

Abstract
Recently, NMDA receptors (NMDARs) have been implicated in a cell contact-dependent suppression of sprouting in culturedXenopus tectal neurons during an early period when neither AMPA/kainate (KA) receptors nor action potentials play a prominent role in cell–cell communication. We asked how the NMDA receptors function in the absence of the depolarizing effect of AMPA/KA receptor activity. We show that type II metabotropic glutamate receptors (mGluRs) can operate synergistically with NMDA receptors in the absence of AMPA/KA receptor function to suppress an early neurite sprouting response of the tectal neurons. Calcium imaging with fluo-3 AM and morphological analyses after exposure to glutamate receptor antagonists show that a combination of AMPA/KA receptor and type II mGluR blockers mimics the decrease in intracellular free calcium in response to glutamate and the structural effects produced by NMDA receptor antagonists in these cultures. Patch-clamp analyses confirmed a decrease in NMDA receptor-mediated currents with type II mGluR blockade, and 8-bromo cAMP application produced a decrease in NMDA receptor-mediated calcium influx. These data suggest that type II mGluRs potentiate NMDA receptor function by decreasing cAMP levels in tectal neurons. We also show that NMDARs exhibit low magnesium sensitivity in tectal neurons during the first few days in culture. Thus both metabotropic and ionotropic glutamate receptors can play a role in the contact-mediated suppression of ongoing sprouting at early neuron–neuron contacts before action potential activity.
Keywords: Xenopus, synaptogenesis, sprouting, AMPA, calcium, cAMP, electrophysiology
Glutamate receptors are the most common excitatory neurotransmitter receptors in the brain. There are three major types of glutamate receptors, each believed to play a somewhat different role in development. AMPA/kainate receptors (AMPA/KARs) are required for rapid depolarization during glutamate-mediated transmission and pass the majority of ions during synaptic events in the mature brain (Hollmann and Heinemann, 1994). NMDA receptors (NMDARs) are slower channels that require glutamate binding and depolarization (initiated by AMPA/KARs) to dislodge a Mg2+ ion. This allows Ca2+ to enter the cell and mediates the role of the NMDAR in synaptic plasticity (Burnashev, 1998). Metabotropic glutamate receptors (mGluRs) are heterotrimeric G-protein-coupled receptors and are believed to modulate transmitter release (Cochilla and Alford, 1998), calcium oscillations (Whittington et al., 1995), and some use-dependent changes in synaptic currents (Rahman and Neuman, 1996; Grover and Yan, 1999).
Electrical activity modulates neuron growth and synapse formation primarily by regulating calcium levels in the cell (Mattson et al., 1988; Constantine-Paton and Cline, 1998). Neuronal calcium levels have been implicated in neurite spine formation (Segal et al., 2000; Wu et al., 2001), growth cone turning (Hong et al., 2000; Zheng, 2000;Gomez et al., 2001), and gene transcription (Hardingham et al., 1999, 2001). Inhibition of the NMDAR increases neurite outgrowth in several systems (Lin and Constantine-Paton, 1998; McKinney et al., 1999), and spontaneous calcium oscillations have also been implicated in the early morphological differentiation of neurons (Gu and Spitzer, 1995, 1997) and axon pathfinding (Gomez and Spitzer, 1999).
In many of the preparations in which Ca2+effects on morphology have been studied, the initial events underlying synapse formation have already occurred. Using a Xenopustectal neuron culture system, we showed previously that cell–cell contacts mediated NMDAR-dependent calcium changes, and NMDARs suppressed sprouting in early cultures before action potentials or glutamatergic synaptic currents are detected (Lin and Constantine-Paton, 1998). The sprout suppression was contact dependent, because NMDAR blockade had no effect on sprouting of isolated cells in the same fields. Unexpectedly, blocking of AMPA/KARs did not alter sprouting in contacted cells, although combined AMPA/KAR and NMDAR blockade completely eliminated the calcium response to glutamate. This raised the question of how NMDAR-mediated Ca2+ currents generated morphological change in young cells when AMPA/KAR blockade was without effect.
Here we show that combined AMPA/KAR and mGluR blockade eliminates the sprout suppression among contacted young neurons, mimicking the response observed with NMDAR antagonists alone. The effect is specific to type II mGluRs, which act by reducing cAMP levels. Type II mGluR blockade decreases NMDAR currents in the tectal cells, and application of the cAMP analog 8-bromo cAMP (8-Br cAMP) to tectal neurons reduces their NMDAR-mediated calcium influx. We also show that NMDARs on some very early neurons have a low sensitivity to magnesium, which disappears within 72 hr of culture. Our results suggest a role for mGluRs in regulating neurite formation and growth at very early stages of neuronal differentiation before synapse formation by modulating glutamate-mediated calcium influx through the NMDAR.
MATERIALS AND METHODS
Dissociated Xenopus tectal cultures.Tectal lobes from anesthetized (0.1% topically applied MS-222) stage 55–60 (Nieuwkoop and Faber, 1967) Xenopus laevis tadpoles were dissected into calcium- and magnesium-free Steinberg's solution (58 mm NaCl, 0.7 mm KCl, 4.6 mm HEPES, and 0.4 mm EDTA). The tissue was transferred to Steinberg's solution plus 1.3 mg/ml trypsin for 30 min at room temperature for digestion. Tissue was then washed three times in Steinberg's solution and once in Xenopus culture medium (Lin and Constantine-Paton, 1998) composed of 50% L-15 medium (Life Technologies) supplemented with 10% fetal calf serum, 5% JSFH salts (240 mm sodium chloride, 9 mm potassium chloride, 21 mm calcium chloride, 21 mmmagnesium sulfate, 400 mm HEPES, and 40 mm sodium bicarbonate), 2% penicillin/streptomycin, 0.1% gentamycin, and 40 μl/100 ml of 25 mg/ml insulin/transferrin sodium selenite (Boehringer Mannheim). Trituration was conducted in Xenopus culture medium supplemented with 0.1 mg/ml DNase I, using a fire-polished Pasteur pipette. Cells were washed three times in culture medium to remove cellular debris and plated onto poly-lysine-coated coverslips at low (10,000 cells/cm2) or high (50,000 cells/cm2) density in the center of a 22 mm coverslip. Appropriate receptor antagonists (if any) were added to the culture medium within 30 min of plating. Cultures were incubated at 18°C in a humidified atmosphere. All chemicals were from Sigma-Aldrich unless noted otherwise.
Neurite end assay. Neuronal cultures were prepared as described above and cultured with appropriate antagonists for 3 d beginning at plating. All drugs were purchased from Tocris. Because many mGluR antagonists in particular have been documented as showing nonspecific effects on various glutamate receptors, special care was taken to use previously tested concentrations of antagonists and, when possible, different antagonists inhibiting the same class of mGluR. For several drugs [RS-α-methyl-4-carboxyphenylglycine (MCPG), nifedipine, RS-α-methylserine-O-phosphate (MSOP), dantrolene], we tested several concentrations and selected the highest concentration that did not have obvious toxic or nonspecific effects on our neuronal cultures. The drug concentrations used were as follows:dl-2-amino-5-phosphonopentanoic acid (AP-5), 100 μm (Lin and Constantine-Paton, 1998); 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 20 μm (Aniksztejn et al., 1991); nifedipine, 10 μm (F. Miskevich, unpublished data); MCPG, 1 mm (S.-Y. Lin, unpublished data);RS-α-ethyl-4-carboxyphenylglycine (E4CPG), 0.5 mm (Flavin et al., 1996); MSOP, 0.2 mm (Cochilla and Alford, 1998) (Miskevich, unpublished data); RS-1-aminoindan-1,5-dicarboxylic acid (AIDA), 0.5 mm (Flavin et al., 1996); 2S-α-ethylglutamic acid (EGLU), 0.2 mm (Flavin et al., 1996); and dantrolene, 20 μm (S. Hill-Feldberg, unpublished data). Images were collected on a Nikon Diaphot 300 microscope using a Cohu 4019 CCD video camera attached to a PowerMac running NIH Image 1.62 analysis software. Neurons were identified on the basis of their small round somata as has been described previously (Lin and Constantine-Paton, 1998). Neurons with this morphology from each set of cultures were counted as isolated if there were no visible processes or cell bodies touching a given cell (including both cell body and all processes). Numbers of free neurite ends were counted for contacted and uncontacted cells independently. Average cell numbers and free neurite ends per cell were then calculated for the contacted and uncontacted neurons subjected to each treatment. The absolute amount of sprouting varied among cultures from different dissociations, making it necessary to confine direct comparisons to sister cultures only. The results were tabulated and analyzed in Excel using two different statistical tests. To compare the number of neurites from isolated and contacted cells on the same coverslip, a two-tailed Student's t test was used to determine whether contacted cells had fewer neurites per cell than isolated ones. Results were considered significantly different at thep < 0.05 level. To compare different treatments within the same dissociation, multiple ANOVA (mANOVA) analysis (Tukeypost hoc test) using Systat 5.2.1 was used to compare the average number of neurites per contacted cell with various treatments, with results considered significant at the p = 0.05 level. mANOVA analysis of free neurite ends of isolated cells did not vary significantly under any of the conditions tested.
Evoked calcium imaging. Cultures were grown in standardXenopus culture medium for 3 d and loaded with 5 μm fluo-3 AM in culture media supplemented with 0.5 mg/ml pluronic F-127 for 45–60 min. Coverslips were then transferred to an imaging chamber and perfused with normal or magnesium-free frog saline solution (FSS) for 15 min. Five to 10 frames collected 10 sec apart were acquired on a Nikon PCM 2000 confocal microscope, and the average baseline fluorescence (Fo) of each cell was calculated. Excitation of the fluo-3 AM fluorophor was set so that the maximum fluorescence of most cells on the coverslip allowed at least a sixfold increase in fluorescence without saturating the image gray scale. Cells were then treated with 20 μm glutamate or 100 μm NMDA + 5 μm glycine for 60 sec, and a second set of 10 frames spaced 10 sec apart was acquired. The average fluo-3 AM intensity in these 10 frames was then taken as the response to that agonist (Fag), and ΔFag was calculated asFag −Fo. The agonist was then washed out by perfusion with FSS for 10 min. This was followed by a 10 min perfusion of FSS containing one or more specific glutamate receptor antagonist(s) or 8-bromo-cAMP. Antagonist concentrations were identical to those used in the chronically treated cultures above, and 8-bromo-cAMP (RBI) was used at 5 or 10 μm concentrations. Finally, as a control for bleaching during the experiment, either glutamate or NMDA was applied along with antagonist or 8-bromo-cAMP to each culture for 60 sec, and a third set of 10 frames spaced 10 sec apart was acquired. Cells with a final response that was <90% of the initial response were not used in this analysis. The response to agonist in the presence of antagonist was calculated as the average of these 10 frames (Fag+ant), and ΔFag+ant was calculated asFag+ant −Fo. The order of antagonist and agonist treatment was varied between coverslips to avoid introducing bias caused by order of presentation. mANOVA analysis was used to determine significant differences between antagonist + agonist treatments. Tukey post hoc tests were used to compare differences between the antagonists, with results considered significant at the p = 0.05 level.
Electrophysiology. Amphotericin-perforated patch-clamp recordings were made from cultured tectal neurons at 3 d in vitro (3 DIV). The cells were perfused with extracellular recording medium consisting of 115 mm NaCl, 2 mm KCl, 2.5 mmCaCl2, 1.5 mmMgCl2, 10 mm glucose, and 10 mm HEPES with pH adjusted to 7.3 using NaOH. Recording pipettes were filled with 110 mmK-gluconate, 10 mm KCl, 5 mm NaCl, 1.5 mmMgCl2, 0.5 mm EGTA, 20 mm HEPES, and 200 μg/ml amphotericin B with pH adjusted to 7.3 by KOH. Amphotericin B was dissolved in DMSO and diluted to the final concentration just before use (Rae et al., 1991). A >1.5 GΩ seal was first obtained on an individual neuron soma, and the progress of tectal membrane perforation was then monitored as a change in access resistance from >1.5 GΩ to ∼25 MΩ. A second pipette was filled with 20 mm glutamate and placed 20 μm away from the patched neurons. A 15 msec 20 psi pressure pulse was delivered to the pipette using a Picospritzer (General Valve). Antagonists were applied in the bathing medium and thoroughly washed out after each application. Currents were acquired using an Axopatch 200B amplifier (Axon Instruments) interfaced to a Digidata 1200 (Axon Instruments) and directly digitized with pClamp 6.0 software for further off-line analysis.
Spontaneous calcium imaging. Spontaneous calcium fluctuations were analyzed using the calcium-sensitive fluorophor fluo-3 AM. Two protocols were applied. Under the first protocol, neurons were grown in normal Xenopus culture medium for 2 or 3 d and loaded with 5 μm fluo-3 AM for 45–60 min in Xenopus culture medium. Pluronic acid F-127 (0.5 mg/ml) was added to facilitate dye loading. Coverslips containing the cultures were then placed in the imaging chamber and perfused for 10–15 min at 2 ml/min with normal FSS (100 mmNaCl, 2 mm KCl, 2.5 mmCaCl2, 3 mmMgCl2, 5 mm glucose, and 5 mm HEPES) supplemented with appropriate antagonists before imaging. Images were collected on a Nikon PCM 2000 confocal microscope at one frame per 30 sec for 30 min using the 488 nm argon laser line and standard FITC filter sets. The images were converted to TIFF files, and transferred to NIH Image for fluorescent intensity measurements using custom-written macros. To identify calcium transients, the average fluorescence intensity of each cell was calculated by averaging all the measured values for that cell. The intensity of each cell at each time point was then normalized against that average value of the cell. Bleaching was minor and compensated for by multiplying the normalized brightness of each measured cell at each time point by the average normalized brightness of all cells at that time point and dividing this value by the average normalized brightness of all cells at all time points. A positive calcium signal was defined as any intensity measurement that exceeded 20% of the average normalized intensity for that individual cell. This criterion is similar to values used by other investigators (Flint et al., 1999). Individual cells were compared across antagonist treatments by superimposing graphs of normalized calcium changes and comparing the duration and frequency of calcium transients.
A second protocol was used to determine whether spontaneous calcium fluctuations were altered by chronic glutamate receptor antagonist treatments. For this analysis, cultures were treated with glutamate antagonists for 3 d from plating and maintained in antagonist through all imaging procedures. Image collection, average baseline, and intensity measurements were the same as above. Calcium fluctuations were grouped into four categories using intensity and temporal characteristics identified by previous investigators (Gu et al., 1994;Flint et al., 1999). Large calcium fluctuations were defined as events that had a maximum intensity at least 50% larger than the average intensity; events were considered small if their maximum intensity was <50% but >20% above average fluorescence. Calcium spikes were events that remained >20% above average for four or fewer consecutive frames (2 min or less). Calcium waves were defined as events that lasted for five or more frames (>2 min). Events were classified as large spikes, small spikes, large waves, or small waves. All events were scored for 60–70 cells under each treatment condition, and the proportions of cells showing each type of calcium transient were compared across treatment groups. With these imaging conditions, our analyses may miss a fraction of the shortest duration calcium spikes yet include all calcium waves.
RESULTS
Glutamate receptors can act cooperatively to regulate neurite sprouting
We wished to determine how NMDARs in early cultures might be activated in the absence of action potentials driving glutamate release. Other classes of glutamate receptors offered likely candidates to modulate NMDAR function because they would respond to the same spontaneous vesicle fusion events that appear to stimulate early NMDARs. The number of neurite sprouts on contacted Xenopusneurons and isolated neurons in the same fields were tabulated separately at 3 DIV after chronic exposure to a range of glutamate receptor antagonists. Consistent with earlier results (Lin and Constantine-Paton, 1998) in all dissociations, the NMDAR antagonist AP-5 caused a significant increase in the number of neurite ends on contacted cells relative to isolated cells (Fig.1A, Exp. 1, Exp. 2, Exp. 4), whereas CNQX applied to sister cultures for the same duration was without effect (Fig. 1A,Exp. 1). Also consistent with previous experiments in which the calcium chelator BAPTA induced contacted cell sprouting (Lin and Constantine-Paton, 1998), the L-type calcium channel blocker nifedipine induced contacted neuron sprouting in these experiments (Fig. 1A, Exp. 3). To test the hypothesis that mGluRs interact with NMDARs in the absence of AMPA/KAR activity to facilitate sprout suppression, tectal cultures were grown in combinations of the AMPA/KAR blocker CNQX and mGluR antagonists. When CNQX and the type I/type II mGluR antagonists E4CPG (Fig. 1A, Exp. 1) or MCPG (Fig.1A, Exp. 2) were applied simultaneously, contacted cells showed numbers of neurite ends that were indistinguishable from uncontacted cells. This is identical to that seen with AP-5 treatment. Sister cultures treated separately with each mGluR antagonist or AMPA/KAR blockade showed normal sprout suppression (Fig. 1A, Exp. 1,Exp. 2). Type III mGluR blockade, with or without CNQX, failed to alter sprout suppression (Fig. 1A,Exp. 3). Also dantrolene, applied alone or with the type I/type II mGluR antagonist E4CPG, failed to alter neurite sprouting among contacted cells (Fig. 1A,Exp. 3). These observations suggest that calcium release from intracellular stores is not critically involved in the response and that mGluR activity normally modulates the NMDAR in the downregulation of neurite sprouting when AMPA/KAR activity is low.
Fig. 1.

Chronically applied AMPA/KAR and type II mGluR antagonists increase sprouting in contacted tectal neurons by 3 DIV. A, Quantification of the average number of free neurite ends on contacted (black bars) and isolated (white bars) cells in the same fields, along with their SEs in the presence of various glutamate receptor antagonists. Each of the four experiments used cells from a single dissociation, and cells from two coverslips in the dissociation were analyzed for each antagonist treatment. A two-tailed Student'st test was performed between contacted and isolated cells on the same coverslip to compare the number of neurites per cell, and mANOVA analysis using a Tukey post hoc test compared differences between the number of neurites per contacted cell under different treatments within a given experiment. Statistics were not applied across experiments because of the dissociation-associated differences in overall sprouting. Multiple ANOVA analysis of free neurite ends among different treatments within a given experiment consistently revealed no changes in sprouting of isolated cells across the control and treatment groups. B, Differential interference contrast (DIC) images of dissociated tectal neurons in cultures treated with 20 μm CNQX and 0.2 mmEGLU show significant sprouting from both isolated and contacted cells. Isolated cells are labeled with i, contacted cells are labeled with c, and free neurite ends are marked with awhite dot. C, Control image showing more free neurite ends on isolated compared with contacted cells. Scale bar, 10 μm.
The neurite sprouting assay was subsequently repeated using specific antagonists for type I (AIDA) and type II (EGLU) mGluRs individually (Fig. 1A, Exp. 4). Neither of these specific mGluR antagonists alone eliminated the difference in neurite ends between contacted and isolated cells. In addition, the type I mGluR antagonist AIDA combined with CNQX had no significant effect on sprouting. In contrast, the type II mGluR antagonist EGLU coapplied with CNQX removed the contact-mediated sprout suppression (Fig. 1A, Exp. 4). This effect is illustrated in the photomicrographs of Figure 1, B and C. Thus, in the absence of AMPA/KAR function, type II mGluRs interact with NMDARs to inhibit neurite sprouting in contacted tectal cells.
A number of investigations studying GABAAreceptor (GABAAR) responses of neurons developingin situ have reported early depolarizing responses in response to GABAAR stimulation (Owens et al., 1996; Ben-Ari, 2001). Thus, we asked whether the 13% of tectal neurons in Xenopus tectal cultures that are GABAergic (Lin and Constantine-Paton, 1998) could produce the GABAAR-mediated depolarizations necessary to relieve the magnesium block on NMDARs. The GABAAR blocker bicuculline methiodide applied chronically from plating at 10 μm consistently failed to eliminate the difference between the number of neurites on isolated or contacted tectal cells when assayed at 3 DIV (n = 430 cells; 1.51 ± 0.10 contacted neurites per cell; 2.87 ± 0.32 isolated neurites per cell; Student's two-tailed t test,p = 0.002). Because the GABAAR antagonist does not appear to increase sprouting in cultures of early tectal neurons, GABA probably does not play a significant role in depolarizing these cells.
Metabotropic glutamate receptors modulate glutamate-induced calcium currents
We used fluo-3 AM imaging of changes in intracellular free calcium (ΔF/Fo) to determine whether inhibition of glutamate-triggered calcium changes was involved in the synergistic effect of type II mGluR and AMPA/KAR antagonists on sprouting. We first determined ΔF/Fo of individual cultured neurons in response to glutamate alone; then we examined the reduction in that calcium response when specific glutamate receptor antagonists were coapplied. Figure2A illustrates this technique in a field of tectal neurons. Figure 2Bshows a typical trace of such a response for the cell marked with anasterisk. Figure 3 summarizes all of the data taken from neurons at 3 DIV for the effects on the calcium response to glutamate in the presence of all antagonists shown to influence the sprouting of contacted cells. As can be seen in the figure, AP-5 and CNQX depressed calcium influx, although to significantly different levels (ANOVA Tukey post hoc test;p = 0.001). Nifedipine decreased calcium influx levels to that of CNQX, suggesting that the AMPA/KAR calcium response to glutamate may be mediated primarily by L-type calcium channels. Type I (AIDA), type II (EGLU), and type I/II (E4CPG) mGluR antagonists did not decrease calcium entry; in fact, consistent increases in the glutamate-induced calcium response of neurons were observed whenever type II mGluRs were blocked and ionotropic receptors were functioning. CNQX plus the type I antagonist AIDA also had no effect on the glutamate-induced calcium flux beyond that seen with AMPA/KAR blockade alone. However, antagonism of type II mGluRs (EGLU and E4CPG) when coapplied with CNQX suppressed calcium influx beyond that seen with CNQX alone (ANOVA Tukey post hoc test;p = 0.0001) to levels indistinguishable from AP-5. These data suggest that type II mGluRs potentiate calcium influx when NMDARs are the only ionotropic glutamate receptors functioning on the tectal cell membranes.
Fig. 2.

Fluo-3 AM imaging of the calcium response to glutamate and its reduction by glutamate receptor antagonists. A, Representative DIC and fluorescent images of Xenopus tectal neurons under various stimulation conditions. CNQX and AP-5 reduce the fluorescence increase in response to glutamate, whereas E4CPG increases the calcium response without permanently altering the baseline fluorescence or impairing the ability of the cell to respond to subsequent applications of glutamate after removal of the antagonists. Scale bar, 10 μm.B, Graph of ΔF/F0 versus time for the single cell marked by the asterisks in Ashowing the quantitation for the glutamate-induced calcium-dependent fluorescence increases in the presence of various glutamate receptor antagonists.
Fig. 3.

Quantitation of the calcium response to glutamate in the presence of glutamate receptor antagonists or calcium channel blockers. The figure shows the calcium response of cells treated with glutamate plus various antagonists (ΔFglu+ant) as a proportion of the cellular response to glutamate alone (ΔFglu) at 3 DIV. Type I (AIDA) mGluR blocker alone does not alter calcium flux (n = 34 cells), although drugs that block the type II mGluR response (EGLU and E4CPG) significantly increase calcium influx (n = 43 and 34 cells, respectively) when applied individually. CNQX and nifedipine decrease calcium flux by 48 and 46% (n = 140 cells each), whereas AP-5 treatment decreases calcium flux by 73% (n = 140 cells). The reduction caused by the type I mGluR blocker (AIDA) plus CNQX is not different from the reduction with CNQX alone (47% decrease; n = 34 cells), whereas CNQX plus either the type I/II (E4CPG) or type II (EGLU) mGluR antagonists show a calcium flux that is decreased by ∼70% compared with glutamate alone (n = 140 and 43 cells, respectively). ANOVA analysis of the various treatment groups shows that AP-5, CNQX + E4CPG, and CNQX + EGLU produce significantly lower calcium fluxes than other treatments (p < 0.02; Tukey post hoc tests). Black bars denote those antagonist treatments that increase the sprouting of contacted tectal neurons at 3 DIV.
Electrophysiological recordings from 3 DIV neurons confirmed a facilitating effect of mGluRs on NMDAR currents. At this age, spontaneous synaptic currents are not detectable. However, robust inward currents can be recorded with perforated-patch recording using a puff of glutamate (Fig.4A). A representative series of traces using this technique is shown in Figure4B, and the electrophysiological results are summarized in Table 1. Blockade of type II mGluRs using EGLU or type I/II mGluRs using E4CPG increased the peak glutamate induced current to an average of 146.7%, confirming the general increase seen using calcium imaging. Blockade of AMPA/KARs with CNQX resulted in an average reduction of the peak glutamate current to 72.3%. Metabotropic receptor blockers affecting type II mGluRs (EGLU or E4CPG) coapplied with CNQX decreased the peak glutamate current to 39.7% and also consistently slowed its decay time (Fig.4B, lowest trace; Table 1). These results confirm the calcium imaging data and suggest that mGluRs can enhance NMDAR function in the absence of AMPA/KAR-mediated depolarization. However, when both ionotropic receptor subtypes are functioning on the cells, type II mGluR blockade actually enhances the glutamate current.
Fig. 4.
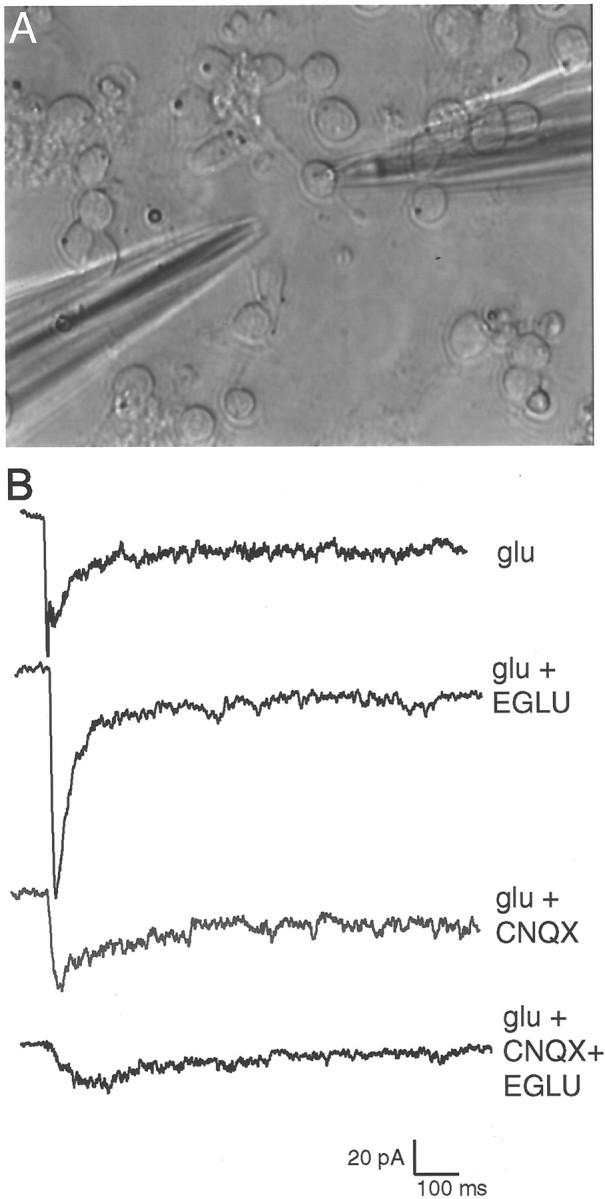
A, A 3 DIV Xenopus culture with a perforated-patch recording electrode (right side) and a pipette containing glutamate to be puffed onto the cell (left side). Spontaneous synaptic currents are not detectable at this stage in culture. B, Electrophysiological recordings from a single cell in response to identically puffed glutamate in the presence of various glutamate receptor blockers. Blockade of type II mGluRs alone using EGLU causes a substantial increase in glutamate response in this neuron, whereas CNQX decreases the glutamate response. Combined type II mGluR and AMPA/KAR blockade (EGLU+CNQX) decreases glutamate response by a larger amount than CNQX alone. These results are consistent with the calcium imaging results reported in Figure 3, and the results of recording from several cells are summarized in Table 1.
Table 1.
Summary of electrophysiological recordings from five cells in response to glutamate in the presence of various glutamate receptor antagonists
| Cell no. | Glutamate | Glu + CNQX | Glu + EGLU | Glu + E4CPG | Glu + CNQX + EGLU | Glu + CNQX + MCPG | Decay time ratio |
|---|---|---|---|---|---|---|---|
| 1 | 29.95 pA | 23.99 pA | 36.98 pA | 17.00 pA | 0.21 | ||
| 80.1% | 123.5% | 56.8% | |||||
| 2 | 47.31 pA | 25.13 pA | 11.1 pA | 0.49 | |||
| 53.1% | 23.5% | ||||||
| 3 | 75.73 pA | 107.09 pA | |||||
| 141.4% | |||||||
| 4 | 43.5 pA | 33.39 pA | 59.13 pA | ||||
| 76.8% | 135.9% | ||||||
| 5 | 91.84 pA | 72.72 pA | 170.78 pA | 35.54 pA | 0.68 | ||
| 79.2% | 186.0% | 39.7% |
Percentages reflect the glutamate current in the presence of antagonists relative to the glutamate current alone. CNQX decreases the cellular response to glutamate. Metabotropic glutamate antagonists that block type II mGluRs increase the glutamate current amplitude when applied individually, but when coapplied with CNQX they decrease the response of the cell to a larger extent than when CNQX is applied alone. The decay times for cells treated with CNQX and mGluR antagonist + CNQX were calculated by first normalizing the amplitudes of the peak glutamate currents between the two treatments and then measuring the time from peak at 0.37 peak amplitude as a single exponential tau estimator. The decay time ratio is defined as the decay time in CNQX alone divided by the decay time in CNQX + mGluR antagonist. Glu, Glutamate.
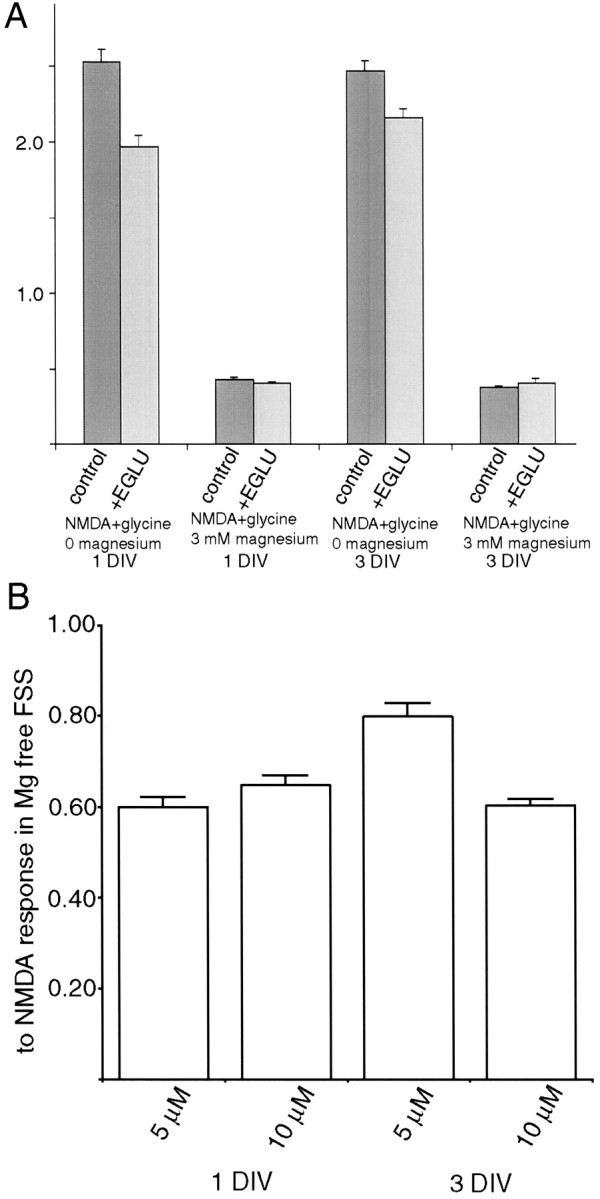
Earlier studies have suggested two ways in which mGluRs may alter NMDAR signaling. Metabotropic receptors could decrease the expression of NMDARs on the surface of the cell (Gomperts et al., 2000) or alter the permeability of the NMDAR channel when small depolarizations or low levels of magnesium allow marginal function (Cho et al., 2000). To distinguish between these possibilities, cells were cultured for 3 d in EGLU + CNQX, and the calcium responses of the neurons to NMDA and glycine in 3 mm magnesium or in magnesium-free FSS were measured using fluo-3. Acute NMDA exposure to both treated and control cells produced the same calcium response at 40 μm and 100 μm NMDA (data not shown). However, the magnitude of the calcium response to this saturating NMDAR stimulation was unchanged from the control after chronic exposure to EGLU + CNQX (Fig.5) regardless of whether magnesium was present in the medium. Thus, the cell surface NMDAR sensitivity of the neurons was not altered by the chronic glutamate receptor antagonist treatment. The magnesium sensitivity of NMDARs on 3 DIV tectal neurons was also unaffected by chronic AMPA/KAR and type II mGluR antagonism.
Fig. 5.

Chronic treatment of dissociatedXenopus tectal neurons with the type II mGluR antagonist EGLU and AMPA/KAR blocker (CNQX) for the first 3 DIV does not change the NMDAR-mediated calcium response of cultured tectal neurons to NMDA and glycine. The two sets of cultures had indistinguishable responses regardless of whether magnesium ions were present in the testing medium.
Spontaneous calcium oscillations do not correlate with an increase in neurite sprouting
As mentioned in the introductory remarks, neurons in culture andin vivo have been shown to have endogenously changing levels of calcium. Consequently, despite the robust acute effects of mGluRs on calcium responses to applied glutamate in tectal neurons, sprouting might be more directly linked to the effects of the antagonists on their spontaneous calcium transients. Using two different experimental paradigms, we tested for positive correlations between glutamate receptor antagonist effectiveness in increasing sprouting among contacted cells and in the effectiveness of the same antagonists in suppressing spontaneous calcium transients using two different experimental paradigms. First, we loaded cells with fluo-3 AM and recorded baseline spontaneous calcium changes every 30 sec for 30 min. The same cells were subsequently recorded during four additional 30 min sessions in the presence of CNQX, AP-5, MCPG, and CNQX + MCPG. The calcium transients were subsequently examined across the various treatment conditions to look for a change from baseline that was consistently associated with exposure to AP-5 or to CNQX + MCPG. None of these acute, short-term treatments consistently altered the frequency of spontaneous calcium transients in the tectal cells (Table2). Of the 108 cells (2 or 3 DIV) showing some activity in any of the treatment conditions, the number of cells exhibiting decreases or increases in spontaneous calcium transients fluctuated widely.
Table 2.
Short-term glutamate antagonist treatment has no consistent effect on calcium transients
| Prevalence of Ca2+ transients compared with FSS | AP-5 (%) | CNQX (%) | MCPG (%) | CNOX + MCPG (%) |
|---|---|---|---|---|
| Larger in antagonist | 24 | 31 | 34 | 46 |
| Smaller in antagonist | 34 | 36 | 23 | 24 |
| Same in antagonist | 42 | 33 | 43 | 30 |
Time-lapse imaging of calcium transients in fields of cells were characterized at 2 and 3 DIV in normal FSS at intervals of 30 sec for 30 min. Glutamate receptor antagonists were then applied for 10 min, and a second set of time-lapse images of the spontaneous calcium transients of the same cells was collected as before. No consistent change in levels of spontaneous activity in individual cells was detected by comparing the calcium transients of antagonist-treated cells with the control images for those same cells.
We next considered the possibility that long-term treatment of tectal neurons with glutamate receptor antagonists (the treatment that actually produced the increase in free neurite ends) might be necessary to discern an effect mediated by spontaneous calcium changes. We recorded the spontaneous calcium transients in cultures that were grown for 3 DIV in the various glutamate receptor antagonists. With this paradigm it was necessary to compare the average response of the population of cells grown under the different conditions. The tabulated results of these comparisons are given in Table3. None of the apparent changes in calcium transients after antagonist treatment suggested any correlation with the increased sprouting found in AP-5- and CNQX + MCPG-treated cultures. For example, neither the average number of waves per cell nor the average number of spikes per cell was similarly altered from control values with CNQX + MCPG and AP-5, as compared with the CNQX or MCPG alone, which does not induce sprouting. Thus, spontaneous calcium oscillations do not appear to be responsible for the sprout suppression mediated by glutamate receptor antagonists.
Table 3.
Changes in spontaneous calcium transients produced by chronic glutamate antagonist treatments for the first 3 d in culture do not correlate with the sprouting effects of the same treatment
| Control | Antagonists | ||||
|---|---|---|---|---|---|
| CNQX | MCPG | CNQX + MCPG | AP-5 | ||
| Number of cells | 77 | 77 | 60 | 74 | 67 |
| % of cells active | 48 | 82 | 63 | 58 | 67 |
| % with waves only | 8 | 3 | 26 | 21 | 13 |
| % with spikes only | 78 | 81 | 42 | 58 | 73 |
| % with spikes and waves | 14 | 16 | 32 | 21 | 14 |
| % with small response only | 84 | 76 | 84 | 72 | 73 |
| % with a large response | 16 | 24 | 16 | 28 | 27 |
| Average number of waves per active cell | 0.19 ± 0.08 | 0.25 ± 0.08 | 0.66 ± 0.10 | 0.45 ± 0.09 | 0.23 ± 0.08 |
| Average number of spikes per active cell | 2.30 ± 0.25 | 3.43 ± 0.26 | 1.71 ± 0.26 | 1.39 ± 0.18 | 2.24 ± 0.29 |
Sister cultures of tectal neurons were grown in normal media and supplemented with glutamate receptor antagonists as shown. Calcium transients were recorded for fields of cells every 30 sec for 30 min; the number and type of observed calcium changes were quantitated for each cell, and then the average response for all neurons in the field was calculated. Antagonists that produce an increased number of free neurite ends among contacted tectal neurons cannot be distinguished from treatments that do not increase free neurite ends in terms of their effects on calcium transients.
Some early Xenopus tectal neurons have a low sensitivity to magnesium ions
The existence of early NMDARs that function in the presence of magnesium would effectively account for the ability of NMDARs to suppress sprouting in the absence of AMPA/KAR function, particularly if mGluR function augmented the NMDAR response. Our previous analysis at 3 DIV had revealed only NMDAR responses with normal magnesium sensitivity (Lin and Constantine-Paton, 1998). However, we reasoned that the cells might change significantly during the 3 d in culture and that neurite sprouting might reflect an earlier responsiveness of NMDARs to endogenous glutamate that was no longer detectable at 3 DIV. Therefore, cultures were loaded with fluo-3 AM at 1 DIV, and changes in their calcium response to NMDA + glycine were measured in the presence and absence of magnesium. Figure6A illustrates the responses of a typical field of cells when first exposed to NMDA + glycine in 3 mm magnesium at 1 DIV, whereas Figure 6B shows the responses of the same cells to multiple applications. Some neurons (Cell A) showed vigorous calcium responses to each pulse of NMDA in 3 mmmagnesium, whereas other neurons (Cells C, E,F) showed fluctuating responses under the same conditions. Other neurons (Cells B, D) never responded to NMDA in 3 mm magnesium. All of the cells responded in magnesium-free recording solution (Fig.6B, far right). Overall, 13 of 45 cells analyzed (29%) showed at least 20% of maximal NMDA response in magnesium-free solution when tested in 3 mmmagnesium solution. This magnesium insensitivity was essentially gone when cells were similarly tested at 3 DIV. Only 1 of 49 cells (2%) examined at 3 DIV showed a significant NMDA current in 3 mm magnesium (data not shown).
Fig. 6.

NMDARs on some early Xenopus tectal neurons have low magnesium sensitivity. A, Frames showing a field of six neurons at 1 DIV after loading with fluo-3 AM. Images collected at 6 sec intervals illustrate the NMDA-induced calcium fluorescence in a recording solution containing 3 mmmagnesium. Two cells (A, C) have significant calcium influx in response to 100 μm NMDA + glycine in 3 mm magnesium, whereas four other cells show little or no response. B, Plot of ΔF/F0 for the same cells over a longer time interval as well as their response to NMDA in magnesium-free solution. The vertical yellow bar shows the frames of the record illustrated in A. Scale bar, 10 μm.
We next classified 1 DIV neurons using the procedure illustrated in Figure 6 as either normal type A (Cells B, D) or reduced magnesium sensitivity type B (Cells A, C,E, F). We then examined the reduction of the calcium response in each type of neuron when antagonists of ionotropic and metabotropic glutamate receptors were present during glutamate application at 1 DIV (Fig. 7). As expected, in the presence of CNQX, type A neurons showed a larger reduction in their calcium response to glutamate than did type B neurons. When MCPG was present along with CNQX, the response of the type A neurons was even more depressed. In contrast, the response of type B neurons in CNQX was unaffected by blocking type I/II mGluRs. Therefore it appears that only the magnesium-sensitive NMDARs are augmented by mGluR-mediated signaling.
Fig. 7.
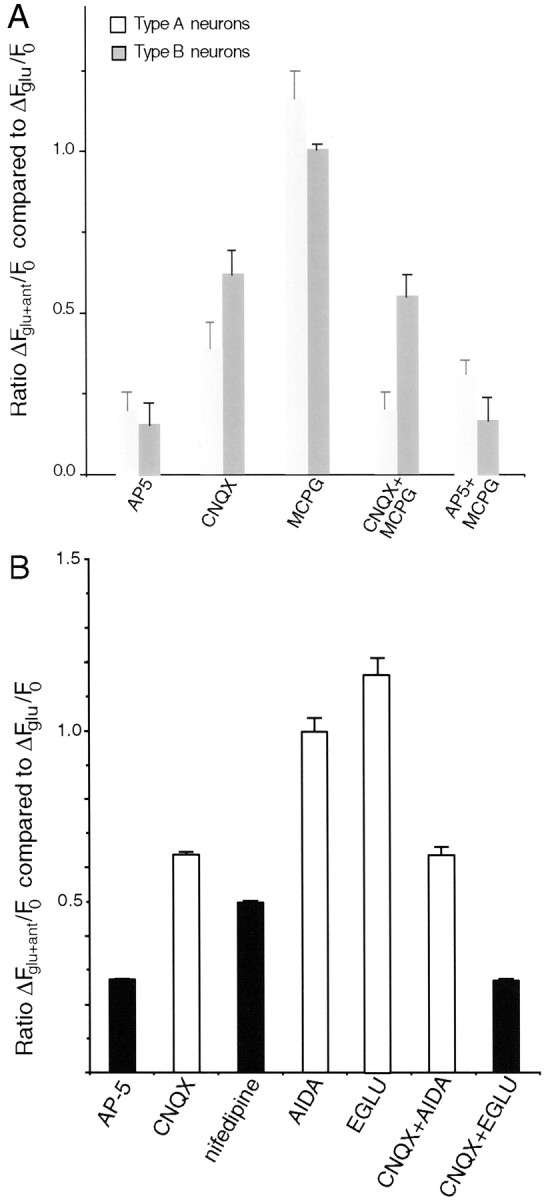
Alterations of the calcium response to glutamate in the presence of glutamate receptor antagonists at 1 DIV.A, Comparison of neurons showing normal (Type A neurons) or reduced (Type B neurons) magnesium sensitivity in response to NMDA + glycine stimulation. As expected, CNQX is less effective at decreasing the calcium influx in cells with low magnesium sensitivity compared with its effect on cells with normal magnesium sensitivity. The effect of type I/II mGluR and AMPA/KAR blockade (CNQX+MCPG) on the calcium response to glutamate is also more pronounced in type A cells showing a normal magnesium sensitivity. MCPG alone actually increases the calcium response to glutamate in these type A neurons. MCPG + AP-5 treatment yields a higher calcium influx through type A neurons compared with AP-5 alone, consistent with a normal weak suppression of calcium entry through AMPA/KAR receptors by type II mGluRs. B, Quantitation of the reduction of the calcium response to glutamate by glutamate receptor and calcium channel antagonists at 1 DIV. CNQX is relatively less effective at 1 DIV (n = 57 cells) than at 3 DIV (Fig. 3) in blocking the glutamate-induced calcium response of tectal neurons. Nifedipine, however, depresses the calcium response to the same extent at both 1 (n = 57 cells) and 3 DIV. The type II mGluR antagonist EGLU (but not the type I antagonist AIDA) in combination with CNQX further decreases the calcium entry in response to glutamate (n = 48 cells). Neither mGluR antagonist alone reduces the calcium response to glutamate, and in fact the calcium response to glutamate when both ionotropic receptor types are functioning is actually increased in the presence of EGLU (ANOVA Tukey post hoc test; p = 0.015).Black columns denote antagonists that resulted in increased free neurite ends among contacted cells after 3 d of chronic application in vitro.
These observations suggested that a combination of magnesium-insensitive NMDAR currents and small magnesium-sensitive NMDAR currents facilitated by mGluRs provide sufficient calcium flux to suppress sprouting in early contacted neurons. To examine this possibility we analyzed calcium responses to glutamate in the presence of specific mGluR antagonists and nifedipine at 1 DIV to compare the calcium response in the total population of neurons at 1 and 3 DIV. As shown by comparison between Figures 7B and 3, the data differ in two respects between 1 and 3 DIV. First, CNQX is relatively less effective at blocking the calcium response to glutamate at the earlier age. Second, by 3 DIV (Fig. 3) the reduction in the calcium response is the same with either CNQX or nifedipine, whereas at 1 DIV (Fig. 7B) nifedipine blocks significantly more calcium influx than CNQX. These differences imply that AMPA/KARs are not the only receptors driving L-type calcium channel activity in very young cultures, but that as the cultures mature, other sources of calcium channel activation disappear. This change may also explain why sprouting of contacted cells, as assayed after 3 d of treatment, is increased after exposure to nifedipine (Fig. 1A,Exp. 3) but not after exposure to CNQX (Fig.1A, Exp. 1). The nifedipine block would eliminate all L-type calcium channel activity for the full duration of the treatment, whereas in very young cultures the CNQX block would only partially decrease calcium channel activity. In short, the three antagonist treatments that effectively eliminate sprout suppression among contacted cells at 3 DIV (AP-5, nifedipine, and CNQX + EGLU) (Figs. 3, 7B, black bars) also consistently block the greatest proportion of the tectal neuron calcium response to glutamate at 1 DIV. Therefore, these treatments, when applied for the first 3 DIV, will produce greater decreases in glutamate-mediated calcium influx than the four antagonist treatments that have no effect on sprouting.
A final observation was that blockade of type I/II mGluRs (Fig.7A, MCPG) or selective blockade of type II mGluRs (Fig. 7B, EGLU) increased the tectal neuron calcium response to glutamate, at least in neurons with normal NMDAR magnesium sensitivity (Fig. 7B) (ANOVA Tukey post hoctest; p = 0.0015). This was most pronounced at 1 DIV, but it was also seen in the calcium imaging data at 3 DIV (Fig. 3,EGLU, E4CPG). Electrophysiological current measurements with either E4CPG or EGLU (Fig. 4B; Table 1) also showed a similar increase. Three observations indicate that this is caused by type II mGluR suppression of AMPA/KAR-mediated calcium influx. First, AP-5 and CNQX block all glutamate-mediated calcium increases in these cells (Lin and Constantine-Paton, 1998). Second, the effect on calcium influx is clearly reversed when AMPA/KARs are inactivated. Third, the presence of MCPG in addition to AP-5 results in a larger glutamate-induced calcium influx than AP-5 alone (Fig. 7A, Type A neurons).
Type I/II mGluR antagonists and cAMP agonists decrease NMDAR response
Finally, we examined the transduction mechanism used by mGluRs to alter NMDAR function during glutamate stimulation. Acute EGLU exposure decreased the calcium response of both 1 and 3 DIV neurons to NMDA in magnesium-free solution but did not change the response of the cells in 3 mm magnesium (Fig.8A). This suggests that the type II mGluR directly affects the calcium entry mediated by the NMDAR but does not change the receptor's sensitivity to magnesium ions.
Fig. 8.
Regulation of NMDAR-mediated calcium influx by acute blockade of type II mGluRs. A, EGLU presented with NMDA + glycine in magnesium-free solution, but not in 3 mmmagnesium, reduces the neuronal calcium response to NMDA. This response is most pronounced at 1 DIV (n = 78 cells;p < 0.0001; Student's t test) but is still significant at 3 DIV (n = 94 cells;p = 0.0039; Student's t test), suggesting that type II mGluR activity normally facilitates calcium influx through the NMDAR using a mechanism that does not alter magnesium sensitivity. B, The cell-permeable cAMP analog 8-Br-cAMP decreases calcium influx through the NMDAR. This decrease is seen at both 1 and 3 DIV, but cells appear to be more sensitive to 8-Br-cAMP at 1 DIV on the basis of the relative responses at each age to 5 μm 8-Br-cAMP (p < 0.0001; ANOVA Tukey post hoc test).n = 104 cells at 1 DIV, 5 μm;n = 107 cells at 1 DIV, 10 μm;n = 88 cells at 3 DIV, 5 μm; andn = 136 cells at 3 DIV, 10 μm.
Type II mGluRs are known to alter cytoplasmic levels of cAMP by regulating adenylate cyclase through trimeric G-proteins (Hollmann and Heinemann, 1994). To examine the involvement of cAMP in the NMDAR-mediated calcium responses of tectal neurons, we used the cell-permeable cAMP analog 8-bromo-cAMP. Acute application of 5 or 10 μm 8-Br-cAMP caused a significant reduction in NMDAR-mediated calcium influx in magnesium-free conditions at both 1 and 3 DIV (Fig. 8B). The effect was most pronounced at 1 DIV, although the effect of 5 μm 8-Br-cAMP at 3 DIV was also significant. These data support a model in which type II mGluR activity depresses adenylate cyclase activity, and the resulting decrease in cAMP levels facilitates calcium influx through the NMDAR. When we experimentally block the type II mGluR, cAMP levels rise, and this increase mediates a reduction in calcium influx through theNMDAR. In combination with the blockade of AMPA/KARs, the reduced NMDAR calcium permeability is sufficient to reduce glutamate-mediated calcium influx to a level that no longer suppresses neurite sprouting.
DISCUSSION
Our results demonstrate that all three classes of glutamate receptors can contribute to an early downregulation of sprouting that occurs during neuron–neuron contact before action potential activity mediates neurotransmission among cultured tectal cells (Lin and Constantine-Paton, 1998). The decreased motility is most likely exerted by calcium influx. Type II mGluR and AMPA/KAR antagonists in combination increase neurite sprouting from contacted cells, similar to that observed with NMDAR antagonists. Our calcium imaging data indicated that both treatments applied over the first 3 DIV would significantly reduce glutamate-mediated calcium influx. Type II mGluR blockade decreases the glutamate-mediated calcium flux through NMDARs by ∼20%, apparently working specifically on receptors with normal magnesium sensitivity. Recordings of glutamate-induced currents in the presence of various receptor antagonists are entirely consistent with the calcium imaging data. We also show that ∼30% of very early Xenopus tectal neurons express NMDARs with a lowered magnesium sensitivity that would allow them to pass some calcium even in the absence of depolarization produced by AMPA/KARs. In addition, we show that neither NMDAR antagonism nor AMPA/KAR and type II mGluR blockade alter spontaneous calcium transients in these tectal cells. Our evidence also suggests that the effect of type II mGluR blockade switches from decreasing to increasing glutamate-mediated calcium influx when AMPA/KAR currents are active.
Type II mGluR regulates ionotropic glutamate receptors through PKA
Type II mGluR regulation of adenylate cyclase activity has been linked to altered levels of cAMP in a number of systems (Prezeau et al., 1992; Flavin et al., 1996; Conn and Pin, 1997), but the direction of the modulation appears to vary by location (Wang et al., 1998;Beaver et al., 1999; Daw et al., 1999). Our data are consistent with a normal role for type II mGluR activation in suppressing the activity of adenylate cyclase and decreasing cAMP levels. Although cAMP can act transcriptionally (for review, see De Cesare and Sassone-Corsi, 2000) or through the cAMP-dependent protein kinase (PKA) (for review, see Hatt, 1999), the short latencies of responses induced by either type II mGluR antagonists or 8-bromo-cAMP suggest that the PKA pathway underlies the phenomena we have observed in tectal neurons. When type II mGluRs are blocked, increased cAMP levels might activate PKA and cause the changes that we observe in ionotropic glutamate receptor function. A PKA-dependent enhancement of AMPA/KAR function similar to that detected in tectal neurons has been documented to occur via direct phosphorylation at mature synapses (Wang et al., 1991; Banke et al., 2000; Snyder et al., 2000). However, the mechanism through which PKA alters NMDAR permeability remains obscure.
PKA has been shown to be a component of a protein complex associating with the NMDAR and the postsynaptic density protein yotiao (Westphal et al., 1999). This could account for rapid PKA effects on NMDARs in mature neurons (Cerne et al., 1993; Nijholt et al., 2000), but the differentiating neurons in our cultures probably do not have fully assembled postsynaptic scaffolding. Although glutamate release among contacted cells has been shown to mediate intracellular calcium increases even at these early stages (Lin and Constantine-Paton, 1998), our recordings have not detected glutamate-mediated synaptic currents in tectal neurons before 5 DIV. Moreover, in more mature systems, PKA activity is associated with increased rather than decreased NMDAR activity (Westphal et al., 1999; Leveque et al., 2000). Perhaps the younger cells that we have examined express an NMDAR subunit or subunit splice variant that allows PKA-mediated phosphorylation to depress the activity of the receptor. It is also possible that PKA is not directly regulating the NMDAR in our system but rather functioning indirectly by regulating the activity of another kinase or phosphatase, as has been suggested by others (Nijholt et al., 2000).
Numerous studies have noted an effect of mGluR activation on facilitating NMDAR function (Aniksztejn et al., 1991; Pisani et al., 1997; Holohean et al., 1999; Martin et al., 1999), yet most of these effects are mediated by type I mGluRs. Metabotropic glutamate receptors have also been linked to the developmental appearance of NMDAR-only synapses in hippocampal micro-island cultures (Gomperts et al., 2000). In Xenopus tectal neurons, type I mGluRs do not produce any effect on sprouting, and type I mGluR blockade has no effect on calcium influx when only NMDA ionotropic receptors are functioning. This difference may again result from the early stage of differentiation of these cells.
Early role of glutamate receptor currents
Our data suggest that two properties of young tectal neurons facilitate a suppression of sprouting during contact with glutamate-releasing neurons. First, an insensitivity to magnesium among a subpopulation of early NMDARs allows activation of that population by small amounts of glutamate. Second, synergism between type II mGluRs and magnesium-sensitive NMDARs facilitates NMDAR-mediated calcium influx when AMPA/KAR receptors are not functional. In the intact brain, these properties are likely to be most prevalent and play a significant role when the neuropil is very immature and synapses with AMPA/KAR function are not yet present. Indeed, the silent synapse hypothesis suggests that the earliest excitatory connections between neurons may be dependent solely on NMDARs (Liao et al., 1995; Wu et al., 1996). Metabotropic glutamate receptor-mediated facilitation combined with lower magnesium sensitivity could explain how NMDARs might function as glutamate-mediated calcium gates even before significant action potential activity is available to evoke transmitter release. However, the suppressive effect of these same metabotropic receptors on glutamate-mediated calcium entry when AMPA/KAR function is present represents a significant reversal of the early type II mGluR role as ionotropic receptor expression matures.
Metabotropic glutamate receptor regulation has been implicated in many of the functions attributed to the NMDA class of receptors. Our data extend the known effects of mGluRs on NMDAR currents by documenting an interaction during the earliest stages of differentiation before action potentials or significant AMPA/KARs play a role in neurotransmitter-mediated neuron–neuron communication. The NMDAR has been shown to be involved in regulating the structure ofXenopus tectal neuron dendritic arbors imaged in intact young Xenopus tadpoles (Rajan and Cline, 1998). In thein vivo retinotectal system, as in culture, NMDAR blockade increases the sprouting of tectal processes (Cline and Constantine-Paton, 1990; Rajan and Cline, 1998). In normal youngXenopus, dendritic motility is most pronounced in caudal tectum (Wu et al., 1999) where synaptic strength and AMPA/KAR function is low (Wu et al., 1996). It has been hypothesized that increasing NMDAR activity is responsible for increased AMPA/KAR function and decreased dendritic motility as tectal neurons mature (Wu et al., 1999;Zou and Cline, 1999). However, the mechanism of NMDAR function in the youngest tectal neurons lacking depolarizations attributable to AMPA/KARs has not been addressed in vivo. Our data suggest that a significant proportion of these young tectal neurons may express NMDARs with low magnesium sensitivity. Although the reason for the minimal magnesium block in tectal cells is unknown, it has been observed in other developing systems in which NR2C and NR2D subunits have been implicated in the effect (Mitani et al., 1998; Kirson et al., 1999). Our data also suggest that the minimal calcium influx through young NMDARs with normal magnesium sensitivity is facilitated by type II mGluRs and that this facilitation can be produced by spontaneous transmitter release onto young membranes where AMPA/KAR expression is low. It is also potentially significant that type II mGluRs appear to decrease AMPA/KAR-mediated calcium in our experiments, possibly suggesting diametrically opposite functions of this class of mGluRs on ionotropic receptor-mediated calcium influx as AMPA/NMDA current ratios increase with maturity.
These observations support a model in which glutamate receptors, by regulating motility, initiate synapse formation before activity-induced transmitter release. In vivo, robust sprouting by newly differentiating neurons would facilitate the cell–cell contacts necessary for synaptogenesis, a condition corresponding to isolated neurons in our cultures. Glutamate released from prospective presynaptic membranes through spontaneous vesicle fusion would drive increased calcium influx and reduce sprouting. This suppression would be most pronounced at those contacts where type II mGluRs and NMDARs colocalize, and further synaptic differentiation would be facilitated at these sites because of their extended lifetimes. Age-associated decreases in dendritic motility in vivo and decreases in free neurite ends from contacted tectal neurons in culture are consistent with this hypothesis. Furthermore, a switch in the role of type II mGluRs from one of enhancing ionotropic receptor-mediated calcium influx to one of suppressing such influx when AMPA/KAR function develops would moderate calcium entry. Suppression of AMPA/KAR-initiated calcium increases by type II mGluRs may also ensure that the NMDAR channel is the dominant route of synaptic calcium entry as evoked release and functional circuits mature.
Footnotes
This work was supported by National Institutes of Health Grants EY06891 (F.M.) and EY06039 (M.C.-P.). We acknowledge Ann Ghormley and Matt Townsend for careful reading of this manuscript.
Correspondence should be addressed to Martha Constantine-Paton, Building 68-380, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307. E-mail:mcpaton@mit.edu.
REFERENCES
- 1.Aniksztejn L, Bregestovski P, Ben-Ari Y. Selective activation of quisqualate metabotropic receptor potentiates NMDA but not AMPA responses. Eur J Pharmacol. 1991;205:327–328. doi: 10.1016/0014-2999(91)90921-c. [DOI] [PubMed] [Google Scholar]
- 2.Banke TG, Bowie D, Lee H-K, Huganir RL, Schousboe A, Traynelis SF. Control of gluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaver CJ, Ji Q, Daw NW. Effect of the group II metabotropic glutamate agonist, 2R,4R-APDC, varies with age, layer, and visual experience in the visual cortex. J Neurophysiol. 1999;82:86–93. doi: 10.1152/jn.1999.82.1.86. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Ari Y. Developing networks play a similar melody. Trends Neurosci. 2001;24:353–360. doi: 10.1016/s0166-2236(00)01813-0. [DOI] [PubMed] [Google Scholar]
- 5.Burnashev N. Calcium permeability of ligand-gated channels. Cell Calcium. 1998;24:325–332. doi: 10.1016/s0143-4160(98)90056-2. [DOI] [PubMed] [Google Scholar]
- 6.Cerne R, Ruskin KI, Randic M. Enhancement of the N-methyl-d-aspartate response in spinal dorsal horn neurons by cAMP-dependent protein kinase. Neurosci Lett. 1993;161:124–128. doi: 10.1016/0304-3940(93)90275-p. [DOI] [PubMed] [Google Scholar]
- 7.Cho K, Kemp N, Noel J, Aggleton JP, Brown MW, Bashir ZI. A new form of long-term depression in the perirhinal cortex. Nat Neurosci. 2000;3:150–156. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- 8.Cline HT, Constantine-Paton M. NMDA receptor agonist and antagonists alter retinal ganglion cell arbor structure in the developing frog retinotectal projection. J Neurosci. 1990;10:1197–1216. doi: 10.1523/JNEUROSCI.10-04-01197.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cochilla AJ, Alford S. Metabotropic glutamate receptor-mediated control of neurotransmitter release. Neuron. 1998;20:1007–1016. doi: 10.1016/s0896-6273(00)80481-x. [DOI] [PubMed] [Google Scholar]
- 10.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 11.Constantine-Paton M, Cline HT. LTP and activity-dependent synaptogenesis: the more alike they are, the more different they become. Curr Opin Neurobiol. 1998;8:139–148. doi: 10.1016/s0959-4388(98)80017-2. [DOI] [PubMed] [Google Scholar]
- 12.Daw NW, Reid SN, Beaver CJ. Development and function of metabotropic glutamate receptors in cat visual cortex. J Neurobiol. 1999;41:102–107. doi: 10.1002/(sici)1097-4695(199910)41:1<102::aid-neu13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 13.De Cesare D, Sassone-Corsi P. Transcriptional regulation by cyclic AMP-responsive factors. Prog Nucleic Acid Res Mol Biol. 2000;64:343–369. doi: 10.1016/s0079-6603(00)64009-6. [DOI] [PubMed] [Google Scholar]
- 14.Flavin HJ, Daw NW, Gregory DS, Reid SN. Glutamate receptors and development of the visual cortex: effect of metabotropic agonists on cAMP. Prog Brain Res. 1996;108:263–272. doi: 10.1016/s0079-6123(08)62545-5. [DOI] [PubMed] [Google Scholar]
- 15.Flint AC, Dammerman RS, Kriegstein AR. Endogenous activation of metabotropic glutamate receptors in neocortical development causes neuronal calcium oscillations. Proc Natl Acad Sci USA. 1999;96:12144–12149. doi: 10.1073/pnas.96.21.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomez TM, Spitzer NC. In vivo regulation of axon extension and pathfinding by growth-cone calcium transients. Nature. 1999;397:350–355. doi: 10.1038/16927. [DOI] [PubMed] [Google Scholar]
- 17.Gomez TM, Robles E, Poo MM, Spitzer NC. Filopodial calcium transients promote substrate-dependent growth cone turning. Science. 2001;291:1983–1987. doi: 10.1126/science.1056490. [DOI] [PubMed] [Google Scholar]
- 18.Gomperts SN, Carroll R, Malenka RC, Nicoll RA. Distinct roles for ionotropic and metabotropic glutamate receptors in the maturation of excitatory synapses. J Neurosci. 2000;20:2229–2237. doi: 10.1523/JNEUROSCI.20-06-02229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grover LM, Yan C. Evidence for involvement of group II/III metabotropic glutamate receptors in NMDA receptor-independent long-term potentiation in area CA1 of rat hippocampus. J Neurophysiol. 1999;82:2956–2969. doi: 10.1152/jn.1999.82.6.2956. [DOI] [PubMed] [Google Scholar]
- 20.Gu X, Spitzer NC. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature. 1995;375:784–787. doi: 10.1038/375784a0. [DOI] [PubMed] [Google Scholar]
- 21.Gu X, Spitzer NC. Breaking the code: regulation of neuronal differentiation by spontaneous calcium transients. Dev Neurosci. 1997;19:33–41. doi: 10.1159/000111183. [DOI] [PubMed] [Google Scholar]
- 22.Gu X, Olson EC, Spitzer NC. Spontaneous neuronal calcium spikes and waves during early differentiation. J Neurosci. 1994;14:6325–6335. doi: 10.1523/JNEUROSCI.14-11-06325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardingham GE, Chawla S, Cruzalegui FH, Bading H. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22:789–798. doi: 10.1016/s0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- 24.Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 25.Hatt H. Modification of glutamate receptor channels: molecular mechanisms and functional consequences. Naturwissenschaften. 1999;86:177–186. doi: 10.1007/s001140050593. [DOI] [PubMed] [Google Scholar]
- 26.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 27.Holohean AM, Hackman JC, Davidoff RA. Mechanisms involved in the metabotropic glutamate receptor-enhancement of NMDA-mediated motoneurone responses in frog spinal cord. Br J Pharmacol. 1999;126:333–341. doi: 10.1038/sj.bjp.0702263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong K, Nishiyama M, Henley J, Tessier-Lavigne M, Poo M. Calcium signaling in the guidance of nerve growth by netrin-1. Nature. 2000;403:93–98. doi: 10.1038/47507. [DOI] [PubMed] [Google Scholar]
- 29.Kirson ED, Schirra C, Konnerth A, Yaari Y. Early postnatal switch in magnesium sensitivity of NMDA receptors in rat CA1 pyramidal cells. J Physiol (Lond) 1999;521:99–111. doi: 10.1111/j.1469-7793.1999.00099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leveque J-C, Macias W, Rajadhyaksha A, Carlson RR, Barczak A, Kang S, Li X-M, Coyle JT, Huganir RL, Heckers S, Konradi C. Intracellular modulation of NMDA receptor function by antipsychotic drugs. J Neurosci. 2000;20:4011–4020. doi: 10.1523/JNEUROSCI.20-11-04011.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 32.Lin S-Y, Constantine-Paton M. Suppression of sprouting: an early function of NMDA receptors in the absence of AMPA/kainate receptor activity. J Neurosci. 1998;18:3725–3737. doi: 10.1523/JNEUROSCI.18-10-03725.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin G, Przewlocki R, Siggins GR. Chronic morphine treatment selectively augments metabotropic glutamate receptor-induced inhibition of N-methyl-d-aspartate receptor-mediated neurotransmission in nucleus accumbens. J Pharmacol Exp Ther. 1999;288:30–35. [PubMed] [Google Scholar]
- 34.Mattson MP, Taylor-Hunter A, Kater SB. Neurite outgrowth in individual neurons of a neuronal population is differentially regulated by calcium and cAMP. J Neurosci. 1988;8:1704–1711. doi: 10.1523/JNEUROSCI.08-05-01704.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinney RA, Luthi A, Bandtlow CE, Gahwiler BH, Thompson SM. Selective glutamate receptor antagonists can induce or prevent axonal sprouting in rat hippocampal slice cultures. Proc Natl Acad Sci USA. 1999;96:11631–11636. doi: 10.1073/pnas.96.20.11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitani A, Watanabi M, Kataoka K. Functional change of NMDA receptors related to enhancement of susceptibility to neurotoxicity in the developing pontine nucleus. J Neurosci. 1998;18:7941–7952. doi: 10.1523/JNEUROSCI.18-19-07941.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin). North-Holland; Amsterdam: 1967. [Google Scholar]
- 38.Nijholt I, Blank T, Liu A, Kugler H, Spiess J. Modulation of hypothalamic NMDA receptor function by cyclic AMP-dependent protein kinase and phosphatases. J Neurochem. 2000;75:749–754. doi: 10.1046/j.1471-4159.2000.0750749.x. [DOI] [PubMed] [Google Scholar]
- 39.Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pisani A, Calabresi P, Centozone D, Bernardi G. Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol. 1997;120:1007–1014. doi: 10.1038/sj.bjp.0700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prezeau L, Manzoni O, Homburger V, Sladeczek F, Curry K, Bockaert J. Characterization of a metabotropic glutamate receptor: direct negative coupling to adenylyl cyclase and involvement of a pertussis toxin-sensitive G protein. Proc Natl Acad Sci USA. 1992;89:8040–8044. doi: 10.1073/pnas.89.17.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 43.Rahman S, Neuman RS. Characterization of metabotropic glutamate receptor-mediated facilitation of N-methyl-d-aspartate depolarization of neocortical neurones. Br J Pharmacol. 1996;117:675–683. doi: 10.1111/j.1476-5381.1996.tb15243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajan I, Cline HT. Glutamate receptor activity is required for normal development of tectal cell dendrites in vivo. J Neurosci. 1998;18:7836–7846. doi: 10.1523/JNEUROSCI.18-19-07836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segal I, Korkotian I, Murphy DD. Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci. 2000;23:53–57. doi: 10.1016/s0166-2236(99)01499-x. [DOI] [PubMed] [Google Scholar]
- 46.Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, Greengard P. Regulation of phosphorylation of the gluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang LY, Salter MW, MacDonald JF. Regulation of kainate receptors by cAMP-dependent protein kinase and phosphatases. Science. 1991;253:1132–1135. doi: 10.1126/science.1653455. [DOI] [PubMed] [Google Scholar]
- 48.Wang XF, Daw NW, Jin X. The effect of ACPD on the responses to NMDA and AMPA varies with layer in slices of rat visual cortex. Brain Res. 1998;812:186–192. doi: 10.1016/s0006-8993(98)01000-2. [DOI] [PubMed] [Google Scholar]
- 49.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 50.Whittington MA, Traub RD, Jefferys JG. Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature. 1995;373:612–615. doi: 10.1038/373612a0. [DOI] [PubMed] [Google Scholar]
- 51.Wu GY, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- 52.Wu G-Y, Zou DJ, Rajan I, Cline H. Dendritic dynamics in vivo change during neuronal maturation. J Neurosci. 1999;19:4472–4483. doi: 10.1523/JNEUROSCI.19-11-04472.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu G-Y, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- 54.Zheng JQ. Turning of nerve growth cones induced by localized increases in intracellular calcium ions. Nature. 2000;403:89–93. doi: 10.1038/47501. [DOI] [PubMed] [Google Scholar]
- 55.Zou D-J, Cline HT. Postsynaptic calcium/calmodulin-dependent protein kinase II is required to limit elaboration of presynaptic and postsynaptic neuronal arbors. J Neurosci. 1999;19:8909–8918. doi: 10.1523/JNEUROSCI.19-20-08909.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]