

Abstract
Multiple sclerosis (MS) is characterized by inflammation within the CNS. This inflammatory response is associated with production of nitric oxide (NO) and NO-related species that nitrosylate thiols. We postulated that MS patients would exhibit an antibody (Ab) response directed against proteins containing S-nitrosocysteine (SNO-cysteine) and showed that anti-NO-cysteine Abs of the IgM isotype are in fact present in the sera of some MS patients (Boullerne et al., 1995). We report here the presence of a seemingly identical Ab response directed against SNO-cysteine in an acute model of MS, experimental autoimmune encephalomyelitis (EAE) induced in Lewis rats with the 68–84 peptide of guinea pig myelin basic protein (MBP68–84). Serum levels of anti-SNO-cysteine Abs peaked 1 week before the onset of clinical signs and well before the appearance of anti-MBP68–84 Abs. The anti-SNO-cysteine Ab peak titer correlated with the extent of subsequent CNS demyelination, suggesting a link between Ab level and CNS lesion formation. In relapsing–remitting MS patients, we found elevated anti-SNO-cysteine Ab at times of relapse and normal values in most patients judged to be in remission. Two-thirds of patients with secondary progressive MS had elevated anti-SNO-cysteine Ab levels, including those receiving interferon β-1b. The data show that a rise in circulating anti-SNO-cysteine Ab levels precedes onset of EAE. Anti-SNO-cysteine Abs are also elevated at times of MS attacks and in progressive disease, suggesting a possible role for these Abs, measurable in blood, as a biological marker for clinical activity.
Keywords: experimental autoimmune encephalomyelitis, multiple sclerosis, autoimmunity, nitric oxide, antibody, clinical marker
Multiple sclerosis (MS), the most common inflammatory demyelinating disease of the CNS, usually begins as a relapsing–remitting process. As one attack follows another, disability accumulates, and usually the illness eventually becomes relentlessly progressive (Noseworthy, 1999). An easily measured biological marker that predicts MS activity would be desirable. None is known, except possibly soluble adhesion molecules (Rieckmann et al., 1998; Trojano et al., 1998), caspase mRNA expression (Furlan et al., 1999), and T-cell activation markers (Khoury et al., 2000).
Nonspecific inflammatory reactants, proposed as contributors to tissue damage in MS, include nitric oxide (NO). NO triggers axon and oligodendrocyte degeneration; both are targets in MS lesions (Boullerne et al., 2001; Smith et al., 2001). NO is volatile, so its production is usually measured indirectly by evaluation of NO synthases or NO-derived products such as nitrosated amino acids, nitrite, and nitrate. Increased nitrite level in CSF of MS patients correlates with clinical activity (Brundin et al., 1999; Svenningsson et al., 1999). In active MS lesions, microglia and macrophages express inducible NO synthase (iNOS; Hooper et al., 1997), and macrophages isolated from an active MS lesion were shown to produce NO (De Groot et al., 1997). Similarly, blood monocytes produce substantial amounts of NO when MS is active (Sarchielli et al., 1997). In experimental autoimmune encephalomyelitis (EAE), an animal model for MS, iNOS mRNA expression within the CNS is enhanced both at onset and peak of clinical signs, with levels returning to normal on remission (Koprowski et al., 1993; Okuda et al., 1995). All the above suggests a relationship between NO production and CNS inflammation.
NO covalently modifies proteins. For example, nitrotyrosine, generated when tyrosine is nitrated by NO-related species, has been detected in MS and EAE lesions (Hooper et al., 1997; Cross et al., 1998). Similarly, cysteine can be nitrosated by NO and NO-related species (Hess et al., 2001), and new epitopes of possible relevance to MS can be generated. We have shown previously that antibodies (Abs) reactive with nitroso-S-cysteine coupled to the protein carrier bovine serum albumin (BSA) are elevated in sera from MS patients compared with sera from healthy controls (Boullerne et al., 1995), suggesting that anti-SNO-cysteine Abs might constitute a biological marker in MS and possibly a predictor of onset and severity of EAE.
We coupled S-nitrosocysteine (SNO-cysteine) to BSA and measured serum levels of anti-SNO-cysteine Ab over the course of actively induced EAE in Lewis rats. We show that an anti-SNO-cysteine-BSA Ab response of the M isotype peaks 6–7 d postimmunization (dpi), i.e., several days before EAE onset, and that titers at this early time point correlate with the extent of subsequent demyelination. We also show that levels of serum anti-SNO-cysteine-BSA Ab are elevated in MS at times of relapse but normal in most patients judged to be in remission. Two-thirds of patients with progressive MS have elevated anti-SNO-cysteine-BSA Abs, including patients receiving interferon β-1b (IFNβ-1b). Anti-SNO-cysteine-BSA Abs may serve as biological markers for disease activity in MS.
MATERIALS AND METHODS
Induction and clinical evaluation of EAE.Female Lewis rats aged 6–10 weeks weighing 145–180 gm were obtained from Charles River France SA. Animal handling conformed to guidelines of the European Community. Each rat received 100 μg of the encephalitogenic 68–84 peptide of guinea pig myelin basic protein (MBP68–84, Y68GSLPQKSQ76R79SQDEN84; Peninsula Laboratories, Belmont, CA) in 50 μl of PBS emulsified with 50 μl of complete Freund's adjuvant (CFA; Difco Laboratories, Detroit, MI) enriched with 1 mg of heat-inactivatedMycobacterium tuberculosis strain H37 RA (Difco; Mannie et al., 1985). The mixture was injected at 50 μl/hind footpad. The total dose of M. tuberculosis received per rat was 1.05 mg: 50 μg in the CFA plus 1 mg of supplement.
In the first experiment, the EAE group comprised seven rats aged 6–7 weeks. The control group was composed of five rats that received 50 μl/hind footpad PBS emulsified in 50 μl of CFA alone, i.e., a total dose of 50 mg of M. tuberculosis/rat. In a second experiment, nine rats aged 9–10 weeks were immunized for EAE following the protocol given above. Controls again consisted of five rats injected with PBS-CFA alone.
A clinical score was assigned daily for 35 d after immunization. The clinical score was graded as 0, no clinical signs; 1, flaccid tail; 2, flaccid tail and hindlimb paresis; and 3, hindlimb paralysis. EAE onset was defined as the day a rat developed a flaccid tail. A cumulative disease severity score (Liedtke et al., 1998) was generated for each rat, summing the daily scores from 11 to 20 dpi, because recovery began on day 19 in the first experiment and on day 21 in the second experiment.
Detection of rat anti-SNO-cysteine antibodies. Tail blood was sampled every 7–9 d. Sera were tested by ELISA for specific Abs directed against SNO-cysteine linked to a BSA carrier via treatment with 0.25% glutaraldehyde for 5 min (SNO-cysteine-BSA) as described previously (Boullerne et al., 1995). For ELISA, 96-well polystyrene plates (Maxisorp; Nunc, Roskilde, Denmark) were coated overnight with SNO-cysteine-BSA (or BSA treated with glutaraldehyde alone, BSA-g, as a control), at a concentration of 10 μg/ml in 0.05 mcarbonate buffer, pH 9.6. Free binding sites were blocked by incubation for 1 hr at 37°C with PBS containing 0.05% Tween 20 (PBS-Tween) plus 10% glycerol and 0.1% BSA (buffer A). After rinsing twice with PBS-Tween, rat sera diluted 1:500 in buffer A were added in duplicate and incubated for 2 hr at 37°C. After two rinses with PBS-Tween, plates were incubated with peroxidase-conjugated goat anti-rat IgM (M-chain-specific; Jackson ImmunoResearch, West Grove, PA) or with goat anti-rat IgG (heavy and light chain-specific; Jackson ImmunoResearch) both diluted to 0.12 μg/ml in PBS-Tween plus 0.1% BSA for 1 hr at 37°C. After three rinses with PBS-Tween, reactivity was revealed with orthophenylenediamine (Sigma, St. Louis, MO) as chromogen. Optical density (OD) was read at 490 nm in a plate reader (MR 5000; Dynatech Laboratories, Ashford, UK). The anti-SNO-cysteine-BSA titer was calculated by subtracting the OD read on BSA-g (blank) from the OD read on SNO-cysteine-BSA.
Specificity of IgM binding to immobilized SNO-cysteine-BSA was assessed by adding SNO-cysteine-BSA to the liquid phase and measuring inhibition. A 7 dpi rat serum sample chosen for its high Ab titer was preincubated at a dilution of 1:500 with 10−10–10−5m SNO-cysteine-BSA in buffer A for 16 hr at 4°C. After centrifugation at 10,000 × g for 30 min, supernatants were tested in ELISA with SNO-cysteine-BSA coated on 96-well plates to determine whether SNO-cysteine-BSA in liquid phase inhibited IgM binding to SNO-cysteine-BSA adsorbed on plates in proportion to its concentration.
Detection of rat anti-myelin basic protein antibodies. Abs directed against MBP68–84 were assayed as described above for anti-SNO-cysteine-BSA Abs but with plates coated with MBP68–84 at 0.5 μg/ml and with rat sera diluted 1:1,000. Each serum was tested in duplicate in two independent experiments for Abs to SNO-cysteine-BSA and to MBP68–84 peptide, and the four OD values were averaged to give a mean Ab titer. Within-plate OD variation was 5–6%; between-plate OD variation was ±26%.
Determination of demyelination. The extent of myelin depletion was determined in cerebellar and C1–C6 cervical spinal cord white matter at 35 dpi. Animals were deeply anesthetized with chloral hydrate (400 mg/kg, i.p.) and perfused transcardially with 150 ml of PBS containing 5 × 104 IU/ml heparin, followed by 500 ml of 4% paraformaldehyde dissolved in 0.1m phosphate buffer, pH 7.4. Brain and spinal cord were removed and immersed in 4% paraformaldehyde for 24 hr at 4°C. C1–C6 cervical spinal cord and cerebellum blocks were washed in 0.1m phosphate buffer for 2 hr at 4°C and then incubated in a 30% sucrose solution in 0.1 m phosphate buffer overnight at 4°C for cryoprotection. Samples were stored at −80°C until processing. Sagittal cerebellum sections of 30 μm were cut using a cryostat (HM500M; Microm). Cerebellum sections were immunostained for MBP to evaluate myelin depletion according to a procedure adapted fromVandenbark et al. (1986). Free-floating adjacent sections of cerebellum were incubated overnight with polyclonal rabbit Ab against MBP diluted at 1:20,000 (kindly provided by Dr. A. Fournier, Victor Segalen University). Sections were then incubated with a biotin-labeled goat anti-rabbit Ab diluted at 1:200 (Dako SA, Trappes, France). Immunoreactivity was visualized by the biotin–streptavidin technique using 3,3′-diaminobenzidine as chromogen (avidin–biotin complex kit; Dako) and incubation for 10 min. Negative controls were performed by omitting the primary Ab and by using Abs unrelated to CNS antigens. The mean staining intensity per defined square surface was measured using a densitometer system (Samba 2640; Alcatel) and expressed in arbitrary units of OD. For each section, MBP labeling was measured in the inner part of the granular layer within three different lobes. Blank values were obtained from the molecular layer of the cerebellum, known to contain primarily unmyelinated fibers. The average value of MBP staining per animal was calculated by measuring the mean value of four sections within lobes 4, 6, and 8 and then subtracting the mean of blank values.
Eleven series of five transverse cervical spinal cord sections (C1–C6) of 30 μm were cut using a cryostat (HM500M). The first section of each series (thus 150 μm apart) was stained for myelin by Spielmeyer's method as described previously (De Castro and Ramon y Cajal, 1933). Myelin appears black after staining by Spielmeyer's method. Free-floating sections were incubated in 2.5% ammonium iron (III) sulfate aqueous solution, washed in water, and stained with hematoxylin. To evaluate the amount of myelin loss in white matter on stained sections, an image analysis system (NIH Image running on an Apple Macintosh computer) coupled to a microscope (Nikon, Tokyo, Japan) was used to measure the area of lesion (clear) and the total area of white matter on four to six sections of each animal. Total length of cervical spinal cord examined was 600–900 μm. The percentage of myelin loss was calculated by dividing the lesion size into the total area of white matter for each section.
Determination of inflammation. From the same series of cervical spinal cord cross sections (C1–C6), the second section of each series (again 150 μm apart) was stained with hematoxylin and eosin. Infiltrations were observed perivascularly, within the parenchyma and in the meninges. An inflammatory index was calculated by averaging the total number of infiltration sites per section. The size of individual infiltration sites was weighted by a factor of 1–3 to reflect the intensity of the pathology. The maximum number of infiltration sites was 5 per section. Five to 11 sections were analyzed for each of nine animals. The total length of cervical spinal cord examined was 750–1650 μm.
Patients. Venous blood was obtained from 6 French MS patients and from 25 American MS patients. All patients gave informed consent, and the protocol was in conformity with French law and had the approval of the University of Chicago Institutional Review Board. Half the American MS patients were being treated with IFNβ-1b. All patients were classified as having definite MS according to the criteria of Poser et al. (1983) and were subclassified into five subgroups: (1) acute relapse, defined as an abrupt neurological deterioration lasting for >24 hr; no patient in relapse was receiving IFNβ-1b; (2, 3) remission, in which sera from relapsing–remitting MS patients in remission were subdivided into those receiving IFNβ-1b (2) and those who were untreated (3); and (4, 5) secondary progressive MS, in which all patients had evident neurological deterioration that had been evolving for >6 months and were classified as treated with IFNβ-1b (4) or untreated (5).
Control sera were from 5 French and 19 American healthy volunteers. Titers of human anti-SNO-cysteine-BSA IgM were determined blind according to a procedure described previously (Boullerne et al., 1995). The method was as outlined above for the EAE experiments, except that serum or plasma was diluted 1:300, and Ab was revealed with peroxidase-conjugated rabbit anti-human IgM (M-chain-specific; Dako, Carpinteria, CA) diluted at 1:1000. The average OD of each healthy donor group was used to determine the relative OD of serum or plasma of each MS patient according to the following calculation: relative OD = ODpatient − mean ODhealthy group/mean ODhealthy group.
To detect any difference in anti-SNO-cysteine-BSA Ab level between serum and plasma of the same individuals, several paired samples from MS patients and controls were tested within the same assay. No difference in relative OD was detected. For this reason, either serum or plasma titers were measured in individual subjects.
Specificity of IgM to SNO-cysteine-BSA. Specificity of IgM binding to immobilized SNO-cysteine-BSA was assessed by adding SNO-cysteine-BSA or competitors to the liquid phase and measuring binding inhibition. Six MS sera chosen for their high Ab titer were preincubated at dilutions of 1:300 and 1:450 with competitors at concentrations of 10−10–10−5m in buffer A for 16 hr at 4°C. After centrifugation at 10,000 × g for 30 min or at 12,000 ×g for 13 min, supernatants were tested by ELISA with SNO-cysteine-BSA coated on 96-well plates to determine whether SNO-cysteine-BSA or any competitor added to the liquid phase interfered, in proportion to its concentration, with IgM binding to SNO-cysteine-BSA adsorbed on plates. Competitors tested included cysteine-BSA (a homolog of SNO-cysteine-BSA) and SNO-cysteine-BSA preincubated with 2 mmHgCl2 (Sigma) in 0.3× PBS, pH 7.4, for 4 hr at 4°C, according to a previously described method (Saville, 1958), to destroy nitrosylation. After centrifugation, the supernatant was recovered, assayed for protein content, and incubated with MS sera.
Additional competitors included BSA-g nitrosylated by incubation with NaNO2 (Sigma) in 0.5 m HCl (NO-BSA-g) as described for SNO-cysteine-BSA (Boullerne et al., 1995), cysteine-BSA incubated with the NO donorS-nitroso-N-acetyl-d,l-penicillamine (SNAP) (Sigma) at 27 mm for 4 hr at 37°C at pH 7–8, cysteine-BSA incubated with the peroxynitrite donor 3-morpholino-sydnonimine (SIN-1) (Sigma) at 29 mmfor 4 hr at 37°C, and cysteine-BSA incubated with H2O2 (Fisher Scientific, Pittsburgh, PA) at 88 mm for 4 hr at 37°C. These competitors, once prepared, were dialyzed for 24 hr at 4°C in 0.01m Tris-HCl buffer, pH 7.4, followed by 2 washes with 0.1× PBS.
Western blot of nitrosylated proteins. Western blots were performed in 10% acrylamide SDS-PAGE with SDS at a concentration of 0.2% in the sample buffer and at 0.02% in the gel. Blots were performed at 4°C to preserve nitrosylation. Nitrosylated proteins loaded on gel were the following: (1) SNO-cysteine-BSA, (2) NO-BSA-g, (3) SNO-cysteine-BSA treated with 10 mm dithiothreitol (DTT, Sigma) for 2 hr at 4°C, (4) NO-BSA-g treated with 10 mm DTT, and (5) SNO-cysteine-BSA treated with 2 mm HgCl2 (as described above). Nitrosylated proteins were transferred onto Immobilon P membrane (Millipore, Bedford, MA). MS serum was added at a dilution of 1:300 in PBS-Tween 20 containing 0.1% BSA buffer, and incubation was continued overnight at 4°C. After three rinses with PBS, peroxidase-conjugated goat anti-human IgM (Sanofi Diagnostics Pasteur) diluted 1:1,000 in Tris-buffered saline, pH 7.4 (TBS), was added for 2 hr at room temperature. After three rinses with TBS, reactivity was revealed with 4-chloro-1-naphtol (Sigma) as chromogen.
Paired CSF–sera. Venous blood and CSF were obtained concomitantly from 25 German MS patients not subclassified into subgroups. The CSF was obtained for diagnostic purposes. All patients gave informed consent, and the protocol was in conformity with German law. All patients were classified as having definite MS according to the criteria of Poser et al. (1983). Sera were tested at a dilution of 1:400, and CSF was tested at a dilution of 1:2 to allow for the low IgM content in CSF. Both sera and CSF were preincubated with SorboTech (a generous gift from Genzyme Virotech, Ruesselsheim, Germany) for 15 min at room temperature before incubation onto the plate to remove all human IgG including rheumatoid factor (IgG antibody to IgM) that might give a false-positive OD. Titers of human anti-SNO-cysteine-BSA IgM were determined blind in paired sera and CSF on the same plate according to the method outlined previously.
Nitrite and nitrate measurements. SNO-cysteine-BSA was tested for nitrite and nitrate content by Griess assay at various times after preparation. The procedure was adapted from that of Drulovic et al. (2001). Briefly, samples were incubated with nitrate reductase fromAspergillus (EC 1.6.6.2), reduced βNADPH, and FAD for 2 hr at 37°C to convert nitrate into nitrite. Excess βNADPH was oxidized by adding lactic dehydrogenase from rabbit muscle (EC1.1.1.27) plus pyruvate (all reagents from Sigma) and incubated for 30 min at 37°C. Samples were assayed before and after nitrate reduction to measure nitrite and combined nitrite and nitrate levels, respectively. The Griess assay was performed in microplates by mixing samples with the Griess reagent, comprising a 1:1 mixture of 0.15% naphthylethylenediamine and 1.5% sulfanilamide in 1N HCl and incubating for 5 min at room temperature. Absorbance was read at 562 nm in a microplate reader. The nitrite concentration was calculated from a NaNO2 standard curve, and complete conversion of nitrate into nitrite was confirmed by including a standard curve of NaNO3 in each test. For samples containing DTT, standard curves of nitrite and nitrate were prepared with DTT at the same concentration. Values of samples processed through nitrate reduction were corrected for the residual level of nitrate present in water by subtracting the background OD.
Purification of serum immunoglobulins. Crude immunoglobulin preparations were obtained from sera of four MS patients in relapse and four healthy donors by precipitation (v/v) with a saturated solution of ammonium sulfate (Merck, Darmstadt, Germany). After 10 dialyses in PBS at 4°C to wash away ammonium sulfate, IgM fractions were purified by exclusion chromatography on a Sephadex G-200 gel filtration column (Amersham Biosciences, Freiburg, Germany). All immunoglobulin fractions were tested at 25 μg/ml for their anti-SNO-cysteine-BSA Ab titer of IgM isotype as described previously (Boullerne et al., 1995).
Statistical analyses. Serum and plasma Ab titers of rats and of MS patients were subjected to one-way ANOVA followed by a post hoc analysis using the Newman–Keuls multiple-comparison test. Values of MBP content in cerebellum were compared with the unpairedt test. The Spearman correlation, chosen because some data were not Gaussian, was used to correlate titers of anti-SNO-cysteine-BSA and anti-MBP68–84 Abs, cumulative disease scores, inflammation in spinal cord, and demyelination in spinal cord and cerebellum.
RESULTS
Circulating anti-SNO-cysteine-BSA antibodies in EAE
In the first experiment, all rats developed EAE. The time course of the clinical scores showed a biphasic pattern (Fig.1A). The mean onset of the first episode occurred at 12.6 dpi, although one rat showed extremely early clinical signs with a score of 3 at 6 dpi. Mean clinical signs peaked at 17 dpi, with an average clinical score of 2.0 ± 0.577 (mean ± SD). Recovery was almost complete at 23 dpi, with an average clinical score of 0.43 ± 0.79. A second clinical episode occurred in four of seven rats with a mean onset at 28 dpi. Relapses lasted for 1 week, followed by complete recovery. All seven rats were killed after a 6-month observation period. No CNS pathological analysis was performed.
Fig. 1.

First series of experiments on young rats aged 6–7 weeks (n = 7). A, Biphasic pattern of clinical course of EAE in 6- to 7-week-old rats. Clinical scores were graded daily for the scores 0–3 over a 35 d follow-up. Daily average clinical scores are plotted (mean ± SD).B, Serum anti-SNO-cysteine-BSA IgM titers peak 1 week after immunization in rats induced for EAE. Values are expressed as mean OD ± SEM of Ab titers. Anti-SNO-cysteine-BSA IgM titers in sera from EAE rats (open bars) were significantly elevated at 7 dpi when compared with other time points (***p < 0.001). IgG titers (closed bars) rose less than IgM titers but were nonetheless elevated at 7 dpi compared with other time points (*p < 0.05). No rise of anti-SNO-cysteine-BSA IgM was observed in the control group immunized with CFA alone (shaded bars).C, IgM binding to immobilized SNO-cysteine-BSA is inhibited by SNO-cysteine-BSA in liquid phase (competitor) in a dose-dependent manner, indicating specific binding of IgM to SNO-cysteine-BSA. Each plot is the mean OD ± SEM of two independent experiments each performed in duplicate.D, Serum Ab response against MBP68–84peptide in rats with EAE. Values are expressed as mean OD ± SEM of Ab titers obtained from two independent analyses each in duplicate. Anti-MBP68–84 IgM titers (open bars) were significantly elevated at 14 and 21 dpi when compared with other time points (*p < 0.05). Anti-MBP68–84 IgG titers (closed bars) were significantly elevated at 21 and 30 dpi (*p < 0.05).
A 10-fold increase above baseline in the IgM response to SNO-cysteine-BSA was observed at 7 dpi. The average OD of sera sampled at 7 dpi was 0.779 ± 0.163 (mean ± SEM; range, 0.332–1.296) compared with 0.081 ± 0.013 (range, 0.044–0.125;p < 0.001, post hoc analysis) on the day before immunization (Fig. 1B). The IgM response at 14, 21, and 30 d was twofold to threefold elevated compared with baseline. The 7 d value was significantly greater than at later time points (p < 0.001, post hocanalysis). Animals that relapsed showed a higher Ab titer at 7 dpi (1.024 ± 0.211, n = 4) than animals with a single episode (0.452 ± 0.070, n = 3) although no secondary rise in Ab response on day 21 preceded relapses.
The specificity of IgM binding to SNO-cysteine-BSA was demonstrated by an inhibition experiment using various concentrations of SNO-cysteine-BSA in the liquid phase. This showed displacement of Ab binding proportional to the amount of SNO-cysteine-BSA added (Fig.1C).
The IgG response to the SNO-cysteine-BSA epitope, although less dramatic than the IgM response, also peaked at 7 dpi (Fig.1B). IgG titers were threefold elevated at 7 dpi (0.288 ± 0.073; range, 0.090–0.543) when compared with baseline (0.064 ± 0.017; range, 0.028–0.150; p < 0.05,post hoc analysis). IgG titers decayed subsequently. The control group immunized with CFA alone showed neither clinical signs nor an Ab response to SNO-cysteine-BSA (Fig. 1B).
Anti-SNO-cysteine-BSA antibodies precede anti-MBP antibodies
Serum levels of anti-MBP68–84 IgM were significantly increased at 14 and 21 dpi in the EAE group when compared with other time points (p < 0.05, post hoc analysis). Anti-MBP68–84 IgG levels were significantly increased at 21 and 30 dpi (p< 0.05, post hoc analysis), as illustrated in Figure1D. No Ab response directed against MBP68–84 was observed in the control group immunized with CFA alone (data not shown).
Clinical signs and anti-SNO-cysteine-BSA antibodies
In the second EAE experiment, using rats aged 9–10 weeks, mean disease onset occurred at 15.7 dpi, 3 d later than in the first experiment. Disease was milder, as expected for older rats, with an average clinical score of 1.0 ± 1.0 (mean ± SD). There was no second episode (Fig.2A). Again, serum levels of anti-SNO-cysteine-BSA IgM at 6 dpi in the EAE group (n = 9) were significantly greater than at baseline (average OD, 0.492 ± 0.052; range, 0.315–0.720; vs average OD before immunization, 0.087 ± 0.008; range, 0.060–0.130;p < 0.001, post hoc analysis). Values were lower than in the first experiment in which disease was more severe. As in the previous study, anti-SNO-cysteine IgM peaked 1 week after immunization (Fig. 2B).
Fig. 2.
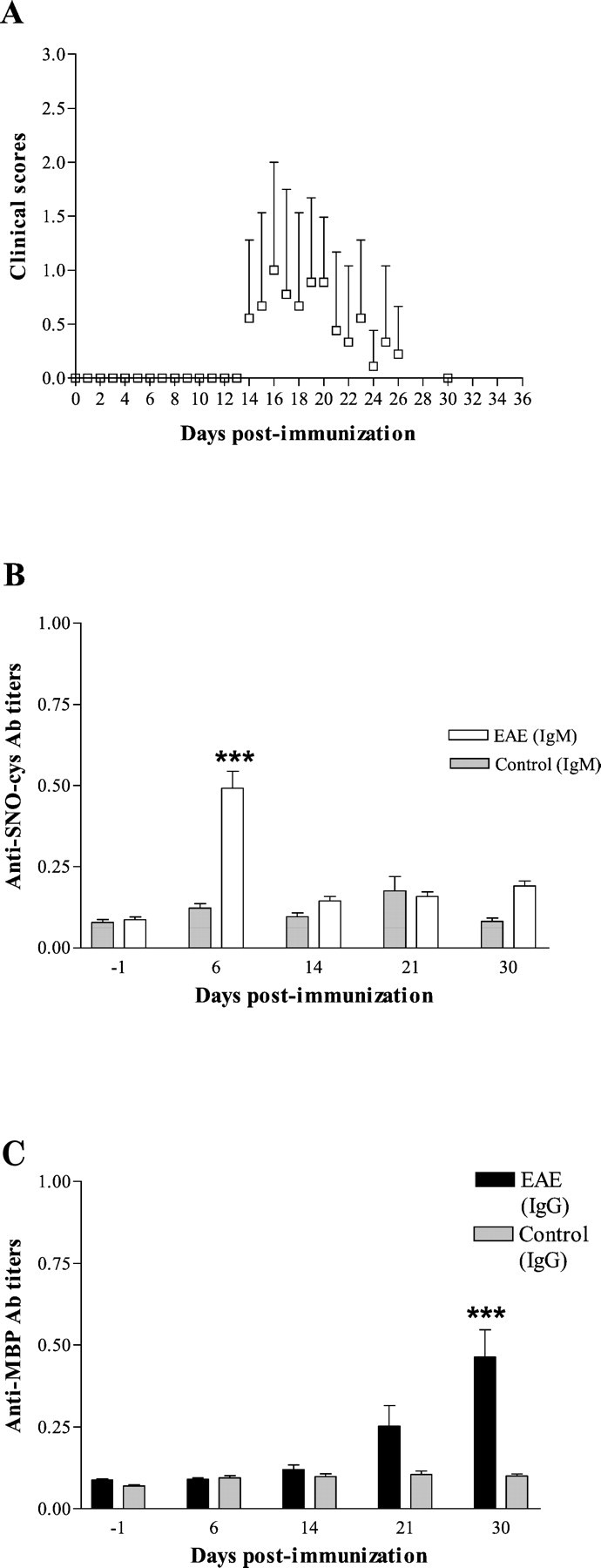
Second series of experiments on older rats aged 9–10 weeks (n = 9). A, Monophasic pattern of clinical course of EAE in 9- to 10-week-old rats showing a milder disease. Clinical scores were graded daily for the scores 0–3 over a 35 d follow-up. Daily average clinical scores are plotted (mean ± SD). B, Anti-SNO-cysteine-BSA IgM titers in sera from EAE rats (open bars) were significantly elevated at 6 dpi when compared with other time points (***p < 0.001). No rise of anti-SNO-cysteine-BSA IgM was observed in the control group immunized with CFA alone at 6 dpi (shaded bars). Values are expressed as mean OD ± SEM of Ab titers obtained from two independent analyses each in duplicate. C, Anti-MBP68–84 IgG titers (open bars) were significantly elevated at 30 dpi when compared with other time points (***p < 0.001). No rise of anti-MBP68–84 IgG titers was observed in the control group immunized with CFA alone at any time (shaded bars). Values are expressed as mean OD ± SEM of Ab titers obtained from two independent analyses each in duplicate.
Again, serum levels of anti-MBP68–84 IgG at 30 dpi in the EAE group (n = 9) were significantly greater than at baseline (average OD, 0.463 ± 0.083; range, 0.100–0.910; vs average OD before immunization, 0.088 ± 0.004; range, 0.070–0.105; p < 0.001, post hoc analysis) but were lower than in the first experiment in which disease was more severe (Fig. 2C).
To determine whether individual values for anti-SNO-cysteine-BSA or anti-MBP68–84 Ab titers correlated with subsequent clinical severity, we compared the cumulative scores of all rats (n = 16) from the two experiments with their Ab titers. There was no correlation between anti-SNO-cysteine-BSA Ab levels at any time point and cumulative clinical severity scores, nor did anti-SNO-cysteine-BSA Ab titers correlate with MBP68–84 Ab titers. There was no correlation between MBP68–84 Ab titers at any time point and cumulative clinical severity scores.
Correlation among demyelination, inflammation, and anti-SNO-cysteine-BSA antibodies
The extent of demyelination at 35 dpi, evaluated in the cervical spinal cord by Spielmeyer staining in EAE rats of the second experiment, varied substantially from rat to rat (Fig.3). A direct relationship was found between the level of anti-SNO-cysteine-BSA Abs at 6 dpi and the extent of demyelination in the spinal cord (Spearman r = 0.93;p = 0.0007; n = 9; Fig.4A) and between the level of anti-SNO-cysteine-BSA Abs at 6 dpi and the extent of inflammation in the spinal cord (r = 0.85;p = 0.006; n = 9; Fig.4B).
Fig. 3.
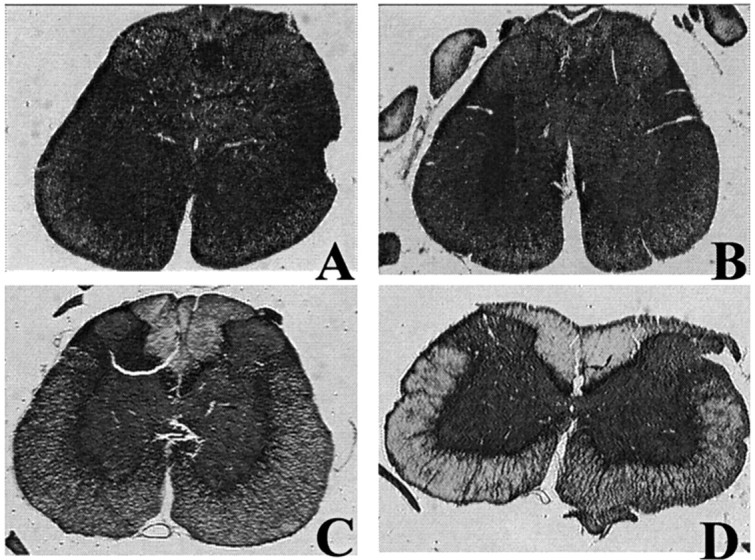
Demyelination in cervical spinal cord (C1–C6) assessed by Spielmeyer staining at 35 dpi varies from one rat to another. The ring of white matter is stained in black by Spielmeyer, whereas the central gray matter appears graybecause of the combined hematoxylin coloration. A control rat immunized with CFA alone shows no myelin loss (A), whereas rats with EAE show mild demyelination (B), moderate demyelination (C), or extensive demyelination (D).
Fig. 4.

Anti-SNO-cysteine-BSA IgM titers at 6 dpi correlate with subsequent demyelination and inflammation in spinal cord and cerebellum in rats with EAE of the second experiment (n = 9). Values of Ab titers are expressed as mean OD of two independent experiments in duplicate. A, Demyelination was assessed by Spielmeyer staining of C1–C6 spinal cord sections at 35 dpi, and demyelination values are expressed as percentage of myelin loss. A direct relationship was found between demyelination and anti-SNO-cysteine-BSA IgM titers for sera of EAE rats at 6 dpi (closed squares) with Spearman coefficientr = 0.93 (p = 0.0007).B, Inflammation was assessed by hematoxylin and eosin staining of C1–C6 spinal cord sections at 35 dpi, and values of inflammation are expressed as an inflammation index. A direct relationship was found between inflammation and anti-SNO-cysteine-BSA IgM titers for sera of rats with EAE at 6 dpi (closed squares) with Spearman coefficient r = 0.85 (p = 0.006). C, Anti-SNO-cysteine-BSA IgM titers at 6 dpi correlate with subsequent demyelination of cerebellum at 35 dpi in rats with EAE assessed by MBP content. Values of MBP content are expressed as mean OD (arbitrary units). A direct inverse relationship was found between MBP content and anti-SNO-cysteine-BSA IgM titers for sera of rats with EAE at 6 dpi (closed squares) with Spearman coefficientr = −0.82 (p = 0.011).D, Demyelination assessed by Spielmeyer staining in C1–C6 spinal cord sections correlates with demyelination of cerebellum assessed by MBP content at 35 dpi for rats with EAE (closed squares). A direct inverse relationship was found, with Spearman coefficient r = −0.87 (p = 0.0045).
The extent of myelin depletion in the cerebellum, as evaluated by quantitative immunostaining for MBP, revealed a 28% decrease in the EAE group (8.04 ± 0.52, mean ± SEM; n = 9) compared with control rats (11.12 ± 1.28; n = 5;p = 0.02, t test). A direct relationship was found between the level of anti-SNO-cysteine-BSA Abs at 6 dpi and subsequent MBP depletion in cerebellum (r = −0.82;p = 0.011; n = 9; Fig. 4C). There was a strong correlation between demyelination in spinal cord and in cerebellum in EAE rats (r = −0.87;p = 0.0045; n = 9; Fig.4D). No correlation was found between anti-MBP68–84 Abs at any time point and CNS demyelination or inflammation at 35 dpi.
Anti-SNO-cysteine-BSA antibodies in MS
Ab titers directed against SNO-cysteine-BSA varied with clinical activity (Fig. 5). Ab titers were of IgM isotype as noted previously (Boullerne et al., 1995). Ab titers are graphed as relative OD to permit for pooling of experiments using different groups of healthy controls and MS patients. All eight samples from six MS patients in relapse (one patient was studied during three relapses) showed significantly elevated titers of anti-SNO-cysteine-BSA IgM (1.879 ± 0.285, mean ± SEM; n = 8), when compared with healthy controls (0.00 ± 0.092;n = 19; p < 0.001) and with both MS groups in remission, one group treated with IFNβ-1b (0.139 ± 0.224; n = 7; p < 0.01; one patient studied twice was negative on both occasions) and the other group untreated (0.521 ± 0.238; n = 11;p < 0.05; one patient studied on four occasions was positive twice, and another was negative on two occasions).
Fig. 5.

Anti-SNO-cysteine-BSA IgM Abs are elevated during clinical activity in MS patients when compared with healthy controls (group 1; closed squares; n = 19). The ordinate gives relative optical density (see Materials and Methods). Secondary progressive MS patients were divided into an untreated group (group 2; open triangles;n = 14) and a group treated with IFNβ-1b (group 3; closed triangles; n = 9). Relapsing–remitting MS patients were classified into patients in acute relapse and untreated (group 4; open diamonds;n = 8) and in remission and receiving IFNβ-1b treatment (group 5; closed circles;n = 7) or in remission without treatment (group 6;open circles; n = 11). Relative ODs were significantly elevated in some MS groups: group 1 versus groups 2 and 4 (p < 0.001) and versus group 3 (p < 0.05); group 2 versus groups 5 (p < 0.001) and 6 (p < 0.01); and group 4 versus groups 5 (p < 0.01) and 6 (p < 0.05).
Two-thirds of secondary progressive MS patient samples (SPMS) showed elevated titers of anti-SNO-cysteine-BSA IgM whether treated with IFNβ-1b (1.107 ± 0.396; n = 9) or untreated (1.847 ± 0.375; n = 14). These elevations were significant when compared with the control group (untreated SPMS,p < 0.001; IFNβ-treated SPMS, p < 0.05) and with the two groups in remission (untreated SPMS vs IFNβ-treated patients in remission, p < 0.001; untreated SPMS vs untreated patients in remission, p < 0.01). Four untreated SPMS patients were studied serially, one on five occasions (patient A), one on three occasions (patient B), and two others twice (patients C and D). Eleven of 14 samples (79%) were positive, including 5 of 5 samples of patient A, 3 of 3 samples of patient B, 1 of 2 samples of patient C, and 0 of 2 samples of patient D. Eight SPMS patients receiving IFNβ were studied; one patient was studied twice. Five of 9 samples (55%) were positive, including both from the patient studied twice.
Because a correlation was found in sera of MS patients (but not in healthy donors) between total IgM levels and anti-SNO-cysteine IgM titers in our previous study (Boullerne et al., 1995), we attempted to rule out a nonspecific increase in anti-SNO-cysteine Ab simply attributable to IgM hyperglobulinemia. We measured anti-SNO-cysteine Ab titers in IgM purified from sera of four MS patients in relapse and from four healthy donors, all at the same protein concentration. A marked increase of anti-SNO-cysteine-BSA Ab titer was found in IgM purified from MS sera compared with IgM purified from healthy donor sera (Fig. 6).
Fig. 6.

Anti-SNO-cysteine-BSA IgM Abs are enriched in the IgM of MS patients. Immunoglobulin fractions were tested at a protein concentration of 25 μg/ml. Anti-SNO-cysteine-BSA IgM is elevated in total IgM purified from the serum of an MS patient during relapse on a gel filtration column (closed squares) compared with a healthy donor (open squares). The mean of two independent experiments in duplicate is plotted. MS and control sera representative of four MS and four healthy donor sera tested are shown.
Specificity of anti-SNO-cysteine-BSA antibodies in MS
To characterize the specificity of circulating IgM toward the nitrosylated cysteine residue linked to the BSA carrier by a five-carbon chain (glutaraldehyde linking), binding inhibition experiments were performed. First, sera from three MS patients were incubated with the following competitors: SNO-cysteine-BSA, SNO-cysteine-BSA treated with HgCl2 to abolish nitrosylation, and cysteine-BSA (a homolog for SNO-cysteine-BSA). Binding of MS serum IgM to SNO-cysteine-BSA absorbed on plates was inhibited by 50% (IC50) after exposure to 4 × 10−7m soluble SNO-cysteine-BSA (Fig. 7A). HgCl2-treated SNO-cysteine-BSA inhibited binding poorly, as did cysteine-BSA, showing that nitrosylation is essential for binding of IgM to SNO-cysteine-BSA.
Fig. 7.
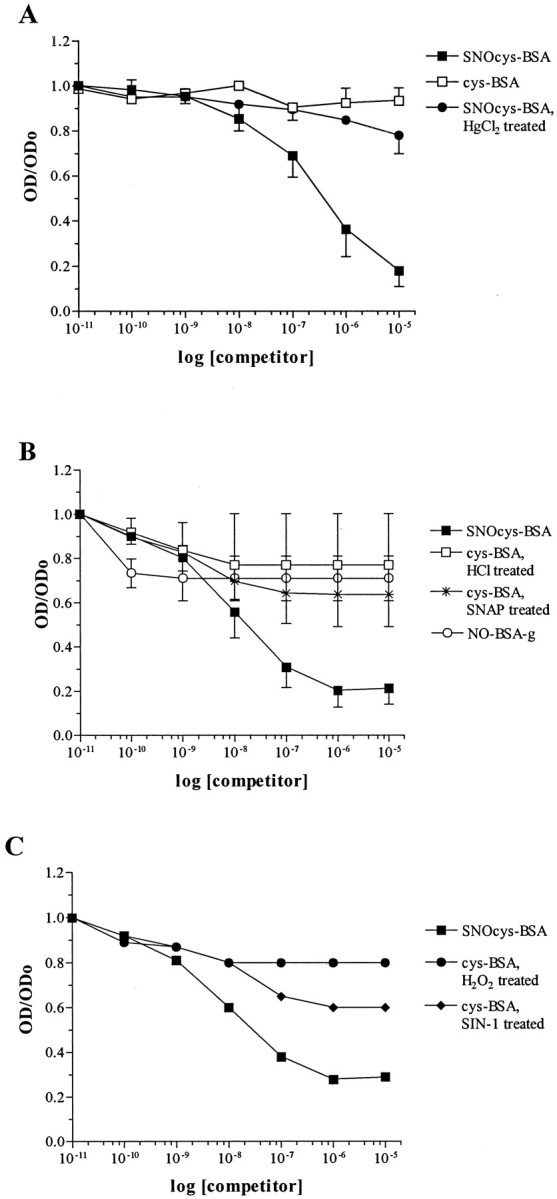
MS sera are specific for the SNO-cysteine epitope. IgM binding to immobilized SNO-cysteine-BSA is inhibited by a competitor in the liquid phase in a dose-dependent manner. Binding inhibition is calculated by dividing OD of each competitor concentration by OD without competitor (ODo). A, Specific binding of IgM to immobilized SNO-cysteine-BSA (closed squares) with IC50 at 4 × 10−7m. Absence of nitrosylation, i.e., with cysteine-BSA (open squares) or SNO-cysteine-BSA treated with HgCl2 (closed circles), prevented IgM binding inhibition. Each plot is the average OD ± SD of three sera tested in two independent experiments, each performed in duplicate. B, Specific binding of IgM to immobilized SNO-cysteine-BSA (closed squares) with IC50 at 2 × 10−8m. Conformational modifications of the BSA carrier induced by acidic nitrosylation (NO-BSA-g; open circles) or cysteine-BSA treated with HCl treatment (open squares) or of cysteine-BSA nitrosylated by SNAP (stars) did not abolish binding. Eachplot is the average OD ± SD of three other sera tested in two independent experiments, each performed in duplicate.C, Other oxidative modifications of cysteine-BSA induced by the peroxynitrite donor SIN-1 (closed diamonds) or by hydrogen peroxide (closed circles) did not inhibit IgM binding to immobilized SNO-cysteine-BSA (closed squares). Each plot is the average OD of one serum tested in two independent experiments, each performed in duplicate.
Additional inhibition experiments were performed using sera from 3 additional MS patients and the following competitors: SNO-cysteine-BSA, cysteine-BSA treated with HCl (a control for acidic nitrosylation by NaNO2), cysteine-BSA treated with the NO donor SNAP, cysteine-BSA treated with the peroxynitrite donor SIN-1, cysteine-BSA treated with H2O2, and BSA-g nitrosylated by NaNO2 (NO-BSA-g). Again, the strongest avidity was observed for SNO-cysteine-BSA, with an IC50 of 2 × 10−8m (Fig.7B,C). The other competitors failed to inhibit binding at >50%, even at 10−5m. The findings indicate a high degree of specific binding to SNO-cysteine-BSA for circulating IgM from MS patients.
One MS serum was blotted on the following nitrosylated molecules: SNO-cysteine-BSA, NO-BSA-g, DTT-treated SNO-cysteine-BSA, DTT-treated NO-BSA-g, and HgCl2-treated SNO-cysteine-BSA. Western Blots were performed under low SDS conditions to preserve nitrosylation. Preferential binding to SNO-cysteine-BSA was observed (Fig. 8). Some staining was observed after treatment of SNO-cysteine-BSA with DTT (2 hr at 4°C), but only faint staining remained after treatment of SNO-cysteine-BSA with HgCl2, the most efficient treatment for destroying nitrosylation. NO-BSA-g was not stained. Again, binding of serum IgM to nitrosylated cysteine residues linked to BSA with glutaraldehyde was highly selective.
Fig. 8.

Western blot of one MS serum on various nitrosylated antigens showed specificity for SNO-cysteine-BSA. Proteins were loaded at 6 μg/lane. SNO-cysteine-BSA (lane 1) was well stained; NO-BSA-g (lane 2) and DTT-treated NO-BSA-g (lane 4) were not recognized; DTT-treated SNO-cysteine-BSA (lane 3) was stained; and HgCl2-treated SNO-cysteine-BSA (lane 5) was stained faintly.
Stability of SNO-cysteine-BSA
Because nitrosylated compounds are often labile, we examined the stability of nitrosylated SNO-cysteine-BSA kept at room temperature and exposed to light over 15 d. We used two methods to evaluate nitrosylation. We assessed nitrosylation directly by scanning a solution of SNO-cysteine-BSA (1.26 mg/ml in 0.1× PBS) from 240 to 500 nm and recording the OD at 400 nm (OD400), the peak of absorbance of nitrosylation. A 35% decrease of OD400 was observed after a 24 hr exposure to room temperature and light (OD decreased from 0.270 to 0.176). OD400 decreased further to 0.135 over the following 14 d. After complete denitrosylation (accomplished by incubation with 10 mm DTT at room temperature for 3 d), OD400 was at background values. The above indicates that there was an ∼50% loss of detectable nitrosylation over 2 weeks at room temperature.
We also used the Griess assay to measure nitrite and nitrate release from the SNO-cysteine-BSA solution described above. An increase of released nitrite and nitrate was observed over a 13 d exposure to room temperature and to light, increasing from 30 μm at day 1 to 49 μm at day 13 (Fig.9). In contrast, complete denitrosylation followed incubation with 10 mm DTT for 3 d at room temperature and resulted in release of 154 μm nitrite and nitrate, close to the maximal expected value of 159 μm. Treatment with 10 mm DTT for 2 hr resulted in only partial release of nitrite and nitrate, in keeping with the Ab binding observed in blotting experiments when SNO-cysteine-BSA was treated for 2 hr with 10 mm DTT at 4°C (Fig. 8). Collectively, these observations indicate that nitrosylation of SNO-cysteine-BSA is adequately conserved during the 1–2 d interval required to coat the ELISA plates and perform the assay.
Fig. 9.

Time course study of nitrite and nitrate release from SNO-cysteine-BSA on exposure to light and room temperature. Nitrate was enzymatically reduced into nitrite before evaluation by Griess assay. Nitrite levels (open bars) and combined nitrite and nitrate levels (NO ; shaded bars) released from an SNO-cysteine-BSA solution rose slowly from day 1 to day 13 (13 d). Incubation of SNO-cysteine-BSA with 10 mm DTT for 2 hr on day 22 (22 d) markedly increased nitrite and nitrate levels. After 3 d exposure to 10 mm DTT (25d), complete denitrosylation was achieved. Nitrite or nitrate levels were not detected at any time point in the cysteine-BSA control.
Origin of anti-SNO-cysteine-BSA antibodies in MS
To assess the origin of anti-SNO-cysteine-BSA antibodies in MS, we tested pairs of CSF and sera from MS patients. Circulating anti-SNO-cysteine-BSA antibody titers were elevated in 10 of 25 sera. Anti-SNO-cysteine-BSA antibodies were detected in only one CSF. The OD in this sample CSF diluted 1:2 was 0.207, and in the serum diluted 1:400 it was 0.219. We conclude that anti-SNO-cysteine-BSA antibody does not originate in the CNS.
DISCUSSION
We report an Ab response against an uncommon epitope, SNO-cysteine-BSA, in sera from Lewis rats with actively induced EAE. A similar Ab response is identified in MS. Titers of IgM Abs to SNO-cysteine-BSA did not correlate with the severity of clinical disease in EAE but did correlate with the extent of demyelination measured after clinical recovery, suggesting a possible role for anti-SNO-cysteine-BSA Abs in the demyelinative process.
A concern related to the stability of SNO-cysteine-BSA. The compound was shown to be stable over time. Generation of SNO-cysteine-BSA introduces conformational changes to the carrier protein BSA; therefore, it became important to establish that the epitope recognized was indeed nitrosylated cysteine. During the first step of SNO-cysteine-BSA generation, cysteine is linked to BSA by glutaraldehyde. This step would be expected to cross-link BSA itself. During the second step, chemical nitrosylation of cysteine might additionally nitrosylate tyrosine residues of the BSA carrier, possibly creating nitrotyrosine. We excluded an Ab response against glutaraldehyde-treated BSA and against BSA nitrosylated directly. Western blots failed to show binding of IgM to the modified BSA carriers. These negative results were reinforced by the failure of glutaraldehyde-treated BSA and nitrosylated BSA to inhibit IgM binding to SNO-cysteine-BSA bound to ELISA plates.
Another concern was that the Ab response might have been directed against some other oxidation product derived from the linked cysteine. Inhibition experiments established that the IgM response was specific for SNO-cysteine-BSA. The above supports the idea that the epitope recognized on SNO-cysteine-BSA conjugates is the nitrosylated cysteine moiety.
Our finding of anti-SNO-cysteine Abs provides evidence for the formation in vivo of S-nitroso complexes and for the development of Abs directed against new epitopes generated by NO-related species-mediated nitrosylation. NO-related species-mediated new epitope formation accounts for the finding of nitrotyrosine on proteins in CNS lesions both in MS and in active EAE (Hooper et al., 1997; Cross et al., 1998). Unlike nitrotyrosine in the CNS, the anti-SNO-cysteine-BSA Abs documented herein in blood peaked well before the onset of clinical signs or CNS inflammation, suggesting a potential role for them as a predictor of subsequent disease activity.
The anti-SNO-cysteine Abs must be directed against a peripherally expressed epitope. We found IgM anti-SNO-cysteine Abs in the CSF in only 1 of 25 MS cases, arguing strongly against a CNS origin for these Abs, at least in humans. Levels of the NO-derived products nitrite and nitrate increase in the plasma within 24 hr of immunization with MBP in rats (Cowden et al., 1998; O'Brien et al., 1999), indicating brisk early peripheral NO production and by extension prompt epitope expression. In contrast, iNOS mRNA in the CNS was first detected 4 d before disease onset in rodents immunized with MBP (Koprowski et al., 1993; Okuda et al., 1995). The peripheral origin of anti-SNO-cysteine IgM Abs is further supported by their detection in sera of rats with adjuvant arthritis and no CNS inflammation, albeit at a lower intensity than in rats with EAE (A. I. Boullerne, unpublished observations). Adjuvant arthritis is preceded by early production of NO, raised plasma levels of nitrite and nitrate, and elevated iNOS expression in the spleen (Fletcher et al., 1998). All of these appear preclinically, just as in EAE.
A second component of the humoral immune response, driven by MBP itself, was observed. IgM Abs directed against the MBP68–84 peptide peaked 2–3 weeks after immunization, as found previously in rats immunized with MBP68–84 peptide or with MBP (Kibler et al., 1977; Fesel and Coutinho, 1998). Note that the anti-SNO-cysteine-BSA IgM response peaked well before the anti-MBP IgM response, suggesting that the former was a natural Ab response (Avrameas and Ternynck, 1993). An IgG response against SNO-cysteine-BSA also occurred conjointly with the IgM response but was less intense, as has been reported previously for natural auto-Abs (Matsiota-Bernard et al., 1993). The early conjoint appearance of anti-SNO-cysteine-BSA IgM and IgG is typical for a natural Ab response known to be both early and chiefly of the IgM isotype, with lesser IgG isotype representation. In mice immunized with bacteria, natural Abs to DNA and to tubulin peak well ahead of specific antibacterial Abs (Matsiota-Bernard et al., 1993), in keeping with the time courses of anti-SNO-cysteine-BSA Abs and anti-MBP Abs observed here.
Anti-MBP68–84 Abs did not correlate with cumulative clinical signs in this study, as in numerous previous studies in EAE. Anti-SNO-cysteine-BSA antibodies similarly did not correlate with cumulative clinical signs but did correlate with demyelination and inflammation.
The role of circulating anti-SNO-cysteine Abs in EAE remains unknown. This stated, the correlation of anti-SNO-cysteine Abs at day 6 with subsequent demyelination argues against an epiphenomenon. Demyelination occurs in the EAE model that we used and has been noted by others using similar immunization protocols within the same CNS areas studied by us (Pender, 1987; Chalk et al., 1994). Anti-SNO-cysteine Abs may contribute to demyelination, provided that they reach the CNS and that cysteine residues on CNS proteins are nitrosylated during inflammation within the CNS. Abs in the blood can reach the CNS parenchyma if there is a breakdown of the blood–brain barrier (BBB). They can also be actively transported by brain endothelial cells. IgM has been found within vesicles in endothelial cells within MS lesions, providing evidence for active transport of circulating IgM across the BBB (Gay and Esiri, 1991). As an example of circulating Ab-mediated central demyelination, we note that peripherally injected monoclonal Abs directed against myelin oligodendrocyte glycoprotein trigger demyelination in Lewis rats in which EAE has been adoptively transferred with MBP-activated T-cells (Linington et al., 1988;Piddlesden et al., 1993).
Alternatively, a protective role can be envisaged for anti-SNO-cysteine IgM over the longer term. Ab-facilitated clearance of damaged myelin may favor subsequent remyelination. Germ line rodent and human (“natural”) IgMκ Abs promote remyelination within the CNS in the Theiler's virus-induced murine model of demyelination (Asakura et al., 1998; Warrington et al., 2000). The Abs recognize various oligodendrocyte antigens, including MBP, the human natural killer-1 epitope, galactocerebroside, and sulfatide. In mice with adoptively transferred EAE, treatment with a germ line IgMκ Ab that recognizes an uncharacterized oligodendrocyte surface antigen lessens both CNS inflammation and demyelination (Miller et al., 1997).
Our MS patients were classified as having relapsing–remitting MS (RRMS) in relapse, RRMS deemed stable clinically, and SPMS. All 8 samples obtained during relapses had elevated SNO-cysteine-BSA Ab titers. In the untreated group of stable RRMS patients, 5 of 11 samples were positive. Only 1 of 7 samples was positive for Abs in the group of stable RRMS patients receiving IFNβ. Stable RRMS patients were less commonly anti-SNO-cysteine-BSA Ab-positive if they were receiving IFNβ (1 of 7 samples, 14%) than if they were untreated (5 of 11 samples, 45%). IFNβ treatment lessens both overt and subclinical attack frequency in RRMS so that this difference might have been anticipated. Overall, 6 of 18 samples (33%) from clinically stable RRMS patients were positive against 8 of 8 samples (100%) from RRMS patients in relapse. The difference between relapsing and clinically stable patients is significant. In our previous study (Boullerne et al., 1995), 38% of MS patients with relapsing–remitting disease in long-term remission tested positive versus 33% in the present study. The other groups in the previous study included primary progressive MS, positive at 50% and not studied here, patients with stabilized previously progressive MS, positive at 24% and not studied here, and a group of patients, some in relapse and others that had had an attack within the preceding months (unlike the relapsing patients studied here who were all in the midst of an attack), positive at 41%.
In SPMS, IFNβ treatment had little effect on anti-SNO-cysteine Ab positivity. The untreated group of SPMS patients had 79% positive samples for Abs, and the group of SPMS patients treated with IFNβ had 55% positive samples. Thus, 16 of 23 samples (70%) from SPMS patients were positive overall. IFNβ treatment fails to slow disability progression in SPMS, so that the failure of IFNβ to favorably affect anti-SNO-cysteine Abs in SPMS is in keeping with clinical experience.
Natural Ab titers rise promptly in humans during infectious illnesses (Avrameas and Ternynck, 1993). Viral infections frequently precede MS relapses (Sibley et al., 1985) and may have driven the anti-SNO-cysteine Ab elevations seen by us during MS relapses. Eighty-six percent of MS patients tested positive for anti-DNA, antinuclear, and anti-organ-specific autoantibodies during acute attacks. Only 30% remained positive during remission, whereas 92% of SPMS patients were autoantibody-positive (Spadaro et al., 1999). Thus, there is precedent for our finding of an antibody that correlates with MS disease activity.
Anti-SNO-cysteine Ab titers are consistently elevated during MS attacks and are elevated in one-third of samples from patients with clinically stable disease and in two-thirds of samples from patients with SPMS. Anti-SNO-cysteine Abs may provide a marker for disease activity in MS.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale, Institut Fédératif de Recherche des Neurosciences Cliniques et Expérimentales, Région d'Aquitaine, Association pour la Recherche Médicale en Aquitaine (K.G.P.), grants from the National Multiple Sclerosis Society and Berlex Inc., the Alan Friend Multiple Sclerosis Research Fund, and gifts from the Mills Family Philanthropic Fund and the Butz Foundation (B.G.W.A.). A.I.B. and M.A.J. are fellows of the National Multiple Sclerosis Society. T.T. is recipient of a PhD fellowship from the Ligue Française contre la Sclerose en Plaques. We thank Dr. C. Gerin (University of Chicago) for thoughtful reading of this manuscript.
Correspondence should be addressed to Anne I. Boullerne, Department of Neurology, University of Chicago, MC2030, 5841 South Maryland Avenue, Chicago, IL 60637. E-mail: abouller@neurology.bsd.uchicago.edu.
REFERENCES
- 1.Asakura K, Miller DJ, Pease LR, Rodriguez M. Targeting of IgMkappa antibodies to oligodendrocytes promotes CNS remyelination. J Neurosci. 1998;18:7700–7708. doi: 10.1523/JNEUROSCI.18-19-07700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avrameas S, Ternynck T. The natural autoantibodies system: between hypotheses and facts. Mol Immunol. 1993;30:1133–1142. doi: 10.1016/0161-5890(93)90160-d. [DOI] [PubMed] [Google Scholar]
- 3.Boullerne AI, Petry KG, Meynard M, Geffard M. Indirect evidence for nitric oxide involvement in multiple sclerosis by characterization of circulating antibodies directed against conjugated S-nitrosocysteine. J Neuroimmunol. 1995;60:117–124. doi: 10.1016/0165-5728(95)00061-6. [DOI] [PubMed] [Google Scholar]
- 4.Boullerne AI, Nedelkoska L, Benjamins JA. Role of calcium in nitric oxide-induced cytotoxicity: EGTA protects mouse oligodendrocytes. J Neurosci Res. 2001;63:124–135. doi: 10.1002/1097-4547(20010115)63:2<124::AID-JNR1004>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 5.Brundin L, Morcos E, Olsson T, Wiklund NP, Andersson M. Increased intrathecal nitric oxide formation in multiple sclerosis; cerebrospinal fluid nitrite as activity marker. Eur J Neurol. 1999;5:585–590. doi: 10.1046/j.1468-1331.1999.650585.x. [DOI] [PubMed] [Google Scholar]
- 6.Chalk JB, McCombe PA, Smith R, Pender MP. Clinical and histological findings in proteolipid protein-induced experimental autoimmune encephalomyelitis (EAE) in the Lewis rat. Distribution of demyelination differs from that in EAE induced by other antigens. J Neurol Sci. 1994;123:154–161. doi: 10.1016/0022-510x(94)90218-6. [DOI] [PubMed] [Google Scholar]
- 7.Cowden WB, Cullen FA, Staykova MA, Willenborg DO. Nitric oxide is a potential down-regulating molecule in autoimmune disease: inhibition of nitric oxide production renders PVG rats highly susceptible to EAE. J Neuroimmunol. 1998;88:1–8. doi: 10.1016/s0165-5728(98)00040-x. [DOI] [PubMed] [Google Scholar]
- 8.Cross AH, Manning PT, Keeling RM, Schmidt RE, Misko TP. Peroxynitrite formation within the central nervous system in active multiple sclerosis. J Neuroimmunol. 1998;88:45–56. doi: 10.1016/s0165-5728(98)00078-2. [DOI] [PubMed] [Google Scholar]
- 9.De Castro F, Ramon y Cajal S. Elementos de técnica micrografica del sistema nervioso. Tipografia Artistica; Madrid: 1933. [Google Scholar]
- 10.De Groot CJ, Ruuls SR, Theeuwes JW, Dijkstra CD, Van der Valk P. Immunocytochemical characterization of the expression of inducible and constitutive isoforms of nitric oxide synthase in demyelinating multiple sclerosis lesions. J Neuropathol Exp Neurol. 1997;56:10–20. doi: 10.1097/00005072-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Drulovic J, Dujmovic I, Mesaros S, Samardzic T, Maksimovic D, Stojsavljevic N, Levic Z, Mostarica Stojkovic M. Raised cerebrospinal fluid nitrite and nitrate levels in patients with multiple sclerosis: no correlation with disease activity. Mult Scler. 2001;7:19–22. doi: 10.1177/135245850100700104. [DOI] [PubMed] [Google Scholar]
- 12.Fesel C, Coutinho A. Dynamics of serum IgM autoreactive repertoires following immunization: strain specificity, inheritance and association with autoimmune disease susceptibility. Eur J Immunol. 1998;28:3616–3629. doi: 10.1002/(SICI)1521-4141(199811)28:11<3616::AID-IMMU3616>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher DS, Widmer WR, Luell S, Christen A, Orevillo C, Shah S, Visco D. Therapeutic administration of a selective inhibitor of nitric oxide synthase does not ameliorate the chronic inflammation and tissue damage associated with adjuvant-induced arthritis in rats. J Pharmacol Exp Ther. 1998;284:714–721. [PubMed] [Google Scholar]
- 14.Furlan R, Filippi M, Bergami A, Rocca MA, Martinelli V, Poliani PL, Grimaldi LM, Desina G, Comi G, Martino G. Peripheral levels of caspase-1 mRNA correlate with disease activity in patients with multiple sclerosis; a preliminary study. J Neurol Neurosurg Psychiatry. 1999;67:785–788. doi: 10.1136/jnnp.67.6.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gay D, Esiri M. Blood-brain barrier damage in acute multiple sclerosis plaques. An immunocytological study. Brain. 1991;114:557–572. doi: 10.1093/brain/114.1.557. [DOI] [PubMed] [Google Scholar]
- 16.Hess DT, Matsumoto A, Nudelman R, Stamler JS. S-Nitrosylation: spectrum and specificity. Nat Cell Biol. 2001;3:E46–E49. doi: 10.1038/35055152. [DOI] [PubMed] [Google Scholar]
- 17.Hooper DC, Bagasra O, Marini JC, Zborek A, Ohnishi ST, Kean R, Champion JM, Sarker AB, Bobroski L, Farber JL, Akaike T, Maeda H, Koprowski H. Prevention of experimental allergic encephalomyelitis by targeting nitric oxide and peroxynitrite: implications for the treatment of multiple sclerosis. Proc Natl Acad Sci USA. 1997;94:2528–2533. doi: 10.1073/pnas.94.6.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kibler RF, Fritz RB, Chou F, Jen Chou C-H, Peacocke NY, Brown NM, McFarlin DE. Immune response of Lewis rats to peptide C1 (residues 68–88) of guinea pig and rat myelin basic proteins. J Exp Med. 1977;146:1323–1331. doi: 10.1084/jem.146.5.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khoury SJ, Guttmann CR, Orav EJ, Kikinis R, Jolesz FA, Weiner HL. Changes in activated T cells in the blood correlate with disease activity in multiple sclerosis. Arch Neurol. 2000;57:1183–1189. doi: 10.1001/archneur.57.8.1183. [DOI] [PubMed] [Google Scholar]
- 20.Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fang Fu Z, Hanlon C, Dietzschold B. In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic diseases. Proc Natl Acad Sci USA. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liedtke W, Cannella B, Mazzaccaro RJ, Clements JM, Miller KM, Wucherpfennig KW, Gearing AJ, Raine CS. Effective treatment of models of multiple sclerosis by matrix metalloproteinase inhibitors. Ann Neurol. 1998;44:35–46. doi: 10.1002/ana.410440110. [DOI] [PubMed] [Google Scholar]
- 22.Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. 1988;130:443–454. [PMC free article] [PubMed] [Google Scholar]
- 23.Mannie MD, Paterson PY, U'Prichard DC, Flouret G. Induction of experimental allergic encephalomyelitis in Lewis rats with purified synthetic peptides: delineation of antigenic determinants for encephalitogenicity, in vitro activation of cellular transfer, and proliferation of lymphocytes. Proc Natl Acad Sci USA. 1985;82:5515–5519. doi: 10.1073/pnas.82.16.5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsiota-Bernard P, Mahana W, Avrameas S, Nauciel C. Specific and natural antibody production during Salmonella typhimurium infection in genetically susceptible and resistant mice. Immunology. 1993;79:375–380. [PMC free article] [PubMed] [Google Scholar]
- 25.Miller DJ, Bright JJ, Sriram S, Rodriguez M. Successful treatment of established relapsing experimental autoimmune encephalomyelitis in mice with a monoclonal natural autoantibody. J Neuroimmunol. 1997;75:204–209. doi: 10.1016/s0165-5728(97)00027-1. [DOI] [PubMed] [Google Scholar]
- 26.Noseworthy JH. Progress in determining the causes and treatment of multiple sclerosis. Nature. 1999;399:A40–A47. doi: 10.1038/399a040. [DOI] [PubMed] [Google Scholar]
- 27.O'Brien NC, Charlton B, Cowden WB, Willenborg DO. Nitric oxide plays a critical role in the recovery of Lewis rats from experimental autoimmune encephalomyelitis and the maintenance of resistance to reinduction. J Immunol. 1999;163:6841–6847. [PubMed] [Google Scholar]
- 28.Okuda Y, Nakatsuji Y, Fujimura H, Esumi H, Ogura T, Yanagihara T, Sakoda S. Expression of the inducible isoform of nitric oxide synthase in the central nervous system of mice correlates with the severity of actively induced experimental allergic encephalomyelitis. J Neuroimmunol. 1995;62:103–112. doi: 10.1016/0165-5728(95)00114-h. [DOI] [PubMed] [Google Scholar]
- 29.Pender MP. Demyelination and neurological signs in experimental allergic encephalomyelitis. J Neuroimmunol. 1987;15:11–24. doi: 10.1016/0165-5728(87)90003-8. [DOI] [PubMed] [Google Scholar]
- 30.Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol. 1993;143:555–564. [PMC free article] [PubMed] [Google Scholar]
- 31.Poser CM, Paty DW, Scheinberg GL, MacDonald I, Davis F, Ebergs G, Johnson K, Sibley WA, Silberberg DH, Tourtelotte WW. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227–231. doi: 10.1002/ana.410130302. [DOI] [PubMed] [Google Scholar]
- 32.Rieckmann P, Altenhofen B, Riegel A, Kallmann B, Felgenhauer K. Correlation of soluble adhesion molecules in blood and cerebrospinal fluid with magnetic resonance imaging activity in patients with multiple sclerosis. Mult Scler. 1998;4:178–182. doi: 10.1177/135245859800400317. [DOI] [PubMed] [Google Scholar]
- 33.Sarchielli P, Orlacchio A, Vicinanza F, Pelliccioli GP, Tognoloni M, Saccardi C, Gallai V. Cytokine secretion and nitric oxide production by mononuclear cells of patients with multiple sclerosis. J Neuroimmunol. 1997;80:76–86. doi: 10.1016/s0165-5728(97)00136-7. [DOI] [PubMed] [Google Scholar]
- 34.Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst. 1958;83:670–672. [Google Scholar]
- 35.Sibley WA, Bamford CR, Clark K. Clinical viral infections and multiple sclerosis. Lancet. 1985;1:1313–1315. doi: 10.1016/S0140-6736(85)92801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith KJ, Kapoor R, Hall SM, Davies M. Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol. 2001;49:470–476. [PubMed] [Google Scholar]
- 37.Spadaro M, Amendolea MA, Mazzucconi MG, Fantozzi R, Di Lello R, Zangari P, Masala G. Autoimmunity in multiple sclerosis: study of a wide spectrum of autoantibodies. Mult Scler. 1999;5:121–125. doi: 10.1177/135245859900500209. [DOI] [PubMed] [Google Scholar]
- 38.Svenningsson A, Petersson A-S, Andersen O, Hansson GK. Nitric oxide metabolites in CSF of patients with MS are related to clinical disease course. Neurology. 1999;53:1880–1882. doi: 10.1212/wnl.53.8.1880. [DOI] [PubMed] [Google Scholar]
- 39.Trojano M, Avolio C, Ruggieri M, Defazio G, Giuliani F, Paolicelli D, Livrea P. Serum soluble intercellular adhesion molecule-I in MS: relation to clinical and Gd-MRI activity and to rIFN beta-Ib treatment. Mult Scler. 1998;4:183–187. doi: 10.1177/135245859800400318. [DOI] [PubMed] [Google Scholar]
- 40.Vandenbark AA, Nilaver G, Konat G, Teal P, Offner H. Chronic neurologic dysfunction and demyelination induced in Lewis rats by repeated injections of encephalitogenic T-lymphocyte lines. J Neurosci Res. 1986;16:643–656. doi: 10.1002/jnr.490160406. [DOI] [PubMed] [Google Scholar]
- 41.Warrington AE, Asakura K, Bieber AJ, Ciric B, Van Keulen V, Kaveri SV, Kyle RA, Pease LR, Rodriguez M. Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc Natl Acad Sci USA. 2000;97:6820–6825. doi: 10.1073/pnas.97.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]