the ΔcfaS mutant had a significant increase in NPN uptake, compared with the wild-type strain, while the complemented strain had NPN uptake similar to that of the wild-type strain (Fig. 7A). Thus, the sensitivity to antibiotics of the ΔcfaS mutant could result from the increased membrane permeability with the loss of CfaS function.
FIG 6.

Susceptibility of H. pylori strains to four antimicrobial agents. The MICs were determined by broth microdilution assays, as described in Materials and Methods. The strains, in strain 26695 (A) and strain HP159 (B) backgrounds, are indicated; 266959+Dioc and HP159+Dioc denote that 50 μM dioctylamine was added to the BHI broth containing FCS for MIC assays.
FIG 7.

Disruption of membrane permeability by inhibition of CfaS function. (A) Uptake of NPN by H. pylori cells. The data are the means, with standard error bars, from three independent determinations, analyzed by Student's t test. **, P < 0.01; ***, P < 0.001; NS, not significant. (B) Morphological alterations of H. pylori strains. TEM was used to investigate the morphology of H. pylori cells after overnight culture. 26695+Dioc-treated denotes the cells grown with 50 μM dioctylamine. Scale bars, 500 nm.
To confirm this, we used transmission electron microscopy (TEM) to examine morphological alterations of H. pylori. Untreated wild-type cells appeared normal, with intact cell membranes and dense cytoplasm (Fig. 7B). The cells exposed to dioctylamine lost their characteristic spiral shape and turned into spherical/coccoid forms, with detachment of the inner membrane from the outer membrane; similar morphological changes occurred in the ΔcfaS mutant cells (Fig. 7B). Note that the complementation of CFA function restored a spiral cell morphology without surface damage. Taking these data together, we demonstrate that loss of CfaS function compromises membrane permeability and damages cell membranes, leading to enhanced drug penetration and thus drug killing.
DISCUSSION
Fatty acid cyclopropanation is a common postsynthetic modification of bacterial membrane lipid bilayers (16). In most cases, the physiological rationale for this cyclopropanation modification remains obscure, although some studies have indicated a substantial role for cyclopropanation in membrane fluidity and permeability (33). The major human pathogen H. pylori is characterized by the presence of C19:0 cyc as one of the major fatty acids (12), which can help to differentiate it from closely related bacteria such as Campylobacter species and Wolinella species or to differentiate among the Helicobacter species. Considering the expense, timing, and extent of the UFA-to-CFA conversion, the CFAs seem to confer some adaptive advantages to H. pylori. Here we demonstrate that loss of the cyclopropanation modification leads to pleiotropic alterations in the cell envelope structure. This could be mainly attributed to alterations in the membrane fatty acid composition and greatly increased levels of the UFA C18:1 but not two saturated fatty acids (C16:0 and C18:0). Indeed, UFAs have been shown to inhibit H. pylori growth through their incorporation into phospholipids and membrane destruction (34). Importantly, loss of the normal morphology of this organism further results into increased acid and drug susceptibility and attenuated abilities to colonize mouse stomachs. Moreover, chemical inhibition of the CFA synthase enzyme (CfaS) causes in vitro phenotypes similar to those of ΔcfaS mutants and blocks bacterial replication in animal models of H. pylori infection. These studies thus provide a strong scientific basis for targeting CfaS as an attractive target for the development of anti-H. pylori drugs.
The major role of CFA formation in protection from extreme acid shock was first shown in E. coli (18). This protection also applies to other bacteria, including H. pylori, based on our acid resistance assays, although Pseudomonas putida and L. lactis subsp. cremoris were two exceptions (19, 20). Our results also reveal a novel physiological role of cyclopropanation in H. pylori. Helicobacter isolates primarily isolated from the gastric mucosa tend to synthesize large amounts of CFA, whereas isolates identified as intestinal colonizers generally do not (31). This finding implies that CFA formation may contribute to gastric colonization for H. pylori. In a NSH57-infected mouse model, CfaS was shown to be required for gastric colonization, a novel finding. The most likely cause of this essentiality of CfaS in gastric colonization is mediation of resistance to host gastric acid secretion. The straightforward effect would be a decrease in proton permeability of the membranes due to conversion of UFAs to CFAs by CfaS in the phospholipid component. Indeed, the hypothesis was confirmed in E. coli via in situ real-time assessment of net H+ transport across the bacterial membrane (35).
Furthermore, we observed that loss of the cyclopropanation modification rendered H. pylori susceptible to antibiotics. The increased susceptibility to antibiotics due to the deletion of CfaS may be the result of increased membrane permeability and disruption of the cell membrane, facilitating antibiotic penetration and cell killing. The cell membrane of Gram-negative bacteria is an essential organelle and a robust permeability barrier that prevents entry of many antibiotics (36). Thus, effective permeabilization of the cell membrane could be a strategy to control bacterial infections by enhancing antibiotic action/delivery. Modification of the lipid A phosphate groups of H. pylori lipopolysaccharide has been shown to be required for polymyxin resistance (37). Similarly, we think that loss of the cyclopropanation modification might affect the cell membrane structure and integrity, thus influencing permeability and eventually increasing the sensitivity of H. pylori to antibiotics. Thus, inhibition of CFA synthesis may render H. pylori defective in establishing successful infections in vivo by decreasing the colonization potential and fitness advantage (resistance to antibiotics).
It would be unlikely for the CfaS inhibitor dioctylamine to be explored as a promising antimicrobial drug, due to its toxicity. However, our data provide strong support for the future exploration of CfaS as a drug target, with CfaS inhibitors presenting an important avenue for the development of new anti-H. pylori drugs. The powerful strategy with this validated target not only would avoid cross-resistance to established targets but also would remove much of the usual uncertainty regarding in vivo clinical efficacy.
MATERIALS AND METHODS
Chemicals, bacterial strains, and growth conditions.
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Sinopharm Chemical Reagent Co. (Shanghai, China) and used as received unless otherwise specified. The bacterial strains and plasmids used in this study are summarized in Table 1. H. pylori strains 26695 (38) and NSH57 (39) were kindly provided by Nina R. Salama (Fred Hutchinson Cancer Research Center, USA). Clinical isolate HP159 was obtained from a human gastric biopsy specimen, using standard protocols. H. pylori strains were routinely cultured either in BHI broth (Becton, Dickinson, Sparks, MD) containing 10% fetal calf serum (FCS) and Dent’s supplement or on Columbia blood agar (Oxoid, Basingstoke, UK) plates containing 5% FCS and Dent’s supplement, in a microaerophilic environment (10% CO2-85% N2-5% O2, with 90% relative humidity), for 2 to 3 days at 37°C in a double-gas CO2 incubator (model CB160; Binder, Germany). Selection of H. pylori mutants was performed using kanamycin at 25 μg/ml or chloramphenicol at 10 μg/ml. E. coli strain DH5α was routinely grown in LB broth or on LB agar supplemented with ampicillin (100 μg/ml), chloramphenicol (30 μg/ml), and/or kanamycin (50 μg/ml) when appropriate.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotypea | Reference or source |
|---|---|---|
| E. coli | ||
| DH5α | Δ(argF-lac)U169 ϕ80dlacZ58(M15) ΔphoA8 glnV44 deoR481 gyrA96 recA1 endA1 hsdR17 | Laboratory stock |
| BL21 (Tuner) | F− ompT hsdSB(rB− mB−) gal dcm lacY1 | Novagen |
| Rosetta(DE3) | F− ompT hsdSB(rB− mB−) gal dcm (DE3) pRARE | Novagen |
| H. pylori | ||
| 26695 | Wild-type | 38 |
| HpS42 | Kmr; 26695 ΔcfaS | This work |
| HpS43 | Kmr Cmr; 26695 ΔcfaS::cfaS | This work |
| Hp159 | Drug-resistant clinical isolate | Laboratory stock |
| HpS44 | Kmr; Hp159 ΔcfaS | This work |
| HpS45 | Kmr Cmr; Hp159 ΔcfaS::cfaS | This work |
| NSH57 | Mouse-adapted derivative of G27 strain | 39 |
| HpS46 | Kmr; NSH57 ΔcfaS | This work |
| HpS47 | Kmr Cmr; NSH57 ΔcfaS::cfaS | This work |
| HpS48 | Kmr; 26695 transformed with plasmid pHel3 | This work |
| HpS49 | Kmr; 26695 transformed with plasmid pBHKP380 | This work |
| Plasmids | ||
| pBluescript SK(+) | Ampr; cloning vector | Stratagene |
| pQE-2 | Ampr; T5 promoter-based expression vector | Qiagen |
| pHel3 | Kmr; E. coli/H. pylori shuttle vector | 43 |
| pBHKP109 | Ampr Kmr; PCR-amplified aphA3 inserted between PstI and BamHI sites of pBluescript SK(+) | This work |
| pBHKP202 | Ampr Cmr; PCR-amplified catGC inserted between PstI and BamHI sites of pBluescript SK(+) | This work |
| pBHKP203 | Ampr Kmr; PCR-amplified HpcfaSup inserted between PstI and EcoRI sites plus PCR-amplified HpcfaSdn inserted between BamHI and XbaI sites of BHKP109 | This work |
| pBHKP252 | Cmr; PCR-amplified Hp0203up inserted between KpnI and XhoI sites plus PCR-amplified Hp0204dn inserted between BamHI and XbaI sites of BHKP202 | This work |
| pBHKP388 | Cmr; PCR-amplified H. pylori cfaS (containing its promoter) inserted between XhoI and PstI sites of BHKP252 | This work |
| pBHKP380 | Kmr; PCR-amplified H. pylori cfaS (containing its promoter) inserted between KpnI and XhoI sites of pHel3 | This work |
| pBHKP292 | Ampr; PCR-amplified E. coli mtn inserted between NdeI and SalI sites of pQE-2 | This work |
| pBHKP338 | Ampr; PCR-amplified B. subtilis luxS inserted between KpnI and SalI sites of pQE-2 | This work |
| pBHKP389 | Ampr; PCR-amplified H. pylori cfaS inserted between NdeI and SalI sites of pQE-2 | This work |
Kmr, kanamycin resistance; Cmr, chloramphenicol resistance; Ampr, ampicillin resistance.
DNA manipulation and construction of H. pylori ΔcfaS mutants.
Plasmid DNA and chromosomal DNA were extracted with the QIAprep spin miniprep kit (Qiagen). Oligonucleotide primers (listed in Table S1 in the supplemental material) were synthesized and the cloned genes were verified by sequencing performed by GeneScript Co. (China). The aphA3 cassette (encoding the kanamycin resistance gene) ∼1.3-kb fragment was amplified by PCR using the P1/P2 primer pair. The PCR product was cloned into the PstI and BamHI sites of pBluescript SK(+) vector to generate BHKP109. H. pylori strain 26695 genomic DNA was used as a template to amplify an ∼500-bp DNA fragment both upstream and downstream of the target H. pylori HP0416 locus. Primers P3 and P4 were used to amplify the upstream region, while primers P5 and P6 were used to amplify the downstream region. The two regions were successively ligated to BHKP109 to generate pBHKP203, carrying a sandwich fusion in which the aphA3 cassette was flanked by the upstream and downstream regions of H. pylori cfaS. This plasmid was then introduced into H. pylori wild-type strains by natural transformation via allelic exchange. Colonies (ΔcfaS) were isolated on Columbia blood agar plates supplemented with kanamycin at 25 μg/ml. The deletion of H. pylori cfaS in the genome of the mutant strain was confirmed by PCR using appropriate primers, followed by sequencing of the PCR products.
Construction of H. pylori complementation strains.
The complemented ΔcfaS::cfaS strain was constructed by inserting a wild-type copy of the cfaS gene in the region between hp0203 and hp0204, where there are untranslated regions of the H. pylori chromosome (30). The catGC cassette (with the chloramphenicol resistance gene) ∼900-bp fragment first was amplified by PCR using the P13/P14 primer pair and then was cloned into the PstI and BamHI sites of the pBluescript SK(+) vector to generate BHKP202. The P9/P10 and P11/P12 primer pairs were then used to amplify the upstream and downstream untranslated regions, respectively, between hp0203 and hp0204. The two regions were successively ligated to BHKP202 to generate pBHKP252, carrying a sandwich fusion in which the catGC cassette was flanked by the upstream and downstream untranslated regions. The H. pylori cfaS gene containing its promoter region was amplified by PCR with the P7/P8 primer pair and was cloned into the PstI and XhoI sites of pBHKP252 to generate pBHKP388. This plasmid was then introduced into H. pylori ΔcfaS strains by natural transformation via allelic exchange. Colonies (ΔcfaS::cfaS) were isolated on Columbia blood agar plates containing kanamycin (25 μg/ml) and chloramphenicol (10 μg/ml). The complementation of H. pylori cfaS in the genome of the complemented strain was confirmed by PCR using appropriate primers, followed by sequencing of the PCR products.
Preparation of fatty acid methyl esters and fatty acid analysis.
The H. pylori cultures were grown at 37°C in 10 ml BHI broth containing FCS, to an optical density at 600 nm (OD600) of 0.4. The phospholipids were extracted from 0.5 g (wet weight) of bacteria and dried under nitrogen by the method of Bligh and Dyer (40); fatty acid methyl esters were then prepared as described previously (41). The resulting methyl esters were redissolved in 100 μl of hexane and analyzed by gas chromatography-mass spectroscopy (GC-MS), using an Agilent system consisting of a 5975C mass selective detector, a 7683B autosampler, and a 7890A gas chromatograph equipped with a ZB-WAX (Phenomenex Inc.) capillary column (30 m by 0.25 mm [inner diameter], with 250-μm film thickness). A 1-μl sample was injected with a split ratio of 5:1. The inlet temperature was 230°C, and the interface temperature was 250°C. Helium as the carrier gas was set at a constant flow rate of 1 ml/min. The temperature program was 140°C for 5 min, 5°C/min to 265°C, and then 265°C for 10 min. The spectra acquired were recorded in the scanning range of m/z 25 to m/z 800 and were processed using AMDIS (National Institute of Standards and Technology, Gaithersburg, MD) and MSD ChemStation E.02.02.1431 (Agilent) software. The fatty acid esters were identified by comparing their retention times with those of standard compounds (Nu-ChekPrep, Elysian, MN) and comparing their mass spectra with reference spectrum libraries NIST08 (National Institute of Standards and Technology) and WILEY8n (Palisade Corp.). The fatty acid compositions were determined as described previously (12).
Protein expression and purification.
The H. pylori cfaS gene amplified from H. pylori strain 26695 genomic DNA, with a N-terminal hexahistidine tag, was inserted into vector pQE-2 to give plasmid pBHKP389. H. pylori CfaS was expressed in E. coli Rosetta(DE3) grown at 37°C in LB medium. The cultures were induced with 0.2 mM isopropyl-β-d-thio-d-galactoside (IPTG) at an OD600 of 0.8 and were grown at 37°C for an additional 3 h prior to harvest. The cells were collected, resuspended in lysis buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, 1 mM dithiothreitol [pH 8.0]), lysed by sonication, and centrifuged. The clarified bacterial supernatant was loaded onto a nickel-ion affinity column (Qiagen). The column was washed with wash buffer (50 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole, 1 mM dithiothreitol [pH 8.0]) to remove contaminating proteins, and the His-tagged H. pylori CfaS protein was eluted in the same buffer (elution buffer) containing 200 mM imidazole. The protein was concentrated by ultrafiltration (10-kDa cutoff) and exchanged into sodium phosphate buffer (50 mM NaH2PO4, 200 mM NaCl, 1 mM dithiothreitol [pH 8.0]). The purity of the samples was monitored by SDS-PAGE. Recombinant S-adenosyl-l-homocysteine nucleosidase from E. coli and LuxS from Bacillus subtilis were purified from an overproducing BL21 (Tuner) strain transformed with pBHKP292 carrying the E. coli mtn gene and from an overproducing E. coli Rosetta(DE3) strain transformed with pBHKP338 carrying the Bacillus subtilis luxS gene, respectively, as described above.
Enzymatic assay for H. pylori CfaS.
The assay was performed in a 96-well polystyrene microtiter plate as described previously (32). Briefly, a standard reaction mixture (100 μl) consisted of 20 mM potassium phosphate buffer (pH 7.5), 750 μM SAM, 24 μg/ml H. pylori CfaS (0.5 μM), 1 mg/ml phospholipids, 3 μg/ml S-adenosyl-l-homocysteine nucleosidase, and 70 μg/ml LuxS. The phospholipids used here were extracted from the strain 26695 ΔcfaS cells by the method of Bligh and Dyer (40). A premixture without H. pylori CfaS was preincubated at 37°C for 5 min; the reaction was then initiated by the addition of H. pylori CfaS, and the mixture was incubated at 37°C for 20 min. The reaction was stopped by the addition of 100 μl of quenching solution, which contained 6 M urea, 400 μM 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB), and 0.5% (wt/vol) Triton X-100 in 20 mM potassium phosphate buffer (pH 7.5). The quenched samples were automatically mixed by the microplate reader and incubated at room temperature for 10 min before being read at 412 nm (2-nitro-5-thiobenzoate [TNB] ε = 13,700 M−1 cm−1) on the microplate reader. Control experiments with all components but without H. pylori CfaS were always carried out simultaneously, and the absorption of these controls was subtracted from the absorption of the real sample. To examine the inhibition by dioctylamine, stock solutions were prepared at 8 mM in dimethyl sulfoxide (DMSO). Working solutions were obtained by dilution in DMSO at appropriate concentrations so that the final concentration of DMSO in the assay did not exceed 5% (vol/vol). An enzymatic mixture without H. pylori CfaS was prepared as described above, and 94 μl of this mixture was distributed into the wells of a 96-well plate. Then, 5 μl of the inhibitor solution, at various concentrations, was added into the wells and the reactions were initiated by the addition of 1 μl of H. pylori CfaS. The assays were performed and treated as described above for the microplate assay. All data points were determined in triplicate. IC50 values were determined as described previously (32).
Assessment of sensitivity to low-pH conditions.
H. pylori strains were grown at 37°C to an OD600 of 0.3, and the cells were suspended at a concentration of 108 cells/ml in buffer (20 mM Tris-HCl, 120 mM NaCl) with different pH values (pH 7.0, 5.0, or 3.0). The cell suspensions were incubated at 37°C, under microaerophilic conditions (5% O2-85% N2-10% CO2), for 30 min or 1 h. The samples were serially diluted and plated on Columbia blood agar plates for CFU counting after 4 days of incubation under microaerophilic growth conditions. The percent cell survival at pH 5.0 or pH 3.0, relative to that at pH 7.0, was calculated.
Antibiotic sensitivity assay.
The sensitivity of H. pylori strains to antibiotics was evaluated by examining MIC values, which were determined by broth microdilution assays, as follows. Twofold serial dilutions of the test compounds were prepared in a 96-well microtiter plate containing 100 μl of BHI broth supplemented with 10% FCS. A 2-day-old H. pylori liquid culture was diluted 10-fold in BHI broth and was inoculated into each well to give a final concentration of 5 × 105 to 1 × 106 CFU/ml. The plates were incubated for 3 days in a microaerophilic atmosphere at 37°C. After incubation, the plates were examined visually, and the MICs were determined to be the lowest concentrations that resulted in no turbidity.
Macrophage killing assay.
The survival of H. pylori cells within macrophages was investigated following published methods (44), with minor modifications. Briefly, RAW 264.7 macrophages were seeded in 24-well plates in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FCS (1 ml) and were incubated for 3 days at 37°C in 5% CO2 (cell density was about 105 cells per well). The medium was replaced with fresh medium to remove the nonadherent cells. For the experiments, RAW 264.7 cells were cocultured with intact H. pylori cells at a multiplicity of infection (MOI) of 100. Phagocytosis was synchronized by centrifugation at 600 × g for 5 min and then allowed to proceed for 1 h in a CO2 gas incubator. Extracellular bacteria were removed by washing and incubation for 1 h at 37°C in 5% CO2 in medium supplemented with gentamicin (100 mg/ml). After three washes to remove the antibiotics, the cells were further incubated in fresh medium for 2 h, 4 h, 8 h, 12 h, and 24 h. After removal of the medium, the macrophages were lysed for 5 min with ice-cold PBS with 0.1% saponin. Appropriate dilutions of the supernatant were plated on Columbia blood agar plates and incubated in a microaerophilic atmosphere at 37°C for 4 days, to count the surviving bacteria. The number of surviving bacteria (CFU per milliliter) was compared with the number of viable bacteria after washing.
Outer membrane permeability assay.
The outer membrane permeabilization was assessed by measuring the uptake of NPN (42). H. pylori strains were grown at 37°C to an OD600 of 0.3. Approximately 1 × 107 bacterial cells were incubated with 100 μM NPN in 200 μl of PBS. PBS was used as the negative control. Fluorescence measurements were taken after shaking at each time point, using a Synergy HTX multimode microplate reader (BioTek Instruments, Winooski, VT) at an excitation wavelength of 350 nm and an emission wavelength of 420 nm. The NPN assays were performed at room temperature and repeated three times, and the results are expressed as relative fluorescence units.
Transmission electron microscopy.
The morphology of H. pylori strain 26695 treated with dioctylamine and the effects of H. pylori cfaS deletion on the structure of H. pylori were examined with TEM. Briefly, overnight cultures of H. pylori strains, supplemented with 50 μM dioctylamine or not, were centrifuged, and bacterial pellets were fixed by resuspension in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4). Bacterial pellets were then embedded in 2% agarose and postfixed with 1% osmium tetroxide overnight at room temperature. After washing, samples were dehydrated multiple times with increasing concentrations of ethanol and were embedded in Durcupan resin (Sigma-Aldrich). Fifty-five-nanometer sections were examined using a JEM-1200 transmission electron microscope (JEOL, Tokyo, Japan) equipped with a 4K Eagle digital camera (FEI, Hillsboro, OR).
Ethics.
Animal studies were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (IACUC approval no. 1707025) and were conducted in accordance with the international standards for animal welfare and institutional guidelines.
Mouse colonization assay.
Six-week-old, specific-pathogen-free, female C57BL/6 mice were used for this study. NSH57 is a mouse-adapted H. pylori strain obtained after multiple passages through mice infected by strain G27 (39). Wild-type NSH57, ΔcfaS mutant, and cfaS-complemented cells were harvested after 48 h of growth in BHI broth containing FCS, in a microaerophilic environment, and were suspended in PBS to an OD600 of 3 (109 CFU/ml). The bacterial suspensions (0.4 ml) were administered to C57BL/6 mice (4 × 108 cells/mouse) through oral gavage every 48 h, repeated four times. Three weeks after the first inoculation, the mice were sacrificed and the stomachs were removed, weighed, and homogenized in BHI broth. Stomach homogenate dilutions were plated on Columbia blood agar plates supplemented with bacitracin (100 μg/ml), vancomycin (10 μg/ml), and amphotericin B (10 μg/ml). The plates were incubated at 37°C under microaerophilic conditions for 5 to 7 days, H. pylori colonies were counted, and the data are expressed as CFU per gram of stomach.
For competition experiments, the mice were inoculated with strains NSH57 and NSH57 ΔcfaS at the same time, four times (2 days intervals), with a dose of 2 × 108 viable cells of each stain in a total of 0.4 ml PBS per animal being administered each time. After inoculation, a portion of the inoculum was plated on Columbia blood agar plates to enumerate the actual numbers of wild-type and mutant bacteria present in the inocula. After 3 weeks, the mice were euthanized and the stomach was removed as described above. Dilutions of the homogenate were plated on Columbia blood agar plates to enumerate total bacteria and on Columbia blood agar plates with added kanamycin (25 μg/ml) to enumerate mutant bacteria. The competitive index was determined as CFU of mutant bacteria/CFU of wild-type bacteria. This competitive index number was corrected by dividing the actual input ratio enumerated by plating the inocula after infection.
Statistical analysis.
Statistical comparisons of the results were made using analysis of variance. Data were expressed as the means ± standard errors of the means. Significant differences between the means for control and treatment groups were analyzed by Student's t test.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (grants 31570053 and 31870029 to H.B.), the National Science Foundation of the Jiangsu Higher Education Institutions of China (grant 18KJA310002 to H.B.), and the Jiangsu Specially Appointed Professor and Jiangsu Medical Specialist Programs of China (to H.B.).
We thank Nina Salama and Karen Ottemann for providing H. pylori strains.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00374-19.
REFERENCES
- 1.Wroblewski LE, Peek RM Jr, Wilson KT. 2010. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cover TL, Blaser MJ. 2009. Helicobacter pylori in health and disease. Gastroenterology 136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malfertheiner P, Megraud F, O’Morain CA, Gisbert JP, Kuipers EJ, Axon AT, Bazzoli F, Gasbarrini A, Atherton J, Graham DY, Hunt R, Moayyedi P, Rokkas T, Rugge M, Selgrad M, Suerbaum S, Sugano K, El-Omar EM. 2017. Management of Helicobacter pylori infection: the Maastricht V/Florence Consensus Report. Gut 66:6–30. doi: 10.1136/gutjnl-2016-312288. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H, Matsuzaki J. 2018. Gastric cancer: evidence boosts Helicobacter pylori eradication. Nat Rev Gastroenterol Hepatol 15:458–460. doi: 10.1038/s41575-018-0023-8. [DOI] [PubMed] [Google Scholar]
- 5.Doorakkers E, Lagergren J, Engstrand L, Brusselaers N. 2018. Helicobacter pylori eradication treatment and the risk of gastric adenocarcinoma in a Western population. Gut 67:2092–2096. doi: 10.1136/gutjnl-2017-315363. [DOI] [PubMed] [Google Scholar]
- 6.Chey WD, Leontiadis GI, Howden CW, Moss SF. 2017. ACG clinical guideline: treatment of Helicobacter pylori infection. Am J Gastroenterol 112:212–239. doi: 10.1038/ajg.2016.563. [DOI] [PubMed] [Google Scholar]
- 7.Alfizah H, Norazah A, Hamizah R, Ramelah M. 2014. Resistotype of Helicobacter pylori isolates: the impact on eradication outcome. J Med Microbiol 63:703–709. doi: 10.1099/jmm.0.069781-0. [DOI] [PubMed] [Google Scholar]
- 8.Hu Y, Zhang M, Lu B, Dai J. 2016. Helicobacter pylori and antibiotic resistance, a continuing and intractable problem. Helicobacter 21:349–363. doi: 10.1111/hel.12299. [DOI] [PubMed] [Google Scholar]
- 9.Sachs G, Scott DR, Weeks DL, Rektorscheck M, Melchers K. 2001. Regulation of urease for acid habitation, p 277–284. In Mobley HLT, Mendz GL, Hazell SL (ed), Helicobacter pylori: physiology and genetics. ASM Press, Washington, DC. [PubMed] [Google Scholar]
- 10.Cabeen MT, Jacobs-Wagner C. 2005. Bacterial cell shape. Nat Rev Microbiol 3:601–610. doi: 10.1038/nrmicro1205. [DOI] [PubMed] [Google Scholar]
- 11.Yang DC, Blair KM, Salama NR. 2016. Staying in shape: the impact of cell shape on bacterial survival in diverse environments. Microbiol Mol Biol Rev 80:187–203. doi: 10.1128/MMBR.00031-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geis G, Leying H, Suerbaum S, Opferkuch W. 1990. Unusual fatty acid substitution in lipids and lipopolysaccharides of Helicobacter pylori. J Clin Microbiol 28:930–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scherer C, Muller KD, Rath PM, Ansorg RA. 2003. Influence of culture conditions on the fatty acid profiles of laboratory-adapted and freshly isolated strains of Helicobacter pylori. J Clin Microbiol 41:1114–1117. doi: 10.1128/jcm.41.3.1114-1117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bi H, Zhu L, Jia J, Zeng L, Cronan JE. 2016. Unsaturated fatty acid synthesis in the gastric pathogen Helicobacter pylori proceeds via a backtracking mechanism. Cell Chem Biol 23:1480–1489. doi: 10.1016/j.chembiol.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen YY, Ganzle MG. 2016. Influence of cyclopropane fatty acids on heat, high pressure, acid and oxidative resistance in Escherichia coli. Int J Food Microbiol 222:16–22. doi: 10.1016/j.ijfoodmicro.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 16.Grogan DW, Cronan JE Jr.. 1997. Cyclopropane ring formation in membrane lipids of bacteria. Microbiol Mol Biol Rev 61:429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cronan JE., Jr. 2002. Phospholipid modifications in bacteria. Curr Opin Microbiol 5:202–205. doi: 10.1016/S1369-5274(02)00297-7. [DOI] [PubMed] [Google Scholar]
- 18.Chang YY, Cronan JE Jr.. 1999. Membrane cyclopropane fatty acid content is a major factor in acid resistance of Escherichia coli. Mol Microbiol 33:249–259. doi: 10.1046/j.1365-2958.1999.01456.x. [DOI] [PubMed] [Google Scholar]
- 19.To TMH, Grandvalet C, Tourdot-Maréchal R. 2011. Cyclopropanation of membrane unsaturated fatty acids is not essential to the acid stress response of Lactococcus lactis subsp. cremoris. Appl Environ Microbiol 77:3327–3334. doi: 10.1128/AEM.02518-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pini CV, Bernal P, Godoy P, Ramos JL, Segura A. 2009. Cyclopropane fatty acids are involved in organic solvent tolerance but not in acid stress resistance in Pseudomonas putida DOT-T1E. Microb Biotechnol 2:253–261. doi: 10.1111/j.1751-7915.2009.00084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor FR, Cronan JE Jr.. 1979. Cyclopropane fatty acid synthase of Escherichia coli: stabilization, purification, and interaction with phospholipid vesicles. Biochemistry 18:3292–3300. doi: 10.1021/bi00582a015. [DOI] [PubMed] [Google Scholar]
- 22.Wang AY, Grogan DW, Cronan JE Jr.. 1992. Cyclopropane fatty acid synthase of Escherichia coli: deduced amino acid sequence, purification, and studies of the enzyme active site. Biochemistry 31:11020–11028. doi: 10.1021/bi00160a011. [DOI] [PubMed] [Google Scholar]
- 23.Kim BH, Kim S, Kim HG, Lee J, Lee IS, Park YK. 2005. The formation of cyclopropane fatty acids in Salmonella enterica serovar Typhimurium. Microbiology 151:209–218. doi: 10.1099/mic.0.27265-0. [DOI] [PubMed] [Google Scholar]
- 24.Palacios-Chaves L, Zúñiga-Ripa A, Gutiérrez A, Gil-Ramírez Y, Conde-Álvarez R, Moriyón I, Iriarte M. 2012. Identification and functional analysis of the cyclopropane fatty acid synthase of Brucella abortus. Microbiology 158:1037–1044. doi: 10.1099/mic.0.055897-0. [DOI] [PubMed] [Google Scholar]
- 25.Saborido Basconcillo L, Zaheer R, Finan TM, McCarry BE. 2009. Cyclopropane fatty acyl synthase in Sinorhizobium meliloti. Microbiology 155:373–385. doi: 10.1099/mic.0.022608-0. [DOI] [PubMed] [Google Scholar]
- 26.Yuan Y, Lee RE, Besra GS, Belisle JT, Barry CE III.. 1995. Identification of a gene involved in the biosynthesis of cyclopropanated mycolic acids in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 92:6630–6634. doi: 10.1073/pnas.92.14.6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glickman MS. 2003. The mmaA2 gene of Mycobacterium tuberculosis encodes the distal cyclopropane synthase of the α-mycolic acid. J Biol Chem 278:7844–7849. doi: 10.1074/jbc.M212458200. [DOI] [PubMed] [Google Scholar]
- 28.Barkan D, Liu Z, Sacchettini JC, Glickman MS. 2009. Mycolic acid cyclopropanation is essential for viability, drug resistance, and cell wall integrity of Mycobacterium tuberculosis. Chem Biol 16:499–509. doi: 10.1016/j.chembiol.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao V, Fujiwara N, Porcelli SA, Glickman MS. 2005. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med 201:535–543. doi: 10.1084/jem.20041668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langford ML, Zabaleta J, Ochoa AC, Testerman TL, McGee DJ. 2006. In vitro and in vivo complementation of the Helicobacter pylori arginase mutant using an intergenic chromosomal site. Helicobacter 11:477–493. doi: 10.1111/j.1523-5378.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haque M, Hirai Y, Yokota K, Mori N, Jahan I, Ito H, Hotta H, Yano I, Kanemasa Y, Oguma K. 1996. Lipid profile of Helicobacter spp.: presence of cholesteryl glucoside as a characteristic feature. J Bacteriol 178:2065–2070. doi: 10.1128/jb.178.7.2065-2070.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guianvarc'h D, Drujon T, Leang TE, Courtois F, Ploux O. 2006. Identification of new inhibitors of E. coli cyclopropane fatty acid synthase using a colorimetric assay. Biochim Biophys Acta 1764:1381–1388. doi: 10.1016/j.bbapap.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Nagamachi E, Shibuya S, Hirai Y, Matsushita O, Tomochika K, Kanemasa Y. 1991. Adaptational changes of fatty acid composition and the physical state of membrane lipids following the change of growth temperature in Yersinia enterocolitica. Microbiol Immunol 35:1085–1093. doi: 10.1111/j.1348-0421.1991.tb01630.x. [DOI] [PubMed] [Google Scholar]
- 34.Khulusi S, Ahmed HA, Patel P, Mendall MA, Northfield TC. 1995. The effects of unsaturated fatty acids on Helicobacter pylori in vitro. J Med Microbiol 42:276–282. doi: 10.1099/00222615-42-4-276. [DOI] [PubMed] [Google Scholar]
- 35.Shabala L, Ross T. 2008. Cyclopropane fatty acids improve Escherichia coli survival in acidified minimal media by reducing membrane permeability to H+ and enhanced ability to extrude H+. Res Microbiol 159:458–461. doi: 10.1016/j.resmic.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Powers MJ, Trent MS. 2018. Phospholipid retention in the absence of asymmetry strengthens the outer membrane permeability barrier to last-resort antibiotics. Proc Natl Acad Sci U S A 115:E8518–E8527. doi: 10.1073/pnas.1806714115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran AX, Whittimore JD, Wyrick PB, McGrath SC, Cotter RJ, Trent MS. 2006. The lipid A 1-phosphatase of Helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J Bacteriol 188:4531–4541. doi: 10.1128/JB.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 39.Baldwin DN, Shepherd B, Kraemer P, Hall MK, Sycuro LK, Pinto-Santini DM, Salama NR. 2007. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect Immun 75:1005–1016. doi: 10.1128/IAI.01176-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 41.Feng Y, Cronan JE. 2009. Escherichia coli unsaturated fatty acid synthesis: complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J Biol Chem 284:29526–29535. doi: 10.1074/jbc.M109.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helander IM, Mattila-Sandholm T. 2000. Fluorometric assessment of Gram-negative bacterial permeabilization. J Appl Microbiol 88:213–219. doi: 10.1046/j.1365-2672.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- 43.Heuermann D, Haas R. 1998. A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Mol Gen Genet 257:519–528. doi: 10.1007/s004380050677. [DOI] [PubMed] [Google Scholar]
- 44.Odenbreit S, Gebert B, Puls J, Fischer W, Haas R. 2001. Interaction of Helicobacter pylori with professional phagocytes: role of the cag pathogenicity island and translocation, phosphorylation and processing of CagA. Cell Microbiol 3:21–31. doi: 10.1046/j.1462-5822.2001.00088.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.