

Abstract
RAS regulation and signaling are largely accomplished by direct protein-protein interactions, making RAS protein dynamics a critical determinant of RAS function. Here, we report a crystal structure of GDP-bound KRASV14I, a mutated KRAS variant associated with the developmental RASopathy disorder Noonan syndrome (NS), at 1.5–1.6 Å resolution. The structure is notable for revealing a marked extension of switch 1 away from the G-domain and nucleotide-binding site of the KRAS protein. We found that this extension is associated with a loss of the magnesium ion and a tilt in the position of the guanine base because of the additional carbon introduced by the isoleucine substitution. Hydrogen-deuterium exchange MS analysis confirmed that this conformation occurs in solution, but also disclosed a difference in kinetics when compared with KRASA146T, another RAS mutant that displays a nearly identical conformation in previously reported crystal structures. This conformational change contributed to a high rate of guanine nucleotide-exchange factor (GEF)-dependent and -independent nucleotide exchange and to an increase in affinity for SOS Ras/Rac GEF 1 (SOS1), which appears to be the major mode of activation for this RAS variant. These results highlight a mechanistic connection between KRASA146T and KRASV14I that may have implications for the regulation of these variants and for the development of therapeutic strategies to manage KRAS variant-associated disorders.
Keywords: Ras protein, GTPase, GTPase Kras (KRAS), guanine nucleotide-exchange factor (GEF), X-ray crystallography, GTPase, Noonan syndrome, nucleotide exchange, RAS, RASopathy
Introduction
RAS mutations are common in disease, including cancer (1) and developmental disorders (2). RAS mutations vary in their properties including their impact on global signal transduction (3) and transforming potency (4, 5). These higher-order characteristics likely relate to differences in fundamental properties such as enzymatic activity and protein dynamics. Moreover, the recent development of covalent inhibitors of KRASG12C, a mutation common in lung cancer, suggests that successful therapeutic interventions against RAS will require a mutation-specific approach (6).
RASopathy syndromes are a collection of developmental disorders characterized by hyperactivity of canonical RAS pathways and hyperproliferation of selected tissues (7). One such syndrome is Noonan syndrome (NS),2 an autosomal dominant genetic disorder characterized by variable expression of short stature, craniofacial dysmorphism, congenital heart defects, and myeloproliferative disorders, such as juvenile myelomonocytic leukemia (8). Many NS patients are highly functional, despite requiring specialty care to manage aspects of their condition. However, available therapies do not address the root cause of NS directly. Mutations in multiple RAS-associated signaling pathway genes can cause NS (9–11) with approximately 5% occurring in KRAS (12). KRAS V14I and T58I substitutions are particularly associated with NS (12). V14I is also seen in a limited number of large bowel tumors (14) and lung tumors (11), although at a much lower level than the classical KRASG12V oncogene. KRASV14I has been extensively characterized from a biological standpoint, given the development of a genetically engineered mouse model that phenocopies key aspects of Noonan syndrome (9–11). Nevertheless, the structural basis for the reported phenotypes has not been investigated.
RAS proteins are GTPases that function as molecular switches, cycling between active, GTP-bound, and inactive, GDP-bound, states, and regulating multiple pathways such as the MAPK pathway (1). RAS proteins contain two mobile structural elements, switch 1 (SW1) and switch 2 (SW2), which, based on their conformation, regulate many biological functions of RAS including its enzymatic activity and other protein-protein interactions (15–17). SW1 primarily interacts with downstream effectors such as RAF and PI3K, whereas SW2 is more involved in GTP hydrolysis, although it also participates in direct interactions with some RAS effectors (16, 18, 19). Guanine exchange factors (GEFs) and GTPase activating proteins (GAPs) engage both SW1 and SW2 during catalysis of nucleotide exchange (20) and GTP hydrolysis (21), respectively. Differences in switch dynamics between disease-associated RAS mutants translate into variable biochemical behaviors such as nucleotide-binding affinity, GTP hydrolysis, and protein-protein interactions (22, 23). Structural evaluation of these RAS variants is crucial to understanding the mechanisms that drive biochemical and disease phenotypes (21, 24, 25).
Many activating RAS mutations, such as the common KRAS mutation G12D, function by impairing GTPase activity. However, KRASV14I displays an intermediate level of intrinsic and GAP-mediated GTPase activity when compared with other oncogenic isoforms (12). Moreover, KRASV14I shows increased nucleotide-exchange activity, which is likely responsible for the accumulation of its active, GTP-bound state (9). Rapid nucleotide exchange is a poorly understood mechanism for RAS activation, but has been hypothesized to be caused by structural changes in RAS itself, particularly in the switch regions (2).
We solved the crystal structure of KRASV14I:GDP and found that nucleotide exchange is facilitated by destabilization and opening of the GTP-binding site, similar to other classes of rapid nucleotide-exchanging RAS mutants such as KRASA146T. Hydrogen-deuterium exchange (HDX)-mass spectrometry (MS) confirmed the open conformation occurs in solution, but with different kinetics than KRASA146T. This open conformation enhances the affinity of KRASV14I for the GEF SOS1. This atypical mechanism of RAS activation may provide new opportunities for targeting open conformations of RAS.
Results
Crystal form 1: extended SW1 conformation
We solved crystal structures of KRASV14I in complex with GDP in two different space groups. The data collection and refinement statistics are in Table 1. The P321 form, form 1, diffracted to ∼1.5 Å, showed a single molecule in the asymmetric unit and demonstrated a remarkable extended SW1 and β2 conformation with respect to KRASWT (Fig. 1, A and B). Electron density is clear and defined for all SW1 residues (Fig. 1C). The SW1 conformation is constrained by few crystal contacts and the refined model demonstrates a low B-factor, suggesting this conformation is not an artifact of crystallization (Fig. 2). In most monomeric RAS structures, SW1 starts just after α1 and extends to β2, participating in nucleotide binding and forming interactions with the base, sugar, and phosphate groups (22, 26, 27). In addition, previous structures showed an antiparallel interaction between β2 and β3. However, in the KRASV14I structure, the C-terminal region of α1 undergoes a large conformational change, forming a new anti-parallel β interaction with what was originally β2 (now β3new). In this conformation, the usual β2–β3 interaction is disrupted and SW1 no longer participates in GDP binding. This conformation is essentially the same as KRASA146T in complex with GDP (28).
Table 1.
Data collection and refinement statistics
| Form 1 | Form 2 | |
|---|---|---|
| Data collection | ||
| Space group | P321 (6MQG) | C2 (6MQN) |
| Cell parameters | ||
| a, b, c (Å) | 77.75, 77.75, 55.93 | 67.71, 84.22, 87.84 |
| α, β, γ (°) | 90.00, 90.00, 120.00 | 90.00, 110.26, 90.00 |
| Resolution (highest shell) (Å) | 50.00–1.50 (1.53–1.50) | 50.00–1.60 (1.63–1.60) |
| No. of unique reflections | 31,559 | 59,738 |
| Redundancy (last shell) | 14.5 (14.5) | 6.5 (4.1) |
| Completeness (last shell) (%) | 100.0 (99.9) | 98.1 (91.3) |
| I/σ(I) (last shell) | 38.05 (3.13) | 28.5 (1.2) |
| Rpim (last shell) | 0.024 (0.255) | 0.025 (0.447) |
| CC1/2 | 0.91 | 0.57 |
| CC (51) | 0.98 | 0.85 |
| Refinement statistics | ||
| Resolution range (Å) | 28.84–1.49 | 42.11–1.60 |
| No. of reflections used | 31,213 | 54,214 |
| No. of protein atoms | 2,925 | 3,928 |
| No. of water molecules | 193 | 381 |
| No. of ligand atoms | 28 | 84 |
| Rworka | 0.15 | 0.18 |
| Rfreeb | 0.19 | 0.22 |
| Root mean square deviation bond lengths (Å) | 0.004 | 0.006 |
| Root mean square deviation bond angles (°) | 0.653 | 0.863 |
| Ramachandran | ||
| Favored/allowed | 98.77/1.23 | 98.68/1.32 |
| Outlier (%) | 00.00 | 00.00 |
| Average B-factors (Å2) | ||
| Protein | 18.15 | 20.64 |
| Ligand | 15.73 | 15.37 |
| Water | 36.44 | 27.94 |
a Rwork = Σ‖Fobs| − |Fcal‖/Σ|Fobs|.
b Rfree = Σ‖Fobs| − |Fcal‖/Σ|Fobs|, where Fobs is from a test set of reflections that are not used in structural refinement.
Figure 1.

Open conformation of KRASV14I:GDP crystal structure. SW1 is in yellow and SW2 is in green. A, KRASV14I:GDP, form 1 (sg:P321), demonstrates an open SW1 and formation of a new antiparallel β strand interaction. B, KRASWT:GDP (PDB 4obe) shows classical closed SW1 conformation as a comparison. C, 2Fo − Fc electron density contoured to 1σ (blue) is well-defined for SW1 residues 25–46. D, Mg2+ ion density is not observed in KRASV14I:GDP crystals. 2Fo − Fc electron density contoured at 1.0 σ in blue. Ile14 is shown in sticks and colored cyan, water molecules are shown as nonbonded spheres and colored red, and GDP is shown in sticks, and E, KRASV14I:GDP, form 2 (sg:C2), shows disorder in SW1 but the β2–β3 interaction is intact.
Figure 2.
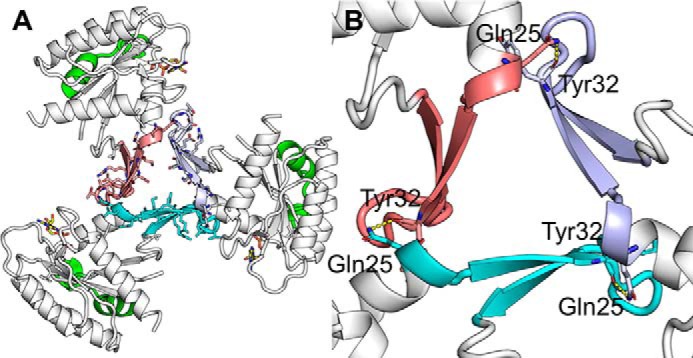
Switch 1 crystal contacts in KRASV14I crystal form 1. A, overall 3-fold symmetry axes, three switch 1 are colored in cyan, orange, and purple/blue for clarity. B, magnification of A. Few hydrogen bonds (yellow) are formed between asymmetric units, hydrogen bonds formed between the side chain of Gln25 and Tyr32.
The conformation of SW2 is also altered; an unsurprising finding given that SW1 and SW2 are immediately adjacent and demonstrate concerted movements in other structures (23). We also note a lack of density for the magnesium ion from the active site (Fig. 1D). In the absence of magnesium and β2–β3 interactions, greater flexibility is observed in SW2, leading to decreased helicity, causing SW2 residues to extend further into the solvent relative to the KRASWT (Fig. 1, A and B). As a result of these changes, the catalytic residue Gln61 and p-loop residues 10GAGGVGKS17 are highly exposed to solvent, with a solvent-accessible area for the protein and GDP of 2621 and 57 Å2, respectively. Both values are higher than in the KRASWT structure (PDB code 4obe) at 2290 and 26 Å2, respectively (29).
As a consequence of SW1 and SW2 movements and the loss of the active-site magnesium, crystal form 1 displays alterations in nucleotide interactions found in prior RAS structures that may explain the high rate of nucleotide exchange observed for KRASV14I (30). In KRASWT:GDP (PDB code 4obe), the main chain carbonyl of Asp30 hydrogen bonds with ribose, and Ser17 bonds to the β-phosphate oxygen (22). In the V14I mutant both interactions are lost. Also, the guanine base is typically sandwiched between Phe28 and Lys117 side chains (31), but this interaction is also lost in KRASV14I (Fig. S1).
Finally, we noted electron density consistent with oxidation of the thiol at Cys118. This modification has previously been shown to increase the nucleotide-exchange rate in RAS (13), suggesting that this may contribute to the observed biochemical properties of this mutant. However, it also suggests that Cys118 is exposed in solution, similar to how Cys118 becomes exposed when RAS is bound to SOS1. This may hold significance for potential therapeutic efforts, given that Cys118 has been targeted by covalent small molecules in RAS:SOS complexes (32).
Crystal form 2: disorder in the switches
KRASV14I also formed crystals in the C2 space group that diffracted to ∼1.6 Å, and included three molecules in the asymmetric unit (data collection and refinement statistics in Table 1). Unlike crystal form 1, the switch regions are generally disordered, with the exception of one molecule where density for SW2 is observable and shows a closed, helical (58–62) conformation (Fig. 1E). The side chain of catalytic residue Gln61 is oriented toward the nucleotide phosphate group and interacts via solvent molecules. These interactions would be predicted to allow transitions between the inactive and active state (33, 34). No electron density was observed for 6–9 residues in SW1, depending on the molecule, but the observable β2–β3 motif formed an antiparallel interaction, similar to closed KRAS structures in the PDB. This feature suggests that GDP-bound KRASV14I may also exist in solution in a more typical closed conformation. The core G-domains of both forms 1 and 2 are similar, with exceptions in Tyr32, a residue previously implicated in rapid nucleotide-exchange mechanisms (35, 36). The position of Tyr32 is disordered in all three monomers. p-Loop residues 10GAGGVGKS17 are also highly exposed to solvent, similar to the form 1 structure, and no magnesium ion density is observed.
Hydrogen-deuterium exchange in solution is consistent with form 1
Protein crystallization requires that relatively stable protein conformations exist in solution for crystals to form. Nevertheless, it is theoretically possible that protein conformations could be driven by crystal contacts themselves. Nevertheless, in this particular case, the atomic displacement parameter, Wilson B-value, of the diffraction data are 13.68 Å2 and the overall protein isotropic B-factor is below 18 Å2 suggesting that the protein conformation is highly stable within the crystalline lattice. Nevertheless, to confirm that the protein conformation observed in crystal form 1 exists in solution, we used HDX-MS to evaluate switch dynamics in RAS, given that HDX-MS has been used previously to measure alterations in RAS switch dynamics (17, 37, 38). Here we directly compared HDX of KRASV14I:GDP versus KRASWT:GDP. We also compared these data with a previously published dataset KRASA146T:GDP (28). The HDX-MS results support the validity of the crystal structures that these structures exist in solution, and that they are not the result of crystal contacts. We observed a faster deuterium exchange in peptides that cover both the p-loop and the N terminus of α1(12–19) as well as areas beneath the p-loop (115–124) when compared with KRASWT (Fig. 3 and Fig. S3). At residues 23–39, the deuterium exchange rate was initially slower than in KRASA146T, but similar to the exchange rate in KRASWT (Fig. S3). We also observed higher exchange in loops beneath the nucleotide ring (132–144), likely related to rapid nucleotide exchange. Although the estimated time that KRASV14I spends in the open state, relative to KRASA146T, cannot be fully estimated by these data, they do support that KRASA146T spends more time overall in the open state. This finding could explain, in part the ability of A146T mutant to form tumors, whereas V14I-associated tumors are less common (39).
Figure 3.

HDX-MS of KRASV14I shows exposure of regions surrounding nucleotide. A, deuterium incorporation into four peptides that demonstrate the altered exchange kinetics in KRASV14I, KRASWT, and KRASA146T. The full HDX-MS dataset can be found in Fig. 4 and Table S1. B, regions of KRASV14I (highlighted in red) demonstrate increased deuterium exchange relative to KRASWT.
GDP displacement leads to SW1 instability in V14I
The mechanism of SW1 destabilization is not immediately apparent, given that residue 14 is not located in the switches. Residue 14 is in the p-loop, opposite the nucleotide relative to SW1. We therefore explored whether the mechanism of SW1 destabilization was due to displacement of the nucleotide because of the substitution at residue 14, which incorporates an extra buried carbon. Previous work has shown that the nucleotide binds to RAS with picomolar affinity, which can be rationalized by multiple, specific interactions between the nucleotide ligand and the protein-binding pocket that highly constrain its position (40, 41). In particular, the position of the guanine base is stabilized by four key residues, namely Phe28, Lys117, Asp119, and Lys147 (Fig. 4A), including a sandwich interaction between Phe28 and Lys117 side chains and the guanine base.
Figure 4.

Guanine nucleotide positioning is altered in KRASV14I. A, superimposed structures of KRASV14I (purple sticks) and KRASWT (green sticks, PDB 4obe). Residues that stabilize GDP are shown. The interaction between GDP and Phe28 is lost in KRASV14I. B, alignment of KRASWT and KRASV14I show the distance change between Cα atoms of residues Ile14 and Phe82 (in β4, just below the p-loop). C, alterations in the position of residue 14 Cα relative to PDB 4obe upon global alignment (blue) and distance between residues 14 and 82 Cα in various KRAS structures (red). Distances are increased in both KRASV14I structures. D, 14 degree tilt of the exposed GDP in KRASV14I relative to KRASWT (in yellow stick).
We posited that addition of a carbon in the side chain at position 14, as seen with the V14I substitution, has the potential to displace the nucleotide. The KRASV14I structures themselves provide evidence of small rearrangements surrounding position 14 that could influence nucleotide position. For example, in V14I we see loss of an interaction between the carbonyl oxygen of Ala11 and the main chain nitrogen of Ile14 relative to WT. To further demonstrate V14I-related changes, we aligned KRASV14I, using the Cα residues excluding the switch regions, with other X-ray structures of KRAS-GDP and compared the distances between structural elements adjacent to the p-loop as well as relative distances between those elements and the nucleotide. We expected that an additional carbon in Ile14 would push the main chain of the p-loop away from structural elements below the p-loop. We compared the change in the distance between Cα for each structure relative to KRASWT (PDB 4obe) and saw an average difference of 0.79 Å (S.D. 0.02), whereas other KRAS structures, including A146T, showed an average difference of 0.20 Å (S.D. 0.02) from WT (Fig. 4, B and C, red bars). The differences were statistically significant (p < 1.2 E-8). The intramolecular distance between Cα atoms of residues Ile14 and Phe82 for each structure also show similar differences from V14I (Fig. 4, B and C, blue bars). The net result of these changes is that the nucleotide base is pushed up from below, altering the pitch of the nucleotide by ∼14 degrees (Fig. 4D). Of note, a similar change in the pitch of the nucleotide base also occurs for KRASA146T, but for different reasons. In KRASA146T, formation of a new hydrogen bond between N7 of the purine base and the side chain O of the threonine substitution at position 146, which sits on the opposite side of the nucleotide relative to position 14, pulls the guanine ring toward Thr146 and SW1 (28). As a result, the purine base is tilted by ∼14 degrees. Altogether, these observations suggest that the remarkable similarities in the extended open conformation of SW1 between KRASA146T and KRASV14I structures, together with the shared rapid nucleotide-exchange phenotype, are evidence of a common underlying atypical mechanism of RAS activation.
Open SW1 leads to enhanced interactions with SOS
The open SW1 conformation exposes the binding interface accessed by the GEF SOS1 (Fig. 5A), suggesting that even though intrinsic nucleotide exchange is elevated for this class of exchange mutants, this protein conformation may further enable interactions with GEFs, leading to additional enhancements in nucleotide exchange. We performed protein-protein affinity measurements to evaluate the interaction between V14I and SOS1. Evaluation of protein affinity between RAS and SOS1 by microscale thermophoresis (MST) demonstrated ∼40-fold increase in binding affinity with SOS1:KRASV14I affinity at 220 nm, whereas SOS1:WT was 8300 nm (Fig. 5B). This demonstrates that although the intrinsic rate of GDP dissociation in KRASV14I is high, the open conformation enhances GEF interactions, further accelerating nucleotide exchange.
Figure 5.

Interaction between KRASV14I and SOS1. A, the binding interface of KRASV14I for SOS1 based on PDB 1NVU. Interacting residues on KRASV14I are highlighted in red. B, SOS1:KRAS binding affinity as measured by microscale thermophoresis. KRASV14I shows enhanced affinity toward SOS1 relative to KRASWT. All measurements were performed in triplicate: Kd for WT is 8.3 ± 0.6 (μm) and V14I is 0.22 ± 0.1 (μm). The SOS1 construct includes the REM and catalytic domains.
Discussion
KRASV14I, together with KRASA146T, constitute a unique class of RAS mutations that are activated primarily by rapid intrinsic nucleotide cycling, driven by altered protein dynamics. We provided the structural basis for rapid nucleotide exchange, specifically that the nucleotide-binding pocket is disrupted by changes in nucleotide positioning due to V14I substitutions, resulting in a major structural reorganization centered around SW1. These changes are accompanied by a loss of the active-site magnesium ion. Both aspects are reminiscent of structures of HRAS bound to the SOS catalytic pocket, suggesting that these structures could provide snapshots of intermediate steps involved in SOS-mediated nucleotide exchange (20).
The biological effects of the altered dynamics of SW1 in V14I, and the functionally similar A146T, also appear to extend beyond nucleotide exchange, to other processes essential for KRAS signaling. It is tempting to speculate from the open KRASV14I:GDP and KRASA146T:GDP structures is that GTP-bound proteins might also present an open SW1, which is incompatible with RAS-RAF interactions (34). However, this does not appear to be the case based on studies of KRASV14I and KRASA146T in biological systems. For example, interrogation of RAS signaling in a genetic mouse model of KRASV14I demonstrated up-regulation of GTP-bound KRASV14I as measured by pull downs using the Ras-binding domain of the c-RAF. These findings are incompatible with an extended SW1, suggesting that KRASV14I adopts a closed conformation when GTP-bound. Nevertheless, these mice showed little up-regulation of pERK and pAKT relative to WT throughout their development (9). KRASA146T:GTP also retains the ability to bind RAF, but KRASA146T mouse models showed modest up-regulation of pERK and pAKT relative to WT, although significantly less than KRASG12D (28). Together, these data demonstrate that nucleotide-exchange mutants such as KRASV14I remain competent to bind RAF, and signal through the MAPK pathway, but are attenuated in signaling, likely explaining the less aggressive clinical phenotypes relative to mutations characterized primarily by GAP-insensitivity such as KRASG12D.
These findings may be explainable by our observations that active site destabilization in KRASV14I and KRASA146T results in increased interactions with GEF (30). Presumably, this is brought about by the GEF-binding surface of KRASV14I being exposed in the open SW1 conformation. Whether this interaction leads to a dependence on GEFs in vivo is unknown. However, recent data on KRASA146T is suggestive (28). In that study, tissues from mice expressing KRASA146T or KRASG12D were analyzed for alterations in specific kinase pathways. Up-regulation of p90RSK activity in KRASA146T was noted. This is potentially significant given that p90RSK is a negative regulator of SOS1 (42). One possibility is that down-regulation of GEF activity via p90RSK is able to suppress nucleotide exchange to some degree, explaining the relatively mild phenotypes seen with nucleotide-exchange mutants. However, it also raises the possibility of discovering synthetic lethal interactions within these regulatory frameworks that could be exploited for therapeutic purposes. Whether other regulators of GEFs play a role in modulating the activity of nucleotide exchangers is another critical question.
Although this study places KRASV14I and KRASA146T in the same functional category, it is unclear if other RAS mutants with rapid exchange phenotypes share similar mechanisms and regulation. For example, KRASG13D has been identified as a rapid exchanger, but the mechanism of exchange is thought to relate to altered electrostatics of the nucleotide-binding pocket, not protein dynamics (22). Given this fundamental difference, it seems likely that regulatory mechanisms of G13D activation, if any, will be different for V14I or A146T. On the other hand, KRASK117N, another nucleotide-exchange mutation found mostly in colorectal cancer (39), may be subject to similar regulatory mechanisms given that the mutation occurs near the guanine base, similar to A146T.
Finally, the open conformation of SW1 in this mutant class raises the possibility of directly and selectively targeting this class of mutants using covalent inhibitors. As noted, we identified that Cys118 is oxidized in our crystal structures, suggesting that this residue is exposed to solvent. Interestingly, this residue has been targeted in the context of the RAS:SOS complex using covalent ligands (32). Selectively targeting this cysteine in un-complexed exchange mutants due to the open SW1 conformation is an intriguing possibility.
Experimental procedures
Protein expression and purification
A bacterial expression construct encoding human KRASV14I lacking the C terminus residues, 170MSKDGKKKKKKSKTKCVIM188 (expressed residues 1–169), that includes an N-terminal His6 tag and tobacco etch virus cleavage sequence was made by site-directed mutagenesis starting from a WT KRAS expression construct. Escherichia coli BL21 (DE3) cells were transformed and grown in Luria-Bertani (LB) medium at 37 °C to an A600 of 0.8–1.0. Protein expression was induced by the addition of 0.5 mm isopropyl β-d-thiogalactopyranoside. Cells were collected by centrifugation after 16 h at 16 °C and lysed in lysis buffer (50 mm Tris, pH 8.0, 500 mm NaCl, 2 mm MgCl2, 5 mm 2-mercaptoethanol, 1 mm benzamidine, and 1 mm PMSF). Lysate was clarified by centrifugation at 51,000 × g for 60 min at 4 °C. The supernatant was applied to an IMAC cartridge (Bio-Rad Laboratories, Inc.) and protein was desalted into crystallization buffer (20 mm Tris, pH 8.0, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT). His-tagged tobacco etch virus protease (1:5, w/w) was added to the elute, and protein was incubated at 4 °C overnight. Finally, the cleaved protein was passed through an IMAC cartridge to isolate from the tag. Further fractionation was done using a Superdex 75 size exclusion column. The peak fractions were analyzed by SDS-PAGE and the fractions containing KRASV14I were pooled and concentrated using an Amicon® Ultra Centrifugal Filter with a molecular mass cutoff of 10,000 Da to 30 mg/ml and stored at −80 °C.
Crystallization and data collection
Concentrated KRASV14I (30 mg/ml) in a buffer composed of 20 mm HEPES and 20 mm NaCl, pH 8.0, was used for crystallization trials. Initial crystallization trials were performed with a Mosquito (TTP LabTech) crystallization robot using the sitting-drop method (100 nl of protein plus 100 nl of crystallization solution). Optimized single protein crystals were grown using hanging drop vapor diffusion at 20 °C in a reservoir solution containing 1.8 m sodium phosphate monobasic monohydrate and potassium phosphate dibasic, pH 8.2. Three-week-old crystals were mounted with 15% glycerol in mother liquor as cryoprotectant and flash cooled in liquid nitrogen. X-ray diffraction data were collected at the Advanced Photon Source beamline 19-BM at 100 K. The crystal belonged to the P321 space group and diffracted to 1.49 Å. The second crystal was grown in the same conditions and 2-week-old crystals were harvested and flash cooled in liquid nitrogen. The crystal belonged to the C2 space group and diffracted to 1.6 Å at the Advanced Photon Source beamline 19-ID at 100 K. The initial diffraction data were indexed, integrated, and scaled using HKL2000/3000 (43).
Structure determination and refinement
Crystal structures were determined by molecular replacement using WT KRAS (PDB 4obe) as an initial search model with Phaser in the CCP4 Suite (44). The models were further built and refined using Phenix (45) and COOT (46) by manual model correction. Solvent molecules were added, and the final refinement performed by phenix.refine. Stereochemical parameters of the final model were analyzed with the assistance of Molprobity (47). Data collection and refinement statistics are listed in Table S1.
HDX-MS of KRASV14I
HDX-MS experiments were performed similarly to previous reports (17, 48). Purified proteins were diluted into sample buffer (10 mm HEPES, pH 8.0, 5 mm MgCl2, 5 μm GDP, 150 mm NaCl) to a concentration of 10 μm prior to labeling. Samples were deuterated for the times described using labeling buffer, and labeling was stopped with an equal volume of quench buffer (Table S1). Quenched samples were immediately analyzed. Deuterium measurement with MS was performed as previously described (17, 48) using a Synapt G2si HDMSE, online digestion Enzymatic column, 1 × 50, 1.8-μm HSS T3 separation, 1.7-μm BEH trap. Deuterium incorporation graphs were generated using DynamX 3.0 software (Waters) by subtracting the centroid of the isotopic distribution at each labeling time point from the centroid of the isotopic distribution of the undeuterated reference species. Because the data were not corrected for back-exchange, each data point represents the relative deuterium level at each time point for each peptide (49).
MST with KRASV14I and SOS1
KRASV14I (100 μm) was incubated with 200 μm GDP in 1× PBS at room temperature for 1 h. The solution was then desalted into 1× PBS with 2 mm MgCl2 and 1% Tween 20 via a ZebaTM (Thermo Fisher) spin desalting column. 16-Point serial dilutions of KRASV14I were prepared and mixed 1:1 with GFP-556SOS1049 to a final volume of 20 μl. The reaction mixtures were loaded into premium-treated capillaries and analyzed by a Monolith NT.115 (Nanotemper Technologies) at 60% MST power and 40% LED power with a laser on-time of 30 s. The KD was calculated by taking the average of triplicate Fnorm measurements at each concentration and fitting the data by using Palmist (50) and plotting in Prism 7. Raw data have been normalized and converted to percent response (fluorescence signal of lowest and highest concentration of KRASV14I are set as 0 and 100%, respectively).
Author contributions
A. K. B., J. L., and K. D. W. conceptualization; A. K. B. and J. L. data curation; A. K. B., J. L., T. E. W., S. G., J. R. E., and K. D. W. formal analysis; A. K. B., J. R. E., and K. D. W. supervision; A. K. B., J. R. E., and K. D. W. funding acquisition; A. K. B., J. L., and K. D. W. writing-original draft; A. K. B., J. L., T. E. W., S. G., D. G., A. N., J. R. E., and K. D. W. writing-review and editing; J. L., T. E. W., S. G., D. G., and A. N. investigation.
Supplementary Material
Acknowledgments
Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by the University of Chicago Argonne, LLC, for the United States Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
This work was supported by National Institutes of Health Grant U54 CA196519, a V Foundation for Cancer Research V Scholar Grant, DOD Grant W81XWH-16-1-0106 (to K. D. W.), and a research collaboration grant with the Waters Corporation (to J. R. E.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S3.
The atomic coordinates and structure factors (codes 6MQG and 6MQN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NS
- Noonan syndrome
- MAPK
- mitogen-activated protein kinase
- GEF
- guanine exchange factor
- SW1
- switch1
- SW2
- switch 2
- GAP
- GTPase-activating protein
- HDX
- hydrogen-deuterium exchange
- MST
- microscale thermophoresis
- PDB
- Protein Data Bank.
References
- 1. Fernández-Medarde A., and Santos E. (2011) Ras in cancer and developmental diseases. Genes Cancer 2, 344–358 10.1177/1947601911411084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tidyman W. E., and Rauen K. A. (2009) The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19, 230–236 10.1016/j.gde.2009.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hammond D. E., Mageean C. J., Rusilowicz E. V., Wickenden J. A., Clague M. J., and Prior I. A. (2015) Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J. Proteome Res. 14, 1535–1546 10.1021/pr501191a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim E., Ilic N., Shrestha Y., Zou L., Kamburov A., Zhu C., Yang X., Lubonja R., Tran N., Nguyen C., et al. (2016) Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov. 6, 714–726 10.1158/2159-8290.CD-16-0160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fasano O., Aldrich T., Tamanoi F., Taparowsky E., Furth M., and Wigler M. (1984) Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 81, 4008–4012 10.1073/pnas.81.13.4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Janes M. R., Zhang J., Li L.-S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S. J., Darjania L., et al. (2018) Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e517 10.1016/j.cell.2018.01.006 [DOI] [PubMed] [Google Scholar]
- 7. Kratz C. P., Franke L., Peters H., Kohlschmidt N., Kazmierczak B., Finckh U., Bier A., Eichhorn B., Blank C., Kraus C., et al. (2015) Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br. J. Cancer 112, 1392–1397 10.1038/bjc.2015.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jongmans M. C., van der Burgt I., Hoogerbrugge P. M., Noordam K., Yntema H. G., Nillesen W. M., Kuiper R. P., Ligtenberg M. J., van Kessel A. G., van Krieken J. H., Kiemeney L. A., and Hoogerbrugge N. (2011) Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur. J. Hum. Genet. 19, 870–874 10.1038/ejhg.2011.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hernández-Porras I., Fabbiano S., Schuhmacher A. J., Aicher A., Cañamero M., Camara J. A., Cussó L., Desco M., Heeschen C., Mulero F., Bustelo X. R., Guerra C., and Barbacid M. (2014) K-RasV14I recapitulates Noonan syndrome in mice. Proc. Natl. Acad. Sci. U.S.A. 111, 16395–16400 10.1073/pnas.1418126111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hernández-Porras I., Jiménez-Catalán B., Schuhmacher A. J., and Guerra C. (2015) The impact of the genetic background in the Noonan syndrome phenotype induced by K-Ras(V14I). Rare Dis. 3, e1045169 10.1080/21675511.2015.1045169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hernández-Porras I., Schuhmacher A. J., Garcia-Medina R., Jiménez B., Cañamero M., de Martino A., and Guerra C. (2016) K-Ras(V14I)-induced Noonan syndrome predisposes to tumour development in mice. J. Pathol. 239, 206–217 10.1002/path.4719 [DOI] [PubMed] [Google Scholar]
- 12. Schubbert S., Zenker M., Rowe S. L., Böll S., Klein C., Bollag G., van der Burgt I., Musante L., Kalscheuer V., Wehner L. E., Nguyen H., West B., Zhang K. Y., Sistermans E., Rauch A., et al. (2006) Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 38, 331–336 10.1038/ng1748 [DOI] [PubMed] [Google Scholar]
- 13. Hobbs G. A., Bonini M. G., Gunawardena H. P., Chen X., and Campbell S. L. (2013) Glutathiolated Ras: Characterization and implications for Ras activation. Free Radic Biol Med. 57, 221–229 10.1016/j.freeradbiomed.2012.10.531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forbes S. A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S., et al. (2014) COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 43, D805–D811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milburn M. V., Tong L., deVos A. M., Brünger A., Yamaizumi Z., Nishimura S., and Kim S. H. (1990) Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic Ras proteins. Science 247, 939–945 10.1126/science.2406906 [DOI] [PubMed] [Google Scholar]
- 16. Vetter I. R., and Wittinghofer A. (2001) The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 10.1126/science.1062023 [DOI] [PubMed] [Google Scholar]
- 17. Harrison R. A., Lu J., Carrasco M., Hunter J., Manandhar A., Gondi S., Westover K. D., and Engen J. R. (2016) Structural dynamics in Ras and related proteins upon nucleotide switching. J. Mol. Biol. 428, 4723–4735 10.1016/j.jmb.2016.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buhrman G., Holzapfel G., Fetics S., and Mattos C. (2010) Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. U.S.A. 107, 4931–4936 10.1073/pnas.0912226107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scheffzek K., Lautwein A., Kabsch W., Ahmadian M. R., and Wittinghofer A. (1996) Crystal structure of the GTPase-activating domain of human p120GAP and implications for the interaction with Ras. Nature 384, 591–596 10.1038/384591a0 [DOI] [PubMed] [Google Scholar]
- 20. Boriack-Sjodin P. A., Margarit S. M., Bar-Sagi D., and Kuriyan J. (1998) The structural basis of the activation of Ras by Sos. Nature 394, 337–343 10.1038/28548 [DOI] [PubMed] [Google Scholar]
- 21. Scheffzek K., Ahmadian M. R., Kabsch W., Wiesmüller L., Lautwein A., Schmitz F., and Wittinghofer A. (1997) The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338 10.1126/science.277.5324.333 [DOI] [PubMed] [Google Scholar]
- 22. Hunter J. C., Manandhar A., Carrasco M. A., Gurbani D., Gondi S., and Westover K. D. (2015) Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 13, 1325–1335 10.1158/1541-7786.MCR-15-0203 [DOI] [PubMed] [Google Scholar]
- 23. Lu J., Bera A. K., Gondi S., and Westover K. D. (2018) KRAS switch mutants D33E and A59G crystallize in the state 1 conformation. Biochemistry 57, 324–333 10.1021/acs.biochem.7b00974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu J., Hunter J., Manandhar A., Gurbani D., and Westover K. D. (2015) Structural dataset for the fast-exchanging KRAS G13D. Data Brief. 5, 572–578 10.1016/j.dib.2015.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hall B. E., Bar-Sagi D., and Nassar N. (2002) The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. U.S.A. 99, 12138–12142 10.1073/pnas.192453199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dharmaiah S., Bindu L., Tran T. H., Gillette W. K., Frank P. H., Ghirlando R., Nissley D. V., Esposito D., McCormick F., and Stephen A. G. (2016) Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. U.S.A. 113, E6766–E6775 10.1073/pnas.1615316113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maurer T., Garrenton L. S., Oh A., Pitts K., Anderson D. J., Skelton N. J., Fauber B. P., Pan B., Malek S., Stokoe D., Ludlam M. J., Bowman K. K., Wu J., Giannetti A. M., Starovasnik M. A., et al. (2012) Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. U.S.A. 109, 5299–5304 10.1073/pnas.1116510109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poulin E. J., Bera A. K., Lu J., Lin Y.-J., Strasser S. D., Paulo J. A., Huang T. Q., Morales C., Yan W., Cook J., Nowak J. A., et al. (2019) Tissue-specific oncogenic activity of K-RasA146T. Cancer Discov. 9, 738–755 10.1158/2159-8290.CD-18-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee B., and Richards F. M. (1971) Interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400 10.1016/0022-2836(71)90324-X [DOI] [PubMed] [Google Scholar]
- 30. Gremer L., Merbitz-Zahradnik T., Dvorsky R., Cirstea I. C., Kratz C. P., Zenker M., Wittinghofer A., and Ahmadian M. R. (2011) Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum. Mutat. 32, 33–43 10.1002/humu.21377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schlichting I., Wittinghofer A., and Rosch P. (1988) Proton NMR-studies of the GDP Mg2+ complex of the Ha-Ras oncogene product-p21. Biochem. Biophys. Res. Commun. 150, 444–448 10.1016/0006-291X(88)90540-2 [DOI] [PubMed] [Google Scholar]
- 32. Winter J. J., Anderson M., Blades K., Brassington C., Breeze A. L., Chresta C., Embrey K., Fairley G., Faulder P., Finlay M. R., et al. (2015) Small molecule binding sites on the Ras: SOS complex can be exploited for inhibition of Ras activation. J. Med. Chem. 58, 2265–2274 10.1021/jm501660t [DOI] [PubMed] [Google Scholar]
- 33. Buhrman G., Wink G., and Mattos C. (2007) Transformation efficiency of RasQ61 mutants linked to structural features of the switch regions in the presence of Raf. Structure 15, 1618–1629 10.1016/j.str.2007.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fetics S. K., Guterres H., Kearney B. M., Buhrman G., Ma B., Nussinov R., and Mattos C. (2015) Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516 10.1016/j.str.2014.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hall B. E., Yang S. S., Boriack-Sjodin P. A., Kuriyan J., and Bar-Sagi D. (2001) Structure-based mutagenesis reveals distinct functions for Ras switch 1 and switch 2 in SOS-catalyzed guanine nucleotide exchange. J. Biol. Chem. 276, 27629–27637 10.1074/jbc.M101727200 [DOI] [PubMed] [Google Scholar]
- 36. White M. A., Nicolette C., Minden A., Polverino A., Van Aelst L., Karin M., and Wigler M. H. (1995) Multiple Ras functions can contribute to mammalian cell transformation. Cell 80, 533–541 10.1016/0092-8674(95)90507-3 [DOI] [PubMed] [Google Scholar]
- 37. Lu J., Harrison R. A., Li L., Zeng M., Gondi S., Scott D., Gray N. S., Engen J. R., and Westover K. D. (2017) KRAS G12C drug development: discrimination between switch II pocket configurations using hydrogen/deuterium-exchange mass spectrometry. Structure 25, 1442–1448.e1443 10.1016/j.str.2017.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gentile D. R., Rathinaswamy M. K., Jenkins M. L., Moss S. M., Siempelkamp B. D., Renslo A. R., Burke J. E., and Shokat K. M. (2017) Ras binder induces a modified switch-II pocket in GTP and GDP states. Cell Chem. Biol. 24, 1455–1466.e1414 10.1016/j.chembiol.2017.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haigis K. M. (2017) KRAS alleles: the devil is in the detail. Trends Cancer 3, 686–697 10.1016/j.trecan.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. John J., Sohmen R., Feuerstein J., Linke R., Wittinghofer A., and Goody R. S. (1990) Kinetics of interaction of nucleotides with nucleotide-free H-ras p21. Biochemistry 29, 6058–6065 10.1021/bi00477a025 [DOI] [PubMed] [Google Scholar]
- 41. Hunter J. C., Gurbani D., Ficarro S. B., Carrasco M. A., Lim S. M., Choi H. G., Xie T., Marto J. A., Chen Z., Gray N. S., and Westover K. D. (2014) In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. U.S.A. 111, 8895–8900 10.1073/pnas.1404639111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saha M., Carriere A., Cheerathodi M., Zhang X., Lavoie G., Rush J., Roux P. P., and Ballif B. A. (2012) RSK phosphorylates SOS1 creating 14-3-3-docking sites and negatively regulating MAPK activation. Biochem. J. 447, 159–166 10.1042/BJ20120938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 44. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B. 3rd, Snoeyink J., Adams P. D., et al. (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lim S. M., Westover K. D., Ficarro S. B., Harrison R. A., Choi H. G., Pacold M. E., Carrasco M., Hunter J., Kim N. D., Xie T., Sim T., Jänne P. A., Meyerson M., Marto J. A., Engen J. R., and Gray N. S. (2014) Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. Engl. 53, 199–204 10.1002/anie.201307387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wales T. E., and Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 10.1002/mas.20064 [DOI] [PubMed] [Google Scholar]
- 50. Scheuermann T. H., Padrick S. B., Gardner K. H., and Brautigam C. A. (2016) On the acquisition and analysis of microscale thermophoresis data. Anal. Biochem. 496, 79–93 10.1016/j.ab.2015.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Karplus P. A., and Diederichs K. (2012) Linking Crystallographic Model and Data Quality. Science 336, 1030–1033 10.1126/science.1218231 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.