

Abstract
Viral infection induces potent cellular immunity and activated intracellular signaling, which may dictate the driver events involved in immune escape and clonal selection of virus-associated cancers, including Epstein-Barr virus (EBV)-positive lymphomas. Here, we thoroughly interrogated PD-L1/PD-L2-involving somatic aberrations in 384 samples from various lymphoma subtypes using high-throughput sequencing, particularly focusing on virus-associated lymphomas. A high frequency of PD-L1/PD-L2-involving genetic aberrations was observed in EBV-positive lymphomas [33 (22%) of 148 cases], including extranodal NK/T-cell lymphoma (ENKTL, 23%), aggressive NK-cell leukemia (57%), systemic EBV-positive T-cell lymphoproliferative disorder (17%) as well as EBV-positive diffuse large B-cell lymphoma (DLBCL, 19%) and peripheral T-cell lymphoma-not otherwise specified (15%). Predominantly causing a truncation of the 3′-untranslated region, these alterations represented the most prevalent somatic lesions in ENKTL. By contrast, the frequency was much lower in EBV-negative lymphomas regardless of histology type [12 (5%) of 236 cases]. Besides PD-L1/PD-L2 alterations, EBV-positive DLBCL exhibited a genetic profile distinct from EBV-negative one, characterized by frequent TET2 and DNMT3A mutations and the paucity of CD79B, MYD88, CDKN2A, and FAS alterations. Our findings illustrate unique genetic features of EBV-associated lymphomas, also suggesting a potential role of detecting PD-L1/PD-L2-involving lesions for these lymphomas to be effectively targeted by immune checkpoint blockade.
Subject terms: Cancer genomics, Immunosurveillance, Tumour virus infections
Introduction
Epstein-Barr virus (EBV) is one of the most prevalent human viruses. The interplay between EBV replication, latency, and immune control can be disrupted, evoking prolonged proliferation of EBV-infected lymphocytes and their malignant transformation [1, 2]. Since its discovery as the first human tumor virus, EBV has been implicated in the development of a wide range of human cancers, including Burkitt lymphoma, EBV-positive T-cell and NK-cell proliferations, and a subset of diffuse large B-cell lymphoma (DLBCL) [1, 2]. The oncogenic potential of EBV-encoded products has been extensively studied; they are known to mimic a variety of cellular factors involved in cell growth, transcription, and apoptosis, to usurp control of the pathways regulating diverse homeostatic cellular functions. However, despite this oncogenic potential, the virus induces lymphoma only in a fraction of EBV-infected people, generally after a long latency period. Thus, EBV is thought to require somatic alterations in the cellular genome to cause lymphoma, whose impacts on the development of EBV-positive lymphomas have not fully been investigated, even though they might substantially differ from those involved in the development of EBV-negative lymphomas.
Immune checkpoint blockade using anti-PD-1/PD-L1 antibodies is a highly promising therapy that can induce a durable anti-tumor response and a long-term remission in a wide variety of cancer types [3–5]. In particular, an excellent response to anti-PD-1 antibodies has been demonstrated for advanced cases with classical Hodgkin lymphoma (cHL), a defining feature of which is frequent copy number gains or amplifications involving PD-L1 and/or PD-L2, suggesting a close link between PD-L1/PD-L2 genetic alterations and the therapeutic response to these agents [6–8]. In addition to cHL, several subtypes of B-cell lymphomas are shown to have structural variations (SVs) involving PD-1 ligands, such as chromosomal translocation causing promoter replacement [9–12]. Moreover, recently we have reported frequent SVs disrupting the 3′-untranslated region (UTR) of PD-L1 in multiple cancers, especially in adult T-cell leukemia/lymphoma (ATL) and DLBCL [13, 14]. These observations point to a potential of diverse PD-L1/PD-L2 somatic alterations as a key driver of lymphomagenesis. However, the comprehensive landscape of these alterations in many subtypes of non-Hodgkin lymphomas (NHLs) remains elusive.
In this study, we first interrogated PD-L1/PD-L2-involving genetic aberrations in various lymphoma subtypes and found their frequent involvement in EBV-associated lymphomas. We further investigated different underlying genetic mechanisms between EBV-positive and -negative tumors.
Materials and methods
Patient samples
A total of 384 lymphoma specimens of different histology types, including 337 samples from Japan and 47 samples from France [TENOMIC consortium of the Lymphoma Study Association (LYSA) group], were enrolled in this study according to the protocols approved by the Institutional Review Boards (IRB, Supplemental Table S1). All samples were collected from patients with informed consent, except for already-collected, anonymized samples whose use was permitted by the IRB. Pathological diagnosis was based on the 2008 World Health Organization (WHO) classification [15], except for EBV-positive DLBCL, for which the restriction to elderly patients was not applied according to the recent revision of the WHO classification [16]. This study was approved by the institutional ethics committees of the Graduate School of Medicine, Kyoto University and other participating institutes.
Detection of PD-L1/PD-L2-involving genetic alterations
SVs and focal copy number alterations (CNAs) affecting PD-1 ligands were explored using targeted-capture sequencing with a custom SureSelect library (Agilent Technologies, Santa Clara, CA, USA) designed for capturing the entire sequences of the PD-L1 and PD-L2 genes, including their exons, introns, and 5′- and 3′-UTRs (Supplemental Figure S1). Sequencing data were obtained using the Illumina HiSeq 2500 platform with a standard 125-bp paired-end read protocol. SVs and focal CNAs were detected using the Genomon pipeline (https://github.com/Genomon-Project/) and the CNACS algorithm, respectively, as previously described [13, 17, 18]. Putative SVs were manually curated and further filtered by removing those (i) with Fisher’s exact P-value >0.1; (ii) with <4 supporting reads in tumor; or (iii) present in any of control samples. SV breakpoints were visually inspected using Integrative Genomics Viewer (IGV). Candidate focal CNAs (< 20 Mb in length) were also manually reviewed and further filtered by removing those with <3 probes.
Detection of EBV genome
For the detection of EBV sequence, after sequencing reads were mapped to the EBV genome (NC_007605), the number of the EBV-aligned reads in proper pairs were enumerated and divided by the number of total reads uniquely mapped to the human reference genome (GRCh37). Then, the obtained ratio was evaluated for the cut-off value of 0.00015%, which was determined so that all EBV-positive cases with DLBCL and extranodal NK/T-cell lymphoma (ENKTL) confirmed by in situ hybridization (ISH) for EBV-encoded small RNA (EBER), LMP1 immunohistochemistry (IHC), or Southern blot were included, while minimizing EBV-negative cases. RNA sequencing (RNA-seq) data from the Cancer Genome Atlas (TCGA) cohort were analyzed in a similar manner, where the detection of EBV reads was considered as positive. Southern blot for EBV genome was performed according to a standard procedure.
Whole-exome analysis of ENKTL
Whole-exome sequencing data for ENKTL (accession number SRP057085) were obtained from the National Center for Biotechnology Information Sequence Read Archive and applied for the detection of somatic alterations using the Genomon pipeline, as previously described [18]. Candidate mutations with (i) Fisher’s exact P-value ≤0.01; (ii) ≥4 variant reads in tumor; and (iii) allele frequency in tumor ≥0.025 were adopted and filtered by excluding (i) synonymous single nucleotide variants (SNVs); (ii) known variants listed in the 1000 Genomes Project (October 2014 release), NCBI dbSNP build 131, National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) 6500, the Human Genome Variation Database (version 2.0), or our in-house single nucleotide polymorphism (SNP) database, unless they were listed in the COSMIC database (v70). SVs and focal CNAs were analyzed in the same manner as described above.
Targeted-capture sequencing of EBV-negative and -positive DLBCLs
Targeted-capture sequencing was performed using a custom SureSelect library (Agilent Technologies) for 140 lymphoma-associated genes (Supplemental Table S2), as previously described [18, 19]. Additional probes for 1999 SNPs were designed to calculate genomic copy numbers [17]. Somatic mutations with (i) ≥5 variant reads in tumor; and (ii) allele frequency in tumor ≥0.025 were adopted and filtered by removing (i) synonymous SNVs; (ii) known variants listed in SNP databases (as described above); (iii) variants only present in unidirectional reads; (iv) variants occurring in repetitive genomic regions. Candidate mutations were further filtered by removing missense SNVs with allele frequency of 0.4–0.6. Finally, mapping errors were removed by visual inspection with IGV. SVs and focal CNAs were analyzed in the same manner as described above.
RNA-seq and expression microarray analysis
RNA-seq data for endemic Burkitt lymphoma [20] (SRP062178), anaplastic large cell lymphoma [21] (ALCL, SRP044708), and cutaneous T-cell lymphoma [22] (CTCL, SRP058948) were obtained from the National Center for Biotechnology Information Sequence Read Archive. RNA-seq data for peripheral T-cell lymphoma [23] (PTCL, phs000689.v1.p1) and primary mediastinal B-cell lymphoma [24] (PMBCL, EGAD00001000692) were obtained through Database of Genotypes and Phenotypes and European Genome-phenome Archive, respectively. Microarray data for normal human cells were obtained from HemaExplorer [25]. For RNA-seq data, expression quantification and detection of fusion transcripts were performed using the Genomon pipeline, as previously described [13, 18].
Analysis of TCGA data sets
As previously reported, we analyzed 10,210 TCGA samples from 33 cancer types, for which RNA-seq data were available from the Cancer Genomic Hub (https://cghub.ucsc.edu), to search for 3′-UTR-disrupted PD-L1 and PD-L2 transcripts [13]. To evaluate the copy number of PD-L1 and PD-L2, the level 3 segmented copy number data (Affymetrix Genome-Wide Human SNP Array 6.0) were downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov) for DLBCL and stomach adenocarcinoma samples.
Immunohistochemical analysis
IHC for PD-L1 was performed on formalin-fixed paraffin-embedded tissue sections using antibodies directed against the N-terminal (E1J2J, Cell Signaling Technology, Beverly, MA, USA) and C-terminal (SP142, Spring Bioscience, Fremont, CA, USA) domains of PD-L1. IHC for Ki-67 was performed using a mouse monoclonal antibody (MIB-1, Dako, Tokyo, Japan). The antigen–antibody complexes were visualized with Histofine Simple Stain MAX PO (Nichirei Bioscience, Tokyo, Japan). Some sections were double-stained with PD-L1 (E1J2J) and CD3, using a mouse monoclonal antibody (F7.2.38, Dako) with PolyView IHC reagent (mouse-AP, Enzo Life Sciences, Farmingdale, NY, USA) (Supplemental Figure S2). A tumor sample was considered positive for PD-L1 expression, when >5% of tumor cells were stained, where PD-L1 expression in tumor cells was discriminated from that in immune cells on the basis of cell morphology and/or cytoplasmic CD3 expression. LMP1 expression was detected using a mouse monoclonal antibody (CS1-4, Dako) with the EnVision system (Dako). EBER-ISH was performed on the Leica Bond-III Automatic Stainer (Leica Microsystems, Wetzlar, Germany) using Bond Ready-to-Use ISH EBER Probe and Bond Ready-to-Use Anti-Fluorescein Antibody (Leica Biosystems, Wetzlar, Germany).
Fluorescent in situ hybridization (FISH)
FISH analyses were performed on unstained slides with bacterial artificial chromosome (BAC) clone-derived DNA probes for the 5′- and 3′-parts of PD-L1 locus. The hybridized slides were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and observed with a fluorescence microscope BX51 (Olympus, Tokyo, Japan).
CRISPR-mediated gene targeting
CRISPR-mediated gene editing was used to generate PD-L1 and PD-L2 3′-UTR disruptions, as previously described [13]. Briefly, human PD-L1 and PD-L2 3′-UTR single guide RNA (sgRNA) targeted sites were designed manually and checked in silico. The pSpCas9(BB)-2A-GFP (pX458) vector expressing Cas9 (Addgene plasmid 48138) was digested with BbsI and ligated to the annealed sgRNA oligonucleotides. HEK293T (obtained from the RIKEN Cell Bank) and T2 (a gift from H. Kawamoto, Kyoto University) cells were transfected with indicated vectors using X-tremeGENE 9 DNA Transfection Reagent (Roche, Basel, Switzerland) and the Neon transfection system (Thermo Fisher Scientific, Waltham, MA, USA), respectively. To validate CRISPR/Cas9-mediated disruption of PD-L2 3′-UTR, genomic PCR flanking the breakpoint region was performed. The sgRNA and primer sequences are listed in Supplemental Tables S3, S4. Cell lines were authenticated by the providers and routinely tested for mycoplasma infection.
Flow cytometric analysis
To detect surface PD-L1 or PD-L2 expression, cells were stained with allophycocyanin (APC)-conjugated anti-CD274 (29E.2A3) and anti-CD273 (24F.10C12) antibodies (BioLegend, San Diego, CA, USA), and analyzed on FACS LSR Fortessa (BD Biosciences, San Jose, CA, USA). The data analyses were performed with FlowJo software (TreeStar, Ashland, OR, USA).
Statistical analysis
Statistical analyses were performed with R3.4.2 software (The R Foundation for Statistical Computing). For functional assays, statistical significance was assessed by Student’s two-tailed t-test with a Welch’s correction. The molecular features between EBV-negative and -positive tumors were compared using the Fisher’s exact test, Cochran–Armitage trend test, or Brunner–Munzel test. P-values were considered statistically significant at <0.05. In box plots, the center line and lower and upper hinges correspond to the median, and the first and third quartiles (25 and 75 percentiles), respectively. The upper and lower whiskers extend from the upper and lower hinges to the largest or smallest values no further than 1.5× inter-quartile range from the hinges.
Code availability
The code for the Genomon pipeline is available at https://github.com/Genomon-Project/.
Results
Analysis of PD-L1/PD-L2 genetic alterations in a variety of B- and T/NK-cell lymphomas
PD-L1/PD-L2-involving genetic abnormalities were interrogated using targeted-capture sequencing in a total of 384 samples from different lymphoma subtypes, including DLBCL (n = 75), follicular lymphoma (FL, n = 49), mucosa-associated lymphoid tissue lymphoma (MALT, n = 20), mantle cell lymphoma (MCL, n = 32), PMBCL (n = 10), PTCL-not otherwise specified (PTCL-NOS, n = 104), and EBV-positive T-cell and NK-cell proliferations (n = 94) (Supplemental Table S1). Overall, SVs affecting PD-L1 and PD-L2 were detected in 25 (7%) and 6 (2%) samples, respectively, while focal gains or amplifications involving these genes were found in 30 (8%) samples (Supplemental Figures S1 and S3 and Supplemental Table S5). These alterations were observed in both B-cell and T/NK-cell lymphomas, with varying frequency (0-57%) depending on histology (Fig. 1a). Regardless of SV types involved (deletion, inversion, tandem duplication, and translocation), most of these SVs resulted in 3′-UTR truncation of the corresponding genes (Fig. 1b, c). Although reported in previous publications [9–12], translocations of the PD-L1 or PD-L2 promoter to an ectopic regulatory locus was rarely observed. Except for Burkitt lymphoma [20] (0 of 20 samples), similar results were obtained from the analysis of publicly available transcriptome sequencing data of PTCL [23] (1 of 35), ALCL [21] (1 of 23), CTCL [22] (1 of 13), and PMBCL [24] (3 of 6), where an marked increase in PD-L1 or PD-L2 mRNA expression was observed in most cases with PD-L1- or PD-L2-involving aberrant transcripts (Supplemental Figure S4A–C and Supplemental Table S6). In addition, a PTCL-NOS case with a PD-L1 3′-UTR truncation showed strong PD-L1 expression in IHC (Supplementary Figure S3D and Supplemental Table S7). Of interest were those cases in which multiple SVs involving PD-L1 and/or PD-L2 were observed in a single tumor (DLBCL58, MCL29, and ANKL7), underscoring the importance of the deregulated expression of these PD-1 ligands in clonal selection and expansion of these tumors.
Fig. 1.
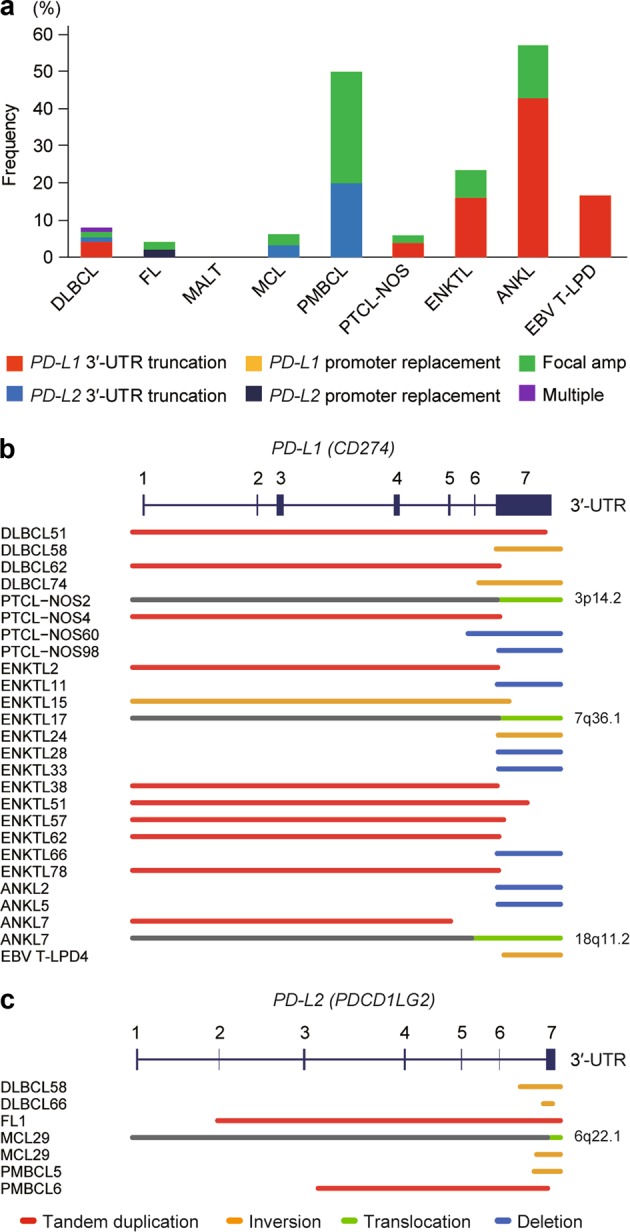
Genetic alterations involving PD-1 ligands in various subtypes of lymphomas. a Frequency of genetic alterations involving PD-L1 and/or PD-L2 in each lymphoma subtype. Type of alterations is indicated by color. Cases harboring both SV and focal CNA affecting PD-L1 and/or PD-L2 are combined into the corresponding SV group. Multiple represents cases harboring both PD-L1 and PD-L2 SVs. DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; MALT, mucosa-associated lymphoid tissue lymphoma; MCL, mantle cell lymphoma; PMBCL, primary mediastinal B-cell lymphoma; PTCL-NOS, peripheral T-cell lymphoma-not otherwise specified; ENKTL, extranodal NK/T-cell lymphoma; ANKL, aggressive NK-cell leukemia; EBV T-LPD, Systemic EBV-positive T-cell lymphoproliferative disorder; Amp, amplification. b, c Different types of SVs affecting PD-L1 (b) and PD-L2 (c) are shown by indicated colors
PD-L2 SV is a characteristic genetic alteration of B-cell lymphomas
In contrast to PD-L1-involving SVs, which were detected in both B- and T/NK-cell lymphomas, those involving PD-L2 were found exclusively in B-cell lymphomas (Figs 1a and 2a). Similarly, while PD-L1 SVs are present in a variety of solid cancers [13], no PD-L2-involving SVs were identified among 10,162 cancer samples from 32 tumor panels, for which RNA-seq data were available from TCGA. These results suggest that PD-L1 is affected in a broad spectrum of human malignancies, whereas PD-L2 SVs are largely restricted to B-cell lymphomas, possibly reflecting the expression pattern of PD-1 ligands (Fig. 2b). Based on these findings, we assessed whether disruption of PD-L2 3′-UTR could induce PD-L2 overexpression using CRISPR/Cas9-mediated gene editing, as was the case with enhanced PD-L1 expression through disruption of its 3′-UTR (Fig. 2c). Although disruption of PD-L2 3′-UTR resulted in a significant elevation of PD-L2 expression in T2 human B and T lymphoblast hybrid cell line (Fig. 2d, e and Supplemental Figure S5), it did not affect PD-L2 expression in HEK293T cells, confirming the restricted relevance of 3′-UTR-mediated regulation of PD-L2 expression to B-cell lineage.
Fig. 2.

CRISPR/Cas9-mediated 3′-UTR disruption induces PD-L2 overexpression. a Proportion of PD-L1 and PD-L2 SVs in B-NHL and T/NK-NHL as well as other tumors. b PD-L1 and PD-L2 mRNA expression in normal human B, CD4+ T, CD8+ T, and NK cells. Expression microarray data were obtained from HemaExplorer [25]. c Positions of targeting sgRNAs used for CRISPR/Cas9-mediated deletions and inversions of PD-L1 and PD-L2 3′-UTR. d, e Representative plots (d) and frequencies (e) of PD-L1+ (top) or PD-L2+ (bottom) cells in green fluorescent protein (GFP)+ fraction by flow cytometry in HEK293T (e, left) or T2 (d, e, right) cells transfected with Cas9 and no, single, or pairwise sgRNAs targeting PD-L1 or PD-L2 3′-UTR (n = 3 biological replicates). Data represent means ± s.d. *P < 0.05, Welch’s t-test
Frequent PD-L1 genetic alterations in EBV-positive T-cell and NK-cell proliferations
Consistent with previous reports [10, 11], PD-L1/PD-L2-involving lesions were frequently observed in PMBCL, accounting for a half of the cases (Fig. 1a). Notably, mainly consisting of its 3′-UTR truncations, PD-L1-involving abnormalities were prevalently found in EBV-positive T-cell and NK-cell proliferations (17–57%), including ENKTL (19 of 81 samples), aggressive NK-cell leukemia (ANKL, 4 of 7), and systemic EBV-positive T-cell lymphoproliferative disorder (EBV T-LPD, 1 of 6) (Fig. 1a, b). Among these, the most common entity is ENKTL, which is an aggressive neoplasm with a predilection for Asian and South American populations [26]. These PD-L1-involving SVs and focal CNAs were confirmed using multi-color FISH in all ENKTL cases by way of positive break-apart and clustered signals, respectively (Fig. 3a and Table 1). In IHC, PD-L1 expression was detected in 6 of 19 ENKTL cases examined, where almost all tumor cells were positive for PD-L1 (Fig. 3b and Table 1). Among them, 4 cases had PD-L1-involving genetic lesions, while all ENKTL cases having PD-L1-involving genetic lesions were positive for PD-L1 expression in IHC, except for one case with a focal PD-L1 amplification. These findings suggest that PD-L1-involving genetic abnormalities are the major cause of PD-L1 overexpression in ENKTL, although other mechanisms, such as induction by EBV-encoded products, might operate, particularly in those cases with positive IHC but lacking detectable PD-L1 genetic lesions [27]. Additionally, samples with PD-L1 3′-UTR truncations showed a higher percentage of Ki-67 positive cells, compared with PD-L1 SV-negative ones, suggesting a possible intrinsic role for PD-L1 in regulating cellular growth (Fig. 3c and Table 1). To determine the relative contribution of PD-L1-involving genetic alterations in NK/T-cell lymphomagenesis, we analyzed published exome data of 25 ENKTL cases for other somatic events than PD-L1 abnormalities [28]. In accordance with a previous report [28], the most frequently mutated genes included DDX3X and TP53 (Fig. 3d). Moreover, we found that these common drivers were also affected by SVs and CNAs, which were overlooked in the previous study. Nevertheless, the most common genetic alterations in ENKTL were represented by those involving PD-L1, which were found in as many as 10 (40%) of ENKTL cases (Fig. 3d, Supplemental Figure S6, and Supplemental Table S8), even though their frequency is likely to be substantially underestimated because of the lack of coverage for intronic and UTR sequences in the exome data to capture the relevant SVs. Analysis of the clonal structure of tumor cells showed that PD-L1 3′-UTR truncations were present in major clones in most cases (Fig. 3e), suggesting that PD-L1 genetic alterations may represent a relatively early event in tumor development.
Fig. 3.

ENKTL harbors frequent PD-L1 SVs, leading to its overexpression. a FISH analysis showing PD-L1 break-apart (middle) and cluster formation (right) in ENKTL cells. Design of the break-apart assay using BAC probes recognizing the 5′-part (green) and 3′-part (red) of PD-L1 loci is shown. Amp, amplification. b PD-L1 IHC (top and middle) and EBER-ISH (bottom) of ENKTL cases with or without PD-L1 genetic alterations. Antibodies specifically detecting N-terminal (top) and C-terminal (middle) domains of PD-L1 were used. c Percentage of Ki-67+ cells in tumor cell fraction in ENKTL cases with or without PD-L1 genetic alterations. Each dot represents a single case. **P < 0.005, Brunner–Munzel test. A summary of the results is shown in Table 1. d Frequently altered genes identified by whole-exome sequencing for 25 ENKTL cases [28]. Type of somatic alterations is indicated by color. e Hierarchy of somatic mutations and PD-L1 SVs is shown with their allele frequencies in 8 ENKTL cases analyzed by whole-exome sequencing. Driver mutations are shown in green, and PD-L1 SVs are shown in red
Table 1.
PD-L1 genetic alterations, its expression, and EBV status in ENKTL
| Case | PD-L1 SV | PD-L1 CNA | PD-L1 FISH | PD-L1 IHC (N) | PD-L1 IHC (C) | EBER-ISH | LMP1 IHC | Ki-67 |
|---|---|---|---|---|---|---|---|---|
| ENKTL10 | − | − | − | − | − | + | − | 30% |
| ENKTL11 | 3′-UTR truncation | Focal amp | NE | NE | NE | + | − | NE |
| ENKTL12 | − | − | − | − | − | + | + | 80% |
| ENKTL13 | − | − | − | − | − | + | − | 100% |
| ENKTL14 | − | − | − | − | − | + | − | 10% |
| ENKTL15 | 3′-UTR truncation | Focal amp | Cluster formation | + | + | − | + | 90% |
| ENKTL16 | − | − | − | − | − | + | + | 60% |
| ENKTL17 | 3′-UTR truncation | − | Split signal | + | + | + | + | 100% |
| ENKTL18 | − | − | − | − | − | − | + | 40% |
| ENKTL19 | − | − | NE | NE | NE | + | − | NE |
| ENKTL20 | − | − | − | − | − | + | − | 100% |
| ENKTL21 | − | − | − | + | + | + | + | 50% |
| ENKTL22 | − | − | − | + | + | + | + | 70% |
| ENKTL23 | − | − | − | − | − | + | − | 80% |
| ENKTL24 | 3′-UTR truncation | Focal amp | Split signal | + | + | + | + | 90% |
| ENKTL25 | − | Focal amp | Cluster formation | − | − | + | − | 60% |
| ENKTL26 | − | − | − | − | − | + | − | 10% |
| ENKTL27 | − | − | − | − | − | − | + | 20% |
| ENKTL28 | 3′-UTR truncation | − | NE | NE | NE | + | − | NE |
| ENKTL29 | − | Focal amp | Cluster formation | + | + | − | + | 50% |
| ENKTL30 | − | − | − | − | − | + | − | 90% |
| ENKTL31 | − | − | − | − | − | + | − | 10% |
Results of targeted-capture sequencing (PD-L1 SV and PD-L1 CNA), PD-L1 FISH, IHC for PD-L1 (with the N-terminal or C-terminal antibody), LMP1, and Ki-67, and EBER-ISH for 22 ENKTL cases are shown
FISH fluorescent in situ hybridization, IHC immunohistochemistry, EBER-ISH in situ hybridization for EBV-encoded small RNA, NE not evaluable
Association of EBV infection with genetic alterations of PD-1 ligands
A high frequency of PD-L1 SVs was previously reported in a unique form of HTLV-1-mediated PTCL, ATL [13, 14], raising a possibility that genetic defects involving PD-1 ligands play a pivotal role in immune escape of virus-related tumors. In this point of view, it would be of interest to see the frequency of PD-L1/PD-L2-involving abnormalities among EBV-associated lymphomas and stomach adenocarcinoma, another EBV-related cancer. Among 75 DLBCL cases in our cohort, 27 cases (DLBCL49-75) were positive for EBV by Southern blot and/or targeted-capture sequencing, and thus considered as EBV-positive DLBCL (Fig. 4a). We observed a significantly higher frequency of PD-L1/PD-L2-involving genetic aberrations in EBV-positive DLBCL (5 of 27) than in EBV-negative DLBCL (1 of 48) (P < 0.05) (Fig. 4a, b). The observation was confirmed by examining DLBCL cases in the TCGA cohort (27% vs. 5%, P = 0.07) (Fig. 4b). Similar trends were also seen for PTCL-NOS (Fig. 4a, c), as well as stomach adenocarcinoma from the TCGA cohort (Fig. 4d). These results suggest a strong relationship between EBV infection and PD-L1/PD-L2-involving genetic abnormalities.
Fig. 4.

PD-L1/PD-L2 genetic alterations associated with EBV infection. a Proportion of EBV DNA reads to total mapped reads according to lymphoma subtype (offset, 0.0001). Each dot represents a single case. Red line indicates the cut-off value (0.00015%) for EBV positivity. Type of genetic alterations involving PD-1 ligands is indicated by color. Multiple represents cases harboring both PD-L1 and PD-L2 SVs. b–d Frequency of genetic alterations involving PD-L1/PD-L2 according to EBV status in DLBCL (b), PTCL-NOS (c), and stomach adenocarcinoma (d). Fisher’s exact test is used for DLBCL and PTCL-NOS, and Cochran–Armitage trend test is applied to stomach adenocarcinoma. EBV high group corresponds to the EBV-positive group in the TCGA classification of stomach adenocarcinoma [38]
Different underlying genetic mechanisms between EBV-negative and -positive DLBCLs
Based on these findings, we hypothesized that EBV-positive tumors may constitute a genetically distinct neoplasm from the EBV-negative counterpart. Thus, we compared frequencies of major driver alterations implicated in lymphomagenesis between EBV-positive and -negative DLBCLs (Supplemental Tables S9–S11). Although the numbers of mutations and CNAs were comparable between EBV-negative and -positive DLBCLs, EBV-positive DLBCLs had a larger number of SVs than EBV-negative tumors (Supplemental Figure S7A). The frequency and type of somatic alterations observed in our cohort were quite similar to previous studies [29, 30] (Supplemental Figure S7B). Conspicuously, compared to EBV-negative DLBCLs, EBV-positive DLBCLs showed substantially higher frequencies of TET2 and DNMT3A mutations, while showing very low frequencies of CD79B and MYD88 mutations as well as CDKN2A loss-of-function alterations (Fig. 5a, b). In addition, despite the common occurrence of PD-L1/PD-L2 somatic alterations, EBV-positive tumors were largely devoid of FAS mutations and deletions, which are also thought to be involved in escape from immune surveillance by T cells [31]. There were no differences in genetic alterations between EBV-positive DLBCLs with and without PD-L1/PD-L2 genetic alterations (Supplemental Figure S7C). Taken together, these observations suggest the presence of distinct genetic mechanisms for the pathogenesis of EBV-positive and -negative DLBCLs.
Fig. 5.

Genetic differences between EBV-negative and -positive DLBCLs. a Frequency and type of somatic alterations identified by targeted-capture sequencing for lymphoma-associated genes in 48 EBV-negative and 27 EBV-positive DLBCL cases. Type of alterations is indicated by color. BCR, B-cell receptor; GCB, germinal center B-cell. *P < 0.05, **P < 0.01, Fisher’s exact test. b Distribution of somatic mutations encoded in MYD88 (NM_002468), CD79B (NM_000626), TET2 (NM_001127208), and FAS (NM_000043) detected in 48 EBV-negative and 27 EBV-positive DLBCL cases. The protein domains were obtained from the NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene/) and the UniProt database (https://www.uniprot.org/)
Discussion
We have revealed a high prevalence of PD-L1/PD-L2-involving SVs and/or focal amplifications and accompanying overexpression of the corresponding PD-1 ligand in a wide variety of lymphomas, suggesting a critical role of evasion from cellular immunity in the development of these lymphomas. Of particular interest in this respect is especially high frequencies of these abnormalities in EBV-associated lymphomas, which were detected in as many as 22% of these lymphomas. Affecting more than 90% of adult individuals, EBV infection represents one of the most prevalent viral infections among human populations, where it induces potent cellular immunity [1, 2]. In fact, up to 5% of cytotoxic T cells in our body are devoted to eliminate EBV-infected cells [1]. Intact immunity to EBV is essential to self-terminate infectious mononucleosis or prevent other lymphoproliferative diseases associated with transplantation or other immunocompromised status [1, 2]. Thus, evasion from anti-viral immunity is thought to be a critical step for EBV-infected B or T/NK cells to achieve neoplastic growth, except for Burkitt lymphoma, which exhibits a restrictive pattern of expression of latent encoded proteins (type 1 latency) [1, 2]. In this context, EBV-infected cells that acquire PD-L1/PD-L2-involving alterations are thought to effectively evade anti-EBV immune surveillance to be clonally selected for further neoplastic outgrowth, which in turn points to a possibility that checkpoint blockade targeting the PD-1/PD-L1 axis might provide effective therapeutics against EBV-related lymphomas otherwise associated with a dismal prognosis with conventional chemotherapy [26, 32]. In agreement with this hypothesis is a recent report describing a remarkable activity of anti-PD-1 antibody against patients with relapsed or refractory ENKTL [33]. Therefore, it would be an emerging question whether or not detection of PD-L1/PD-L2-involving alterations helps identify those lymphomas in which an excellent response to PD-L1/PD-L2 blockade is expected, even though the overall efficacy may not be remarkable [34].
Oncogenic viral infections are closely related with the development of various cancers, such as hepatitis B- and C-virus-associated hepatocellular carcinoma, human papilloma virus-associated head and neck cancers, polyomavirus-associated Merkel cell carcinoma, and HTLV-1-associated ATL. In contrast to somatic mutations or SVs of the tumor genome, which can generate a limited number of antigenic peptides for T cell recognition, the entire protein product expressed from viral genes represents non-self, thereby conferring strong neoantigenecity [3]. Therefore, a deregulated PD-1 and PD-L1 axis might be relevant to immune evasion in virus-associated cancers, and thus the presence of the causative virus could represent a predictive biomarker for response to immune checkpoint blockade. In accordance with this theory, substantial efficacy of anti-PD-1 therapy among several virus-related tumors have been noted, although further studies are warranted [3].
Another finding of significant importance is a distinct pattern of somatic alterations in EBV-positive DLBCL, providing new insight into the genetic basis of virus-induced lymphomas. EBV gene products, such as LMP1, have been reported to strongly activate NF-κB pathway in a ligand-independent manner, and hinder p16 (encoded by CDKN2A) -Rb pathway, which can explain the absence of CD79B, MYD88, and CDKN2A alterations in EBV-positive DLBCL, although EBV-positive DLBCL is characterized by an activated B-cell (ABC) immunophenotype and prominent NF-κB activation [32, 35]. Frequent TET2 and DNMT3A mutations in EBV-positive DLBCL indicate the possible involvement of deregulated DNA methylation and demethylation processes in this disease [36]. In addition to PD-L1 3′-UTR truncation, which is associated with Ki-67 positivity in ENKTL, TET2 disruption is also reported to cause aberrant proliferation in an antigen receptor-dependent manner [37]. These alterations can contribute to tumor growth, which may explain the paucity of mutations in B-cell receptor (BCR)/NF-κB pathway-associated molecules. Furthermore, an inverse relationship of PD-L1/PD-L2 and FAS alterations suggests different roles of these molecules in escape from T-cell mediated immune surveillance against virally infected cells.
Taken together, our findings contribute to understand the pathogenesis of EBV-associated lymphomas and exploit a new diagnostic strategy to identify patients who are likely to respond to PD-1/PD-L1 blockade therapy by detection of EBV and/or PD-L1/PD-L2 genetic alterations in NHLs.
Supplementary information
Acknowledgements
We thank Miki Sagou and Takeshi Shirahari for technical assistance and all the members of the TENOMIC program. This work was supported by Grant-in-Aid from the Japan Agency for Medical Research and Development [Project for Cancer Research and Therapeutic Evolution (16cm0106501) and Practical Research for Innovative Cancer Control (17ck0106261)], National Cancer Center Research and Development Funds (29-E-3 and 30-A-1), the Naito Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Princess Takamatsu Cancer Research Fund, the Fondation ARC pour la recherche sur le cancer (20151203514), the France Lymphome Espoir association, and the GEFLUC Paris-Ile de France. LC was sponsored by a grant from ITMO Cancer AVIESAN (Alliance Nationale pour les Sciences de la Vie et de la Santé/National Alliance for Life Sciences and Health) within the framework of the Cancer Plan and by a fellowship awarded by the European Hematology Association and the Japanese Society of Hematology. The supercomputing resources were provided by the Human Genome Center, the Institute of Medical Science, The University of Tokyo.
Compliance with ethical standards
Conflict of interest
KK and SO have a patent application pending related to findings from this study. The remaining authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41375-019-0380-5) contains supplementary material, which is available to authorized users.
References
- 1.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–68. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Grywalska E, Markowicz J, Grabarczyk P, Pasiarski M, Rolinski J. Epstein-Barr virus-associated lymphoproliferative disorders. Post Hig Med Dosw. 2013;67:481–90. doi: 10.5604/17322693.1050999. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 5.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–61. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311–9. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol : Off J Am Soc Clin Oncol. 2016;34:2690–7. doi: 10.1200/JCO.2016.66.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol. 2016;34:3733–39. [DOI] [PMC free article] [PubMed]
- 9.Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471:377–81. doi: 10.1038/nature09754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong LC, Twa DD, Mottok A, Ben-Neriah S, Woolcock BW, Zhao Y, et al. Comprehensive characterization of programmed death ligand structural rearrangements in B-cell non-Hodgkin lymphomas. Blood. 2016;128:1206–13. doi: 10.1182/blood-2015-11-683003. [DOI] [PubMed] [Google Scholar]
- 11.Twa DD, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–5. doi: 10.1182/blood-2013-10-535443. [DOI] [PubMed] [Google Scholar]
- 12.Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood 2015. [DOI] [PMC free article] [PubMed]
- 13.Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016;534:402–6. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 14.Kogure Y, Kataoka K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. 2017;108:1719–25. doi: 10.1111/cas.13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swerdllow S, Campo E, Harris NL. WHO classification of tumours of haematopoietic and lymphoid tissues. France: IARC Press; 2008. [Google Scholar]
- 16.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshizato T, Nannya Y, Atsuta Y, Shiozawa Y, Iijima-Yamashita Y, Yoshida K, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129:2347–58. doi: 10.1182/blood-2016-12-754796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47:1304–15. doi: 10.1038/ng.3415. [DOI] [PubMed] [Google Scholar]
- 19.Kataoka K, Iwanaga M, Yasunaga JI, Nagata Y, Kitanaka A, Kameda T, et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood. 2018;131:215–25. doi: 10.1182/blood-2017-01-761874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abate F, Ambrosio MR, Mundo L, Laginestra MA, Fuligni F, Rossi M, et al. Distinct viral and mutational spectrum of endemic Burkitt lymphoma. PLoS Pathog. 2015;11:e1005158. doi: 10.1371/journal.ppat.1005158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27:516–32. doi: 10.1016/j.ccell.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47:1011–9. doi: 10.1038/ng.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–70. doi: 10.1038/ng.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gunawardana J, Chan FC, Telenius A, Woolcock B, Kridel R, Tan KL, et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet. 2014;46:329–35. doi: 10.1038/ng.2900. [DOI] [PubMed] [Google Scholar]
- 25.Bagger FO, Rapin N, Theilgaard-Monch K, Kaczkowski B, Thoren LA, Jendholm J, et al. HemaExplorer: a database of mRNA expression profiles in normal and malignant haematopoiesis. Nucleic Acids Res. 2013;41(Database issue):D1034–1039. doi: 10.1093/nar/gks1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tse E, Kwong YL. How I treat NK/T-cell lymphomas. Blood. 2013;121:4997–5005. doi: 10.1182/blood-2013-01-453233. [DOI] [PubMed] [Google Scholar]
- 27.Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O'Donnell E, et al. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res. 2012;18:1611–8. doi: 10.1158/1078-0432.CCR-11-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47:1061–6. doi: 10.1038/ng.3358. [DOI] [PubMed] [Google Scholar]
- 29.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24:679–90. doi: 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med. 2018;378:1396–407. doi: 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Afshar-Sterle S, Zotos D, Bernard NJ, Scherger AK, Rodling L, Alsop AE, et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat Med. 2014;20:283–90. doi: 10.1038/nm.3442. [DOI] [PubMed] [Google Scholar]
- 32.Ok CY, Papathomas TG, Medeiros LJ, Young KH. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328–40. doi: 10.1182/blood-2013-03-489708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwong YL, Chan TSY, Tan D, Kim SJ, Poon LM, Mow B, et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood. 2017;129:2437–42. doi: 10.1182/blood-2016-12-756841. [DOI] [PubMed] [Google Scholar]
- 34.Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol : Off J Am Soc Clin Oncol. 2016;34:2698–704. doi: 10.1200/JCO.2015.65.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montes-Moreno S, Odqvist L, Diaz-Perez JA, Lopez AB, de Villambrosia SG, Mazorra F, et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod Pathol. 2012;25:968–82. doi: 10.1038/modpathol.2012.52. [DOI] [PubMed] [Google Scholar]
- 36.Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–99. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsagaratou A, Gonzalez-Avalos E, Rautio S, Scott-Browne JP, Togher S, Pastor WA, et al. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat Immunol. 2017;18:45–53. doi: 10.1038/ni.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.