

Abstract
Cerebellar Purkinje neurons express voltage-gated, tetrodotoxin (TTX)-sensitive sodium channels that not only open and inactivate rapidly during depolarization but also reopen during repolarization, carrying an unusual “resurgent” sodium current. Expression of NaV1.6 α subunits appears necessary but not sufficient to generate this component of current; Purkinje cells without NaV1.6 lack resurgent current, but resurgent current is absent from many other NaV1.6-expressing neurons. These observations raise the question of how modulation or modification of the NaV1.6 subunit may lead to production of resurgent current. Previous studies have suggested that sodium channels of Purkinje neurons are subject to a rapid, voltage-dependent, open channel block by an endogenous particle whose unbinding allows resurgent current to flow. To investigate the nature of this block, we recorded TTX-sensitive sodium currents in outside-out patches from Purkinje cells acutely isolated from mice. In all patches, step depolarizations evoked transient current, and step repolarizations evoked resurgent current. The amplitudes of the transient and resurgent currents were highly correlated across patches (R2 > 0.99), suggesting that the blocking agent is closely associated with the channel. Intracellular protease eliminated fast inactivation, indicating that the blocking element, like the fast inactivation gate, may be proteinaceous. Intracellular application of alkaline phosphatase abolished resurgent current and significantly slowed inactivation of transient current. The phosphatase inhibitor vanadate reduced these effects. Together, the results suggest that constitutive phosphorylation of the sodium channel complex of Purkinje neurons is necessary to maintain a functional blocking element and produce resurgent sodium current.
Keywords: NaV1.6, med, open channel block, inactivation, phosphorylation, alkaline phosphatase, cerebellum
Cerebellar Purkinje neurons express voltage-gated tetrodotoxin (TTX)-sensitive sodium currents with unusual properties. As in most neurons, depolarization from negative potentials evokes transient sodium currents that rapidly activate and inactivate. During repolarization, however, the inactivated sodium channels of Purkinje neurons reopen, producing a brief surge of current, called resurgent sodium current (Raman and Bean, 1997). These kinetic properties apparently require expression of NaV1.6 α subunits (formerly Scn8a or NaCh6) (Burgess et al., 1995; Schaller et al., 1995; Goldin et al., 2000), because Purkinje neurons from mutant mice lacking NaV1.6 expression exhibit little or no resurgent current (Raman et al., 1997). Several cells expressing NaV1.6, however, produce voltage-gated sodium currents without a resurgent component (Raman and Bean, 1997; Smith et al., 1998; Pan and Beam, 1999), raising the question of what changes occur in Purkinje neurons that permit NaV1.6 expression to correlate with the flux of resurgent current.
The kinetics of resurgent current qualitatively resemble those of voltage-gated sodium channels that have been exposed to intracellular polycyclic cations, e.g., pancuronium ions,N-methyl-strychnine, or thiazine dyes, all of which act as voltage-dependent open channel blockers (Yeh and Narahashi, 1977;Cahalan and Almers, 1979; Armstrong and Croop, 1982). These observations raise the possibility that Purkinje neurons contain an endogenous open channel blocker (Raman and Bean, 2001), which may explain why resurgent current exists in only a subset of NaV1.6-expressing neurons. Figure1 depicts the mechanism by which open channel block may transform a “conventional” sodium channel (see Fig. 1A) into one that produces resurgent current (see Fig. 1B). In this scheme, Irepresents the normal fast inactivation gate, and Brepresents a second “blocking” inactivation gate, which blocks the open channel in a voltage-dependent manner. During depolarization to positive potentials, B binds more quickly than I, leading to a rapid decay of transient current. During repolarization,B unbinds, allowing resurgent current to flow beforeI binds (−40 mV) or the channel deactivates (−90 mV).
Fig. 1.
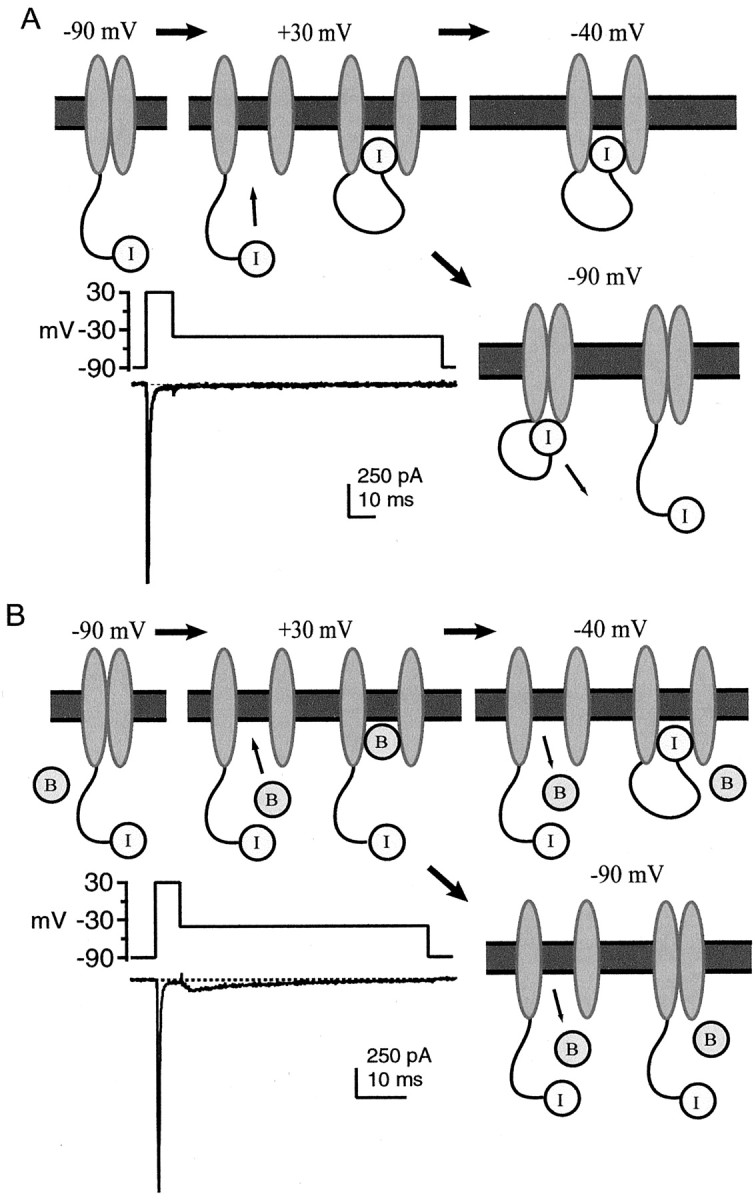
Two mechanisms of inactivation of sodium channels. In these diagrams, I represents the gate underlying normal fast inactivation, and B represents the putative blocking element, which provides a second mechanism of inactivation. The changing conformations of the channels (gray ellipses) are shown during a series of step changes in membrane voltage. A, Conventional sodium channel inactivation. At −90 mV, the channel is closed, andI is unbound (leftmost panel). During depolarization to +30 mV (middle panel), the channel opens (left) and then stops conducting asI binds (right). No additional change occurs during repolarization from +30 to −40 mV (rightmost panel). Repolarization from +30 to −90 mV (bottom panel) leads to channel deactivation (left), followed by recovery from inactivation asI unbinds (right). Inset, Illustration of conventional sodium current kinetics. Whole-cell TTX-sensitive sodium currents evoked from an isolated CA3 neuron, in response to the voltage protocol shown (method of Raman and Bean, 2001). B, Model for generation of resurgent current. At −90 mV, the channel is closed, and I, as well asB, are unbound (leftmost panel). During depolarization to +30 mV (middle panel), the channel opens (left), and rapid, voltage-dependent binding of B terminates current flow (right). Binding of B andI are thought to be mutually exclusive. During repolarization from +30 to −40 mV (rightmost panel), B unbinds, and the channel passes resurgent current (left). The channel stops conducting as I binds (right). Repolarization from +30 to −90 mV (bottom panel) leads to expulsion of B and a brief phase of resurgent current (left), followed by channel deactivation (right). Although B is depicted as a free cytoplasmic element, it may well be tethered to the α subunit or to another membrane-bound protein. Inset, Illustration of kinetics of a channel that carries resurgent sodium current. Whole-cell TTX-sensitive sodium currents evoked from an isolated Purkinje neuron, in response to the voltage protocol shown (method of Raman and Bean, 2001). Note the different time scales inA and B, reflecting the faster time course of inactivation of sodium current at +30 mV in the Purkinje cell compared with the CA3 cell.
Although much biophysical evidence supports this model (Raman and Bean, 2001), the physical nature of the second inactivating mechanism is unknown. Possibilities include the following: (1) intracellular inorganic cations that behave like Mg2+ or polyamines blocking various glutamate receptors (Nowak et al., 1984;Bowie and Mayer, 1995); (2) a proteinaceous gate that is part of the α subunit itself, like the fast inactivation gate (Bezanilla and Armstrong, 1977; Hoshi et al., 1990; West et al., 1992); or (3) a distinct peptide or subunit, like the β subunits of some potassium channels (Rettig et al., 1994; Wallner et al., 1999; Gulbis et al., 2000). In any case, the element that behaves like an open channel blocker must arise from a Purkinje-specific condition to account for the absence of resurgent sodium current in many other NaV1.6-expressing cells.
To investigate the identity of the putative open channel blocker, we recorded TTX-sensitive sodium currents in outside-out patches from isolated cerebellar Purkinje neurons. The results provide evidence that a proteinaceous blocking element is closely associated with the sodium channel complex and that its function is regulated by phosphorylation.
MATERIALS AND METHODS
Cell preparation. Cerebellar Purkinje neurons were acutely isolated from 13- to 17-d-old mice (Regan, 1991; Raman et al., 1997). Experiments were performed on C57BL/6 mice (Charles River Laboratories, Wilmington, MA) or med mice (The Jackson Laboratory, Bar Harbor, ME), which lack expression of the NaV1.6 α subunit (Burgess et al., 1995). The affected med mice were identified by their severe ataxia, which was evident after postnatal day 12 (Burgess et al., 1995; Raman et al., 1997). In accordance with institutional guidelines, mice were anesthetized with methoxyflurane or halothane before decapitation. The superficial layers of the cerebellum were removed and minced in ice-cold, oxygenated dissociation solution containing (in mm): 82 Na2SO4, 30 K2SO4, 5 MgCl2, 10 HEPES, 10 glucose, and 0.001% phenol red (buffered to pH 7.4 with NaOH). The tissue was incubated for 7 min in 10 ml of dissociation solution containing 3 mg/ml protease XXIII at 31°C (pH readjusted), with 100% oxygen blown over the surface of the fluid. The tissue was then washed in warmed, oxygenated dissociation solution containing 1 mg/ml bovine serum albumin and 1 mg/ml trypsin inhibitor (pH readjusted), in which it was microdissected. The pieces were then transferred to Tyrode's solution containing (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose (buffered to pH 7.4 with NaOH) at room temperature. The tissue was triturated with a series of fire-polished Pasteur pipettes to liberate individual neurons. Cells were allowed to settle on poly-d-lysine-coated glass coverslips in the recording chamber. Purkinje cell bodies were identified by their large size and characteristic tear shape. Recordings were made between 1 and 6 hr after trituration.
Electrophysiological recording. Borosilicate pipettes (3–6 MΩ; A-M Systems Inc., Carlsborg, WA) were wrapped with parafilm to minimize capacitance and filled with an internal solution containing (in mm): 120 HCH3SO3, 10 NaCl, 2 MgCl2, 10 HEPES, 1 EGTA, 53 sucrose, 14 Tris-creatinePO4, 4 MgATP, and 0.3 TrisGTP (buffered to pH 7.4 with CsOH). Pronase E (5147; Sigma, St. Louis, MO) and alkaline phosphatase (purified from dog intestine; 8639; Sigma) were added directly to the internal solution as needed. For the experiments of Figure 7, 10 mmNa3VO4 (sodium orthovanadate, referred to as “vanadate”) was substituted for NaCl and sucrose. All drugs were from Sigma, except Rp-8-cpt-cGMP and Rp-8-cpt-cAMP, which were from Biolog (San Diego, CA), and vanadate, which was from Calbiochem (San Diego, CA).
Fig. 7.
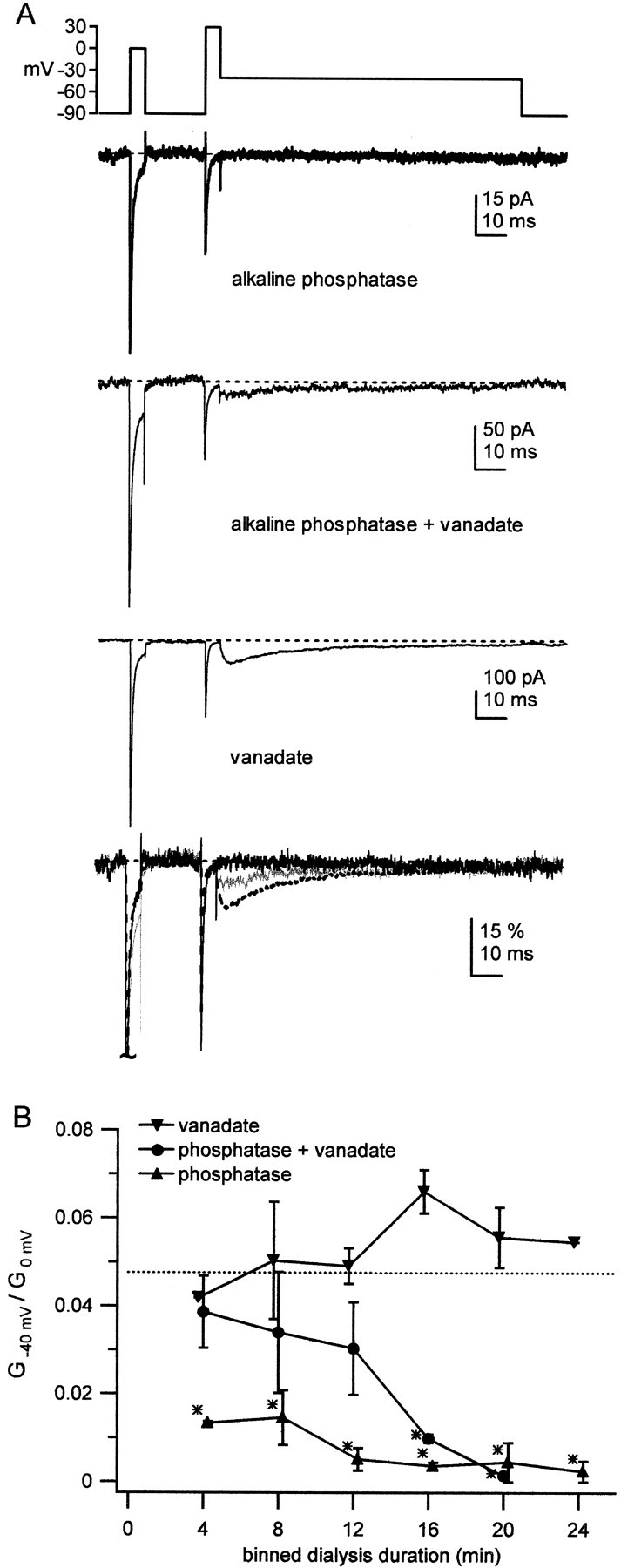
Effect of vanadate on alkaline phosphatase-mediated changes in resurgent sodium current.A, TTX-sensitive sodium currents from an outside-out patch from a normal Purkinje neuron were evoked by the voltage protocol shown. The recording pipette contained intracellular solution with alkaline phosphatase only (0.3 mg/ml; top trace), alkaline phosphatase plus vanadate (10 mm; second trace), or vanadate only (third trace). The illustrated traces were recorded 8–12 min after patch excision. Traces are superimposed and normalized to the peak transient current at 0 mV to illustrate relative differences in resurgent current amplitude (bottom traces; phosphatase alone, solid black trace; vanadate alone, dotted black trace; both, solid gray trace).Vertical scale bar indicates percentage of the peak normalized current. B, Time course of effects of alkaline phosphatase (upward triangles;n = 6), alkaline phosphatase with vanadate (circles; n = 5), or vanadate (inverted triangles; n = 3) on the resurgent current. The ratio of peak conductance at −40 mV to peak transient conductance at 0 mV (G−40 mV/G0 mV) is plotted against time after patch excision. Data were sorted into 4 min bins, and points are slightly offset for clarity. Data are plotted as mean ± SE. Not all patches were held for the entire duration of 24 min.Asterisks represent a significant difference from the mean control conductance ratio (dashed line).
Voltage-clamp recordings were made with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Data were recorded with an InstruTech (Great Neck, NY) ITC-18 interface and PULSE software (Heka Electronik, Lambrecht, Germany). After establishment of the whole-cell configuration, outside-out patches were pulled. Patches were positioned in front of a pair of gravity-driven flow pipes, which contained the different extracellular solutions. Recordings were first made with patches exposed to a control solution, which consisted of Tyrode's solution to which 10 mm TEACl and 3 μmCdCl2 had been added to reduce potassium and calcium currents. For the experiments of Figure 7, in which the Na3VO4 brought the intracellular Na+ concentration to 30 mm, the control solutions consisted of Tyrode's solution to which 10 mm NaCl, 1 mm TEACl, and 3 μm CdCl2 had been added, to maximize the driving force on sodium currents. All recordings were repeated in solutions that were identical to the control but that included 900 nm TTX. In both the control and the TTX solutions, 15–25 sweeps were recorded and averaged to improve the signal-to-noise ratio. Subtractions of the averaged records revealed TTX-sensitive sodium current. This averaging and subtraction procedure allowed resolution of current changes on the order of 1 pA. All recordings were obtained at room temperature.
Spectrophotometry. Spectrophotometric measurements were made with a Cary 500 SCAN UV-VIS-NIR spectrophotometer (Varian Instruments, Walnut Creek, CA) in the Keck Biophysics Laboratory of Northwestern University. Activity of alkaline phosphatase was assayed with 5 mm p-nitrophenyl phosphate (pNPP) as the substrate and the internal solution used for electrophysiological recording as the buffer, with or without 10 mm vanadate. As a control, measurements were made with 10 mm vanadate, without phosphatase, included in the buffer. Absorbance at 410 nm byp-nitrophenolate (dephosphorylated pNPP) was recorded for 20 min in each condition, and baseline absorbance in the absence of phosphatase was subtracted from the data. The initial change in absorbance (min−1) was measured for 0.3 mg of enzyme. From this value, enzyme activity (moles of dephosphorylated substrate per minute per milligram of enzyme) was calculated, given that absorbance is εbc, in which the molar absorptivity for p-nitrophenolate (ε) was 17,500m/cm, the light path length (b) was 1 cm, and the p-nitrophenolate concentration (c) was given in moles per 1 ml of buffer.
Analysis. Data were analyzed with IGOR software (WaveMetrics Inc., Lake Oswego, OR). Conductance values (Fig. 7) were calculated by dividing the measured current by the driving force obtained from the estimated Na+ equilibrium potential. Data are reported as mean ± SDs, except as noted, and statistical significance was assessed with Student's unpaired two-tailedt tests.
RESULTS
To test the possibility that a diffusible intracellular blocker is responsible for the open channel block that results in resurgent current, we recorded TTX-sensitive sodium currents in outside-out patches from Purkinje neurons. Sodium currents were elicited by a step protocol designed to evoke both transient currents, with a depolarization from −90 to 0 mV and resurgent currents with a repolarization from +30 to −40 mV. As shown in Figure2A, transient sodium currents elicited in patches retained the kinetics seen in whole-cell recordings (Raman and Bean, 1997), decaying with a time constant of 0.39 ± 0.036 msec (n = 8) at 0 mV and inactivating by 98.5 ± 0.79%. In all eight patches, the repolarizing step to −40 mV evoked resurgent sodium current (Fig.2A). The current had the slow rise and decay times characteristic of resurgent current in whole-cell recordings (Raman and Bean, 1997), with the peak resurgent current occurring 4.1 ± 1.2 msec after repolarization to −40 mV. As shown in Figure2B, the peak transient current at 0 mV ranged widely, from −17 to −600 pA. A linear fit to the plot of peak resurgent current against peak transient current gave a slope of 6% andR2 > 0.99, indicating that the relative amplitudes of these two components of sodium current are tightly regulated. These results suggest that the putative blocking element is closely associated with the sodium channel complex, providing evidence against the hypothesis of a diffusible blocker.
Fig. 2.

Resurgent sodium current in outside-out patches from Purkinje neurons. A, TTX-sensitive sodium currents from an outside-out patch from a Purkinje neuron were evoked by the voltage protocol shown. Dotted line indicates 0 pA intraces in all figures. B, For each patch (n = 8), peak resurgent current measured at −40 mV is plotted against peak transient current measured at 0 mV. Linear regression over the points (dashed line) gives a correlation coefficient (R2) of 0.998, a slope of 0.059, and an intercept of 0.75 pA.
The surprisingly high correlation between the transient and resurgent current amplitudes raises the question of the molecular identity of the channels. Purkinje neurons are known to express NaV1.6, as well as other sodium channel α subunits, including NaV1.1 and NaV1.2, (de Miera et al., 1997; Felts et al., 1997). The latter two subunits carry transient current (Smith et al., 1998) but appear unlikely to produce resurgent current (Raman et al., 1997). If the channels in our somatic patches consist of variable relative densities of these three channel-forming subunits, the ratio of resurgent to transient current should vary more widely. The nearly invariant ratio that we observed could be explained by nonrandom relative densities of different subunits, by patches consisting almost exclusively of NaV1.6 or by channels in addition to NaV1.6 producing resurgent current in the outside-out patches. It was important to test the latter possibility, given the premise that resurgent sodium current results from a Purkinje-specific modification of NaV1.6. To investigate the extent to which NaV1.6 was required for the resurgent current measured in Figure 2, we made recordings from outside-out patches pulled from Purkinje neurons isolated from med mice, which lack expression of NaV1.6.
In four of four outside-out patches from med Purkinje neurons, transient current was present, but resurgent current was undetectable (Fig. 3A). To make a quantitative comparison between these patches and those from normal cells, we measured current amplitude at the mean time of peak resurgent current in normal patches, i.e., at 4.1 msec after repolarization to −40 mV (Fig. 3B). In patches frommed neurons, this current was well within the level of the noise (Fig. 3B, dotted line). The ratio of the current at −40 mV to the peak transient current at 0 mV was 0.86 ± 0.43% (n = 4), significantly lower than in normal Purkinje neurons (7.52 ± 0.81%; n = 8;p < 0.001). Although we cannot exclude the possibility that NaV1.6 expression is simply a permissive factor, the lack of resurgent current in patches from medmice supports the hypothesis that NaV1.6 forms the primary pore that conducts resurgent sodium current. The presence of transient sodium currents in somatic patches from medmice is consistent with the expression of NaV1.1 and/or NaV1.2 (de Miera et al., 1997; Felts et al., 1997). In conjunction with the high correlation of resurgent and transient current amplitudes in normal patches, however, these data raise the possibility that these other subunits generally have a low somatic expression. Different subcellular localization of sodium channel α subunits has been reported in other preparations (Westenbroek et al., 1989).
Fig. 3.

Absence of detectable resurgent current in patches from med mice. A, Recording from an outside-out patch from a Purkinje neuron lacking NaV1.6 expression. TTX-sensitive sodium currents were evoked by the voltage protocol shown. B, Bar graph illustrating the ratio of current measured at 4.1 msec after repolarization to −40 mV to the peak transient current at 0 mV (I−40 mV/I0 mV) in normal (n = 8) and med(n = 4) mice. The ratios are significantly different (*p < 0.001). As an indicator of the current noise, the current was measured at 94.1 msec after repolarization to −40 mV in each med patch. Each noise measurement was normalized to the peak transient current in each patch, and the mean ratio is plotted for reference (dotted line).
If the putative blocking element is a nondiffusible factor, it may be either an additional protein subunit or a peptide in close association with (or part of) NaV1.6. If so, it may be subject to proteolytic cleavage, like the inactivation particle of sodium channels (Armstrong and Bezanilla, 1977; Bezanilla and Armstrong, 1977; Cota and Armstrong, 1989). To address this possibility, sodium currents were recorded in outside-out patches from normal Purkinje neurons, with 1 mg/ml pronase E included in the intracellular solution. In three of three patches, pronase E abolished fast inactivation at all potentials tested (Fig.4), suggesting that any blocking agents, as well as inactivation gates, were disrupted. Although we cannot exclude the possibility that only a binding site on the α subunit was cleaved, this result is consistent with the hypothesis that the element responsible for open channel block, like the conventional fast inactivation gate, is proteinaceous.
Fig. 4.

Effect of pronase E on inactivation. Pronase E (1 mg/ml) was included in the recording pipette, and TTX-sensitive sodium currents from an outside-out patch from a normal Purkinje neuron were evoked by the voltage protocol shown. Fast inactivation was abolished. A tail current is evident on the repolarizing step to −40 mV.
Such a proteinaceous blocker might be part of the α subunit itself or might be a distinct protein. In either case, some specific regulation or modification of the sodium channel must occur so that Purkinje cells produce resurgent sodium current but spinal motoneurons, for example, do not (Pan and Beam, 1999). Because many ion channels, including sodium channels, can be targets of kinases and phosphatases (Levitan, 1994), we considered the possibility that resurgent sodium current might be regulated by phosphorylation. Previous research has demonstrated that NaV1.6 subunits in expression systems do not exhibit resurgent sodium current (Smith et al., 1998). We reasoned that, in these expression systems, channels would more likely be in dephosphorylated states (but see Murakoshi et al., 1997). Purkinje cells express a variety of kinases, including cAMP-dependent protein kinase (PKA), protein kinase C (PKC), protein kinase G (PKG), and protein tyrosine kinases (Ito, 2001). In initial experiments, we attempted to reduce resurgent current by interfering with phosphorylation, but resurgent current was not abolished by preincubating cells for 1 hr in membrane-permeant kinase inhibitors, such as Rp-8-cpt-cGMP (50 μm; n = 2) or Rp-8-cpt-cGMP plus Rp-8-cpt-cAMP (50 μm each;n = 2; data not shown). We cannot necessarily interpret these data, however, to exclude a role for PKA and PKG. If mechanisms exist to maintain phosphatase activity at a low level in Purkinje cells, then inhibiting the relevant kinase may have little measurable effect. Notably, omitting phosphate from the pipette solution did not change resurgent current in eight of eight patches. In these patches, the resurgent sodium current was 6.9 ± 2.3% of the transient current at 0 mV, not significantly different from control (p > 0.5; data not shown). This result is consistent with a low net activity of endogenous phosphatases near the sodium channels.
We therefore tested the effects of direct application of alkaline phosphatase, a broad-spectrum phosphatase, on resurgent current. As shown in Figure 5A, 3.0 mg/ml alkaline phosphatase included in the intracellular solution completely abolished resurgent current in six of seven patches. In Figure5B, current amplitude measured 4.1 msec after repolarization to −40 mV is plotted against the peak transient current, illustrating that the loss of resurgent current occurs across all amplitudes of transient current. Currents from patches with a low current density are shown at higher gain in the inset to Figure 5B. In addition, alkaline phosphatase slowed inactivation of the transient sodium current at 0 mV to 0.70 ± 0.06 msec (n = 7) (Fig. 5A, inset), a value significantly larger than for the control data (p < 0.005). This slowing of inactivation is predicted by the loss of an ultra-fast open channel block. Consistent with this idea, CA3 neurons, which express NaV1.6 but lack resurgent current, have sodium currents that inactivate more slowly at positive potentials than do sodium currents of Purkinje cells (Raman and Bean, 1997, 2001). This difference in inactivation kinetics at +30 mV is evident in theinsets of Figure 1, A and B.
Fig. 5.

Effects of alkaline phosphatase on transient and resurgent sodium current. A, Alkaline phosphatase (3 mg/ml) was included in the recording pipette, and TTX-sensitive sodium currents from an outside-out patch from a normal Purkinje neuron were evoked by the voltage protocol shown. Resurgent current is undetectable. Inset, Trace A and control from Figure 2A (gray) are normalized to the peak current at 0 mV and superimposed, illustrating the slowing of transient current inactivation at 0 mV, which correlates with the loss of resurgent current. Vertical scale bar indicates percentage of the peak normalized current. B, For each patch treated with 3 mg/ml alkaline phosphatase (n = 7), the current measured after 4.1 msec at −40 mV is plotted against peak transient current measured at 0 mV (upward triangles). The control data and the fit from Figure 2B are included for comparison (open symbols and dotted line).Dashed line indicates zero resurgent current.Inset shows data from patches with the lowest current densities at high gain. Currents at −40 mV are plotted against peak transient current at 0 mV. In addition to the data from patches exposed to control and to 3 mg/ml alkaline phosphatase-containing solutions, data from six patches exposed to 0.3 mg/ml alkaline phosphatase are shown (inverted triangles).
Although the alkaline phosphatase had an unequivocal effect on the sodium current, we were concerned that the high concentration of enzyme, which was purified from intestine, might contain other factors that were responsible for the result. To verify that the effect of alkaline phosphatase was attributable to dephosphorylation, we first assayed the enzymatic activity of the phosphatase with spectrophotometry. The concentration of alkaline phosphatase was decreased 10-fold, to 0.3 mg/ml, for a more sensitive assay, and the time course of dephosphorylation of pNPP was measured. The initial slope of the absorbance was 0.51/min, from which we estimated the initial rate of substrate dephosphorylation to be 97 nmol · min−1 · mg−1phosphatase (Fig. 6, solid line). To test the sensitivity of the reaction to a classical inhibitor of alkaline phosphatase, we included 10 mm vanadate in the buffer. The initial slope of absorbance was 0.039/min, corresponding to an initial rate of substrate dephosphorylation of 7.43 nmol · min−1 · mg−1enzyme (Fig. 6, dotted line). This concentration of vanadate, therefore, led to a 13-fold decrease in enzyme activity, slowing, but not completely inhibiting, dephosphorylation by alkaline phosphatase.
Fig. 6.

Assay of alkaline phosphatase activity in the presence and absence of vanadate. Dephosphorylation of 5 mmpNPP by alkaline phosphatase was measured by spectrophotometry. Absorbance is plotted as a function of time for 0.3 mg/ml alkaline phosphatase alone (solid line), for 0.3 mg/ml alkaline phosphatase with 10 mm vanadate (dotted line), and for 10 mm vanadate alone (dashed line). Including vanadate with the phosphatase led to a 13-fold slowing of enzyme activity. Calculations of enzyme activities are given in Results.
Having measured biochemically the inhibition of alkaline phosphatase by vanadate, we next tested whether vanadate could reduce the effects of the phosphatase on resurgent current. In these experiments, the pipette solution included 0.3 mg/ml alkaline phosphatase, 0.3 mg/ml alkaline phosphatase with 10 mm vanadate, or 10 mmvanadate alone. Sodium currents were measured at ∼4 min intervals after establishment of the whole-cell configuration and patch excision. Traces from the three conditions recorded after 8 min are shown in Figure 7A. The lower concentration of alkaline phosphatase (0.3 mg/ml) effectively abolished resurgent sodium current in six of six cells (Fig. 7A,top trace) (see also Fig. 5B, inset). Including vanadate with the phosphatase preserved resurgent sodium current in five of five cells (Fig. 7A, second trace), whereas vanadate alone did not measurably change resurgent sodium current in three of three cells (Fig. 7A, third trace). For all three conditions, the amplitude of resurgent sodium current relative to the transient current at 0 mV is shown in the bottom trace of Figure 7A.
The time course of these effects is shown in Figure 7B. Data are plotted as the mean ratio of the resurgent to transient sodium conductances, at −40 and 0 mV, respectively. Conductance rather than current ratios were calculated because of the different sodium equilibrium potentials with and without internal vanadate (see Materials and Methods). The control conductance ratio of 0.0476 from the data of Figure 2 is plotted for reference (dashed line). Within 4 min of patch excision, alkaline phosphatase alone significantly reduced the conductance ratio relative to control (Fig.7B, triangles). In the condition including both alkaline phosphatase and vanadate, conductance ratios did not differ significantly from control data for the first 12 min. After 12 min of dialysis, the ratio was significantly decreased, consistent with the spectrophotometric observations of a slow dephosphorylation by the phosphatase in the presence of vanadate (Fig. 7B,circles). Conductance ratios from recordings made in the presence of vanadate alone did not differ significantly from controls at any time during the 24 min of data acquisition (Fig. 7B,inverted triangles). These data are consistent with the idea that alkaline phosphatase abolishes resurgent sodium current via dephosphorylation. Together, these results suggest that resurgent sodium current in Purkinje neurons requires constitutive phosphorylation of some element in, or in close association with, the sodium channel.
DISCUSSION
In addition to conventional fast inactivation, the channels that produce resurgent sodium current include a second inactivating mechanism (Raman and Bean, 2001), which is qualitatively similar to that induced by open channel block by polycyclic cations. These blockers speed inactivation of sodium currents evoked by depolarization and allow channel reopening during repolarization (Yeh and Narahashi, 1977; Cahalan and Almers, 1979; Armstrong and Croop, 1982). Although the physical structure of the endogenous blocking element of Purkinje neurons is unknown, the important features of the blocker include binding during depolarization, producing the ultra-fast inactivation of transient current, as well as unbinding during repolarization, producing resurgent current. For convenience, we refer to this second inactivation gate as a “blocking element.”
These experiments contribute two major pieces of information to our understanding of the mechanisms that maintain resurgent sodium current. First, the putative blocking element appears to be a protein in close association with the pore-forming subunit. Second, constitutive phosphorylation is necessary for the sodium channel complex to produce resurgent current. A loss of open channel block is predicted to slow the decay of the transient current, as well as abolish the resurgent current. In the presence of alkaline phosphatase, both of these changes were observed. Phosphorylation, therefore, appears to play a permissive role for resurgent current by either making the blocking element or binding site functional, or maintaining a close association between the element and the channel.
Molecular modifications to NaV1.6 that may permit resurgent sodium current.
Our results from med mice suggest strongly that resurgent sodium current requires NaV1.6. Nevertheless, because expression of NaV1.6 by many neurons produces channels that lack resurgent current, some transformation must occur in Purkinje cells to produce a sodium channel with a functional blocking element. A straightforward possibility is that an inorganic diffusible cation blocks the open channel. Three observations, however, are difficult to reconcile with this idea: the high correlation between resurgent and transient current amplitudes, the abolition of fast inactivation by internal protease, and the loss of resurgent current after dephosphorylation. Instead, the results are more consistent with the hypothesis that channel block is mediated by a protein closely affiliated with the α subunit. The protein may be (1) part of NaV1.6 itself, after post-translational modification, (2) part of a Purkinje-specific splice variant of NaV1.6, or (3) a separate protein, closely associated with NaV1.6. Phosphorylation at any of these levels may be required to produce a functional open channel block.
Modulation of NaV1.6
One possibility is that most neurons express the same NaV1.6 protein but that direct phosphorylation of NaV1.6 in Purkinje cells induces part of the subunit to act as a blocking element. Kinases, as well as phosphatases, can be localized to sodium channel α subunits and can directly regulate sodium currents (Tibbs et al., 1998; Cantrell et al., 1999b;Ratcliffe et al., 2000). For instance, activation of PKA modulates current amplitudes through NaV1.1 and Nav1.2 channels, and these effects can be intensified by PKC (Smith and Goldin, 1996, 1998, 2000; Cantrell et al., 1999a). NaV1.6, however, has fewer PKA or PKC consensus sites than NaV1.1 or NaV1.2, suggesting that it may be a relatively poor target for modulation by these kinases (Rossie et al., 1987;Konratyuk and Rossie, 1997; Plummer et al., 1998; Cantrell et al., 1999a). Consistent with this hypothesis, PKA has a proportionately smaller effect on currents in normal cortical neurons, which express NaV1.1, NaV1.2, and NaV1.6, than in cortical cells frommed mutants (Maurice et al., 2001). Our experiments with alkaline phosphatase predict that a kinase-mediated induction of a blocking element should speed inactivation. PKA and PKC, however, do not measurably affect the time course of inactivation in cortical or hippocampal neurons, all of which express NaV1.1, NaV1.2, and NaV1.6 (Cantrell et al., 1997, 1999a ; Maurice et al., 2001). Therefore, although currents evoked by repolarization have not been examined, a direct phosphorylation of the α subunit by PKA and/or PKC seems unlikely to be sufficient to produce resurgent sodium current.
Alternatively, a tyrosine kinase may act directly on sodium channel α subunits. Tyrosine phosphorylation and dephosphorylation can modulate sodium currents, and NaV1.2 can interact directly with a receptor protein tyrosine phosphatase β (Ratcliffe et al., 2000). Although no corresponding studies have been done on NaV1.6, this subunit has at least one potential tyrosine phosphorylation site (Plummer et al., 1998) that does not appear to exist on NaV1.1 or NaV1.2. This observation leaves open the possibility that tyrosine phosphorylation can modulate NaV1.6.
In addition, cell-specific enzymes may modulate NaV1.6. One such enzyme that regulates phosphorylation is G-substrate, a protein apparently expressed only by Purkinje cells. When phosphorylated by PKG, G-substrate inhibits phosphatase-1 (Detre et al., 1984; Endo et al., 1999; Hall et al., 1999). Whether G-substrate or phosphatase-1 has a direct influence on sodium channel function has not been explored. Nevertheless, such phosphorylation-dependent interactions suggest that a variety of enzymes may participate in maintaining the tightly regulated kinase and phosphatase activity that are apparently required for resurgent sodium current.
Splice variants
A second possibility is that resurgent sodium current is a consequence of a splice variant of NaV1.6 expressed specifically in Purkinje cells. Although three exons of NaV1.6 can be alternatively spliced, only two splice variants exist in the brains of mature animals (Plummer et al., 1997, 1998; Dietrich et al., 1998). The major splice variant of NaV1.6 in the brain includes a cytoplasmic segment of 10 amino acids, which is lacking in the minor variant (Plummer et al., 1998). At present, it is not known whether these splice variants are differentially expressed across brain regions.
Roles for additional proteins
The third possibility for generation of resurgent current is that a separate blocking protein interacts with the NaV1.6 subunits. In this case, direct phosphorylation of the α subunit, or of the blocking protein itself, may be additionally required. One piece of evidence supporting the idea of a separate protein comes from whole-cell recordings from NaV1.6 null Purkinje neurons. Although any residual resurgent sodium current in our med patches was below our detection threshold, whole-cell recordings from NaV1.6 null Purkinje cells reveal a tiny phase of resurgent current (∼10% of control) (Raman et al., 1997). No such phase exists in CA3 cells from wild-type animals (Raman and Bean, 1997), raising the possibility that a blocking element expressed in Purkinje cells, but not in CA3 cells, has a low affinity for the other sodium channel α subunits.
In most neurons, sodium channel complexes are heteromers containing α, as well as β, subunits (Isom et al., 1994). Purkinje neurons express β1 and β2 subunits, both of which modulate the kinetics of currents through expressed α subunits (Isom et al., 1994;Smith et al., 1998; Morgan et al., 2000; Shah et al., 2001). Coexpression of NaV1.6 with β1 or β2, however, does not yield resurgent sodium current (Smith et al., 1998). These data make it appear unlikely that β1 or β2 acts directly as a blocking element, at least in the absence of constitutive phosphorylation. This result may not be surprising, given their widespread expression (Shah et al., 2001). Interestingly, in addition to coassembling with and modulating α subunits, β1 can recruit other proteins to the sodium channel complex by acting as a cell adhesion molecule (Malhotra et al., 2000, 2001; Isom, 2001). The possible β1-mediated recruitment of blocking proteins to the NaV1.6 has not been investigated.
Whether or not a blocking protein distinct from the α subunit exists, the presence of resurgent current in other neurons that express NaV1.6 will depend on the following: (1) whether the blocking element is present in the neuron, (2) whether the appropriate kinase is expressed in the neuron, and (3) whether the conditions are met for constitutive activity of the kinase (or inhibition of phosphatases) to permit resurgent current. Resurgent sodium current is not, in fact, unique to Purkinje neurons. Unipolar brush cells of the cerebellar cortex show high densities of the current (Mossadeghi and Slater, 1998), and some cerebellar granule cells may also exhibit resurgent sodium currents (D'Angelo et al., 2001) (Z. M. Khaliq and I. M. Raman, unpublished observations).
Physiological consequences of modulation of resurgent sodium current
In outside-out patch recordings, resurgent sodium current was consistently elicited. It is possible, however, that the level of phosphorylation varies in the intact cerebellum. Such variation may occur, for example, as a consequence of kinase-activating or -inhibiting neurotransmitters released in the cerebellum, including acetylcholine, cannabinoids, serotonin, and glutamate (Jaarsma et al., 1997; Moldrich and Wenger, 2000; Dieudonné, 2001; Hansel et al., 2001; Kreitzer and Regehr, 2001). Neurotransmitter-evoked changes in kinase or phosphatase activity, therefore, may change the density of resurgent sodium current. Because the kinetics of the channels producing resurgent current appear to be associated with high-frequency firing of Purkinje cells (Raman and Bean, 1999), kinase-mediated modulation of the NaV1.6 channel may contribute to regulating the excitability of Purkinje neurons.
Footnotes
This work was supported by the Sloan Foundation, the Searle Foundation, and National Institutes of Health Grant NS39395 (I.M.R.). We thank Dr. Michael Kennedy for his help with spectrophotometry, and we gratefully acknowledge the Keck Biophysics Facility at Northwestern University, where the enzymatic assays were performed. We also thank Drs. Jim Surmeier, Tom Otis, Petra Telgkamp, and Zayd Khaliq for helpful comments on this manuscript.
Correspondence should be addressed to Indira M. Raman, Department of Neurobiology and Physiology, 2153 North Campus Drive, Northwestern University, Evanston, IL 60208. E-mail: i-raman@northwestern.edu.
REFERENCES
- 1.Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong CM, Croop RS. Simulation of Na channel inactivation by thiazine dyes. J Gen Physiol. 1982;80:641–662. doi: 10.1085/jgp.80.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 5.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease.”. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 6.Cahalan MD, Almers W. Block of sodium conductance and gating current in squid giant axons poisoned with quaternary strychnine. Biophys J. 1979;27:57–73. doi: 10.1016/S0006-3495(79)85202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel α subunit. J Neurosci. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantrell AR, Scheuer T, Catterall WA. Voltage-dependent neuromodulation of Na+ channels by D1-like dopamine receptors in rat hippocampal neurons. J Neurosci. 1999a;19:5301–5310. doi: 10.1523/JNEUROSCI.19-13-05301.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cantrell AR, Tibbs VC, Westenbroek RE, Scheuer T, Catterall WA. Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci 19 1999b. RC21(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cota G, Armstrong CM. Sodium channel gating in clonal pituitary cells. The inactivation step is not voltage dependent. J Gen Physiol. 1989;94:213–232. doi: 10.1085/jgp.94.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Angelo E, Nieus T, Maffei A, Armano S, Rossi P, Taglietti V, Fontana A, Naldi G. Theta-frequency bursting and resonance in cerebellar granule cells: experimental evidence and modeling of a slow K+-dependent mechanism. J Neurosci. 2001;21:759–770. doi: 10.1523/JNEUROSCI.21-03-00759.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Miera EVS, Rudy B, Sugimori M, Llinás R. Molecular characterization of the sodium channel subunits expressed in mammalian cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1997;94:7059–7064. doi: 10.1073/pnas.94.13.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Detre JA, Nairn AC, Aswad DW, Greengard P. Localization in mammalian brain of G-substrate, a specific substrate for guanosine 3′,5′-cyclic monophosphate-dependent protein kinase. J Neurosci. 1984;4:2843–2849. doi: 10.1523/JNEUROSCI.04-11-02843.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dietrich PS, McGivern JG, Delgado SG, Koch BD, Eglen RM, Hunter JC, Sangameswaran L. Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J Neurochem. 1998;70:2262–2272. doi: 10.1046/j.1471-4159.1998.70062262.x. [DOI] [PubMed] [Google Scholar]
- 15.Dieudonné S. Serotonergic neuromodulation in the cerebellar cortex: cellular, synaptic, and molecular basis. The Neuroscientist. 2001;7:207–219. doi: 10.1177/107385840100700306. [DOI] [PubMed] [Google Scholar]
- 16.Endo S, Suzuki M, Sumi M, Nairn AC, Morita R, Yamakawa K, Greengard P, Ito M. Molecular identification of human G-substrate, a possible downstream component of the cGMP-dependent protein kinase cascade in cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1999;96:2467–2472. doi: 10.1073/pnas.96.5.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG. Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res. 1997;145:71–82. doi: 10.1016/s0169-328x(96)00241-0. [DOI] [PubMed] [Google Scholar]
- 18.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 19.Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic β subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289:123–127. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- 20.Hall KU, Collins SP, Gamm DM, Massa E, DePaoli-Roach AA, Uhler MD. Phosphorylation-dependent inhibition of protein phosphatase-1 by G-substrate. A Purkinje cell substrate of the cyclic GMP-dependent protein kinase. J Biol Chem. 1999;274:3485–3495. doi: 10.1074/jbc.274.6.3485. [DOI] [PubMed] [Google Scholar]
- 21.Hansel C, Linden DJ, D'Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci. 2001;4:467–475. doi: 10.1038/87419. [DOI] [PubMed] [Google Scholar]
- 22.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 23.Isom LL. Sodium channel β subunits: anything but auxiliary. The Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- 24.Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 25.Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- 26.Jaarsma D, Ruigrok TJ, Caffe R, Cozzari C, Levey AI, Mugnaini E, Voogd J. Cholinergic innervation and receptors in the cerebellum. Prog Brain Res. 1997;114:67–96. doi: 10.1016/s0079-6123(08)63359-2. [DOI] [PubMed] [Google Scholar]
- 27.Konratyuk T, Rossie S. Depolarization of rat brain synaptosomes increases phosphorylation of voltage-sensitive sodium channels. J Biol Chem. 1997;272:16978–16983. doi: 10.1074/jbc.272.27.16978. [DOI] [PubMed] [Google Scholar]
- 28.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endo-genous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 29.Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- 30.Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000;275:11383–11388. doi: 10.1074/jbc.275.15.11383. [DOI] [PubMed] [Google Scholar]
- 31.Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Hortsch M, Isom LL. Modulation of sodium channel β1 subunit-mediated ankyrin recruitment by an intracellular tyrosine. Soc Neurosci Abstr. 2001;27:46.26. [Google Scholar]
- 32.Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moldrich G, Wenger T. Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study. Peptides. 2000;21:1735–1742. doi: 10.1016/s0196-9781(00)00324-7. [DOI] [PubMed] [Google Scholar]
- 34.Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. Beta 3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci USA. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mossadeghi B, Slater NT. Persistent and resurgent sodium currents in cerebellar unipolar brush cells. Soc Neurosci Abstr. 1998;24:1078. [Google Scholar]
- 36.Murakoshi H, Shi G, Scannevin RH, Trimmer JS. Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol Pharmacol. 1997;52:821–828. doi: 10.1124/mol.52.5.821. [DOI] [PubMed] [Google Scholar]
- 37.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 38.Pan F, Beam KG. The absence of resurgent sodium current in mouse spinal neurons. Brain Res. 1999;849:162–168. doi: 10.1016/s0006-8993(99)02060-0. [DOI] [PubMed] [Google Scholar]
- 39.Plummer NW, McBurney MW, Meisler MH. Alternative splicing of the sodium channel SCN8A predicts a truncated two-domain protein in fetal brain and non-neuronal cells. J Biol Chem. 1997;272:24008–24015. doi: 10.1074/jbc.272.38.24008. [DOI] [PubMed] [Google Scholar]
- 40.Plummer NW, Galt J, Jones JM, Burgess DL, Sprunger LK, Kohrman DC, Meisler MH. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics. 1998;54:287–296. doi: 10.1006/geno.1998.5550. [DOI] [PubMed] [Google Scholar]
- 41.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium current and disrupted firing patterns in Purkinje Neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 45.Ratcliffe CF, Qu Y, McCormick KA, Tibbs VC, Dixon JE, Scheuer T, Catterall WA. A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase beta. Nat Neurosci. 2000;3:437–444. doi: 10.1038/74805. [DOI] [PubMed] [Google Scholar]
- 46.Regan LJ. Voltage-dependent calcium currents in Purkinje cells from rat cerebellar vermis. J Neurosci. 1991;11:2259–2269. doi: 10.1523/JNEUROSCI.11-07-02259.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 48.Rossie S, Gordon D, Catterall WA. Identification of an intracellular domain of the sodium channel having multiple cAMP-dependent phosphorylation sites. J Biol Chem. 1987;262:17530–17535. [PubMed] [Google Scholar]
- 49.Schaller KL, Krzemien DM, Yarowsky PJ, Krueger BK, Caldwell JH. A novel, abundant sodium channel expressed in neurons and glia. J Neurosci. 1995;15:3231–3242. doi: 10.1523/JNEUROSCI.15-05-03231.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah BS, Stevens EB, Pinnock RD, Dixon AK, Lee K. Developmental expression of the novel voltage-gated sodium channel auxiliary subunit beta 3, in rat CNS. J Physiol (Lond) 2001;534:763–776. doi: 10.1111/j.1469-7793.2001.t01-1-00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18:6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith R, Goldin A. Phosphorylation of brain sodium channels in the I–II linker modulates channel function in Xenopus oocytes. J Neurosci. 1996;16:1965–1974. doi: 10.1523/JNEUROSCI.16-06-01965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith R, Goldin A. Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci. 1998;18:811–820. doi: 10.1523/JNEUROSCI.18-03-00811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith R, Goldin A. Potentiation of rat brain sodium channel currents by PKA in Xenopus oocytes involves the I-II linker. Am J Physiol Cell Physiol. 2000;278:C638–C645. doi: 10.1152/ajpcell.2000.278.4.C638. [DOI] [PubMed] [Google Scholar]
- 55.Tibbs VC, Gray PC, Catterall WA, Murphy BJ. AKAP15 anchors cAMP-dependent protein kinase to brain sodium channels. J Biol Chem. 1998;273:25783–25788. doi: 10.1074/jbc.273.40.25783. [DOI] [PubMed] [Google Scholar]
- 56.Wallner M, Meera P, Toro L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane beta-subunit homolog. Proc Natl Acad Sci USA. 1999;96:4137–4142. doi: 10.1073/pnas.96.7.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydro-phobic residues required for fast sodium channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- 59.Yeh JZ, Narahashi T. Kinetic analysis of pancuronium interaction with sodium channels in squid axon membranes. J Gen Physiol. 1977;69:293–323. doi: 10.1085/jgp.69.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]