

Abstract
Chronic systemic complex I inhibition caused by rotenone exposure induces features of Parkinson's disease (PD) in rats, including selective nigrostriatal dopaminergic degeneration and formation of ubiquitin- and α-synuclein-positive inclusions (Betarbet et al., 2000). To determine underlying mechanisms of rotenone-induced cell death, we developed a chronic in vitro model based on treating human neuroblastoma cells with 5 nm rotenone for 1–4 weeks. For up to 4 weeks, cells grown in the presence of rotenone had normal morphology and growth kinetics, but at this time point, ∼5% of cells began to undergo apoptosis. Short-term rotenone treatment (1 week) elevated soluble α-synuclein protein levels without changing message levels, suggesting that α-synuclein degradation was retarded. Chronic rotenone exposure (4 weeks) increased levels of SDS-insoluble α-synuclein and ubiquitin. After a latency of >2 weeks, rotenone-treated cells showed evidence of oxidative stress, including loss of glutathione and increased oxidative DNA and protein damage. Chronic rotenone treatment (4 weeks) caused a slight elevation in basal apoptosis and markedly sensitized cells to further oxidative challenge. In response to H2O2, there was cytochrome c release from mitochondria, caspase-3 activation, and apoptosis, all of which occurred earlier and to a much greater extent in rotenone-treated cells; caspase inhibition provided substantial protection. These studies indicate that chronic low-grade complex I inhibition caused by rotenone exposure induces accumulation and aggregation of α-synuclein and ubiquitin, progressive oxidative damage, and caspase-dependent death, mechanisms that may be central to PD pathogenesis.
Keywords: α-synuclein, cytochrome c, glutathione, caspase-3, carbonyls, ubiquitin
Parkinson's disease (PD) is marked by selective nigrostriatal dopaminergic degeneration and formation of ubiquitin- and α-synuclein-positive cytoplasmic inclusions known as Lewy bodies (Spillantini et al., 1997; Wooten, 1997). Although the cause of sporadic PD is unknown, genetic and environmental factors may both be important. Epidemiological studies highlight involvement of pesticide exposure in PD pathogenesis (Gorell et al., 1998; Menegon et al., 1998), and there are systemic reductions in activity of complex I of the mitochondrial electron transfer chain in PD patients (Mizuno et al., 1989; Parker et al., 1989; Schapira et al., 1989).
We developed a novel in vivo model of PD, integrating the involvement of pesticide exposure and systemic mitochondrial defects in PD etiology (Betarbet et al., 2000). Rats were systemically exposed to the pesticide rotenone for 1–5 weeks. Rotenone, a naturally occurring compound, is used as an insecticide and to kill nuisance fish in lakes. Rotenone is also a well characterized, specific inhibitor of complex I. Chronic systemic rotenone exposure reproduced many features of PD, including nigrostriatal dopaminergic lesions and development of α-synuclein-positive cytoplasmic inclusions in nigral neurons (Betarbet et al., 2000).
Oxidative stress may contribute to the neurodegeneration observed in PD (Jenner, 1998). Brains of PD patients have decreased levels of reduced glutathione (GSH), and there is oxidative damage to DNA, lipids, and protein (Dexter et al., 1989; Sian et al., 1994; Alam et al., 1997;Pearce et al., 1997; Floor and Wetzel, 1998). Reactive oxygen species (ROS) responsible for this oxidative damage may be produced during dopamine metabolism or during oxidative phosphorylation (Hasegawa et al., 1990; Lotharius and O'Malley, 2000). Within complex I, upstream from the rotenone binding site, is a site of electron leak that can enhance ROS formation (Hensley et al., 1998). Reduced complex I activity, as produced by rotenone andN-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and observed in PD, increases ROS production (Turrens and Boveris, 1980;Hasegawa et al., 1990; Cassarino et al., 1997; Votyakova and Reynolds, 2001). Oxidative stress may also contribute to α-synuclein pathology in PD. Oxidatively modified α-synuclein is more prone to aggregation than native protein (Souza et al., 2000). Furthermore, elevated α-synuclein expression can itself cause oxidative stress (Hsu et al., 2000).
Brains from PD patients show evidence of apoptosis, including fragmented nuclei and caspase activation (Tatton et al., 1998; Hartmann et al., 2000; Tatton, 2000). However, the role of apoptosis in PD remains controversial, and other studies of end-stage PD patients demonstrate little evidence of apoptosis (Burke and Kholodilov, 1998;Wüllner et al., 1999).
Here we report development of an in vitro model to examine mechanisms through which chronic complex I inhibition causes or potentiates neural cell death. Previous studies have examined the acute impact (1–3 d) of rotenone exposure, at relatively high doses, which may not be relevant to the progressive nature of PD (Leist et al., 1999; Taylor et al., 2000; King et al., 2001; Lee et al., 2002b). We exposed human neuroblastoma cells to a low concentration of rotenone for up to 4 weeks and found accumulation of α-synuclein, progressive oxidative damage, and increased basal and H2O2-induced caspase-dependent cell death.
MATERIALS AND METHODS
Cell culture. SK-N-MC neuroblastoma cells were cultured in minimum essential medium (MEM) with Earle's salts containing 5 mm glucose (Mediatech, Herndon, VA), 15% fetal bovine serum (Invitrogen, Carlsbad, CA), 50 U/ml penicillin and streptomycin, 5 mm sodium pyruvate, and nonessential amino acid solutions for MEM (Mediatech). Media were supplemented with 5 nm rotenone (Sigma, St. Louis, MO) or solvent (ethanol) for up to 4 weeks. The concentration of rotenone was chosen on the basis of a pilot study and previous observations (Sherer et al., 2001b). For routine culture, cells were grown in 100 mm plates, fed three times per week with control medium or medium supplemented with 5 nmrotenone, and passaged approximately once a week on reaching confluence. Rotenone (5 nm) did not alter cellular morphology or growth kinetics over the 4 week exposure period.
Mitochondrial respiration. For studies of oxygen consumption, cells were grown in 12 150 cm2 cell culture flasks. Mitochondria were isolated using a previously published protocol (Trounce et al., 1996) without BSA, because BSA binds rotenone nonspecifically. Oxygen consumption was measured polargraphically as described previously (Panov and Scaduto, 1996) using an Instech minichamber (Instech Laboratories, Plymouth Meeting, PA) equipped with a magnetic stirrer and oxygen electrode (Yellow Spring Instrument Co., Yellow Springs, OH) connected to a chart recorder. The following medium was used: 125 mm KCl, 10 mm 4-morpholinepropanesulfonic acid, 2 mmKH2PO4, 2 mmMgCl2, 10 mm NaCl, 0.7 mmCaCl2, 1 mm EGTA, 0.5 μm carbonyl cyanidep-trifluoromethoxyphenylhydrazone, 20 mm glutamate, and 2 mmmalate.
Determination of α-synuclein and ubiquitin levels. Levels and distributions of α-synuclein and ubiquitin were determined by immunocytochemistry and protein dot blots. For immunocytochemistry, control and rotenone-treated cells were grown on 18 mm coverslips coated with 0.1% gelatin (Sigma) and fixed for 15 min with 4% paraformaldehyde. Cells were incubated in primary antibody for 24 hr, followed by 1 hr incubation with biotinylated secondary antibody. The avidin–biotin complex method was used to detect antigen signal (ABC Elite kit; Vector Laboratories, Burlingame, CA), and 3,3′diaminobenzidine tetrachloride was used to visualize the final product. The primary antibodies used were polyclonal rabbit antibody against α-synuclein (1:100; a gift from Mark Cookson, Bethesda, MD) and polyclonal rabbit antibody against ubiquitin (1:1000; Dako, Carpinteria, CA). Secondary antibody used was biotinylated goat anti-rabbit IgG (1:200; Jackson ImmunoResearch, West Grove, PA). We examined images using bright-field microscopy. Images were captured using Zeiss (Thornwood, NY) Axiovision 3.0 software. For final output, images were processed simultaneously and identically using Adobe Photoshop 5.5 software (Adobe Systems, San Jose, CA).
For dot blots, control and rotenone-treated cells were grown on 100 mm plates. Cells were washed two times with PBS, pH 7.4, and incubated in 900 μl of cell lysis buffer (Promega, Madison, WI) containing 0.5 mg/ml benzamidine, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 0.75 mm phenylmethylsulfonyl fluoride, 700 U/ml DNase I, and 1% β-mercaptoethanol (all from Sigma) for 15 min at room temperature. Cells were scraped, and the cell lysate was centrifuged at 12,000 × g for 2 min. The supernatant was collected as the soluble protein fragment. The insoluble pellet was resuspended in 12% SDS. Protein assays were done using a Bio-Rad protein assay and Bio-Rad Dc protein assay (Bio-Rad, Hercules, CA) according to the manufacturer's protocol. Protein (15–20 μg) was combined with an equal volume of 2× loading buffer with β-mercaptoethanol and heated to 65°C for 10 min. Protein was added to an Immunobilon P transfer membrane (Millipore, Bedford, MA) and cross-linked using a UV Stratalinker (Stratagene, La Jolla, CA). The membrane was washed twice for 5 min in PBS and then twice for 10 min in PBS. The membrane was blocked with 1:10 milk diluent/water for 1 hr and incubated overnight at 4°C in primary antibody. The membrane was washed twice for 5 min in PBS and Tween 20 and then twice for 10 min in PBS and Tween 20 and incubated in secondary antibodies conjugated to horseradish peroxidase (1:7500; ICN Pharmaceuticals, Costa Mesa, CA), for 1.5 hr at room temperature. After two 5 min and two 10-min washes in PBS, the blot was detected using Supersignal West Dura extended duration substrate (Pierce, Rockford, IL). The blot was developed and analyzed using Eastman Kodak (Rochester, NY) Digital Science 1D image analysis software version 3.0.2. Primary antibodies were mouse anti-α-synuclein (1:400; Zymed, South San Francisco, CA) and rabbit anti-ubiquitin (1:500; Dako).
Reverse transcription, primer design, and quantitative reverse transcription-PCR. First-strand cDNA was synthesized by priming 2 μg of total RNA with oligo-dT and using a Superscript II enzyme according to the manufacturer's instructions (Invitrogen, Rockville, MD). Primers were designed using the Primer Express program (Applied Biosystems, Foster City, CA). Both primer pairs cross exon boundaries. The following primers were used: β-actin forward, tcaccatggatgatgatatcgcc; β-actin reverse, ccacacgcagctcattgtagaagg; α-synuclein forward, aggactttcaaaggccaagg; and α-synuclein reverse, tcctccaacatttgtcacttgc.
Real-time quantitative PCR was performed using the Applied Biosystems 7900HT system. The amount of double-stranded PCR product synthesized in each cycle was measured using SyBr green I dye, with fluorescence being measured at the end of the annealing step of each cycle to monitor amplification. Reactions were performed in a 20 μl volume with 5 pmol each of forward and reverse primers. Expression levels for each gene for each template were calculated for four simultaneous reactions at each of three different dilutions (undiluted and 1:2 and 1:4 of the original cDNA sample). Average threshold cycle (Ct) values from the replicate PCRs were normalized to the average Ct values for the β-actin control from the same cDNA preparations. The ratio of expression of each gene was calculated as 2(meanΔΔCt), where ΔΔCt is the difference Ct(test gene) − Ct(β-actin). Ratios of rotenone versus mean control were calculated.
Glutathione levels. Control and rotenone-treated cells were grown on 100 mm culture plates. Cells were scraped and collected by centrifugation. The cell pellet was homogenized in 1 ml of PBS containing 1 mm EDTA. The supernatant was collected after centrifugation at 10,000 × g for 15 min at 4°C. The supernatant was deproteinated by adding an equal volume of 10% metaphosphoric acid (Sigma), incubating at room temperature for 5 min, and centrifuging for 5 min at 5,000 × g. Total glutathione was determined using a glutathione assay kit (Cayman Chemical Co., Ann Arbor, MI), which is based on the glutathione reductase enzymatic recycling method. Glutathione was normalized to cellular protein and expressed as a percentage of glutathione levels in control cells at each weekly time point.
Detection of protein carbonyls. For determination of protein carbonyls, control and rotenone-treated cells were grown on 100 mm plates. Soluble and insoluble protein fractions were collected as described above for dot blots. Protein carbonyls were assayed with the Oxyblot protein oxidation detection kit (Intergen, Purchase, NY). Briefly, 10 μg of protein was mixed with an equal volume of 12% SDS and 2 volumes of 1× 2,4-dinitrophenylhydrazine (DNPH) solution. Control reactions used 1× derivatization control solution instead of the DNPH solution. The reaction proceeded for 15 min at room temperature and was stopped by the addition of 1.5 volumes of neutralization solution. Protein (3 μg) was added to an Immunobilon P transfer membrane (Millipore) and cross-linked using a UV Stratalinker (Stratagene). The membrane was washed twice for 5 min in PBS and then twice for 10 min in PBS. The membrane was blocked with 1:10 milk diluent/water for 1 hr and incubated overnight at 4°C in 1:150 rabbit anti-2,4-dinitrophenylhydrazone antibody (Intergen). The membrane was washed twice for 5 min in PBS and Tween 20 and then twice for 10 min in PBS and Tween 20 and incubated in goat-anti-rabbit IgG (1:300; Intergen) for 1.5 hr at room temperature. After two 5-min and two 10-min washes in PBS, the blot was detected using the Supersignal West Dura extended duration substrate (Pierce). The blot was developed and analyzed using Kodak Digital Science 1D image analysis software version 3.0.2.
Oxidative DNA damage. Oxidative DNA damage was determined using anti-8-hydroxydeoxyguanosine (8-oxo-dG) antibodies, according to the manufacturer's protocol (1:300; Trevigen, Gaithersburg, MD). Control and rotenone-treated cells were grown on eight-well Labtek chamber permanox slides coated with 0.1% gelatin (Fischer Scientific, Pittsburgh, PA) and treated with 300 μmH2O2 for 6 hr. Cells were fixed with 4% paraformaldehyde for 15 min. Alexa-488 anti-mouse IgG (1:200; Molecular Probes, Eugene, OR) was used as the secondary antibody. Fluorescence images were captured on a Leitz microscope (Leica, Nussloch, Germany) linked to a microcomputer imaging device (MCID) image analysis system (Imaging Research, St. Catharines, Ontario, Canada) with selective filters to visualize FITC and UV. For final output, images were processed identically and simultaneously using Adobe Photoshop 5.5 software.
Cell death assays. Rates and levels of cell death were determined at 1-week intervals during exposure to rotenone or vehicle. Cell death assays used the cell-impermeable dye Sytox green (Molecular Probes), which intercalates into the DNA of dead cells and fluoresces; it was detected with excitation at 485 nm and emission at 538 nm with a fluorescence microplate system (Molecular Devices, Sunnyvale, CA). Control and rotenone-treated cells were grown at similar cell densities in 96-well plates, loaded with 1 μm Sytox green for 10 min, and then treated with H2O2 at final concentrations of 10, 100, and 300 μm. Control wells received equivalent volumes of fresh medium. In some experiments, cells were preincubated with the caspase inhibitor z-Asp-Glu-Val-fluoromethyl ketone (Z-DEVD-FMK) (150 μm; Calbiochem, San Diego, CA) for 2 hr before addition of H2O2. Fluorescence readings were taken once per hour and normalized to initial plating density after fixation with 4% paraformaldehyde for 45 min at 4°C.
Detection of apoptosis. Control and rotenone-treated cells were grown on eight-well Labtek chamber permanox slides (Fischer Scientific) coated with 0.1% gelatin. Cells were exposed to 300 μm H2O2 for 24 hr and fixed with 4% paraformaldehyde for 15 min. Apoptotic nuclei were detected using an Apoptag Plus fluorescein in situapoptosis detection kit (Intergen) according to the manufacturer's protocol. Nuclei were stained with bisbenzimide (1:1000; Sigma). Fluorescence images were captured on a Leitz microscope (Leica) linked to an MCID image analysis system (Imaging Research) with selective filters to visualize FITC and UV. For final output, images were processed using Adobe Photoshop 5.5 software. The percentage of cells showing apoptotic nuclei was determined.
Cytochrome c distribution and caspase-3 activation. Determination of cytochrome c distribution and caspase-3 activation used immunofluorescence. For double labeling of cytochrome c and activated caspase-3, control and rotenone-treated cells were grown on 18 mm coverslips coated with 0.1% gelatin (Sigma). Cells were exposed to 300 μmH2O2 for 2–4 hr and fixed with 4% paraformaldehyde for 15 min. Cells were washed three times for 5 min with Tris-buffered saline (TBS), blocked with 10% normal goat serum for 30 min, and then incubated overnight in the appropriate primary antibody. After three 5 min washes in TBS, cells were incubated in 1:200 secondary antibody (Alexa anti-mouse IgG or Cy3 anti-rabbit IgG; Molecular Probes) for 1 hr. Cells were washed three times for 5 min in TBS, and then nuclei were stained with bisbenzimide (1:1000; Sigma) for 5 min and washed in TBS three times for 5 min. Cells were coverslipped with Aquamount (Lerner Laboratories, Pittsburgh, PA). Cytochrome c was detected using a mouse monoclonal antibody (1:500; PharMingen, San Diego, CA). Active caspase-3 was detected using a polyclonal rabbit antibody (1:500; Cell Signaling, Beverly, MA). For controls, primary antibodies were omitted. Cells were imaged using aZeiss laser scanning microscope 510 with nonlinear optics. For final output, images were processed using Adobe Photoshop 5.5 software.
Live imaging of caspase activation. Control and rotenone-treated SK-N-MC cells were plated on 25 mm coverslips. After a 6 hr exposure to 300 μmH2O2, cells were simultaneously labeled with 4.5 μm bisbenzimide (Sigma) and 25 μm rhodamine 110 bis-l-aspartic acid amide (Molecular Probes) for 15 min. The presence of activated caspases is indicated by cytoplasmic green fluorescence, because activated caspases cleave side chains from the nonfluorescent rhodamine 110 bis-l-aspartic acid amine to generate green fluorescent rhodamine 110. Cells were imaged using a Zeiss laser scanning microscope 510 with nonlinear optics. For final output, images were processed using Adobe Photoshop 5.5 software.
Statistical analysis. Statistical analysis used multivariate ANOVA or Student's t test for independent samples. Significance was set at p ≤ 0.05. Values shown represent mean ± SEM.
RESULTS
In vitro model of chronic rotenone exposure
In vivo, chronic rotenone exposure caused α-synuclein aggregation and selective nigrostriatal dopaminergic degeneration (Betarbet et al., 2000). Here, SK-N-MC neuroblastoma cells were grown for up to 4 weeks in medium supplemented with 5 nm rotenone, a sublethal dose of rotenone. Oxygen consumption studies showed that 5 nm rotenone caused no reduction in mitochondrial respiration, whereas 15–30 nm rotenone caused 20–30% reductions in mitochondrial respiration. Thus, as in the in vivo model (Betarbet et al., 2000), we found no evidence for a rotenone-induced bioenergetic defect per se. Up to 4 weeks of exposure to 5 nm rotenone did not affect cell density or cell morphology, as determined by phase contrast microscopy and bisbenzimide staining (data not shown); however, as described below, a small proportion of cells (∼5%) began to undergo apoptosis by this time point.
Rotenone exposure increased α-synuclein and ubiquitin levels
To determine whether this in vitro model mimicked the effects of in vivo rotenone infusion, we determined α-synuclein protein levels in control and rotenone-treated cells. After 1 week of rotenone exposure, soluble α-synuclein levels were elevated (41 ± 16% increase; p < 0.05) (Fig.1A). After 4 weeks of rotenone exposure, soluble α-synuclein levels remained elevated (40 ± 15% increase; p < 0.05), but there was also an increase in insoluble α-synuclein as well (29 ± 9% increase; p < 0.05) (Fig. 1A). Levels of α-synuclein mRNA, determined by quantitative reverse transcription-PCR, were not altered significantly despite the increase in α-synuclein protein levels. Immunocytochemistry revealed accumulation of α-synuclein immunoreactivity in the cytoplasm after 4 weeks of rotenone exposure (Fig. 1B,C).
Fig. 1.
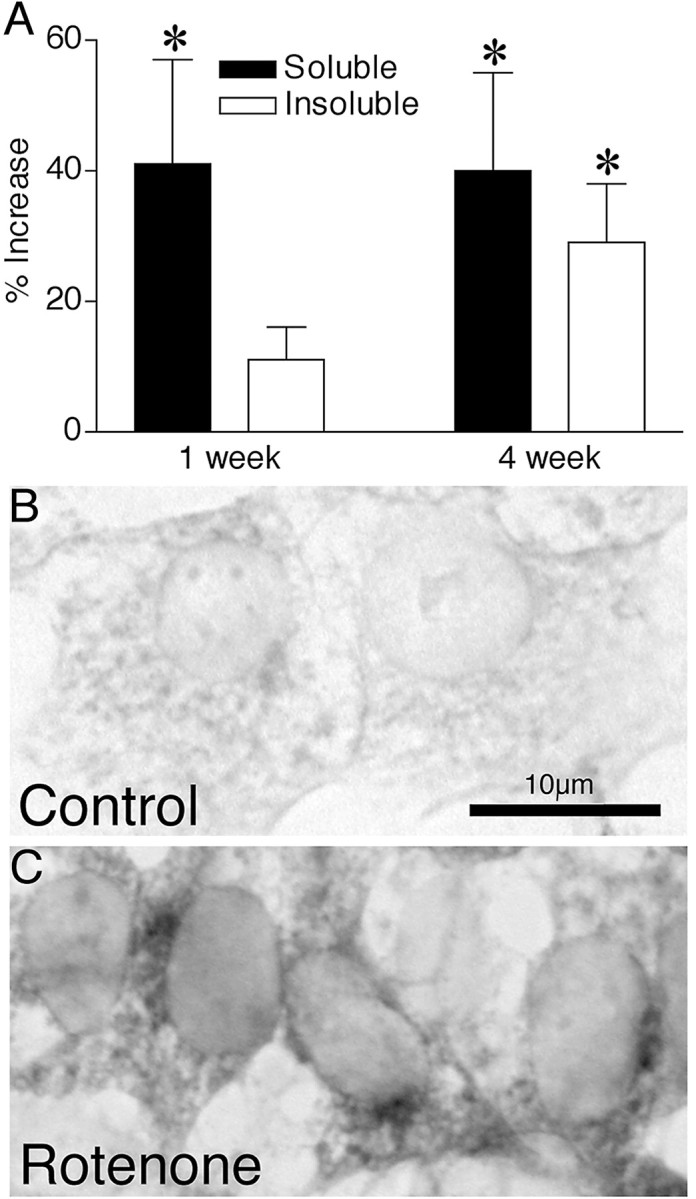
Chronic rotenone exposure increased α-synuclein protein levels. A, One week of rotenone treatment increased soluble α-synuclein levels, whereas 4 weeks of rotenone treatment elevated α-synuclein levels in both soluble and insoluble protein fractions. Results are expressed as percent increase from control and represent mean ± SEM of three or four independent experiments. *p < 0.05 compared with control cells. Compared with control cells (B), chronic rotenone exposure (C) caused cytoplasmic α-synuclein accumulation, as determined by immunocytochemistry. Scale bar, 10 μm. Similar results were observed in four independent experiments.
The Lewy bodies seen in PD brains contain ubiquitin in an insoluble form, and chronic in vivo rotenone exposure reproduced these ubiquitin-positive inclusions (Betarbet et al., 2000). Therefore, we assessed ubiquitin in control and rotenone-treated cells. Short-term rotenone exposure (1 week) did not alter levels of ubiquitin immunoreactivity. However, after 4 weeks of rotenone exposure, ubiquitin levels were elevated in the insoluble protein fraction (87 ± 14% increase; p < 0.05). Immunocytochemistry revealed an elevation in cytoplasmic ubiquitin staining in cells grown chronically (4 weeks) in rotenone (Fig.2).
Fig. 2.
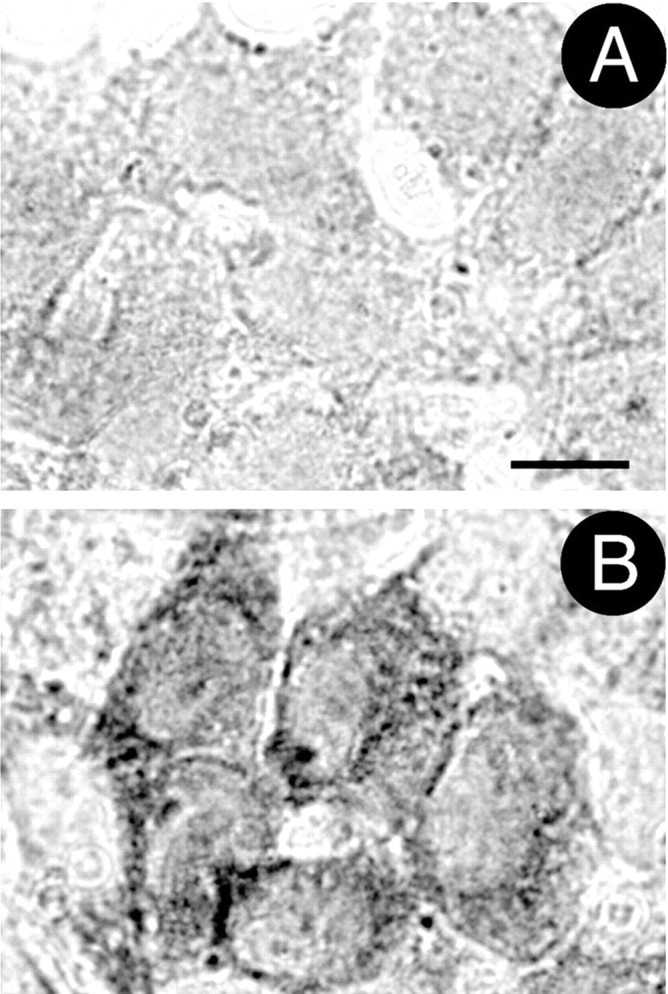
Chronic rotenone exposure increased levels of ubiquitin immunoreactivity. A, B, Chronic rotenone exposure resulted in elevated cytoplasmic ubiquitin levels, as determined by immunocytochemistry. A, Control cells.B, Cells treated with rotenone for 4 weeks. Scale bar, 10 μm. Similar results were observed in four independent experiments.
These results demonstrate that chronic in vitro rotenone treatment caused α-synuclein and ubiquitin accumulation. Elevated α-synuclein levels may cause oxidative stress (Hsu et al., 2000) as well as increased sensitivity to oxidative challenges (Kanda et al., 2000; Ko et al., 2000). Furthermore, rotenone itself induces oxidative damage (Votyakova and Reynolds, 2001; Zhang et al., 2001). Thus, we examined levels of basal and H2O2-induced oxidative damage and cell death after prolonged rotenone exposure.
Chronic rotenone exposure reduced glutathione levels
Pilot studies showed that rotenone treatment caused parallel reductions in oxidized and reduced GSH; after 4 weeks, glutathione disulfide was decreased by 40%, and GSH was decreased by 44%. Therefore, subsequent studies measured total cellular GSH. Short-term rotenone exposure (1–2 weeks) did not alter GSH levels, although there was a nonsignificant trend toward increased levels during the second week of exposure. In contrast, chronic rotenone treatment (3–4 weeks) caused a 50% reduction in cellular GSH levels (p < 0.05) (Fig.3A).
Fig. 3.

Chronic rotenone treatment caused delayed oxidative damage. A, Chronic rotenone exposure (5 nm) decreased cellular GSH. Control values at 1 week were 4.97 ± 1.2 nmol/mg of protein. Results are expressed as percentages of levels in control cells at each time point and represent mean ± SEM of four independent experiments at each time point.B, Delayed oxidative protein damage in rotenone-treated cells. Protein carbonyl levels are expressed as percentages of levels in control cells at each time point. Results show mean ± SEM of four independent experiments at each time point. *p< 0.05 compared with control.
Chronic rotenone exposure caused oxidative protein damage
Rotenone-induced loss of GSH raised the possibility of oxidative damage. Reactive carbonyls, another index of oxidative damage, are found on DNA and protein (Alam et al., 1997) and can be detected using the DNPH reaction. We analyzed protein carbonyl levels using dot blots of both soluble and insoluble protein isolated from control and rotenone-treated cells. Protein was isolated in the presence of DNase I to remove DNA from the lysate. Exposure of cells to rotenone for 1–2 weeks did not alter protein carbonyl levels, but exposure for 3–4 weeks caused a large increase in carbonyls in the insoluble fraction (223 ± 29% of control; p < 0.05) (Fig.3B). Elevated carbonyls were not found in the soluble protein fraction.
Chronic rotenone exposure causes oxidative DNA damage
To assess oxidative DNA damage in control and rotenone-treated cells, we used antibodies to 8-oxo-dG. Short-term rotenone exposure (1–2 weeks) did not induce oxidative DNA damage, but there was a marked increase in 8-oxo-dG immunoreactivity in cells exposed to rotenone for 4 weeks (Fig.4B). Interestingly, after a 6 hr exposure to 300 μmH2O2, an oxidative challenge, cells grown in rotenone for 4 weeks showed increased oxidative DNA damage compared with control cells (Fig. 4). Nuclear staining with bisbenzimide showed that many rotenone-treated cells with oxidative DNA damage also had nuclear condensation or fragmentation characteristic of apoptosis (Figs. 4B,D). Thus, chronic rotenone exposure increased basal levels of oxidative DNA damage.
Fig. 4.

Chronic rotenone treatment caused oxidative DNA damage. Control cells (A, C) and cells treated with rotenone for 4 weeks (B, D) were stained with antibodies against 8-oxo-dG, a marker of oxidative DNA damage (top panels). The same cells were also labeled with bisbenzimide for nuclear morphology (bottom panels). Rotenone-treated cells showed increased 8-oxo-dG immunoreactivity before H2O2 exposure (B). H2O2 increased 8-oxo-dG staining to a greater extent in rotenone-treated cells (D) than in control cells (C). Many cells with oxidative DNA damage showed fragmented nuclear morphology characteristic of apoptosis (B, D). Similar results were observed in four replicates.
Chronic rotenone exposure potentiated H2O2-induced cell death and oxidative protein damage
Because chronic rotenone treatment caused progressive oxidative damage, and rotenone-treated (4 weeks) cells were much more vulnerable to oxidative DNA damage resulting from H2O2 exposure, we examined whether chronic rotenone treatment elevated the rates of basal and oxidant-induced death. At weekly intervals, cell death was assayed using Sytox green fluorescence. Sytox green intercalates into the DNA of cells in which the plasma membrane has been perturbed, and as such, it measures both necrotic and apoptotic cells. We chose H2O2 as an oxidative stressor because dopaminergic cells are normally exposed to H2O2 during dopamine synthesis and catabolism. Pilot studies showed that 10 and 100 μm H2O2 did not induce consistent death in control SK-N-MC cells, whereas 300 μm H2O2caused progressive cell death over 24 hr. For this reason, all subsequent comparisons between control and rotenone-treated cells focused on responses to 300 μmH2O2 during the 24 hr after exposure. We did not detect any differences in rates of basal cell death resulting from chronic rotenone treatment (up to 4 weeks) by this measure (Fig. 5A,B). Although short-term exposure to rotenone (1–2 weeks) did not alter cell death in response to H2O2, chronic rotenone exposure (3–4 weeks) markedly and progressively potentiated and accelerated oxidant-induced cell death (p < 0.05) (Fig. 5A,B). The rate of cell death, calculated as the slope of the linear portion of cell death curves, was 92% higher in cells treated chronically with rotenone (p < 0.05).
Fig. 5.

Chronic rotenone treatment sensitized cells to H2O2-induced death and oxidative protein damage. Cells were grown in medium supplemented with 5 nmrotenone for 1–4 weeks before exposure to 300 μmH2O2. Cell death was then monitored over 24 hr. Cells treated with rotenone for 1 week (A) showed responses to H2O2 similar to those of control cells. However, after exposure to rotenone for 4 weeks (B), cells were markedly sensitized to H2O2. Results show mean ± SEM of three independent experiments at each time point. *p < 0.05. A.U., Arbitrary units.
Chronic rotenone exposure also potentiated oxidative protein damage after exposure to H2O2. Although a 6 hr exposure to H2O2 did not increase protein carbonyl levels in control cells, soluble protein carbonyl levels were significantly elevated in cells that had been grown in the presence of rotenone for 4 weeks (246 ± 30% of baseline;p < 0.05). These results demonstrate that chronic rotenone treatment markedly sensitized cells to subsequent oxidative stress.
Chronic rotenone exposure increased apoptotic death
Because Sytox green fluorescence (Fig. 5) did not differentiate necrotic or apoptotic cell death, we used terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling (TUNEL) staining to detect the DNA strand breaks characteristic of apoptosis. At baseline, cells treated with rotenone for 4 weeks had a small but significant increase in apoptosis compared with control cells (p < 0.05) (Fig.6A,C,E). This slight increase in apoptotic death was not detected with the Sytox green assay (Fig. 5B, bottom curves). After exposure to H2O2, there was substantially more apoptosis in rotenone-treated cells than in control cells (p < 0.05) (Fig.6B,D,E), and the rate of apoptotic death was substantially faster in cells treated chronically with rotenone (1.44 ± 0.02% vs 0.38 ± 0.07% apoptosis/hr;p < 0.05).
Fig. 6.

Chronic rotenone treatment increased apoptotic cell death before and after H2O2 exposure. Cells were analyzed for DNA fragmentation using TUNEL staining before and 24 hr after H2O2 exposure. Control cells showed little evidence of apoptosis (A), whereas cells treated with rotenone for 4 weeks showed a small but significant increase in apoptosis before H2O2 exposure (C, E). Cultures treated with rotenone for 4 weeks showed more TUNEL-positive cells after H2O2exposure (D) compared with control cultures (B). Arrows indicate some TUNEL-positive cells. E, Quantification of apoptotic cells. Cultures treated with rotenone for 4 weeks showed elevated apoptosis before and 24 hr after H2O2 exposure. Results are expressed as mean ± SEM of four experiments. *p < 0.05 compared with control.
Cytochrome c redistribution and activation of caspase-3
Under some conditions, mitochondrial impairment may cause cytochrome c release, caspase-3 activation, and apoptosis. We conducted a time course study to examine the role of cytochromec redistribution and caspase-3 activation in the differential cell death observed in rotenone-treated cells. At specific time points before and after H2O2 treatment, cells were triple-labeled with bisbenzimide and antibodies against cytochromec and activated caspase-3. In control cells, cytochromec had a punctate distribution reflecting its mitochondrial localization, and there was no caspase-3 activation (Fig.7A). After 2 hr exposure to H2O2, there was redistribution of cytochrome c such that staining was less punctate and less intense, consistent with cytosolic redistribution (Fig. 7B). After 4 hr, scattered cells began to show evidence of caspase-3 activation, but nuclear morphology remained normal (Fig. 7C). In cells treated with rotenone for 4 weeks, there was generally a normal cytochrome cdistribution, but there were occasional cells with apparent cytochromec redistribution or caspase-3 activation associated with nuclear fragmentation (Fig. 7D). After a 2 hr exposure to H2O2, rotenone-treated cells showed extensive activation of caspase-3, and many cells had fragmented nuclei (Fig. 7E). At 4 hr, there was further caspase-3 activation, and many cells were in advanced stages of apoptosis, based on nuclear morphology (Fig. 7F). At baseline and 2 and 4 hr after H2O2 treatment, more rotenone-treated cells showed caspase-3 activation than control cells (p < 0.05) (Fig. 7G).
Fig. 7.
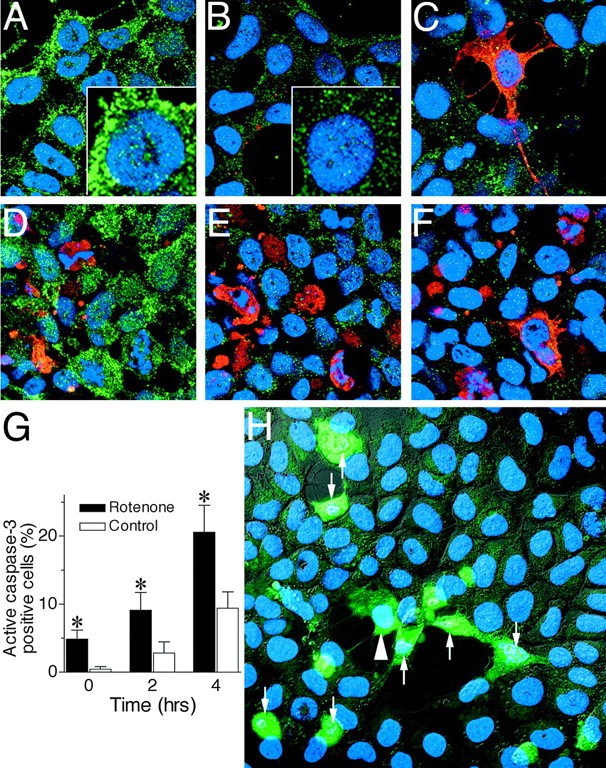
Time course of cytochromec redistribution and caspase-3 activation in control and rotenone-treated cells after H2O2 exposure. Control cells (A–C) and cells treated with rotenone for 4 weeks (D–F) were triple-stained for nuclear morphology (bisbenzimide; blue), cytochromec (green), and activated caspase-3 (red) before (A, D) and 2 hr (B, E) and 4 hr (C, F) after H2O2 exposure. Under basal conditions, both control and rotenone-treated cells showed punctate cytochromec staining, consistent with its mitochondrial localization (A, D). After H2O2exposure (B, C, E, F), there was release of cytochrome c from mitochondria to cytosol, resulting in loss of punctate staining and a more diffuse, less intense staining pattern. A, B, insets, Cytochromec redistribution at higher power. D, In occasional rotenone-treated cells under basal conditions, there was redistribution of cytochrome c such that staining was less punctate and less intense, consistent with cytosolic distribution. Increased caspase-3 activation was evident 2 and 4 hr after H2O2 exposure but occurred to a greater extent in rotenone-treated cells (E, F) than in control cells (B, C). Many of the rotenone-treated cells expressing activated caspase-3 contained fragmented nuclei characteristic of apoptosis, indicating that rotenone treatment was associated with more advanced stages of apoptosis. Similar results were observed in four independent experiments. G, Quantification of cells with caspase-3 activation after H2O2 treatment. More rotenone-treated cells expressed activated caspase-3 before and 2–4 hr after H2O2 exposure. *p < 0.05 compared with controls. H, Caspase activation in live cells after H2O2 exposure. Cells treated with rotenone for 4 weeks were exposed to 300 μmH2O2 for 6 hr and then imaged simultaneously for cell morphology using phase-contrast microscopy (gray), nuclear morphology using bisbenzimide (blue), and caspase activation using rhodamine 110 bis-l-aspartic acid amide (green). H2O2 caused some cells to retract their processes, round up, and undergo nuclear fragmentation (arrows). Some cells showed caspase activation before nuclear fragmentation (arrowhead). Similar results were observed in four independent experiments.
To confirm caspase activation after H2O2 exposure, we monitored caspase substrate cleavage in living cells. Cells were treated for 6 hr with H2O2 and loaded with rhodamine-110 bis-l-aspartic acid amide, a cell permeant, nonfluorescent substrate that fluoresces on cleavage by caspase-3 or -7. Cells were simultaneously loaded with bisbenzimide to analyze nuclear morphology. After H2O2 treatment, rotenone-treated cells retracted their processes and activated caspase proteases, as indicated by rhodamine 110 green fluorescence (Fig.7H, arrows). At this time point, many cells that showed caspase activation also demonstrated evidence of the nuclear fragmentation characteristic of apoptosis (Fig. 7H,arrows), but some cells expressed activated caspases before nuclear fragmentation (Fig. 7H, arrowheads). Thus, live cell imaging confirmed caspase activation after H2O2 exposure in rotenone-treated cells.
To further assess the involvement of caspase activation in H2O2-induced cell death, we exposed cells to H2O2 in the presence or absence of 150 μm Z-DEVD-FMK, which inhibits caspase-3 as well as caspase-6, -7, -8, and -10. Treatment with Z-DEVD-FMK delayed and reduced H2O2-induced cell death (Fig. 8).
Fig. 8.

Caspase inhibition delayed and reduced H2O2-induced death. Cells treated with rotenone for 4 weeks were incubated with vehicle or caspase inhibitor (Z-DEVD-FMK) for 2 hr before and throughout the exposure to H2O2. Cell death was analyzed as described. Results are expressed as mean ± SEM. Similar results were obtained in three independent experiments. *p < 0.05 compared with cells treated with rotenone and H2O2 alone.
DISCUSSION
The goal of the current study was to develop a novel in vitro system to model the pathogenesis of PD. Although PD is a progressive, chronic neurological disease, most in vitrostudies, including ours, have examined pathogenic mechanisms over 24–48 hr (Seaton et al., 1998; Fukuhara et al., 2001; Zhang et al., 2001; Stephans et al., 2002). Moreover, PD is marked by a modest systemic complex I dysfunction, an ∼25% loss of activity (Langston et al., 1983; Mizuno et al., 1989; Parker et al., 1989; Schapira et al., 1989); however, many studies that examine mechanisms of 1-methyl-4-phenylpyridinium (MPP+) and rotenone toxicity use concentrations of rotenone or MPP+ that are more than sufficient to inhibit complex I completely (Leist et al., 1999; King et al., 2001;Zhang et al., 2001). In contrast, the concentration of rotenone we used (5 nm) (1) is low relative to its IC50 (20–30 nm), (2) did not inhibit respiration in mitochondria isolated from these cells, and (3) produced a level of complex I inhibition similar to what is seen in PD (Sherer et al., 2001b). Damage developed over a protracted time course, and features of PD pathogenesis, such as α-synuclein accumulation and aggregation and oxidative damage, were reproduced, even in the absence of α-synuclein mutations or overexpression. This chronic model system may provide an accurate means of studying toxic mechanisms and compensatory cellular responses that occur over time in PD.
It is important to note that our model used human neuroblastoma cells rather than dopaminergic neurons. Unfortunately, it is extremely difficult to keep sufficient numbers of primary dopaminergic neurons alive long enough to perform the types of experiments described here. Despite this limitation, there are advantages of our system. First, cells can be cultured in the presence of rotenone for at least 1 month. Second, the cells express endogenous human α-synuclein, and chronic rotenone treatment increased its levels and caused it to become insoluble. Finally, we can simulate the oxidative stress inherent to dopaminergic neurons by adding exogenous H2O2. Tyrosine hydroxylase and monoamine oxidase, two enzymes involved in dopamine metabolism, produce H2O2 as a normal byproduct. Additionally, auto-oxidation of dopamine results in H2O2 formation and, subsequently, ·OH, an extremely reactive ROS (Lotharius and O'Malley, 2000). Thus, H2O2 is a relevant oxidative stressor in the context of dopaminergic degeneration that occurs in PD.
This in vitro system allowed us to begin to define the sequence of cellular responses to chronic complex I inhibition. Although 1 week of rotenone exposure increased soluble α-synuclein levels, chronic rotenone treatment resulted in cytoplasmic accumulation of insoluble α-synuclein and ubiquitin. Similarly, chronic in vivo rotenone induced formation of cytoplasmic inclusions containing α-synuclein and ubiquitin (Betarbet et al., 2000). Other studies have also suggested a role for complex I dysfunction in regulating α-synuclein levels. MPTP administration increased α-synuclein immunoreactivity in brain (Kowall et al., 2000; Vila et al., 2000), but it is unclear whether the accumulated α-synuclein in these studies was soluble or insoluble. Although rotenone exposure has been reported to cause α-synuclein aggregation, these studies used high concentrations of rotenone (100 nm to 100 μm) and short periods (hours to days) and were conducted in either artificial cell-free systems or cells that overexpressed α-synuclein, thereby making aggregation much more likely (Uversky et al., 2001; Lee et al., 2002a,b). Also, at rotenone concentrations of >1 μm, rotenone may have nonspecific effects (Barrientos and Moraes, 1999).
In our study, because there was no change in α-synuclein mRNA levels, the increased α-synuclein protein levels most likely reflect retarded degradation. α-Synuclein may be a substrate for the ubiquitin–proteasome system (UPS), and it has been suggested that impairment of UPS function may be central to PD pathogenesis (Bennett et al., 1999; McNaught et al., 2001; Rideout et al., 2001). Whether chronic rotenone impairs the UPS is currently under investigation. Nevertheless, our in vitro model demonstrates that chronic low-grade complex I inhibition can increase levels of endogenous human α-synuclein and eventually cause it to become insoluble.
Although Lee et al. (2002b) found that 100 nm rotenone could induce formation of inclusions containing α-synuclein, they used an overexpression system. Because α-synuclein aggregation is concentration-dependent, overexpression of the protein makes aggregation much more likely. It is also worth noting that, in contrast to our results, these authors did not find that rotenone treatment increased the level of α-synuclein protein, perhaps because of the acute nature of their experiments.
How chronic rotenone causes α-synuclein to become insoluble is unclear but might be related to oxidative damage. Interestingly, the elevation in soluble α-synuclein levels preceded other changes associated with chronic rotenone treatment. Elevated α-synuclein expression alone can both cause oxidative damage and sensitize cells to exogenous oxidative challenges, and rotenone itself can produce oxidative stress (Hsu et al., 2000; Kanda et al., 2000; Ko et al., 2000; Seyfried et al., 2000; Zhang et al., 2001). Additionally, oxidative damage and cytochrome c can cause α-synuclein to aggregate (Hashimoto et al., 1999; Giasson et al., 2000). Therefore, we examined in rotenone-treated cells the progression of oxidative damage, cytochrome c redistribution, and cell death at baseline and after exogenous oxidative stress.
Chronic rotenone treatment caused delayed oxidative damage, which correlated temporally with α-synuclein aggregation. Thus, after 3 weeks of rotenone exposure, cells had a loss of GSH and oxidative DNA and protein damage. Oxidative damage, perhaps resulting from decreased complex I dysfunction, may be important in PD pathogenesis. Brains from PD patients demonstrate loss of GSH and oxidative DNA and protein damage (Alam et al., 1997; Floor and Wetzel, 1998; Jenner, 1998). In the in vivo rotenone model, chronic rotenone infusion induced selective oxidative damage in striatum (Sherer et al., 2001a). Other studies have also demonstrated rotenone-induced ROS synthesis and oxidative damage. However, these studies typically used excessive concentrations of rotenone (5–100 μm) or examined acute effects (15 min to 1 hr) of complex I inhibition in cells or isolated mitochondria (Hensley et al., 1998; Bailey et al., 1999; Boldyrev et al., 1999; Zhang et al., 2001). Another acute study found that a low concentration of rotenone (20 nm) did not cause persistent superoxide production over a 24 hr period (Nakamura et al., 2000). In contrast to these studies, Barrientos and Moraes (1999) found that 5 nm rotenone, a concentration that inhibited complex I by ∼50% and respiration by only 20%, induced a marked increase in ROS production and lipid peroxidation. However, these phenomena could only be observed after 2–3 d of rotenone exposure. This suggests that mild complex I inhibition may cause a slow but steady production of ROS that is below the limit of sensitivity of conventional acute assays. Unlike these acute model systems, our chronic rotenone model allowed analysis of cumulative oxidative damage over time, and the temporal correlation between oxidative protein damage and α-synuclein aggregation suggests a possible causal relationship (Giasson and Lee, 2000).
After 4 weeks, rotenone-treated cells began to show cytochromec redistribution, caspase activation, and apoptosis, as assessed by TUNEL staining and nuclear morphology. This small but significant elevation in basal apoptotic death (5.6 vs 1.4%) was undetectable by our plate-reader assay and only became apparent with TUNEL staining. Interestingly, we have found numerous nigral dopaminergic neurons expressing activated caspase-3 after in vivo rotenone infusion; however, we have not found convincing evidence of apoptosis in vivo (Sherer et al., 2001a). Previous studies determined that acute exposure (4–48 hr) to extremely high doses of rotenone (1–100 μm) resulted in caspase activation and both apoptotic and necrotic cell death (Seaton et al., 1998; Leist et al., 1999; King et al., 2001; Zhang et al., 2001). Our results demonstrate that chronic, low-level complex I inhibition leads to delayed activation of the apoptotic pathway, a mechanism that may be relevant to late-onset PD.
Dopaminergic neurons are thought to exist in a state of constant oxidative stress, in large part because of generation of H2O2. In our system, H2O2 induced cytochromec redistribution, activation of caspases, and DNA fragmentation, as described by others (Jiang et al., 2001; Zhuang et al., 2001). Compared with control cultures, however, rotenone-treated cultures showed H2O2-induced caspase activation and apoptosis earlier and in a larger percentage of cells. Although a similar process occurs after H2O2 exposure in control and rotenone-treated cells, more rotenone-treated cells entered and progressed more quickly through a caspase-dependent apoptotic pathway. Moreover, the rate of cell death was two to three times as fast in cells treated chronically with rotenone (Fig. 5B). An analogous situation may occur in PD, in which there is a normal, age-related loss of dopaminergic neurons (Ma et al., 1999), possibly related to ongoing oxidative stress, and an additional insult, possibly a mild complex I defect. The modest cell death induced by a chronic complex I defect superimposed on the age-related decline in dopaminergic cells means the threshold for development of parkinsonian symptoms will be reached much earlier in PD.
The mechanisms responsible for the sensitization of rotenone-treated cells to H2O2 are not clear. Studies from Chinopoulos and Adam-Vizi (2001) indicate that mitochondria with mild complex I defects depolarize acutely in response to H2O2. We do not know whether this phenomenon is important in our system; however, sensitization required ∼3 weeks of rotenone exposure despite the immediate effects of rotenone on complex I. Because H2O2-induced death reportedly requires mitochondrial-derived ROS (Dumont et al., 1999), progressive oxidative damage resulting from chronic rotenone treatment may render cells more vulnerable to this oxidative challenge. In addition, the rotenone-induced loss of GSH probably contributes to increased vulnerability.
The in vivo rotenone model of PD substantiated the involvement of chronic systemic complex I defects in PD pathogenesis (Betarbet et al., 2000). The in vitro model of chronic rotenone exposure provides a novel system, which links a number of events implicated in PD pathogenesis, including altered α-synuclein metabolism, decreased GSH, progressive oxidative damage, and apoptosis. This model system may provide an improved understanding of mechanisms of cell death in PD and an opportunity to screen potential therapeutic strategies.
Footnotes
This work was supported by National Institutes of Health Grants NS38399 (J.T.G.) and F32NS11132 (T.B.S.).
Correspondence should be addressed to Dr. J. Timothy Greenamyre, Center for Neurodegenerative Diseases, Emory University, Whitehead Biomedical Research Building, Room 505M, 615 Michael Street, Atlanta, GA 30322. E-mail: jgreena@emory.edu.
REFERENCES
- 1.Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- 2.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 3.Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of α-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- 5.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 6.Boldyrev AA, Carpenter DO, Huentelman MJ, Peters CM, Johnson P. Sources of reactive oxygen species production in excitotoxin-stimulated cerebellar granule cells. Biochem Biophys Res Commun. 1999;256:320–324. doi: 10.1006/bbrc.1999.0325. [DOI] [PubMed] [Google Scholar]
- 7.Burke RE, Kholodilov NG. Programmed cell death: does it play a role in Parkinson's disease? Ann Neurol. 1998;44:S126–S133. doi: 10.1002/ana.410440719. [DOI] [PubMed] [Google Scholar]
- 8.Cassarino DS, Fall CP, Swerdlow RH, Smith TS, Halvorsen EM, Miller SW, Parks JP, Parker WD, Jr, Bennett JP., Jr Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson's disease. Biochim Biophys Acta. 1997;1362:77–86. doi: 10.1016/s0925-4439(97)00070-7. [DOI] [PubMed] [Google Scholar]
- 9.Chinopoulos C, Adam-Vizi V. Mitochondria deficient in complex I activity are depolarized by hydrogen peroxide in nerve terminals: relevance to Parkinson's disease. J Neurochem. 2001;76:302–306. doi: 10.1046/j.1471-4159.2001.00060.x. [DOI] [PubMed] [Google Scholar]
- 10.Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 11.Dumont A, Hehner SP, Hofmann TG, Ueffing M, Droge W, Schmitz ML. Hydrogen peroxide-induced apoptosis is CD95-independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-κB. Oncogene. 1999;18:747–757. doi: 10.1038/sj.onc.1202325. [DOI] [PubMed] [Google Scholar]
- 12.Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem. 1998;70:268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 13.Fukuhara Y, Takeshima T, Kashiwaya Y, Shimoda K, Ishitani R, Nakashima K. GAPDH knockdown rescues mesencephalic dopaminergic neurons from MPP+-induced apoptosis. NeuroReport. 2001;12:2049–2052. doi: 10.1097/00001756-200107030-00051. [DOI] [PubMed] [Google Scholar]
- 14.Giasson BI, Lee VM. A new link between pesticides and Parkinson's disease. Nat Neurosci. 2000;3:1227–1228. doi: 10.1038/81737. [DOI] [PubMed] [Google Scholar]
- 15.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 16.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- 17.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC. Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc Natl Acad Sci USA. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasegawa E, Takeshige K, Oishi T, Murai Y, Minakami S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem Biophys Res Commun. 1990;170:1049–1055. doi: 10.1016/0006-291x(90)90498-c. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto M, Takeda A, Hsu LJ, Takenouchi T, Masliah E. Role of cytochrome c as a stimulator of α-synuclein aggregation in Lewy body disease. J Biol Chem. 1999;274:28849–28852. doi: 10.1074/jbc.274.41.28849. [DOI] [PubMed] [Google Scholar]
- 20.Hensley K, Pye QN, Maidt ML, Stewart CA, Robinson KA, Jaffrey F, Floyd RA. Interaction of α-phenyl-N-tert-butyl nitrone and alternative electron acceptors with complex I indicates a substrate reduction site upstream from the rotenone binding site. J Neurochem. 1998;71:2549–2557. doi: 10.1046/j.1471-4159.1998.71062549.x. [DOI] [PubMed] [Google Scholar]
- 21.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. α-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jenner P. Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov Disord. 1998;13:24–34. [PubMed] [Google Scholar]
- 23.Jiang D, Jha N, Boonplueang R, Andersen JK. Caspase 3 inhibition attenuates hydrogen peroxide-induced DNA fragmentation but not cell death in neuronal PC12 cells. J Neurochem. 2001;76:1745–1755. doi: 10.1046/j.1471-4159.2001.00151.x. [DOI] [PubMed] [Google Scholar]
- 24.Kanda S, Bishop JF, Eglitis MA, Yang Y, Mouradian MM. Enhanced vulnerability to oxidative stress by α-synuclein mutations and C-terminal truncation. Neuroscience. 2000;97:279–284. doi: 10.1016/s0306-4522(00)00077-4. [DOI] [PubMed] [Google Scholar]
- 25.King TD, Bijur GN, Jope RS. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3β and attenuated by lithium. Brain Res. 2001;919:106–114. doi: 10.1016/s0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- 26.Ko L, Mehta ND, Farrer M, Easson C, Hussey J, Yen S, Hardy J, Yen SH. Sensitization of neuronal cells to oxidative stress with mutated human α-synuclein. J Neurochem. 2000;75:2546–2554. doi: 10.1046/j.1471-4159.2000.0752546.x. [DOI] [PubMed] [Google Scholar]
- 27.Kowall NW, Hantraye P, Brouillet E, Beal MF, McKee AC, Ferrante RJ. MPTP induces α-synuclein aggregation in the substantia nigra of baboons. NeuroReport. 2000;11:211–213. doi: 10.1097/00001756-200001170-00041. [DOI] [PubMed] [Google Scholar]
- 28.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 29.Lee HJ, Choi C, Lee SJ. Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002a;277:671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 30.Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ. Formation and removal of α-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem. 2002b;277:5411–5417. doi: 10.1074/jbc.M105326200. [DOI] [PubMed] [Google Scholar]
- 31.Leist M, Single B, Naumann H, Fava E, Simon B, Kuhnle S, Nicotera P. Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Exp Cell Res. 1999;249:396–403. doi: 10.1006/excr.1999.4514. [DOI] [PubMed] [Google Scholar]
- 32.Lotharius J, O'Malley KL. The parkinsonism-inducing drug 1-methyl-4-phenylpyridinium triggers intracellular dopamine oxidation: a novel mechanism of toxicity. J Biol Chem. 2000;275:38581–38588. doi: 10.1074/jbc.M005385200. [DOI] [PubMed] [Google Scholar]
- 33.Ma SY, Ciliax BJ, Stebbins G, Jaffar S, Joyce JN, Cochran EJ, Kordower JH, Mash DC, Levey AI, Mufson EJ. Dopamine transporter-immunoreactive neurons decrease with age in the human substantia nigra. J Comp Neurol. 1999;409:25–37. doi: 10.1002/(sici)1096-9861(19990621)409:1<25::aid-cne3>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 34.McNaught KS, Olanow CW, Halliwell B, Isacson O, Jenner P. Failure of the ubiquitin-proteasome system in Parkinson's disease. Nat Rev Neurosci. 2001;2:589–594. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 35.Menegon A, Board PG, Blackburn AC, Mellick GD, Le Couteur DG. Parkinson's disease, pesticides, and glutathione transferase polymorphisms. Lancet. 1998;352:1344–1346. doi: 10.1016/s0140-6736(98)03453-9. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem Biophys Res Commun. 1989;163:1450–1455. doi: 10.1016/0006-291x(89)91141-8. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura K, Bindokas VP, Marks JD, Wright DA, Frim DM, Miller RJ, Kang UJ. The selective toxicity of 1-methyl-4-phenylpyridinium to dopaminergic neurons: the role of mitochondrial complex I and reactive oxygen species revisited. Mol Pharmacol. 2000;58:271–278. doi: 10.1124/mol.58.2.271. [DOI] [PubMed] [Google Scholar]
- 38.Panov AV, Scaduto RC., Jr Substrate specific effects of calcium on metabolism of rat heart mitochondria. Am J Physiol. 1996;270:H1398–H1406. doi: 10.1152/ajpheart.1996.270.4.H1398. [DOI] [PubMed] [Google Scholar]
- 39.Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 40.Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson's disease. J Neural Transm. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- 41.Rideout HJ, Larsen KE, Sulzer D, Stefanis L. Proteasomal inhibition leads to formation of ubiquitin/α-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem. 2001;78:899–908. doi: 10.1046/j.1471-4159.2001.00474.x. [DOI] [PubMed] [Google Scholar]
- 42.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 43.Seaton TA, Cooper JM, Schapira AH. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res. 1998;809:12–17. doi: 10.1016/s0006-8993(98)00790-2. [DOI] [PubMed] [Google Scholar]
- 44.Seyfried J, Soldner F, Kunz WS, Schulz JB, Klockgether T, Kovar KA, Wüllner U. Effect of 1-methyl-4-phenylpyridinium on glutathione in rat pheochromocytoma PC 12 cells. Neurochem Int. 2000;36:489–497. doi: 10.1016/s0197-0186(99)00156-4. [DOI] [PubMed] [Google Scholar]
- 45.Sherer TB, Betarbet R, Greenamyre JT. The rotenone model of Parkinson's disease in vivo: selective striatal oxidative damage and caspase-3 activation in nigrostriatal neurons. Soc Neurosci Abstr. 2001a;27:653.2. [Google Scholar]
- 46.Sherer TB, Trimmer PA, Borland K, Parks JK, Bennett JP, Jr, Tuttle JB. Chronic reduction in complex I function alters calcium signaling in SH-SY5Y neuroblastoma cells. Brain Res. 2001b;891:94–105. doi: 10.1016/s0006-8993(00)03203-0. [DOI] [PubMed] [Google Scholar]
- 47.Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 48.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 49.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 50.Stephans S, Miller GW, Levey AI, Greenamyre JT (2002) Acute mitochondrial and chronic toxicological effects of 1-methyl-4-phenylpyridinium in human neuroblastoma cells. Neurotoxicology, in press. [DOI] [PubMed]
- 51.Tatton NA. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease. Exp Neurol. 2000;166:29–43. doi: 10.1006/exnr.2000.7489. [DOI] [PubMed] [Google Scholar]
- 52.Tatton NA, Maclean-Fraser A, Tatton WG, Perl DP, Olanow CW. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson's disease. Ann Neurol. 1998;44:S142–148. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- 53.Taylor SC, Shaw SM, Peers C. Mitochondrial inhibitors evoke catecholamine release from pheochromocytoma cells. Biochem Biophys Res Commun. 2000;273:17–21. doi: 10.1006/bbrc.2000.2894. [DOI] [PubMed] [Google Scholar]
- 54.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 55.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of α-synuclein fibril formation: a possible factor in Parkinson's disease. FEBS Lett. 2001;500:105–108. doi: 10.1016/s0014-5793(01)02597-2. [DOI] [PubMed] [Google Scholar]
- 57.Vila M, Vukosavic S, Jackson-Lewis V, Neystat M, Jakowec M, Przedborski S. α-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J Neurochem. 2000;74:721–729. doi: 10.1046/j.1471-4159.2000.740721.x. [DOI] [PubMed] [Google Scholar]
- 58.Votyakova TV, Reynolds IJ. ΔΨm-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 59.Wooten GF. Neurochemistry and neuropharmacology of Parkinson's disease. In: Watts RL, Koller WC, editors. Movement disorders: neurologic principles and practice. McGraw-Hill; New York: 1997. pp. 153–160. [Google Scholar]
- 60.Wüllner U, Kornhuber J, Weller M, Schulz JB, Loschmann PA, Riederer P, Klockgether T. Cell death and apoptosis regulating proteins in Parkinson's disease: a cautionary note. Acta Neuropathol (Berl) 1999;97:408–412. doi: 10.1007/s004010051005. [DOI] [PubMed] [Google Scholar]
- 61.Zhang JG, Tirmenstein MA, Nicholls-Grzemski FA, Fariss MW. Mitochondrial electron transport inhibitors cause lipid peroxidation-dependent and -independent cell death: protective role of antioxidants. Arch Biochem Biophys. 2001;393:87–96. doi: 10.1006/abbi.2001.2486. [DOI] [PubMed] [Google Scholar]
- 62.Zhuang S, Demirs JT, Kochevar IE. Protein kinase C inhibits singlet oxygen-induced apoptosis by decreasing caspase-8 activation. Oncogene. 2001;20:6764–6776. doi: 10.1038/sj.onc.1204867. [DOI] [PubMed] [Google Scholar]