

Abstract
Alzheimer's Disease (AD) is a neurodegenerative disorder that is characterized by extracellular deposits of amyloid-β peptide (Aβ) and a severe depletion of the cholinergic system, although the relationship between these two events is poorly understood. In the neocortex, there is a loss of cholinergic fibers and receptors and a decrease of both choline acetyltransferase (ChAT) and acetylcholinesterase enzyme activities. The nucleus basalis of Meynert (NBM), which provides the major cholinergic input to the neocortex, undergoes profound neuron loss in AD. In the present study, we have examined the cholinergic alterations in amyloid precursor protein transgenic mice (APP23), a mouse model of cerebral β-amyloidosis. In aged APP23 mice, our results reveal modest decreases in cortical cholinergic enzyme activity compared with age-matched wild-type mice. Total cholinergic fiber length was more severely affected, with 29 and 35% decreases in the neocortex of aged APP23 mice compared with age-matched wild-type mice and young transgenic mice, respectively. However, there was no loss of cholinergic basal forebrain neurons in these aged APP23 mice, suggesting that the cortical cholinergic deficit in APP23 mice is locally induced by the deposition of amyloid and is not caused by a loss of cholinergic basal forebrain neurons. To study the impact of cholinergic basal forebrain degeneration on cortical amyloid deposition, we performed unilateral NBM lesions in adult APP23 mice. Three to 8 months after lesioning, a 38% reduction in ChAT activity and significant cholinergic fiber loss were observed in the ipsilateral frontal cortex. There was a 19% decrease in Aβ levels of the ipsilateral compared with contralateral frontal cortex with no change in the ratio of Aβ40 to Aβ42. We conclude that the severe cholinergic deficit in AD is caused by both the loss of cholinergic basal forebrain neurons and locally by cerebral amyloidosis in the neocortex. Moreover, our results suggest that disruption of the basal cholinergic forebrain system does not promote cerebral amyloidosis in APP23 transgenic mice.
Keywords: Alzheimer's disease, APP, amyloid, basal forebrain, cholinergic system, ChAT, AChE, neurodegeneration, nucleus basalis of Meynert, neocortex, lesion, mouse, stereology, hippocampus, aging
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that affects a large proportion of elderly people. Although genetic factors seem to strongly contribute to disease susceptibility, only a small number of cases are caused by dominant mutations (Selkoe, 1999). To date, all such mutations alter processing of the amyloid precursor protein (APP), leading to changes in the production or fibrillization of amyloid-β (Aβ), the major constituent of amyloid plaques found in AD brain (Hardy, 1997; Selkoe, 1997; Haass and Steiner, 2001).
Besides the extracellular deposition of Aβ, the AD brain is characterized by intracellular neurofibrillary tangles and profound changes in the cholinergic system (Bartus et al., 1982; Coyle et al., 1983; Goedert, 1993). In both neocortex and hippocampus of AD brain, a loss of cholinergic fibers and terminals, decreases in cholinergic receptors and/or signal transduction, and significant reductions in choline acetyltransferase (ChAT) and acetylcholinesterase (AChE) enzyme activities have been reported (Coyle et al., 1983; Perry et al., 1992; Ransmayr et al., 1992; Bierer et al., 1995; Jope et al., 1997; Geula et al., 1998; Ladner and Lee, 1998).
The major cholinergic innervation to the cerebral cortex originates from the nucleus basalis of Meynert (NBM), together with the horizontal limb of the diagonal band of Broca, the ventral pallidum, the magnocellular preoptic area, the substantia innominata, and the nucleus of the ansa lenticularis (hereafter referred to as the NBM complex). Cholinergic innervation to the hippocampus is mainly provided by the medial septum (MS) and the vertical limb of the diagonal band of Broca (VDB) (McKinney et al., 1983; Gaykema et al., 1990; Kitt et al., 1994). In AD brain, a profound loss of these cholinergic basal forebrain neurons has been reported (Whitehouse et al., 1982; Vogels et al., 1990; Jope et al., 1997; Cullen and Halliday, 1998). This neuron loss may be caused secondarily as a result of Aβ neurotoxicity to the cholinergic terminals followed by retrograde degeneration. Alternatively, degeneration of cholinergic basal forebrain neurons may be the primary lesion with subsequent loss of cortical cholinergic innervation.
The relationship between cerebral amyloidosis and cholinergic depletion in AD remains poorly understood (Roberson and Harrell, 1997; Auld et al., 1998; Geula et al., 1998). It has been demonstrated that stimulation of muscarinic acetylcholine receptor subtypes increases non-amyloidogenic APP processing (Nitsch et al., 1992). It has also been reported that AChE accelerates the assembly of Aβ into insoluble amyloid fibrils (Inestrosa et al., 1996). Accordingly, dysfunction of the cholinergic system may influence cerebral amyloidosis. Vice versa, it has been demonstrated that Aβ is neurotoxic to cholinergic neurons and that low concentrations of Aβ can directly inhibit cholinergic signaling (Auld et al., 1998; Pettit et al., 2001). Thus, increased Aβ levels may contribute physiologically and/or pathologically to the cholinergic changes in AD brain.
Several APP transgenic mouse models have been generated that exhibit age-related Aβ deposition in plaques and vessels predominantly in the neocortex and hippocampus (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997). The amyloid deposits display major characteristics of human AD plaques and human cerebral amyloid angiopathy including congophilic Aβ cores, amyloid-associated cell death, dystrophic neurites, activated microglia, and reactive astrocytes (Masliah et al., 1996; Frautschy et al., 1998; Calhoun et al., 1999; Phinney et al., 1999; Stalder et al., 1999; Van Dorpe et al., 2000; Winkler et al., 2001; Bondolfi et al., 2002). These mouse models offer the opportunity to study cholinergic alterations that result from, or lead to, cerebral amyloidosis. To this end we have used biochemical and morphological techniques to assess cholinergic changes in neocortex and basal forebrain of APP23 transgenic mice. Moreover, to test the hypothesis that cortical cholinergic depletion has an effect on amyloid plaque formation we have lesioned the NBM in APP23 mice.
MATERIALS AND METHODS
Animals. The generation of the B6,D2-TgN(Thy1-APPSwe) transgenic mouse line (APP23) is described elsewhere (Sturchler-Pierrat et al., 1997). In brief, APP751 cDNA with the Swedish mutation (K670N-M671L) was inserted into an expression cassette comprising a murine Thy-1.2 gene construct, and mice were generated by pronuclear injection. The founder mice were then back-crossed with C57BL/6 mice. APP23 mice of generations F5-F10 and corresponding wild-type mice were used. The wild-type control mice were either littermates or nontransgenic age-matched mice from another litter of the same generation of backcrossing.
ChAT and AChE enzyme activities. Mice were killed by decapitation, and the frontal cortex was dissected on ice. The tissue was weighed, snap-frozen on dry ice, and stored at −80°C until analysis. Tissue was homogenized 1:50 (w/v) in 10 mm EDTA containing 0.5% Triton X-100 at pH 7.4. ChAT enzyme activity was measured by a slight modification (Kelly and Moore, 1978) of the method of Fonnum (1975). In brief, tissue homogenates were incubated in a water bath at 37°C in an incubation mixture containing (in mm): 300 NaCl, 8 choline iodide, 20 EDTA, 0.1 eserine hemisulfate, and 0.2 acetyl-[1-14C]-coenzyme A in 50 mm phosphate buffer, pH 7.4 (final concentrations). After stopping the production of radioactively labeled acetylcholine with ice-cold 10 mm sodium dihydrogen phosphate, pH 7.4, the solutions were transferred into scintillation vials, and sodium tetraphenylboron (0.5% in acetonitrile; Merck, Darmstadt, Germany) and the scintillant (0.05% PPO in toluene) were added. Labeled acetylcholine was determined by liquid scintillation counting in the biphasic aqueous, toluene scintillation solution mixture. ChAT enzyme activity was expressed in micromoles per 100 mg of protein per hour. Protein concentration was measured using the Bio-Rad (Munich, Germany) protein assay.
For AChE activity, samples of the same homogenates were sonicated, and aliquots were assayed for AChE activity by colorimetric determination by the method of Ellman et al. (1961) and Cutler et al. (1998). In short, after centrifugation for 15 min (15,000 rpm, 4°C), aliquots from the clear supernatant were used as enzyme source. Twenty microliters of supernatant (quadruplicates) were placed in a 96-well flat-bottom micro-test plate. The reaction was started by adding the following substrate–reagent mixture, using an automatic plate dispenser (Titertek Autodrop): 0.5 mm acetylthiocholine iodide (Fluka, Buchs, Switzerland), 1 mmtetraisopropylpyrophosphoramide (iso-OMPA; Sigma, Buchs, Switzerland), and 0.25 mm 5,5′-dithiobis-2-nitrobenzoate (Fluka) in 0.1m phosphate buffer, pH 7.4. The plate was then placed in the automatic Micro-Reader (Molecular Devices, Palo Alto, CA; UVmax) that recorded the occurrence of the yellow reaction product at 405 nm. The data were processed by a program (Softmax; Molecular Devices) controlled by the plate reader. The calculation of the enzymatic activity (performed by the computer program) is based on a change in optical density in the linear range over time using the molar extinction coefficient of the reaction product (13.3 cm2/mmol). AChE enzyme activity was expressed in nanomoles per milligram of protein per minute.
Immunohistochemistry. Perfusion- or immersion-fixed brains were paraffin embedded according to a previously published procedure (Calhoun et al., 1998a) that involved post-fixation in increasing alcohol concentrations and clearing with Cedarwood oil and methyl salicylate (Aldrich, Buchs, Switzerland). Serial coronal sections (25 μm) were cut with a microtome, and immunohistochemistry was performed using the avidin–biotin–peroxidase method (Calhoun et al., 1998a). In brief, paraffin sections were deparaffinized in xylene, then placed in 100% ethanol for 10 min followed by 30 min in methanol with 0.3% H2O2. Sections were rinsed in PBS and incubated for 1 hr in 5% goat or horse serum (Vector Laboratories, Burlingame, CA) in a humid chamber. Sections were then incubated overnight with a primary antibody in PBS with 3% serum. After rinsing, sections were incubated for 1 hr with biotinylated secondary antibody (Vector Laboratories) diluted 1:200 in PBS. Sections were rinsed again and incubated with the avidin–biotin–peroxidase complex (1:50; ABC Elite kit; Vector Laboratories) in PBS. Finally, sections were reacted with 3′,3-diaminobenzidine-dihydrochloride (DAB; Sigma; 0.08%) and 0.03% hydrogen peroxide in PBS, rinsed, dehydrated, cleared in xylene, and coverslipped. On NBM-lesioned tissue (see below) immunohistochemistry was done on free-floating, fixed-frozen sections according to a previously published protocol (Phinney et al., 1999), similar to the procedure described above. The following antibodies were used: polyclonal anti-Aβ (NT12; 1:500 and 1:2000 for paraffin and frozen sections, respectively) (Schrader-Fischer and Paganetti, 1996) and polyclonal anti-ChAT (AB144P; 1:500 for paraffin sections;Chemicon, Temecula, CA).
AChE histochemistry. Mice were deeply anesthetized with an intraperitoneal injection of an overdose of pentobarbital (50 mg/ml Nembutal) and transcardially perfused with 4% paraformaldehyde in PBS. Brains were removed, post-fixed for 24 hr in 4% paraformaldehyde, cryoprotected in 30% sucrose in PBS, frozen in isopentane at −25°C, and serially sectioned at 40 μm on a freezing–sliding microtome. AChE immunohistochemistry was performed according to a previously published method (Hedreen et al., 1985). Some sections were also immunostained with a polyclonal antibody to AChE (Marsh et al., 1984) (gift of J. Massoulie, Paris, France). No qualitative difference was noted between the histochemical and immunohistochemical reaction confirming previous findings in the rat (Jucker et al., 1996). Histochemical AChE staining was however more reliable and more distinct and was taken for quantitative analysis of cholinergic fiber length (see below).
To study the localization of AChE in more detail, some of the tissue was fixed with paraformaldehyde plus 1% glutaraldehyde, cut on a vibratome (60 μm), stained for AChE, and plastic-embedded according to a previously published protocol (Stalder et al., 1999). Semithin (0.5 μm) and ultrathin (80 nm) sections were cut on a ultramicrotome. Sections were collected on Formvar-coated nickel grids and viewed under high-power light microscopy or electron microscopy.
Stereological assessment of cholinergic fiber length.Stereological techniques were used to estimate the total length of AChE-positive fibers in the neocortex (Table1). To this end, every 16th section throughout the neocortex was histochemically stained for AChE according to the above described protocol. The volume of the neocortex was estimated by superimposing a point-grid on each section and counting the points over neocortex, according to the Cavalieri principle (West et al., 1991; Calhoun et al., 1998b). AChE-positive fiber density was then estimated by superimposing a system of test lines, and intersections of test lines with fibers were counted within the volume of disectors systematically spaced throughout the neocortex using the 100× objective (2756× final magnification) with a numerical aperture of 1.3 (Stocks et al., 1996). This number of intersections between fibers and test lines of a known length produces a result directly related to the length of the fibers themselves within the disector volume (Howard et al., 1992). Because length measurement was performed on coronal sections in all cases, it is assumed that no overall directional orientation with respect to the plane of section exists for cholinergic fibers as they innervate the cortex. AChE-positive fiber length was calculated by multiplying the region volume by the fiber length density (West, 1993). Stereological analysis was performed with the aid of Stereologer software and a motorized x-y-z stage coupled to a video microscopy system (Systems Planning and Analysis, Alexandria, VA). Post-processing section thickness was measured using a focus drive accurate to ±0.1 μm (Applied Scientific Instrumentation, Eugene, OR). Coefficient of error was calculated according to West et al. (1991) and was well below biological variability. Region definitions of neocortex were based on a recent mouse brain atlas (Franklin and Paxinos, 1997).
Table 1.
Stereological parameters for cholinergic fiber, amyloid load, and cholinergic basal forebrain neuron quantification
| Sections | Disector or fractionator | Objects counted1-b | Results | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Type | Thickness1-a | Sample | Area | Height | Spacing | Value1-c | CE | ||
| AChE fiber length in neocortex | 40 μm frozen | 15.6 ± 0.3 μm | 1 /16 | 1900 μm2 | 8 μm | 800 μm | 525 ± 25 | 591.93 ± 19.2 m | 0.047 ± 0.008 |
| Amyloid load neocortex | 25 μm paraffin | 27.0 ± 0.3 μm | 1 /20 | 19000 μm2 | 0 μm | 800 μm | 514 ± 86 | 18.4 ± 3.8% | 0.038 ± 0.001 |
| Cholinergic neurons in NBM complex | 25 μm paraffin | 23.2 ± 1.1 μm | 1 /8 | 8300 μm2 | 18 μm | 160 μm | 120 ± 8 | 4015 ± 209; 1700 ± 72 μm3 | 0.096 ± 0.003 |
| Cholinergic neurons in MS/VDB | 25 μm paraffin | 23.6 ± 0.9 μm | 1 /4 | 8300 μm2 | 14 μm | 91 μm | 154 ± 12 | 1196 ± 80; 1100 ± 120 μm3 | 0.086 ± 0.003 |
Values are mean ± SEM.
Section thickness measured to ± 0.1 μm after all processing/coverslipping was complete.
Objects counted refers to the average number of intersections of fibers with test lines, number of points hitting amyloid, or number of cholinergic cells counted per subject.
Value represents total length of AChE-positive fibers, amyloid load, or total number and volume of ChAT-positive neurons per hemisphere.
Stereological assessment of amyloid load. Amyloid load was assessed on every 20th paraffin section throughout the neocortex immunostained with NT12. The percentage of neocortical volume occupied by amyloid (amyloid load) was determined by sampling through the entire neocortex with a 20× objective (numerical aperture, 0.45) and counting the percentage of points from a superimposed point grid that hit amyloid (Table 1) (Calhoun et al., 1998b). Stereological analysis was performed with the Stereologer software described above.
Number and volume of ChAT-positive basal forebrain neurons.For estimation of the number (West et al., 1991) and volume (Jensen and Gundersen, 1993) of ChAT-positive cells in the basal forebrain, systematic random series of ChAT-stained paraffin sections were analyzed using a fractionator sampling scheme. For quantitative analysis ChAT-positive neurons in the NBM complex that provide the major cholinergic innervation to the neocortex were combined. Similarly cholinergic neurons in the MS and VDB, that provide the cholinergic innervation to the hippocampus were combined for analysis. Every eighth section throughout the NBM complex and every fourth section throughout the MS–VDB were collected. On each section a point grid was placed randomly over the region to determine the systematic-random placement of the optical disectors. The total number of ChAT-positive neurons within the three-dimensional optical disectors throughout the region were then counted. Objects were counted by focusing through the counting frame using a 63× objective with a numerical aperture of 1.25. The nuclei of ChAT-positive cells were the selected objects for counting (Table 1). Additionally, the volume of each counted cell was estimated. For practical reasons, the tissue could not be randomly rotated during sectioning, and it was thus assumed that there is no change in preferred orientation of the cells in the basal forebrain (Gundersen et al., 1988). A vertical line with three perpendicular grid lines were superimposed, and intersections of grid lines with the cell soma were identified. The mean length of these lines is proportional to neuron volume (Jensen and Gundersen, 1993), although in this case the value is orientation-dependent. Stereological analysis was performed with the Stereologer software described above.
NBM Lesions. Animals were deeply anesthetized using a combination of ketamine (10 mg/kg body weight; Ketalar; Parke-Davis, Ann Arbor, MI) and xylazine (20 mg/kg body weight; Rompun, Bayer, Germany) in saline administered intraperitoneally. Using stereotaxic surgery, the scalp was cut, a hole was drilled, and an electrode was lowered into the NBM [anteroposterior (bregma), −0.2 mm; lateral, 1.5 mm; dorsoventral (dura), −4.5 mm]. A current flow of 0.5 mA was passed for 15 sec. Half of the mice received an additional sham lesion on the contralateral side, in which the electrode was lowered, but no current was passed through. The lesion side was alternated evenly over the groups.
Mice were killed 3–8 months later by decapitation. Brains were removed, and a small piece of the ipsilateral and contralateral frontal neocortex was dissected and combined with a small piece from the ipsilateral and contralateral motor–somatosensory cortex as previously described (Kelly and Moore, 1978). The dissected tissue was weighed, snap-frozen on dry ice, and assayed for ChAT enzyme activity (described above) and APP/Aβ (see below). The rest of the brain was immersion-fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, and frozen. Serial coronal 40 μm sections were cut throughout the frontal cortex, and alternate sections were stained for Aβ and AChE or double-stained for both. Amyloid load was then determined stereologically on the Aβ-stained sections (see above), discarding the sections missing the dissected pieces. This yielded a total of 5–15 Aβ-stained sections through the frontal cortex per mouse. Percentage of amyloid load was determined for both hemispheres separately.
Aβ load and APP processing using Western blotting. It has previously been demonstrated that amyloid cores in APP23 mice are completely soluble in SDS (Kuo et al., 2001). Thus, the tissue samples used to determine ChAT activities (see above) were diluted 1:3 (weight/volume) in 1.5× SDS-sample buffer (containing 1% SDS) and separated using a 10% SDS-polyacrylamide gel containing 8m urea (Klafki et al., 1996). For amyloid quantification 2 μl was loaded from the unlesioned and lesioned side. After separation, proteins were transferred to Immoblin-P (Millipore, Bedford, MA) with a Bio-Rad semidry transfer apparatus for 1 hr at 25 V, according to Wiltfang et al. (1997). Membranes were blocked in 5% nonfat milk in PBS with 0.1% Tween 20 (PBS-T) and reacted overnight at 4°C with Aβ antibody 6E10 (Signet, Dedham, MA) at 1:2000 in PBS-T followed by a goat anti-mouse peroxidase conjugate (Chemicon, Temecula, CA) at 1:2500 in PBS-T for 30 min at room temperature. Bands were visualized using Supersignal (Pierce, Rockford, IL) and developed onto Kodak X-OMAT AR film (Rochester, NY). Different exposures of the film were digitized, and band density measurements for Aβ40 and Aβ42 were made using NIH Image version 1.61 (National Institutes of Health, Bethesda, MD). Each sample pair was run at least three times, and the mean was taken. Only bands within the linear range of the film were analyzed.
To analyze APP processing in more detail, the β-secretase cut, the N terminal secreted fragment (sAPPβ), and the corresponding C terminal fragment C99 were additionally analyzed. For the analysis of sAPPβ, proteins were separated on a 8% SDS-polyacrylamide gel and transferred to Immobilon-P membranes as described previously (Andra et al., 1996). The blot was reacted with rabbit neoepitope antiserum 852 against the C terminus of sAPPβ (P. Paganetti and M. Staufenbiel, unpublished observations). As an internal control, an antibody to tubulin was used.
Statistics. All statistical analysis was done using StatView 5.0.1. The mean ± SEM is indicated. Significance level was set atp < 0.05.
RESULTS
Disruption and loss of cholinergic fibers in neocortex of aged APP23 mice
To study the impact of cerebral amyloidosis on the cholinergic system, we have analyzed AChE-positive fiber length and ChAT-positive boutons in neocortex of APP23 mice. Three age groups with similar numbers of male and female APP23 mice were examined: young (6 months;n = 6), adult (15 months; n = 5), and aged (24 months; n = 7). Corresponding controls were young (6 months; n = 6), adult (15 months;n = 8), and aged (24 months; n = 5) wild-type mice. Quantitative analysis of the amyloid load revealed no amyloid deposition in the young APP23 mice, 4.0 ± 1.8% in adult APP23 mice, and 28.6 ± 1.4% in aged APP23 mice (Fig.1A,B). Consistent with previous reports, the majority of the amyloid deposition in APP23 mice was compact in nature and congophilic (Calhoun et al., 1998b; Stalder et al., 1999). Amyloid deposits were also found in vessels and as extracellular diffuse amyloid (Fig. 1B). Neocortical volume did not differ between the groups at any age (mean volume for all mice = 17.4 mm3; one hemisphere).
Fig. 1.

Amyloid deposits and cholinergic disruption in neocortex of aged APP23 mice. A, No amyloid is detected in 24-month-old wild-type mice. B, Immunostaining for Aβ reveals numerous compact amyloid plaques, diffuse amyloid (arrowheads), and amyloid deposits in vessels (arrow) in neocortex of a 24-month-old APP23 mouse.C, D, Histochemical staining for AChE reveals a disruption and a decrease in cholinergic fiber density in neocortex of 24-month-old APP23 mice (D) compared with 24 month-old control mice (C). The disruption is most evident around plaques but occurs throughout the neocortex. Scale bars, 120 μm.
Histochemical staining for AChE revealed a dense laminated network of cholinergic fibers throughout the neocortex of wild-type mice and young APP23 mice. In adult and aged APP23 mice, however, there was a loss and often dramatic disruption of the cholinergic fiber network (Fig.1C,D). In particular in the aged APP23 mice, a loss of AChE-positive fibers throughout the entire neocortex was evident with intense staining of dystrophic structures at the plaque periphery and diffuse staining of the amyloid cores (Fig.2A). High-power microscopic analysis identified these dystrophic structures as fibers that often formed loops ending in dystrophic boutons (Fig.2A). Semithin and ultrathin sectioning through the diffusely stained AChE-positive amyloid cores demonstrated that AChE immunoreactivity was associated with amyloid fibrils (Fig.2B).
Fig. 2.
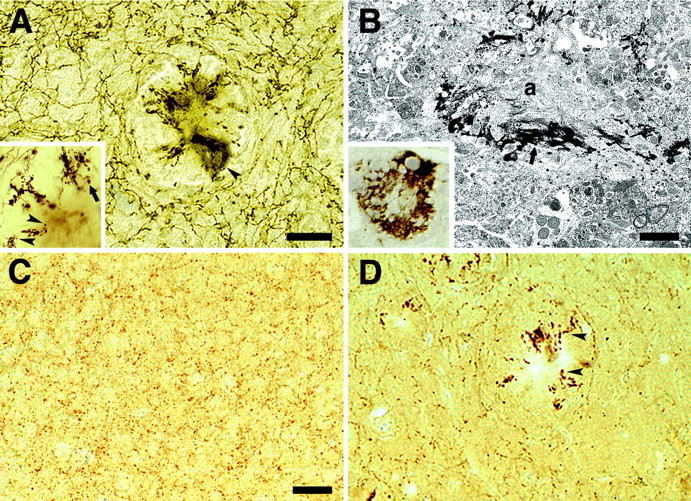
Cholinergic disruption and dystrophy in APP23 neocortex. A, AChE-positive fibers around an amyloid plaque with diffuse staining of the amyloid cores in the neocortex of an aged APP23 mouse (arrowhead).Inset, High-power analysis of the abnormal and often swollen AChE-positive fibers with terminal large boutons in vicinity of the amyloid plaque (arrowheads). Fibers frequently grow toward the amyloid but then form loops or sharply turn around to grow away from the amyloid to turn later again toward the amyloid (arrow). B, High-power analysis of diffuse AChE-staining associated with the amyloid cores.Inset, AChE staining in semithin sections suggests an association of AChE with the amyloid core. Subsequent ultrastructural analysis confirms that AChE reactivity (arrow) is associated with amyloid fibers (a).C, Immunostaining for ChAT reveals dense punctate staining of cholinergic boutons in the frontal cortex of an aged wild-type mouse. D, In contrast, a loss of cholinergic boutons is apparent in aged APP23 mice. Dystrophic ChAT-positive boutons and neuritic structures are present around amyloid plaques (arrowheads). Scale bars: A, 50 μm;B, 1.6 μm; C, D, 50 μm.
ChAT immunohistochemistry revealed punctate staining of cholinergic boutons throughout the neocortex (Fig. 2C,D). Punctate staining was often very dense revealing the shape of individual cholinergic fibers with their immunostained boutons. No qualitative difference in staining was noted between wild-type and young APP23 mice. In contrast, in adult and aged APP23 mice a reduction of ChAT-positive boutons was observed in neocortical areas with a high amyloid burden, such as entorhinal and frontal cortex (Fig.2D). Large dystrophic ChAT-positive structures were present at the periphery of the amyloid plaques and resembled the AChE-positive dystrophic fibers described above.
For quantification of cholinergic fiber length, AChE histochemistry was chosen because of robust staining and significant penetration of the stain into the tissue sections, both prerequisites for stereological analysis. Results for total length of AChE-positive fibers in neocortex revealed the astonishing length of 600 m per hemisphere and was similar for all three groups of wild-type mice (Fig.3). In contrast to this lack of age effect in wild-type mice, there was a clear reduction in fiber length with aging in the APP23 mice that reached 35% compared with young APP23 mice and 25% compared with adult APP23 mice. In comparison with age-matched wild-type mice there was a 29% reduction. Consistently, ANOVA revealed a significant age effect (F(2,31) = 6.40; p < 0.01), a significant transgene effect (F(1,31) = 4.59; p < 0.05), and a significant age × transgene interaction (F(2,31) = 4.30; p < 0.05). Newman–Keuls post hoc analysis indicated that the fiber loss in aged APP23 mice was statistically significant compared with aged wild-type mice (p < 0.01) and to young (p < 0.01) and adult (p < 0.05) APP23 mice (Fig. 3A). A significant negative correlation between AChE-positive fiber length and amyloid load was apparent when adult and aged APP23 mice were combined (R2 = 0.46; p< 0.05) (Fig. 3B). Males and females were combined for AChE-fiber length analysis because previous ANOVA analysis did not reveal any significance for, or interaction with, gender.
Fig. 3.

AChE-positive fiber length and amyloid load in neocortex of APP23 mice. A, Total length of AChE-positive fibers in neocortex of young (6 months), adult (15 months), and aged (24 months) APP23 mice (black bars) and age-matched wild-type mice (open bars). Aged transgenic mice had a significant loss of fiber length compared with aged wild-type mice (**p < 0.01) and with young transgenic mice (**p < 0.01). Indicated is the mean ± SEM for one hemisphere only. B, A significant negative correlation between AChE-positive fiber length and amyloid load was apparent when adult (circles) and aged (squares) APP23 mice were combined.
ChAT and AChE activities in the frontal cortex of APP23 mice
ChAT and AChE enzyme activities were measured in the frontal cortex (Table 2). Three age groups consisting of similar numbers of both sexes of APP23 mice (6 months,n = 8; 19 months, n = 11; 24 months,n = 6) and age-matched wild-type control mice (6 months, n = 8; 19 months, n = 9; 24 months, n = 7) were used. Because of logistical issues, the young, adult, and aged mice were analyzed separately, thus preventing comparison between age groups. Because no difference between males and females was apparent, male and females were combined. Results revealed a modest but significant decrease of ChAT activity in the frontal cortex of aged APP23 mice as compared with age-matched controls (t(11) = 2.27; p < 0.05) (Table 2). Differences in AChE activities did not reach significance, although there was a trend toward decreased activities in transgenic mice in all three groups (Table 2).
Table 2.
ChAT and AChE enzyme activities in the frontal cortex
| Wild-type | APP23 | Decrease (%) | |
|---|---|---|---|
| Mean ChAT activity ± SEM (μmol/100 mg protein/hr) | |||
| Young (6 months) | 17.12 ± 0.6 | 16.57 ± 0.4 | 3.2 |
| Adult (19 months) | 13.79 ± 0.7 | 12.67 ± 0.5 | 8.1 |
| Aged (24 months) | 14.12 ± 0.3 | 12.5 ± 0.7 | 11.4* |
| Mean AChE activity ± SEM (nmol/mg protein/min) | |||
| Young (6 months) | 59.41 ± 2.5 | 56.20 ± 0.8 | 5.4 |
| Adult (19 months) | 62.48 ± 2.4 | 56.56 ± 2.5 | 9.5 |
| Aged (24 months) | 64.51 ± 1.5 | 59.75 ± 4.3 | 7.4 |
Aged transgenic mice showed a significant loss of ChAT activity as compared with age-matched wild-type controls (*p < 0.05). No significant decrease of AChE activity was detected. Because of logistical issues the young, the adult, and the aged mice were analyzed separately, thus preventing comparison among age groups.
Number and volume of ChAT-positive neurons in the basal forebrain
Two age groups of APP23 mice (young: 8 months, n = 8; aged: 27 months, n = 8) and corresponding wild-type mice (young: 8 months, n = 7; aged: 27 months,n = 6) were used with sex equally balanced within groups and treated as one group. ANOVAs for ChAT-positive neuron number were calculated separately for the NBM complex and the MS–VDB (Fig.4). Results did not reveal a significant effect of age or transgene on neuron number (p> 0.05) (Fig. 5A,B). There was also no change in volume of ChAT-positive cells in the NBM complex (Fig. 5C). Rather unexpectedly, a significant transgene effect (F(1,25) = 8.76;p < 0.05) with a nonsignificant age × transgene interaction was found for the volume of ChAT-positive cells in the MS–VDB, indicating that both young and aged APP23 mice have smaller cholinergic neurons in the MS–VDB (Fig. 5D).
Fig. 4.

ChAT-positive neurons in the basal forebrain of APP23 mice. A, B, No apparent difference in neuron number was observed in the NBM of 27-month-old APP23 mice (A) compared with aged-matched wild-type mice (B). C, D, Similarly, no apparent difference in ChAT-positive neuron number in the MS was noted between 27-month-old APP23 (C) mice and age-matched control mice (D). For quantification, see Figure5. Scale bar, 50 μm.
Fig. 5.

Number and volume of cholinergic neurons in the basal forebrain of APP23 mice. Number (A) and volume (C) of ChAT-positive neurons in the NBM complex in APP23 mice (black bars) and wild-type mice (open bars). No significant difference between transgenic and wild-type mice was noted. A similar analysis was done for the number (B) and volume (D) of ChAT-positive neurons in the MS–VDB in APP23 mice (black bars) and wild-type mice (open bars). For neuron volume, a significant reduction of 38 and 42% was found in the 8-month-old and 27-month-old APP23 mice, respectively (*p < 0.05). Data are mean ± SEM. Indicated values are for one hemisphere only.
Lesions of the nucleus basalis of Meynert reduce neocortical Aβ load
Groups of male and female mice were lesioned at 5, 7, 10, and 13 months of age and were killed at 9, 15, 13, and 20 months of age, respectively. Only mice with a complete lesion of the NBM complex and a >20% ChAT decrease in the ipsilateral compared with contralateral frontal cortex were included in the analysis (n = 11; mean ChAT decrease: 38 ± 4%). Histologically, in these mice there was a considerable loss of AChE-positive fibers in the ipsilateral frontal cortex (Fig. 6).
Fig. 6.

Amyloid load in the frontal cortex after NBM lesions. A, B, Double labeling for AChE (brown) and Aβ (blue) in a 13-month-old APP23 mouse 3 months after a unilateral NBM lesion reveals a considerable loss of cholinergic fibers in the ipsilateral (B) compared with contralateral (A) frontal cortex. Stereological assessment revealed a 22% reduction of amyloid deposition in the ipsilateral compared with contralateral side. Scale bar: A,B, 90 μm. C, Western blotting of cortex homogenates. Samples were run in pairs, i.e., contralateral side (c) versus ipsilateral side (i). Shown are three mice. Results for all the mice revealed a 19% decrease of Aβ40 between the two sides. No difference in the ratio between Aβ40 and Aβ42 was found between contralateral and ipsilateral side.
Stereological analysis of the amyloid load on Aβ-immuno-stained sections revealed a mean decrease in the amyloid load of 22 ± 8% in the ipsilateral versus contralateral frontal cortex (Fig.6A,B). The individual changes ranged from +14 to −63% with a decrease in 8 of 11 mice. This difference did not reach statistical significance (p = 0.11, Wilcoxon signed rank test). In contrast, a significant decrease was observed when Aβ40 was analyzed using densitometry of Western blots. The mean decrease was 19 ± 9% with a range from +30% to −61% (Fig.6C). Eight of 11 mice revealed a decrease (p = 0.04, Wilcoxon signed rank test). The ratio between Aβ42 and Aβ40 was not significantly different between the ipsilateral (0.29) and contralateral side (0.26).
To further study APP processing we have analyzed sAPPβ and C99, the two cleavage products of β-secretase. No significant difference was found for sAPPβ between ipsilateral and contralateral side (−7%; p = 0.15; paired t test), and this difference was identical to the one found for tubulin. Consistently, no apparent difference for C99 between ipsilateral versus contralateral was found (results not shown). These results indicate that cholinergic denervation does not lead to a globally detectable shift of APP metabolism to the amyloidogenic pathway in APP23 mice.
DISCUSSION
Cerebral amyloidosis is a hallmark lesion of AD, and genetic analysis has demonstrated that Aβ is central to AD pathogenesis (Selkoe, 1999). Similarly, depletion of the cholinergic system is a robust finding in AD and correlates with cognitive impairment (Collerton, 1986; DeKosky et al., 1992; Bierer et al., 1995). Yet, the link between cerebral amyloidosis and the cholinergic deficit remains poorly understood.
The present study was undertaken to investigate alterations in the cholinergic system in the APP23 mouse model of cerebral amyloidosis. The mice develop amyloid plaques and cerebrovascular amyloid throughout the neocortex and hippocampus with only modest amyloid deposition in the basal forebrain (Sturchler-Pierrat et al., 1997; Calhoun et al., 1998b). Individual amyloid deposits in APP23 mice are also morphologically similar to those in AD brain and include congophilic amyloid cores, amyloid-associated dystrophic neurites, astrocytosis, and microgliosis (Jucker et al., 2001). A difference between amyloid plaques in mouse compared with AD brain is the lack of paired helical filaments in plaque-associated dystrophic neurites (Sturchler-Pierrat et al., 1997). Moreover, there is a difference in solubility and chemical composition of the amyloid (Kuo et al., 2001).
Results of the present study reveal a robust 30% decrease in cholinergic fiber length with distorted and dystrophic cholinergic fibers surrounding the amyloid very similar to that in AD brain (Geula et al., 1998). Because fiber loss in the mice correlated with cortical Aβ load and because we did not find a loss or shrinkage of cholinergic neurons in the NBM, our results suggest that the cortical cholinergic deficit in APP23 mice is locally induced by Aβ deposition. These results are also consistent with earlier observations that retrograde degeneration in the NBM only occurs after more severe cortical tissue damage (Sofroniew et al., 1983; Liberini et al., 1994). In other transgenic mouse models, a lack of neuron loss in the basal forebrain has also been reported. However, in these mouse models, no significant cortical cholinergic deficits have been observed possibly because of the age of the mice and/or the lower neocortical amyloid burden (Wong et al., 1999; Bronfman et al., 2000; Hernandez et al., 2001; Jaffar et al., 2001), although in one plaque-burdened mouse line a decrease in vesicular acetylcholine transporter-positive bouton area and density in frontal cortex was reported (Wong et al., 1999).
Rather unexpectedly, our results reveal that cholinergic neurons in the MS/VDB of APP23 mice are significantly smaller compared with wild-type mice. Amyloid deposition is unlikely to account for this observation, because a decrease in neuron volume was also found in the 8-month-old APP23 mice that exhibited negligible amyloid deposition in hippocampus and no deposition in the MS–VDB. It has been reported that in SN56 cells (N18TG2 neuroblastoma cells fused with mouse primary septal neurons), low concentrations of Aβ can induce long-lasting downregulation of the cholinergic activities without evidence of neurotoxicity (Pedersen et al., 1996; Auld et al., 1998). Thus, it is possible that Aβ levels in the 8-month-old APP23 mice are high enough to induce cholinergic hypoactivity and shrinkage of MS–VDB neurons, although it is unclear why this region would be preferentially affected. Interestingly, in another APP transgenic mouse model, a selective decrease in size of MS cholinergic neurons, but not NBM cholinergic neurons, has also been reported (Bronfman et al., 2000). In this model however, shrinkage was observed in aged transgenic mice but not in young transgenic mice without amyloid deposition. This difference may be explained by the much lower levels of APP expression/Aβ in this latter model. In AD a shrinkage of basal forebrain cholinergic neurons has also been reported (Vogels et al., 1990).
Cholinergic fiber loss in AD neocortex displays considerable regional variability with reductions exceeding 50% in some cortical areas (Mesulam, 1996; Geula et al., 1998). In addition, there is a poor correlation between amyloid plaques and fiber loss, suggesting that amyloid deposition in neocortex cannot be the exclusive cause of the cholinergic loss. Thus, it is likely that in AD brain the significant loss of cholinergic basal forebrain neurons contributes to the cortical cholinergic deficit and accounts for the more pronounced fiber loss as compared with APP23 mice. NBM cholinergic neurons are among the first neuronal groups with neurofibrillary tangle formation and among the first neurons that are lost in AD (Mesulam, 1996; Cullen and Halliday, 1998; Sassin et al., 2000).
In contrast to the significant fiber loss in APP23 mice, our results revealed only a modest loss of ChAT and AChE enzyme activities. Correlative analysis of ChAT and AChE enzyme activities and amyloid load in AD brain have revealed conflicting findings. Some studies have found negative correlations (Perry et al., 1981; Mountjoy et al., 1984;Zubenko et al., 1989; Beach et al., 2000a), whereas others have found no relation (Wilcock et al., 1982; DeKosky et al., 1992; Geula et al., 1998). In AD and in APP23 mice, a significant amount of ChAT and AChE staining is associated with dystrophic neurites surrounding amyloid plaques (Benzing et al., 1993; Moran et al., 1993). Moreover, in both AD and APP23 mice, AChE is a component of amyloid-containing plaques, and it has been suggested that AChE accelerates amyloid fibril formation (Gomez-Ramos et al., 1992; Mesulam et al., 1992; Inestrosa et al., 1996). It is possible that this notable accumulation of ChAT and AChE within and around plaques in APP23 mice accounts for the only modest overall decrease in enzyme activities.
It has been hypothesized that cholinergic depletion in AD contributes to cerebral amyloidosis. This hypothesis is based on the observation that activation of protein kinase C through muscarinic receptor binding stimulates the nonamyloidgenic pathway of APP processing by increasing sAPPα production and reducing Aβ generation (Buxbaum et al., 1992; Nitsch et al., 1992; Hung et al., 1993). Thus, the loss of the cholinergic innervation may lead to increased production of Aβ and amyloid deposition. In vivosupport for altered APP processing has been provided in both NBM-lesioned rats and in rats after muscarinic agonist treatment (Rossner et al., 1997; Lin et al., 1999). However, Aβ levels were not assessed in these studies. In contrast, Aβ levels were assessed 6 months after NBM lesions in rabbits and resulted in a 2.5- and 8-fold increase in neocortical Aβ40 and Aβ42, respectively, however no deposition of amyloid was found (Beach et al., 2000b). Based on these findings we initiated similar NBM lesions in APP23 mice that develop cerebral amyloidosis with aging.
ChAT reduction after NBM lesions in APP23 mice was 38% and is similar to the reduction reported in NBM-lesioned mice, rats, and rabbits (Smith, 1988; Beach et al., 2000b). However, we failed to see increased Aβ levels or deposition in the lesioned hemisphere in APP23 mice. Perhaps it is difficult to further shift the processing of transgenic APP toward the β-secretase pathway because the Swedish double mutation in APP23 mice already greatly favors this pathway (Citron et al., 1992).
The observation that NBM lesions in APP23 mice actually result in a decrease in neocortical amyloid deposition may be explained by the finding that AChE accelerates the assembly of amyloid peptide into amyloid fibrils (Inestrosa et al., 1996). Accordingly, the cholinergic deficit in APP23 mice may result in reduced amyloid formation. However, the observations of amyloid plaque reduction after NBM lesions may also be the result of enhanced clearing mechanisms in a lesioned–deafferented brain tissue and not be specific to the cholinergic system. It has been reported that traumatic brain injury results in a 30% regression of amyloid burden in the hippocampus of PDAPP transgenic mice (Nakagawa et al., 2000). Moreover, entorhinal cortex lesions appear to inhibit amyloid plaque formation in the deafferented dentate gyrus (S. Sisodia, personal communication). The mechanism is not clear but may involve enhanced microglia clearance of amyloid in the denervated–lesioned brain areas.
In conclusion, our results suggest that the cerebral amyloidosis in neocortex of APP23 mice causes significant cholinergic fiber loss and severe disruption of the cholinergic fiber network. Because the mice do not lose basal cholinergic forebrain neurons, these results suggest that the cholinergic deficit in APP23 mice is locally induced by the amyloid in neocortex. In light of the role and importance of the neocortical cholinergic system for cognition (Winkler et al., 1995), the cholinergic changes in APP23 mice may contribute to the recently described cognitive impairment of these mice (Kelly et al., 2002). Importantly, our lesion results suggest that in the APP23 mouse model, loss of cholinergic forebrain neurons and a subsequent loss of cortical cholinergic activity does not promote amyloid deposition.
S.B. and M.E.C. contributed equally to this work.
This work was supported by grants from the Swiss National Science Foundation and the VerUm Foundation (Munich, Germany). We thank J. Massoulie for providing antibodies, P. Mouton for helpful comments regarding stereological procedures, D. Hunziker for technical assistance, and T. Schürch and H. Zysset for assistance in photography.
Correspondence should be addressed to Mathias Jucker, Institute of Pathology, University of Basel, Schönbeinstrasse 40, CH-4003 Basel, Switzerland. E-mail: mjucker@uhbs.ch.
M. E. Calhoun's present address: Kastor Neurobiology of Aging Laboratories, Mount Sinai School of Medicine, New York, NY 10029.
A. L. Phinney's present address: University of Toronto, Center for Research in Neurodegenerative Diseases, Toronto, Ontario M5S 3H2, Canada.
REFERENCES
- 1.Andra K, Abramowski D, Duke M, Probst A, Wiederhold KH, Burki K, Goedert M, Sommer B, Staufenbiel M. Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol Aging. 1996;17:183–190. doi: 10.1016/0197-4580(95)02066-7. [DOI] [PubMed] [Google Scholar]
- 2.Auld DS, Kar S, Quirion R. Beta-amyloid peptides as direct cholinergic neuromodulators: a missing link? Trends Neurosci. 1998;21:43–49. doi: 10.1016/s0166-2236(97)01144-2. [DOI] [PubMed] [Google Scholar]
- 3.Bartus RT, Dean RLd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 4.Beach TG, Kuo YM, Spiegel K, Emmerling MR, Sue LI, Kokjohn K, Roher AE. The cholinergic deficit coincides with Abeta deposition at the earliest histopathologic stages of Alzheimer disease. J Neuropathol Exp Neurol. 2000a;59:308–313. doi: 10.1093/jnen/59.4.308. [DOI] [PubMed] [Google Scholar]
- 5.Beach TG, Potter PE, Kuo YM, Emmerling MR, Durham RA, Webster SD, Walker DG, Sue LI, Scott S, Layne KJ, Roher AE. Cholinergic deafferentation of the rabbit cortex: a new animal model of Abeta deposition. Neurosci Lett. 2000b;283:9–12. doi: 10.1016/s0304-3940(00)00916-2. [DOI] [PubMed] [Google Scholar]
- 6.Benzing WC, Mufson EJ, Armstrong DM. Immunocytochemical distribution of peptidergic and cholinergic fibers in the human amygdala: their depletion in Alzheimer's disease and morphologic alteration in non-demented elderly with numerous senile plaques. Brain Res. 1993;625:125–138. doi: 10.1016/0006-8993(93)90145-d. [DOI] [PubMed] [Google Scholar]
- 7.Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. Neurochemical correlates of dementia severity in Alzheimer's disease: relative importance of the cholinergic deficits. J Neurochem. 1995;64:749–760. doi: 10.1046/j.1471-4159.1995.64020749.x. [DOI] [PubMed] [Google Scholar]
- 8.Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bronfman FC, Moechars D, Van Leuven F. Acetylcholinesterase-positive fiber deafferentation and cell shrinkage in the septohippocampal pathway of aged amyloid precursor protein london mutant transgenic mice. Neurobiol Dis. 2000;7:152–168. doi: 10.1006/nbdi.2000.0283. [DOI] [PubMed] [Google Scholar]
- 10.Buxbaum JD, Oishi M, Chen HI, Pinkas-Kramarski R, Jaffe EA, Gandy SE, Greengard P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc Natl Acad Sci USA. 1992;89:10075–10078. doi: 10.1073/pnas.89.21.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calhoun ME, Kurth D, Phinney AL, Long JM, Hengemihle J, Mouton PR, Ingram DK, Jucker M. Hippocampal neuron and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol Aging. 1998a;19:599–606. doi: 10.1016/s0197-4580(98)00098-0. [DOI] [PubMed] [Google Scholar]
- 12.Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998b;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 13.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci U S A. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 15.Collerton D. Cholinergic function and intellectual decline in Alzheimer's disease. Neuroscience. 1986;19:1–28. doi: 10.1016/0306-4522(86)90002-3. [DOI] [PubMed] [Google Scholar]
- 16.Coyle JT, Price DL, DeLong MR. Alzheimer's disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- 17.Cullen KM, Halliday GM. Neurofibrillary degeneration and cell loss in the nucleus basalis in comparison to cortical Alzheimer pathology. Neurobiol Aging. 1998;19:297–306. doi: 10.1016/s0197-4580(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 18.Cutler NR, Polinsky RJ, Sramek JJ, Enz A, Jhee SS, Mancione L, Hourani J, Zolnouni P. Dose-dependent CSF acetylcholinesterase inhibition by SDZ ENA 713 in Alzheimer's disease. Acta Neurol Scand. 1998;97:244–250. doi: 10.1111/j.1600-0404.1998.tb00645.x. [DOI] [PubMed] [Google Scholar]
- 19.DeKosky ST, Harbaugh RE, Schmitt FA, Bakay RA, Chui HC, Knopman DS, Reeder TM, Shetter AG, Senter HJ, Markesbery WR. Cortical biopsy in Alzheimer's disease: diagnostic accuracy and neurochemical, neuropathological, and cognitive correlations. Intraventricular Bethanecol Study Group. Ann Neurol. 1992;32:625–632. doi: 10.1002/ana.410320505. [DOI] [PubMed] [Google Scholar]
- 20.Ellman GL, Courtney KD, Andres VJ, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 21.Fonnum F. A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem. 1975;24:407–409. doi: 10.1111/j.1471-4159.1975.tb11895.x. [DOI] [PubMed] [Google Scholar]
- 22.Franklin KBJ, Paxinos G. The mouse brain atlas in stereotaxic coordinates. Academic; San Diego, CA: 1997. [Google Scholar]
- 23.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 24.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 25.Gaykema RP, Luiten PG, Nyakas C, Traber J. Cortical projection patterns of the medial septum-diagonal band complex. J Comp Neurol. 1990;293:103–124. doi: 10.1002/cne.902930109. [DOI] [PubMed] [Google Scholar]
- 26.Geula C, Mesulam MM, Saroff DM, Wu CK. Relationship between plaques, tangles, and loss of cortical cholinergic fibers in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:63–75. doi: 10.1097/00005072-199801000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer's disease. Trends Neurosci. 1993;16:460–465. doi: 10.1016/0166-2236(93)90078-z. [DOI] [PubMed] [Google Scholar]
- 28.Gomez-Ramos P, Mufson EJ, Moran MA. Ultrastructural localization of acetylcholinesterase in neurofibrillary tangles, neuropil threads and senile plaques in aged and Alzheimer's brain. Brain Res. 1992;569:229–237. doi: 10.1016/0006-8993(92)90634-l. [DOI] [PubMed] [Google Scholar]
- 29.Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- 30.Haass C, Steiner H. Protofibrils, the unifying toxic molecule of neurodegenerative disorders? Nat Neurosci. 2001;4:859–860. doi: 10.1038/nn0901-859. [DOI] [PubMed] [Google Scholar]
- 31.Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 32.Hedreen JC, Bacon SJ, Price DL. A modified histochemical technique to visualize acetylcholinesterase-containing axons. J Histochem Cytochem. 1985;33:134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- 33.Hernandez D, Sugaya K, Qu T, McGowan E, Duff K, McKinney M. Survival and plasticity of basal forebrain cholinergic systems in mice transgenic for presenilin-1 and amyloid precursor protein mutant genes. NeuroReport. 2001;12:1377–1384. doi: 10.1097/00001756-200105250-00018. [DOI] [PubMed] [Google Scholar]
- 34.Howard CV, Cruz-Orive LM, Yaegashi H. Estimating neuron dendritic length in 3D from total vertical projections and from vertical slices. Acta Neurol Scand [Suppl] 1992;137:14–19. doi: 10.1111/j.1600-0404.1992.tb05033.x. [DOI] [PubMed] [Google Scholar]
- 35.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 36.Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe DJ. Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J Biol Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- 37.Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 38.Jaffar S, Counts SE, Ma SY, Dadko E, Gordon MN, Morgan D, Mufson EJ. Neuropathology of Mice Carrying Mutant APP(swe) and/or PS1(M146L) transgenes: alterations in the p75(NTR) cholinergic basal forebrain septohippocampal pathway. Exp Neurol. 2001;170:227–243. doi: 10.1006/exnr.2001.7710. [DOI] [PubMed] [Google Scholar]
- 39.Jensen EB, Gundersen HLG. The rotator. J Microsc. 1993;170:35–44. [Google Scholar]
- 40.Jope RS, Song L, Powers RE. Cholinergic activation of phosphoinositide signaling is impaired in Alzheimer's disease brain. Neurobiol Aging. 1997;18:111–120. doi: 10.1016/s0197-4580(96)00205-9. [DOI] [PubMed] [Google Scholar]
- 41.Jucker M, D'Amato F, Mondadori C, Mohajeri H, Magyar J, Bartsch U, Schachner M. Expression of the neural adhesion molecule L1 in the deafferented dentate gyrus. Neuroscience. 1996;75:703–715. doi: 10.1016/0306-4522(96)00276-x. [DOI] [PubMed] [Google Scholar]
- 42.Jucker M, Calhoun M, Phinney A, Stalder M, Bondolfi L, Winkler D, Herzig M, Pfeifer M, Boncristiano S, Tolnay M, Probst A, Deller T, Abramowski D, Wiederhold K-H, Sturchler-Pierrat C, Sommer B, Staufenbiel M. Pathogenesis and mechanism of cerebral amyloidosis in APP transgenic mice. Neurodegenerative disorders: loss of function through gain of function (Beyreuther K, Christen Y, and Masters CL, eds), pp 87–95. Springer; Berlin: 2001. [Google Scholar]
- 43.Kelly PH, Moore KE. Decrease of neocortical choline acetyltransferase after lesion of the globus pallidus in the rat. Exp Neurol. 1978;61:479–484. doi: 10.1016/0014-4886(78)90263-7. [DOI] [PubMed] [Google Scholar]
- 44.Kelly PH, Hunziker D, Schlecht HP, Carver K, Abramowski D, Wiederhold KH, Sturchler-Pierrat C, Bondolfi L, Jucker M, Staufenbiel M, Sommer B (2002) Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Submitted. [DOI] [PubMed]
- 45.Kitt CA, Hohmann C, Coyle JT, Price DL. Cholinergic innervation of mouse forebrain structures. J Comp Neurol. 1994;341:117–129. doi: 10.1002/cne.903410110. [DOI] [PubMed] [Google Scholar]
- 46.Klafki HW, Wiltfang J, Staufenbiel M. Electrophoretic separation of betaA4 peptides (1–40) and (1–42). Anal Biochem. 1996;237:24–29. doi: 10.1006/abio.1996.0195. [DOI] [PubMed] [Google Scholar]
- 47.Kuo YM, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC, Kalback WM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Roher AE. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer's disease brains. J Biol Chem. 2001;276:12991–12998. doi: 10.1074/jbc.M007859200. [DOI] [PubMed] [Google Scholar]
- 48.Ladner CJ, Lee JM. Pharmacological drug treatment of Alzheimer disease: the cholinergic hypothesis revisited. J Neuropathol Exp Neurol. 1998;57:719–731. doi: 10.1097/00005072-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 49.Liberini P, Pioro EP, Maysinger D, Cuello AC. Neocortical infarction in subhuman primates leads to restricted morphological damage of the cholinergic neurons in the nucleus basalis of Meynert. Brain Res. 1994;648:1–8. doi: 10.1016/0006-8993(94)91897-x. [DOI] [PubMed] [Google Scholar]
- 50.Lin L, Georgievska B, Mattsson A, Isacson O. Cognitive changes and modified processing of amyloid precursor protein in the cortical and hippocampal system after cholinergic synapse loss and muscarinic receptor activation. Proc Natl Acad Sci USA. 1999;96:12108–12113. doi: 10.1073/pnas.96.21.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marsh D, Grassi J, Vigny M, Massoulie J. An immunological study of rat acetylcholinesterase: comparison with acetylcholinesterases from other vertebrates. J Neurochem. 1984;43:204–213. doi: 10.1111/j.1471-4159.1984.tb06698.x. [DOI] [PubMed] [Google Scholar]
- 52.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKinney M, Coyle JT, Hedreen JC. Topographic analysis of the innervation of the rat neocortex and hippocampus by the basal forebrain cholinergic system. J Comp Neurol. 1983;217:103–121. doi: 10.1002/cne.902170109. [DOI] [PubMed] [Google Scholar]
- 54.Mesulam MM. The systems-level organization of cholinergic innervation in the human cerebral cortex and its alterations in Alzheimer's disease. Prog Brain Res. 1996;109:285–297. doi: 10.1016/s0079-6123(08)62112-3. [DOI] [PubMed] [Google Scholar]
- 55.Mesulam M, Carson K, Price B, Geula C. Cholinesterases in the amyloid angiopathy of Alzheimer's disease. Ann Neurol. 1992;31:565–569. doi: 10.1002/ana.410310517. [DOI] [PubMed] [Google Scholar]
- 56.Moran MA, Mufson EJ, Gomez-Ramos P. Colocalization of cholinesterases with beta amyloid protein in aged and Alzheimer's brains. Acta Neuropathol. 1993;85:362–369. doi: 10.1007/BF00334445. [DOI] [PubMed] [Google Scholar]
- 57.Mountjoy CQ, Rossor MN, Iversen LL, Roth M. Correlation of cortical cholinergic and GABA deficits with quantitative neuropathological findings in senile dementia. Brain. 1984;107:507–518. doi: 10.1093/brain/107.2.507. [DOI] [PubMed] [Google Scholar]
- 58.Nakagawa Y, Reed L, Nakamura M, McIntosh TK, Smith DH, Saatman KE, Raghupathi R, Clemens J, Saido TC, Lee VM, Trojanowski JQ. Brain trauma in aged transgenic mice induces regression of established abeta deposits. Exp Neurol. 2000;163:244–252. doi: 10.1006/exnr.2000.7375. [DOI] [PubMed] [Google Scholar]
- 59.Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- 60.Pedersen WA, Kloczewiak MA, Blusztajn JK. Amyloid beta-protein reduces acetylcholine synthesis in a cell line derived from cholinergic neurons of the basal forebrain. Proc Natl Acad Sci USA. 1996;93:8068–8071. doi: 10.1073/pnas.93.15.8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perry EK, Blessed G, Tomlinson BE, Perry RH, Crow TJ, Cross AJ, Dockray GJ, Dimaline R, Arregui A. Neurochemical activities in human temporal lobe related to aging and Alzheimer-type changes. Neurobiol Aging. 1981;2:251–256. doi: 10.1016/0197-4580(81)90032-4. [DOI] [PubMed] [Google Scholar]
- 62.Perry EK, Johnson M, Kerwin JM, Piggott MA, Court JA, Shaw PJ, Ince PG, Brown A, Perry RH. Convergent cholinergic activities in aging and Alzheimer's disease. Neurobiol Aging. 1992;13:393–400. doi: 10.1016/0197-4580(92)90113-c. [DOI] [PubMed] [Google Scholar]
- 63.Pettit DL, Shao Z, Yakel JL. beta-Amyloid(1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21:RC120. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ransmayr G, Cerrera P, Hirsch EC, Berger W, Fischer W, Agid Y. Alzheimer's disease: is the decrease of the cholinergic innervation of the hippocampus related to intrinsic hippocampal pathology? Neuroscience. 1992;47:843–851. doi: 10.1016/0306-4522(92)90034-y. [DOI] [PubMed] [Google Scholar]
- 66.Roberson MR, Harrell LE. Cholinergic activity and amyloid precursor protein metabolism. Brain Res Brain Res Rev. 1997;25:50–69. doi: 10.1016/s0165-0173(97)00016-7. [DOI] [PubMed] [Google Scholar]
- 67.Rossner S, Ueberham U, Yu J, Kirazov L, Schliebs R, Perez-Polo JR, Bigl V. In vivo regulation of amyloid precursor protein secretion in rat neocortex by cholinergic activity. Eur J Neurosci. 1997;9:2125–2134. doi: 10.1111/j.1460-9568.1997.tb01379.x. [DOI] [PubMed] [Google Scholar]
- 68.Sassin I, Schultz C, Thal DR, Rub U, Arai K, Braak E, Braak H. Evolution of Alzheimer's disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol (Berl) 2000;100:259–269. doi: 10.1007/s004019900178. [DOI] [PubMed] [Google Scholar]
- 69.Schrader-Fischer G, Paganetti PA. Effect of alkalizing agents on the processing of the beta-amyloid precursor protein. Brain Res. 1996;716:91–100. doi: 10.1016/0006-8993(96)00002-9. [DOI] [PubMed] [Google Scholar]
- 70.Selkoe DJ. Alzheimer's disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 71.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 72.Smith G. Animal models of Alzheimer's disease: experimental cholinergic denervation. Brain Res. 1988;472:103–118. doi: 10.1016/0165-0173(88)90016-1. [DOI] [PubMed] [Google Scholar]
- 73.Sofroniew MV, Pearson RC, Eckenstein F, Cuello AC, Powell TP. Retrograde changes in cholinergic neurons in the basal forebrain of the rat following cortical damage. Brain Res. 1983;289:370–374. doi: 10.1016/0006-8993(83)90045-8. [DOI] [PubMed] [Google Scholar]
- 74.Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stocks EA, McArthur JC, Griffen JW, Mouton PR. An unbiased method for estimation of total epidermal nerve fibre length. J Neurocytol. 1996;25:637–644. doi: 10.1007/BF02284830. [DOI] [PubMed] [Google Scholar]
- 76.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van Den Haute C, Mercken M, Moechars D, Laenen I, Kuiperi C, Bruynseels K, Tesseur I, Loos R, Vanderstichele H, Checler F, Sciot R, Van Leuven F. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am J Pathol. 2000;157:1283–1298. doi: 10.1016/S0002-9440(10)64644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vogels OJ, Broere CA, ter Laak HJ, ten Donkelaar HJ, Nieuwenhuys R, Schulte BP. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer's disease. Neurobiol Aging. 1990;11:3–13. doi: 10.1016/0197-4580(90)90056-6. [DOI] [PubMed] [Google Scholar]
- 79.West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- 80.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 81.Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 82.Wilcock GK, Esiri MM, Bowen DM, Smith CC. Alzheimer's disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J Neurol Sci. 1982;57:407–417. doi: 10.1016/0022-510x(82)90045-4. [DOI] [PubMed] [Google Scholar]
- 83.Wiltfang J, Smirnov A, Schnierstein B, Kelemen G, Matthies U, Klafki HW, Staufenbiel M, Huther G, Ruther E, Kornhuber J. Improved electrophoretic separation and immunoblotting of beta-amyloid (A beta) peptides 1–40, 1–42, and 1–43. Electrophoresis. 1997;18:527–532. doi: 10.1002/elps.1150180332. [DOI] [PubMed] [Google Scholar]
- 84.Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Winkler J, Suhr ST, Gage FH, Thal LJ, Fisher LJ. Essential role of neocortical acetylcholine in spatial memory. Nature. 1995;375:484–487. doi: 10.1038/375484a0. [DOI] [PubMed] [Google Scholar]
- 86.Wong TP, Debeir T, Duff K, Cuello AC. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J Neurosci. 1999;19:2706–2716. doi: 10.1523/JNEUROSCI.19-07-02706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zubenko GS, Moossy J, Martinez AJ, Rao GR, Kopp U, Hanin I. A brain regional analysis of morphologic and cholinergic abnormalities in Alzheimer's disease. Arch Neurol. 1989;46:634–638. doi: 10.1001/archneur.1989.00520420054022. [DOI] [PubMed] [Google Scholar]