

Abstract
Pancreatic ductal adenocarcinoma (PDAC) arises through accumulation of multiple genetic alterations. However, cancer cells also acquire and depend on cancer-specific epigenetic changes. To conclusively demonstrate the crucial relevance of the epigenetic programme for the tumourigenicity of the cancer cells, we used cellular reprogramming technology to reverse these epigenetic changes. We reprogrammed human PDAC cultures using three different techniques – (1) lentivirally via induction of Yamanaka Factors (OSKM), (2) the pluripotency-associated gene OCT4 and the microRNA mir-302, or (3) using episomal vectors as a safer alternative without genomic integration. We found that induction with episomal vectors was the most efficient method to reprogram primary human PDAC cultures as well as primary human fibroblasts that served as positive controls. Successful reprogramming was evidenced by immunostaining, alkaline phosphatase staining, and real-time PCR. Intriguingly, reprogramming of primary human PDAC cultures drastically reduced their in vivo tumourigenicity, which appeared to be driven by the cells’ enhanced differentiation and loss of stemness upon transplantation. Our study demonstrates that reprogrammed primary PDAC cultures are functionally distinct from parental PDAC cells resulting in drastically reduced tumourigenicity in vitro and in vivo. Thus, epigenetic alterations account at least in part for the tumourigenicity and aggressiveness of pancreatic cancer, supporting the notion that epigenetic modulators could be a suitable approach to improve the dismal outcome of patients with pancreatic cancer.
Subject terms: Cancer stem cells, Pancreatic cancer
Introduction
Pancreatic cancer, including the most frequent subtype pancreatic ductal adenocarcinoma (PDAC), is currently the third leading cause of cancer-related deaths in the United States [1] and predicted to become the second most frequent cause of cancer-related death by 2030 [2]. It is characterized by extensive desmoplasia, rapid metastasis, and pronounced resistance to chemotherapy and radiation [3]. Due to a paucity of biomarkers for early-stage detection of the disease, PDAC is usually detected at advanced stages with limited therapeutic options [4], rendering this disease a major unmet priority for public health care [5]. A major obstacle for developing more effective therapies relies in our still limited understanding of the molecular mechanisms of PDAC progression. Whole-genome sequencing studies have identified inactivating mutations of chromatin modifiers as frequent genetic events in PDAC tumours [6], further corraborating the notion that epigenetic alterations play an important role in this disease.
Although cancer is generally believed to develop through accumulation of multiple genetic mutations, there is increasing evidence that cancer cells also acquire epigenetic abnormalities during tumour development and progression [7]. Moreover, mutual interactions between genetic mutations and epigenetic alterations still remain to be uncovered. In this context, cellular reprogramming technology can be used to actively modify the epigenome without affecting the underlying genomic sequences and is already applied in regenerative medicine and now increasingly also for disease modelling. It could also be a powerful tool for experimentally dissecting the role of the cancer epigenome during the initiation and progression of cancer. Understanding its specific role and the potential modulation by small chemical compounds may contribute to the development of more effective cancer treatment strategies [8].
While somatic cells have been reprogrammed successfully, cancer cells are extremely difficult to reprogram and few reports exist to date on the successful reprogramming of cancer cell using either nuclear transfer (NT) or viral insertion of pluripotency genes [9]. Specifically, epigenetic reprogramming using nuclear transfer has been used to suppress the cancer phenotype in certain medulloblastoma, RAS-induced melanoma, embryonal carcinoma, and renal cancer cell lines [10–13], but primary cancer cells have not yet been reprogrammed by this method. iPSC technology [14] has also been used to reprogram cancer cell lines into iPSCs, independent of oocytes and blastocysts [15–17]. Still, generating iPSC using epithelial cancer cells from primary human adenocarcinomas has been challenging. In 2013, it was reported that a single iPSC line had been reprogrammed from a late-stage human pancreatic cancer to a near pluripotent state, although it could only be maintained under low-level expression of the exogenous 4 reprogramming factors [3].
Here we explored different cell reprogramming technologies to force primary pancreatic cancer cells to exit from their epigenetically determined tumourigenicity programme. Specifically, we reprogrammed primary human PDAC cells by (1) lentiviral induction of the four Yamanaka Factors (OSKM), (2) lentiviral induction the pluripotency-associated gene OCT4 and the microRNA mir-302, and (3), as a safer alternative, introduction of the latter without genomic integration using episomal vectors. The aim of our study was to identify the most efficient and safest method to reprogram PDAC cells and subsequently study potential alterations of their tumourigenic phenotype.
Materials and methods
Cell culture
Patient-derived xenograft cells were obtained from PDAC patients and xenografted in immunocompromised mice, kindly provided by Dr. Hidalgo [18, 19]. PDAC cells (PDAC-247, -253 and -354) and PANC-1 cells (PDAC cell established line) were cultured in RPMI, 10% FBS, and 50 units/ml penicillin/streptomycin. Human Foreskin Fibroblast (HFF-5) and Human dermal fibroblast (HDF) were cultured in DMEM (Life bioscience) supplemented with 10% FBS (Invitrogen). Mouse embryonic fibroblasts (MEFs) were isolated from day 12.5 embryos of C57BL/6 mice. All mice used in this study were bred and sacrificed appropriately following the code of ethics of the animal research committee at the Royan Institute, Tehran and the CNIO, Madrid. MEF were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine and 50 units and 50 mg ml−1 penicillin and streptomycin, respectively. Reprogrammed cells were cultured using DMEM:F12 medium with 20% KOSR, 1% NEAA, 2 mM Glutamine, 100 ng/ml bFGF, and 0.1 mM β-mercaptoethanol. The plates were coated with BD Matrigel™ hESC-qualified Matrix (BD Biosciencies, 542777), and a monolayer of mitomycin C–treated MEF cells as a feeder. For passaging, cells were harvested using trypsin and replated at 1:30 to 1:100 dilutions in medium containing 10 μM ROCK inhibitor (Y-27632, Stemgent).
Virus production and cell induction
TetO-FUW-OSKM
The inducible polycistronic TetO-FUW-OSKM (Addgene, 20321) lentivirus production was performed according to the manufacturer’s protocol. One day before transfection for needed vectors, we placed 5 × 106 293T cells separately into 10 cm culture dishes. The next day transfection was performed according to the protocol described in the manual. Briefly, the cell media was changed with 5 ml complete media and 2% FBS without antibiotics. In two separate 5 ml tubes we added 1.5 ml of basal medium followed by the addition of 36 μl lipofectamine. To the first tube we added 12 μg of packaging vector and the desired vector to the other tube. After a 5-min incubation period, the tubes were combined and allowed to incubate for another 20 min. We added the complete 3 ml mix to the culture plates in a drop-wise manner. From this step, the cells must be maintained in a Biosafety level II culture room and manipulated as biohazard materials. The next day the medium was replaced by complete medium that contained 10% FBS. This medium was collected after 48 h, filtered through 0.45 μm pore-size filter (BD Biosciences), then centrifuged at 20,000 r.p.m. (Centrifuge, Sigma-Aldrich 3K30) for 2 h at 4 °C after which the pellets were dissolved in 300 μl PBS, aliquoted and maintained at –80 °C.
Reprogramming was initiated by direct induction of cells (HDF, HFF-5, PANC-1, PDAC-247, -253 and -354 cells) with TetO-OSKM and M2rtTA concentrated virus. At day 2 we added Doxycycline, the cells were cultured 5 days in RPMI + 10% FBS. Afterwards, cells were trypsinised and seeded in MEF-layered, Matrigel-coated plates with RPMI + 10% FBS. After 72 h, medium was changed to defined iPS medium. Media was renewed every other day.
Mir302-OCT4
Replication-incompetent lentiviral particles were produced by calcium-phosphate transfection of 293 T cells using the packaging plasmids pMD2.G (VSV-G) and pPAX2, as well as mir302-OCT4 as the shuttle vector. The medium was replaced with fresh DMEM complete 6 h after transfection, and 48 h afterwards the medium was collected, cleared by low-speed centrifugation, filtered through 0.45 μm pore-size filter (BD Biosciences) and ultracentrifuged for 2 h at 20,000 r.p.m. (4 °C). The pellet was then resuspended overnight at 4 °C with 300 μL of PBS, and the viral concentrate was stored in aliquots at –80 °C. The lentiviral vector was composed of CMV-OCT4-mir-302 as reprogramming factors using mCherry as a red fluorescent reporter. Concentrated virus supernatants were used to infect cells and the cells were cultured 5 days in RPMI + 10% FBS. Afterwards, for miR302-OCT4 we sorted for mCherry + cells and seeded them in MEF-layered, Matrigel-coated plates with RPMI + 10% FBS. After 72 h, medium was changed to defined iPS medium. Media was renewed every other day.
Episomal vectors and cell infection
The episomal vectors were obtained from Addgene (#20924, 20925, 20926, 20927, 28213, and 28220; Cambridge, MA, http://www.addgene.org/). Cells were cultured in RPMI supplemented with 10% FBS. 45 micrograms of expression plasmid mixtures were electroporated into 106 cells according to the manufacturer’s instructions. Conditions used were 250 mV, 500 mF, one pulse for all cells. The cells were seeded in gelatin-coated 100-mm dishes. Five days after transfection cells were trypsinized and 4 × 104 cells were re-plated onto 60-mm dishes covered with MEF feeder layer and Matrigel. After 72 h, medium was changed to defined iPS medium. Media was renewed every other day.
Fluorescent Activated Cell Sorting
Single fibroblasts and cancer cells positive for mCherry were sorted using a BD influx FACS sorter. Sorting buffer was composed of PBS with 3 mM EDTA, 3% FCS and 1:1000 DAPI 2.5 mg/ml. Parental cells were used as control. Dead cells were excluded as DAPI + cells. FACS data was analysed using Flowjo version 9 (Tree star).
RNA preparation and quantitative real-time PCR
RNA was purified from parental and reprogrammed cells following standard protocols using Trizol as previously described [20]. Reprogrammed cells were harvested when they reached about 50% confluency and preplated on nongelatinized T25 flasks for 45 min to remove feeder cells. cDNA was produced by the RevertAid First Strand cDNA Synthesis Kit (Fermantas, K1632) according to the manufacturer’s instructions. qRT-PCR was performed using Fast SYBR® Green Master Mix (Invitrogen). Relative and absolute expression was determined after adjusting for β-ACTIN housekeeping gene. Samples were run in a thermocycler 7500 Fast Real Time PCR system (Applied Biosystems). ΔΔCt analysis was performed and standardized to low passage parental cell lines.
Alkaline Phosphatase Staining
Alkaline phosphatase (AP) staining was performed with an Alkaline Phosphatase substrate kit (Vector laboratories) according to manufacturer’s recommendations.
Immunofluorescence
Cells were seeded in 12-well plates, for reprogrammed cells plates were previously coated with BD Matrigel™ hESC-qualified Matrix. Cells were washed with PBS and then fixed in 4% paraformaldehyde for 20 min at RT. After washing, the cells were permeabilized with 0.1% Triton X-100 for 10 min, then blocked in 10% secondary antibody host serum and 0.5% BSA for 1–2 h at RT with agitation. After washing with PBS-Tween, the cells were incubated overnight with primary antibodies (OCT4, SOX2, NANOG, TRA-1-60, TRA-1-81, E-Cadherin and CK19) at 4 °C with shaking. Following the incubation period, cells were washed and incubated with secondary antibodies for 1 h at 4 °C. The nuclei were counterstained with DAPI (Sigma-Aldrich; D8417). We observed the plates under a fluorescent microscope (IX71; Olympus).
Histology and immunohistochemistry
For histopathological analysis, FFPE blocks were serially sectioned (6 μm thick) and stained with haematoxylin and eosin (H&E). Additional serial sections were probed with antibodies against human E-cadherin and Pan-Cytokeratin (Abcam). Following incubation with primary antibodies, samples were incubated with HRP-conjugated secondary antibodies (Dako), and positive cells were visualized using 3,3-diaminobenzidine tetrahydrochloride plus (DAB + ) as a chromogen.
Fluorescent detection of Nanog mRNA in live cells
A total of 247-parental and reprogrammed cell suspensions were seeded into 6-well plates (Thermo Scientific) at a concentration of 106 cells per well, in 3 ml of media. Then 3 μl of SmartFlare probe (EMD Millipore) prediluted into 50 μl of PBS were added to the following wells: scramble-Cy3 background control probe and one well of the mRNA probe specific for Nanog-Cy3. Cells were incubated overnight for a minimum of 16 h at 37 °C and 5% CO2. After incubation, cells were observed using a fluorescent microscope (IX71; Olympus).
Cell proliferation assay
Cells (2 × 103) were seeded in RPMI culture medium with 10% FBS in 96 wells plates. Trypan blue exclusion test was used to assess cell proliferation during 5 days after plating. Results were presented as mean ± SD from three independent experiments each with triplicate wells.
Flow cytometry
Cell cycle analysis was performed using propidium iodide (PI) DNA staining. 106 cells were trypsinised, washed in PBS, centrifuged, and pellets were fixed in 200 µl of 70% ethanol for 2 h at 4 °C, washed with PBS, pelleted and stained with propidium iodide (50 µg/ml) in the presence of RNase A (100 µg/ml) (Sigma-Aldrich, Germany) for 15 min at 37 °C. Flow cytometry analysis was performed on the BD FACS Calibur (San Jose, CA) and Flowing software version 2.5.1 was used for data analysis (www.flowingsoftware.btk.fi).
The level of CD133 expression in 247-parental and reprogrammed cells was determined via flow cytometry using PE-CD133 antibody (1:20, BD Biosciences, USA). PE Mouse IgG1 (1:100, BD Biosciences, USA) was used as negative control. The CD133 labeled cells were analysed by using BD FACS Calibur Flow Cytometry System (San Jose CA, USA).
Sphere formation assay
Cells were trypsinised into single cells and 3 × 104 cells/mL were suspended and cultured in ultra-low attachment 6-well plates (Corning) using serum-free DMEM/F12 supplemented with B27 (1:50, Invitrogen), 20 ng/mL bFGF 20 ng/ml (Royan Biotech), 50 U/mL penicillin/streptomycin and 2% FBS. Spheres > 40 µm were considered for enumeration using an inverted microscope at 40x magnification.
Invasion assay
BioCoat Matrigel invasion chambers with 8.0 µm PET membranes in 24-well plates (Corning Inc. Life Sciences, MA) were used. At total of 25,000 cells was resuspended in 500 µl of serum-free DMEM and added to the upper chamber. In the lower chamber, 800 µl of DMEM with 10% FBS was used as a chemoattractant. After 16 h, the upper well containing membrane was scrubbed carefully with a cotton swab soaked in PBS to remove all non-invaded cells. The lower membrane of the well was rinsed with PBS. Transmigrated cells were stained with Crystal Violet (Sigma Aldrich) for 1–2 min in the dark. The invasion index was calculated as the average number of cells per 10x microscopic field, as determined by ImageJ software.
In vivo tumourigenicity assay
To assess in vivo tumourigenicity of reprogrammed versus parental cells, male 4-6-week-old BALB/C mice were used. Graded doses of cells (0.5 × 106, 1 × 106, 2.5 × 106, 5 × 106, 7 × 106 and 10 × 106) was prepared in 100 μl Matrigel/PBS (0.34 mg/ml BD Biosciences, San Jose, CA) and injected subcutaneously into to each flank of nude mice (n = 6 injections per group). Mice were housed according to institutional guidelines and all experiments were approved by the Animal Experimental Ethics Committee of the Royan Institute. Tumour formation was evaluated after 2 months. Any formed tumours were extracted, weighed, and processed for histology. In addition, the liver and lungs of each mouse were assessed for metastasis. Tumours were fixed in 10% formalin in PBS, embedded in paraffin, and processed for H&E staining.
Statistical analysis
We used the non-parametrical Kruskal-Wallis test, followed by post-hoc pairwise comparison using the Mann-Whitney test (GraphPad Prism 6, GraphPad Software, San Diego, CA). Data are presented as mean ± SD.
Results
Reprogramming of PDAC Cells
To elucidate the possibility of reprogramming human PDAC cancer cells into a epigenetically distinct and hopefully less tumourigenic state, we tested three somatic cell reprogramming methods on our PDAC primary cancer cultures (PDAC-247, -253 and -354). The established human pancreatic cancer cell line PANC-1 and human fibroblast cells (HDF and HFF-5) served as controls. We treated our cells with the following reprogramming plasmids, (1) the doxycycline-inducible TetO-FUW-OSKM [21] plasmid containing OCT4, SOX2, KLF4, and c-MYC that was delivered by lentiviral gene transfer, (2) the CMV-OCT4-miR-302 plasmid with OCT4 and the reprogramming miRNA mir-302 also requiring lentiviral transduction, and (3) the episomal vectors (OCT4, SOX2, KLF4, LMYC and LIN28A, combined with P53 knockdown (shP53)) that do not integrate into the genome and are delivered by virus-free electroporation (Fig. 1a).
Fig. 1.

Reprogramming of PDAC cells using three different methods. a Schematic and timeline of cell reprogramming in human pancreatic cancer cells. b PDAC primary cancer cultures (PDAC-247, -253 and -354), the established human pancreatic cancer cell line PANC-1, and human fibroblast cells (HDF and HFF-5) as a control were treated with three reprogramming methods. The morphology of parental and reprogrammed cells is exhibited 4–8 weeks after infection. We observed distinct types of colonies with round shapes different from the wild type. Referential morphologies are exhibited by HDF and HFF-5; scale bar: 100 μm. Inserts show the red fluorescence of OCT4-mir302-mCherry+ cells. The corresponding colonies are shown as brightfield microscopy images
Growth of iPS-like colonies occurred after only 12 days following gene transfer with episomal vectors in the human fibroblasts cell lines HDF and HFF-5, whereas iPS-like colonies with OSKM or OCT4-miR-302 took 60 days following transduction (Fig. 1b). Successful reprogramming in HFF-5 and HDF fibroblasts was confirmed by induction of endogenous pluripotency-associated genes such as NANOG and its downstream target REX1 (Fig. 2a), and further corroborated by immunostaining with NANOG and increased alkaline phosphatase activity in iPS cells induced with the episomal vectors (Fig. 2b, c). In HFF-5 fibroblasts, episomal vector reprogramming provoked significantly higher levels of NANOG and REX1 as compared to fibroblasts transduced with OSKM. Moreover, we did not detect alkaline phosphatase activity by infection with OSKM or OCT4-miR302. Thus, induction with the episomal vectors appears to be the more efficient method to reprogram our fibroblast cells into iPS cells.
Fig. 2.

Characterization of reprogrammed fibroblasts and PDAC cells. a Expression of pluripotency markers in parental and reprogrammed cells by real-time PCR. b Immunofluorescence staining of pluripotency markers NANOG and OCT4 in the parental and reprogrammed HDF cells. DAPI was used for nuclear counterstaining; scale bar: 50 μm. c Alkaline phosphatase-positive colonies from reprogrammed HDF cells generated by the episomal vectors method
Next, we attempted to reprogram pancreatic cancer cells, first using the established pancreatic cancer cell line PANC-1 and followed by primary cultures of PDAC cells. However, reprogramming of PANC-1 generated epithelial cell aggregates without any sharp border. Because of the epithelial morphology of parental PDAC 247, 253, and 354 cells, it was impossible to define whether they were successfully reprogrammed into iPS cells based on morphology (Fig. 1b). Therefore, we analysed the expression of a set of pluripotency-associated and epigenetic modifier genes. Our data showed that reprogramming by episomal vectors did not result in the upregulation of pluripotency-associated genes such as NANOG in PANC-1 and PDAC-253 and -354 cells compared with their parental cells (Fig. 2a), suggesting that these PDAC cells had not reprogrammed properly following the iPS-inducing procedures.
In contrast, PDAC-247 primary cultures were the only group, which exhibited dramatically high cell death rates, particularly following gene transfer with episomal vectors. PDAC-247 primary cultures started to grow colonies at about 21–50 days following infection, showing morphological changes with increased nuclei to cytoplasm ratio (Fig. 3a). Therefore, we followed this group to further evaluate whether they were indeed reprogrammed into a distinct epigenetic state.
Fig. 3.

Characterization of reprogrammed PDAC cells generated by transfection with episomal vectors. a Cells from PDAC-247 were reprogrammed and different passages of the iPS-like clones are shown. b ALP activity was only observed in a few of the screened colonies from 247- reprogrammed cells; scale bar: 50 μm. c Immunofluorescence staining of pluripotency markers NANOG, TRA-1-81, SOX2, OCT4 and TRA-1-60 in the 247-parental and reprogrammed cells (upper panel). Both parental and reprogrammed cells were negative for SOX2, OCT4 and TRA-1-60 (lower panel). DAPI was used for nuclear counterstaining; scale bar: 100 μm. d Expression of pluripotency markers and epigenetic modifier genes in parental and reprogrammed PDAC-247 cells as assessed by real-time PCR. Gene expression levels were normalized to bACTIN; *p ≤ 0.0001; mean ± SD; n = 3. e Detection of NANOG mRNA expression using SmartFlare mRNA probe for NANOG in live parental and reprogrammed PDAC-247 cells. Inside a single colony, expression of NANOG is more pronounced in some areas. The circular binding pattern of the SmartFlare mRNA probe is typical for live imaging of NANOG; scale bar: 100 μm. f Expression of CD133 in parental and reprogrammed 247 cells by real-time PCR. Gene expression levels were normalized to bACTIN; *p ≤ 0.0001, mean ± SD; n = 3. g Representative flow cytometry images for CD133 staining
Phenotypic characterization of reprogrammed PDAC cells
First, we characterized the PDAC 247-reprogrammed cells (247-REP) by RT-PCR, immunostaining and ALP staining (Fig. 3a–g). Immunostaining showed that some colonies derived from PDAC 247 expressed NANOG and TRA-1-81 as compared to the parental cells (Fig. 3c). However, levels of expression substantially varied between cells of individual colonies. The heterogeneity of NANOG expression in 247-REP colonies was further validated using a SmartFlare mRNA probe to detect NANOG expression (Fig. 3e). Inside a single colony, we noticed that cells with a higher nucleus-to-cytoplasma ratio also showed higher expression of NANOG. To further corroborate our finding, we assessed ALP activity that could only be observed in a few of the screened colonies (Fig. 3b). In addition, gene expression data indicated that 247-REP colonies show a consistent upregulation of the pluripotency marker NANOG and the epigenetic modifier gene TET1, while CMYC, SIRT1, DOT1L and TET2 were actually downregulated (Fig. 3d). We also checked for CD133 expression in our reprogrammed cells and observed an increase in the percentage of CD133 ± cells following induction of reprogramming (Fig. 3f, g).
On the basis of the above data, 247-REP cells appeared to have not been completely reprogrammed into iPS cells, but still showed remarkable changes as compared to their parental cells.
In vitro tumourigenicity and phenotype of reprogrammed PDAC cells
We next asked whether the in vitro tumourigenic potential of 247-REP cells was decreased or even lost after reprogramming. For this reason, we evaluated the proliferative capacity of reprogrammed 247-REP cells over 5 days (Fig. 4a).
Fig. 4.

Tumourigenicity, proliferative and invasive capacity of reprogrammed PDAC cells in vitro. a Cell proliferation of parental and reprogrammed PDAC-247 cells during 5 days after plating; * P ≤ 0.05; n = 3; mean ± SD. b Sphere formation efficiency in first- and second-generation of parental and reprogrammed PDAC-247 cells. Quantification (left) and representative pictures (right); *P < 0.05; n = 3. c Cells were seeded onto the filters of 0.8 µm pore transwells coated with Matrigel and after 16h transmigrated cells were stained with Crystal Violet and quantified. Photos are representative of one of three performed experiments; scale bar: 100 µm, (right). Quantification of cell invasion; *P < 0.05, n = 3, mean ± SD, (left). d Expression of EMT markers in parental and reprogrammed 247 cells by real-time PCR. Gene expression levels were normalized to bACTIN; *p ≤ 0.0001, mean ± SD; n = 3
To further explore this surprising finding of enhanced proliferation for 247-REP cells, we next measured sphere formation capacity of 247-REP cells as an in vitro surrogate for the cells’ tumour-initiating capacity. Interestingly, the sphere formation capacity of 247-REP cells was significantly reduced comparing to their parental cells (Fig. 4b). Notably, 247-REP-derived spheres were also significantly smaller than those produced by their parental cells. Therefore, while reprogramming of PDAC 247 cells enhanced the cells’ proliferative capacity, it still significantly impaired their in vitro tumourigenic potential. These data are in line with the notion that the tumourigenic capacity of cancer cells does not rely exclusively on cell proliferation.
The invasive properties also represent an important feature of PDAC cells and contribute to their aggressiveness. Analysis of the invasive capacity of PDAC 247-REP cells revealed a significant reduction as compared to their parental cells (Fig. 4c). Consistently, gene expression data showed a significant downregulation of EMT genes such as CDH2, SNAIL, and ZEB1 (Fig. 4d). Finally, immunostaining also demonstrated increased E-CADHERIN and Cytokeratin-19 expression, consistent with a more differentiated phenotype of reprogrammed PDAC cells (Fig. 5).
Fig. 5.

Reprogrammed PDAC cells show a more differentiated phenotype. Immunofluorescence staining of the epithelial differentiation markers cytokeratin 19 and E-cadherin in 247-parental and reprogrammed cells; scale bar: 100 µm
In vivo tumourigenicity of reprogrammed PDAC cells
Finally, we assessed the in vivo tumourigenicity of the reprogrammed cells. We injected increasing numbers of both parental and the reprogrammed cells (0.5, 1.0, 2.5, 5.0, 7.0, and 10.0 × 106) subcutaneously into BALB/C nude mice. Parental cells formed tumours within 8–10-weeks and all mice had to be killed three months after injection. In contrast, their reprogrammed counterparts did not form any tumours within 3 months (Fig. 6a). Consistent results were obtained for murine PDAC cells reprogrammed by lentiviral transduction with the OCT4 mir-302 mCherry construct, suggesting that our findings are species-independent (Suppl Fig. 1) [22]. Histological analysis of tumours formed by parental cells revealed that cancer cells were irregularly shaped and contained hyperchromatic nuclei and a high nuclear to cytoplasmic ratio (Fig. 6b). Tumours formed by parental cells showed low expression of E-cadherin and pan-cytokeratin, consistent with a less differentiated state. Thus, our results clearly demonstrate that direct reprogramming decreases the aggressiveness of PDAC cancer cells and subsequent loss of in vivo tumourigenicity, despite their increased in vitro proliferative capacity.
Fig. 6.

Tumourigenicity in vivo following reprogramming. a Subcutaneous injections of increasing numbers of parental and reprogrammed PDAC-247 cells. Parental cells formed tumours after 8–10-weeks and all mice had to be killed three months after injection (upper panel). Images of one representative tumour per group are shown. Tumour weight was measured. Data are shown as box plots with whiskers indicating the range of the data and the box representing the interquartile range (lower panel). b Haematoxylin and eosin staining and immunostaining for the differentiation marker E-CAD and PanCK for tumours derived from PDAC-247 parental cells
Discussion
In the present study, we have shown that resetting the epigenetic profile of PDAC cells via episomal vectors facilitated their reprogramming by induction of pluripotency and resulted in reduced sphere formation in vitro and tumourigenicity in vivo. Several lines of evidence show that cancer progression can be elicited by both reversible epigenetic changes and irreversible mutations in oncogenic and tumour suppressor genes [23]. While cancer cell nuclei have been suggested not to be amenable to the reprogramming process [11], multiple groups have generated putative iPS cells from various types of malignancies [15, 16, 24]. However, reprogramming efficiencies were reported to be low and only a subset of cancers is amenable to reprogramming and, similarly, we only obtained one reprogrammed line (247-REP) from PDAC cells.
Here, we demonstrated that PDAC cells and normal fibroblasts can be reprogrammed using episomal vectors. Episomal reprogramming seems particularly well-suited for clinical translation because it is integration-free, works reliably with patient fibroblasts and blood cells, and is based on a very simple reagent (plasmid DNA) that can easily be generated using current good manufacturing practice (cGMP)-compatible processes [25].
To develop safe and efficient iPS generation methods towards clinical applications, we used non-integrative episomal-based reprogramming of PDAC cells on feeder conditions. Importantly, in comparison to two other lentiviral methods, this episomal method can more efficiently reprogram fibroblast and PDAC cells resulting in both a shorter time (<14 days) and higher quality of the reprogramming process. In addition, reprogrammed PDAC cells exhibit loss of tumourigenicity both in vitro and in vivo. Our results showed that induction with the episomal vectors is the most efficient method to reprogram our fibroblast and PDAC cells. In contrast to other reports, our reprogrammed tumour cells did not remain dependent on the ectopic overexpression of the reprogramming factors.
Certain mutations might make cancer cells refractory to reprogramming so that they are not sensitive to the reset of the epigenome. For instance, PDAC epithelial cells from patients pre-treated with radiation did not produce any iPS colonies, perhaps due to senescence induced by irradiation and DNA damage. Given that many cancer patients are treated with chemotherapy and irradiation prior to surgical resection, such treatments may prevent the reprogramming of these cells [26]. In this study, all PDAC primary cultures harbour KRAS mutations. However, only PDAC 247 could overcome this genetic barrier and lost their tumour-initiating potential during the reprogramming process. Interestingly, OCT4 expression was not upregulated, suggesting that tumour cells harbour barriers impeding the reactivation of OCT4, which might prevent the acquisition of a stable pluripotent state.
Global epigenetic remodelling processes are well-known hallmarks of tumour development and also play an import role in tumourigenesis [27]. Here, we demonstrated that resetting the epigenetic profile of PDAC cells induced by nuclear reprogramming can facilitate their differentiation and reduces the tumour-initiating potential. Reprogramming toward pluripotency induces a stepwise increase in the developmental potential. This allows tumour cells to acquire a terminal differentiation other than its origin [17].
Our study revealed that a PDAC epigenome could be reprogrammed resulting in significantly impaired tumourigenicity in vitro and in vivo. We could also show that reprogramming of PDAC cells was consistent with subsequent expression of differentiation markers such as E-cadherin and cytokeratin-19, while EMT genes were reduced. Therefore, we demonstrated a direct correlation between the tumourigenic potential of a cancer cell and its differentiation status.
Apart from expression of pluripotency-associated genes to confirm the pluripotent state, we showed that epigenetic modulators such as TET2, SIRT1, and DOT1L were decreased, while TET1 was upregulated. Reprogrammed cells also showed an upregulation of NANOG at the protein level. However, considering the expression of the surface reprogramming marker TRA-1-80, our reprogrammed colonies may only have reached partial reprogramming. Interestingly, partial reprogramming might be also sufficient or even advantageous. Partial reprogramming had been reported to facilitate further differentiation with a reduced propensity for tumourigenicity in vivo (Fig. 7) [28].
Fig. 7.
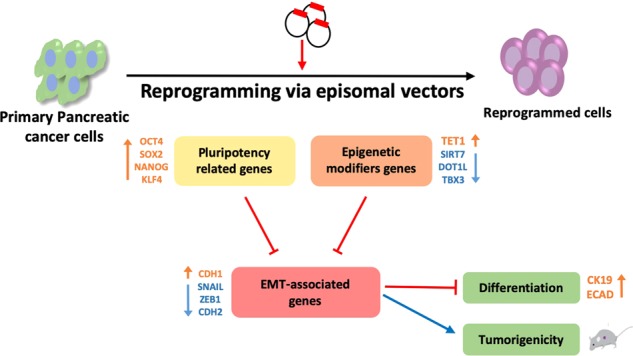
Model of cancer reprogramming and subsequent differentiation in 247-REP cells. Schematic illustration of the correlation between epigenetic modifiers, stemness and EMT genes following reprogramming in PDAC 247 cells. The sequential introduction of the episomal vectors leads to reprogramming of 247-PDAC cells and subsequent differentiation and loss of tumourigenicity (see discussion for details)
Regarding the level of expression of different chromatin modifiers, a strong change in the expression pattern could be observed. This indicates that the reprogrammed cells have a different chromatin modifier expression profile than the parental cells. The SIRT family has been reported playing a significant role in cancer development, tumourigenesis and metastasis, in particular its deacetylase activity plays a key function. Thus, SIRT family members promoted the EMT process through two different ways, as a reader of the EMT transcription factor SNAIL and by suppression of TET1 having EMT repressing capacities [29]. SIRT1 has also been reported to facilitate iPS reprogramming and is required for efficient post reprogramming telomere elongation and the maintenance of pluripotency [30]. Our 247- REP cells also could not maintain pluripotency which could be explained by SIRT1 downregulation in these cells. It seems that DOT1L only plays a role in the early stages of reprogramming, which is in line with the reversal of our downregulated DOT1L levels. Inhibition of DOT1L has also been reported to be effective against leukaemia and is currently tested in clinical studies (Suppl Fig. 2) [31].
Our data showed a positive correlation between TET1 upregulation and TBX3 downregulation and their effect on cell proliferation, sphere formation, EMT gene expression, and invasion potentials in 247-REP cells. This notion is also consistent with previous findings that the tumour development is associated with a decrease of TET gene expression, while ectopic expression of TET1 inhibited colony formation, cell migration, and invasion in ovarian cancer cells [32]. Furthermore, TET1 overexpression reversed the epithelial-mesenchymal transition (EMT) process in colon cancer cells [33, 34]. Furthermore, it has been found that TET1 bound to the promoter of ZEB2 and inhibited cancer cell proliferation, colony formation and invasion (Fig. 7) [35].
It has been also revealed that expression of TBX3, a T-box transcription factor repressor, can promote progression in a model of early breast cancer by altering cell properties involved in cell survival/colony formation and invasiveness, as well as key regulatory and EMT/invasiveness-related gene expression [36]. Consistently, our reprogrammed cells also showed significant downregulation of TBX3 and EMT related genes and also reduced their migratory capacities.
Collectively, our study showed that pluripotency factors and the pluripotency state can suppress features of the cancer phenotype, restore differentiation potential, and alter cancer-related gene expression. Finally, cellular reprogramming may provide a model to study how epigenetic abnormalities may be pivotal to the origins of cancer and whether reprogramming might play a role in the formation of key subpopulations of cancer cells [37]. Hence, targeting these epigenetic alterations will provide effective therapeutic approaches for the treatment of PDAC and will be pursued in future studies. Further work is necessary for more in-depth understanding of the molecular regulatory mechanisms of PDAC reprogramming and the development of novel therapies in the regenerative arena.
Highlights
Primary human pancreatic ductal adenocarcinoma (PDAC) cells were successfully reprogrammed.
Induction with episomal vectors was most efficient to reprogram PDAC cells.
Reprogrammed PDAC cells were functionally distinct from parental cells with drastically reduced tumourigenicity in vitro and in vivo.
Reprogrammed PDAC cells more readily differentiated in vivo resulting in rapid loss of stemness.
Supplementary information
Acknowledgements
We thank Dr Manuel Hidalgo for providing the PDAC samples (CNIO Material Transfer Agreement 1204090835CHMH). We would like to acknowledge members of the Department of Stem Cells and Developmental Biology laboratories at Royan Institute for their constructive advice.
Funding
The research was supported by the ERC Advanced Investigator Grant (Pa-CSC 233460, to C.H.), the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n° 602783 (CAM-PaC, to CH) and funding provided by the Royan Institute (#91000413 to ME).
Author contributions
Conception and design: CH, ME, RK, AA. Development of methodology: CH, ME, RK, AA. Acquisition of data and key reagents: RK, AA, JCR, RT. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): RK, MT, AA. Writing, review, and/or revision of the manuscript: RK, AA, HB, CH, ME.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alexandra Aicher, Email: aicher_a@yahoo.com.
Marzieh Ebrahimi, Email: Mebrahimi@royaninstitute.org.
Christopher Heeschen, Email: christopher.heeschen@icloud.com.
Supplementary information
The online version of this article (10.1038/s41388-019-0871-x) contains supplementary material, which is available to authorized users.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Sainz B, Jr, Martín B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014;74:7309–20. doi: 10.1158/0008-5472.CAN-14-1354. [DOI] [PubMed] [Google Scholar]
- 4.Cioffi M, Trabulo S, Hidalgo M, Costello E, Greenhalf W, Erkan M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res. 2015;21:2325–37. doi: 10.1158/1078-0432.CCR-14-1399. [DOI] [PubMed] [Google Scholar]
- 5.Zagorac S, Alcala S, Fernandez Bayon G, Bou Kheir T, Schoenhals M, Gonzalez-Neira A, et al. DNMT1 inhibition reprograms pancreatic cancer stem cells via upregulation of the miR-17-92 cluster. Cancer Res. 2016;76:4546–58. doi: 10.1158/0008-5472.CAN-15-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada Y, Haga H, Yamada Y. Concise review: dedifferentiation meets cancer development: proof of concept for epigenetic cancer. Stem Cells Transl Med. 2014;3:1182–7. doi: 10.5966/sctm.2014-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semi K, Yamada Y. Induced pluripotent stem cell technology for dissecting the cancer epigenome. Cancer Sci. 2015;106:1251–6. doi: 10.1111/cas.12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuda Y, Semi K, Yamada Y. Application of iPS cell technology to cancer epigenome study: uncovering the mechanism of cell status conversion for drug resistance in tumor. Pathol Int. 2014;64:299–308. doi: 10.1111/pin.12180. [DOI] [PubMed] [Google Scholar]
- 10.Blelloch RH, Hochedlinger K, Yamada Y, Brennan C, Kim M, Mintz B, et al. Nuclear cloning of embryonal carcinoma cells. Proc Natl Acad Sci USA. 2004;101:13985–90. doi: 10.1073/pnas.0405015101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochedlinger K, Blelloch R, Brennan C, Yamada Y, Kim M, Chin L, et al. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004;18:1875–85. doi: 10.1101/gad.1213504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li L, Connelly MC, Wetmore C, Curran T, Morgan JI. Mouse embryos cloned from brain tumors. Cancer Res. 2003;63:2733–6. [PubMed] [Google Scholar]
- 13.McKinnell RG, Deggins BA, Labat DD. Transplantation of pluripotential nuclei from triploid frog tumors. Science. 1969;165:394–6. doi: 10.1126/science.165.3891.394. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 15.Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, Camargo FD, et al. Generation of iPSCs from cultured human malignant cells. Blood. 2010;115:4039–42. doi: 10.1182/blood-2009-07-231845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, Tanaka F, et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci USA. 2010;107:40–5. doi: 10.1073/pnas.0912407107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency-based reprogramming. Oncogene. 2013;32:2249–60. doi: 10.1038/onc.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9:433–46. doi: 10.1016/j.stem.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–61. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 20.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 21.Carey BW, Markoulaki S, Hanna J, Saha K, Gao Q, Mitalipova M, et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci USA. 2009;106:157–62. doi: 10.1073/pnas.0811426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hermann PC, Sancho P, Canamero M, Martinelli P, Madriles F, Michl P, et al. Nicotine promotes initiation and progression of kras-induced pancreatic cancer via gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology. 2014;147:1119–33. doi: 10.1053/j.gastro.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 24.Kumano K, Arai S, Hosoi M, Taoka K, Takayama N, Otsu M, et al. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood. 2012;119:6234–42. doi: 10.1182/blood-2011-07-367441. [DOI] [PubMed] [Google Scholar]
- 25.Schlaeger TM, Daheron L, Brickler TR, Entwisle S, Chan K, Cianci A, et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33:58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Zaret KS. Reprogramming of human cancer cells to pluripotency for models of cancer progression. EMBO J. 2015;34:739–47. doi: 10.15252/embj.201490736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JJ, Murphy GF, Lian CG. Melanoma epigenetics: novel mechanisms, markers, and medicines. Lab Invest. 2014;94:822–38. doi: 10.1038/labinvest.2014.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsui T, Takano M, Yoshida K, Ono S, Fujisaki C, Matsuzaki Y, et al. Neural stem cells directly differentiated from partially reprogrammed fibroblasts rapidly acquire gliogenic competency. Stem Cells. 2012;30:1109–19. doi: 10.1002/stem.1091. [DOI] [PubMed] [Google Scholar]
- 29.Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med. 2015;21:335–43. doi: 10.1038/nm.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Bonis ML, Ortega S, Blasco MA. SIRT1 is necessary for proficient telomere elongation and genomic stability of induced pluripotent stem cells. Stem Cell Rep. 2014;2:690–706. doi: 10.1016/j.stemcr.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–25. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan H, Yan Z, Chen W, Wu Y, Han J, Guo H, et al. TET1 inhibits EMT of ovarian cancer cells through activating Wnt/beta-catenin signaling inhibitors DKK1 and SFRP2. Gynecol Oncol. 2017;147:408–17. doi: 10.1016/j.ygyno.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Morandi A, Taddei ML, Chiarugi P, Giannoni E. Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front Oncol. 2017;7:40. doi: 10.3389/fonc.2017.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Z, Zhang HS, Liu Y, Zhang ZG, Du GY, Li H, et al. Loss of TET1 facilitates DLD1 colon cancer cell migration via H3K27me3-mediated down-regulation of E-cadherin. J Cell Physiol. 2018;233:1359–69. doi: 10.1002/jcp.26012. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, An X, Yu H, Zhang S, Tang B, Zhang X, et al. MiR-29b/TET1/ZEB2 signaling axis regulates metastatic properties and epithelial-mesenchymal transition in breast cancer cells. Oncotarget. 2017;8:102119–33. doi: 10.18632/oncotarget.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krstic M, Macmillan CD, Leong HS, Clifford AG, Souter LH, Dales DW, et al. The transcriptional regulator TBX3 promotes progression from non-invasive to invasive breast cancer. BMC Cancer. 2016;16:671. doi: 10.1186/s12885-016-2697-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–89. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.