

Abstract
Respiratory long-term facilitation (LTF) is a form of serotonin-dependent plasticity induced by intermittent hypoxia. LTF is manifested as a long-lasting increase in respiratory amplitude (and frequency) after the hypoxic episodes have ended. We tested the hypotheses that LTF of phrenic amplitude requires spinal serotonin receptor activation and spinal protein synthesis. A broad-spectrum serotonin receptor antagonist (methysergide) or protein synthesis inhibitors (emetine or cycloheximide) were injected intrathecally in the cervical spinal cord of anesthetized rats. Control rats, injected with vehicle (artificial CSF), exhibited an augmented phrenic burst amplitude after three 5 min episodes of hypoxia (78 ± 15% above baseline, 60 min after hypoxia; p < 0.05), indicating LTF. Pretreatment with methysergide, emetine, or cycloheximide attenuated or abolished phrenic LTF (20 ± 4, 0.2 ± 11, and 20 ± 2%, respectively; allp > 0.05). With protein synthesis inhibitors, phrenic LTF differed from control by 15 min after intermittent hypoxia. As an internal control against unintended drug distribution, we measured respiratory LTF in hypoglossal (XII) motor output. At 60 min after intermittent hypoxia, all treatment groups exhibited similar XII LTF (artificial CSF, 44 ± 10%; methysergide, 40 ± 5%; emetine, 35 ± 9%; and cycloheximide, 57 ± 29%; allp < 0.05), suggesting that drugs were restricted at effective doses to the spinal cord. We conclude that phrenic LTF requires spinal serotonin receptor activation and protein synthesis. Serotonin receptors on phrenic motoneuron dendrites may induce new protein synthesis, thereby giving rise to phrenic LTF.
Keywords: control of breathing, serotonin, plasticity, intermittent hypoxia, motoneuron, long-term facilitation
Serotonin-dependent plasticity is widely observed in invertebrate and vertebrate systems. InAplysia sensorimotor synapses, serotonin receptor activation covalently modifies existing proteins and initiates transcription and translation of new proteins, thereby enhancing synaptic transmission (Carew, 1996; Abel and Kandel, 1998). In vertebrates, serotonin receptor activation influences some forms of synaptic plasticity, such as long-term potentiation in the visual cortex (Kojic et al., 1997;Edagawa et al., 2001) and hippocampus (Mongeau et al., 1997; Tecott et al., 1998; Sarnyai et al., 2000). There is also evidence for serotonin-dependent spinal plasticity (Calejesan et al., 1998; Li and Zhuo, 1998).
Respiratory long-term facilitation (LTF) is a serotonin-dependent increase in respiratory motor output after intermittent (but not continuous) exposures to low oxygen (hypoxia) (Millhorn et al., 1980;Baker and Mitchell, 2000; Mitchell et al., 2001). In anesthetized rats, respiratory LTF is most frequently observed as an increase in the amplitude of respiratory nerve bursts in, for example, the phrenic and hypoglossal nerves (phrenic and hypoglossal LTF) (Fuller et al., 2000;Mitchell et al., 2001). Some studies also report long-lasting increases in respiratory frequency (frequency LTF) (Bach and Mitchell, 1996;Baker and Mitchell, 2000), although this finding is inconsistent (Hayashi et al., 1993; Kinkead et al., 1998; Kinkead and Mitchell, 1999; Fuller et al., 2001a,b; Zabka et al., 2001a,b). Pretreatment with a systemic serotonin type 2 (5-HT2) receptor antagonist blocks phrenic and hypoglossal LTF (Fuller et al., 2001b), but the location of the relevant 5-HT2 receptors is unknown. LTF may require 5-HT2 receptor activation near medullary premotoneurons that generate inspiratory drive, on respiratory motoneurons, or both. There is suggestive evidence that serotonin receptors associated with respiratory motoneurons are critical to LTF. For example, although serotonergic neurons in caudal raphe nuclei project to many regions involved in respiratory control (Bianchi et al., 1995; Bonham, 1995; McCrimmon et al., 1995), the greatest density of serotonergic terminals is within motor nuclei (cf Pilowsky et al., 1990; Voss et al., 1990). Furthermore, sensory denervation of cervical spinal segments associated with the phrenic nucleus is associated with increased serotonin terminal density near phrenic motoneurons and enhanced phrenic LTF (Kinkead et al., 1998). Thus, we hypothesize that the serotonin receptors necessary for respiratory amplitude LTF are located on or near respiratory motoneurons (Mitchell et al., 2001).
Although serotonin receptor activation is required to initiate LTF (Fuller et al., 2001b), little is known regarding the cellular processes that maintain it. Because other forms of neuroplasticity with similar time domains require new protein synthesis (Bailey et al., 1996; Steward and Schuman, 2001), we hypothesize that episodic serotonin receptor activation during intermittent hypoxia initiates new protein synthesis in or near respiratory motoneurons, thereby maintaining LTF.
To test the hypotheses that the amplitude component of respiratory LTF requires serotonin receptor activation and protein synthesis on or near respiratory motoneurons, we took advantage of the anatomical separation between cranial hypoglossal and spinal phrenic motoneurons. If serotonin receptor activation and protein synthesis within the respective motor nuclei are required, then phrenic and hypoglossal LTF should be affected differentially by spinal drug application. Thus, we injected a serotonin receptor antagonist or protein synthesis inhibitors intrathecally to the cervical spinal cord before intermittent hypoxia while monitoring phrenic and hypoglossal LTF.
MATERIALS AND METHODS
Experiments were conducted on adult male Sasco Sprague Dawley rats (colony K62; Charles River Laboratories, Wilmington, MA), weighing between 325 and 480 gm. The Animal Care and Use Committee at the University of Wisconsin-Madison approved all experimental procedures.
Surgical procedures. Rats were anesthetized initially with isoflurane (2.5%) in 50% O2 (balance N2) and then slowly converted to urethane anesthesia (1.6 gm/kg, i.v.). The adequacy of anesthesia was assessed periodically by testing blood pressure responses to toe pinch; supplemental doses of urethane were given as necessary. One hour after beginning surgery, an intravenous infusion of a 1:4 solution of sodium bicarbonate and standard lactated Ringer's solution was initiated to maintain an acid–base balance (5 ml · kg−1 · hr−1).
Rats were vagotomized, paralyzed with pancuronium bromide (2.5 mg/kg), and pump-ventilated (tidal volume, 2–2.5 ml; Rodent Respirator model 683; Harvard Apparatus, South Natick, MA). Using a dorsal approach, the left phrenic and hypoglossal (XII) nerves were dissected, cut distally, and desheathed. For intrathecal injections, a laminectomy was performed over C2–C3, and a small hole was cut in the dura at the cranial edge of C3. A small catheter (silicone tubing, 2 French; Access Technologies, Skokie, IL) was fed through the hole such that the tip of the catheter lay over C4–C5. The catheter was connected to a 50 μl glass syringe (Hamilton, Reno, NV) filled with artificial CSF (aCSF) or solutions of methysergide, emetine, or cycloheximide in aCSF. Artificial CSF consisted of the following (in mm): 120 NaCl, 3 KCl, 2 CaCl, 2 MgCl, 23 NaHCO3, and 10 glucose.
Measurements. Phrenic and XII nerves were isolated, desheathed, submerged in mineral oil, and placed on bipolar silver recording electrodes. Nerve activity was amplified (gain, 10,000; A-M systems, Everett, WA), bandpass-filtered (100 Hz to 10 kHz), and integrated (CWE 821 filter; Paynter, Ardmore, PA; time constant, 50 msec). The signal was then digitized, recorded, and analyzed using the WINDAQ data acquisition system (DATAQ Instruments, Akron, OH). After surgery, at least 1.5 hr was allowed for electroneurograms and blood pressure to stabilize.
To establish steady baseline nerve activity, rats were ventilated with hyperoxia (FIO2, 0.5; PaO2, >120 mmHg), with CO2 added to the inspired gas so that PaCO2 was 2–3 mmHg above the CO2 apneic threshold (Bach and Mitchell, 1996). After establishment of baseline conditions, 10–15 μl of drug or vehicle was injected intrathecally (C4–C5) over 1.5–2 min. Previous studies in rats using bromophenol blue dye indicate that 10 μl intrathecal injections produce a relatively restricted dye distribution near the injection site (Yaksh and Rudy, 1976). To further restrict cephalad flow of CSF in the spinal cord after intrathecal injections, the head of the rat was elevated throughout the protocol. After intrathecal injections, 35–40 min was allowed for drugs to penetrate the spinal cord and for the electroneurograms to stabilize before beginning the protocol.
End-tidal CO2 was monitored throughout the protocol using a flow-through capnograph with sufficient response time to measure end-tidal CO2 in rats (Novametrix, Wallingford, CT). Blood gases and arterial pressure were monitored and corrected as necessary to ensure that posthypoxia values were maintained near baseline levels. PaCO2 was adjusted by manipulating inspired CO2 or ventilator frequency; any negative base excess values were corrected with intravenous sodium bicarbonate, and changes in blood pressure were offset by increasing the venous infusion of lactated Ringer's solution. Experiments were included in the analysis only if (1) PaO2 during hypoxia was between 35 and 45 mmHg; (2) PaO2 during the hyperoxic baseline and recovery periods was >120 mmHg; and (3) PaCO2remained within 1 mmHg of baseline throughout the protocol.
Protocol. Peak integrated phrenic and XII amplitudes were measured before, during, and after three 5 min episodes of hypoxia (FIO2, 0.11) separated by 5 min of isocapnic hyperoxia. These measurements were made in rats with intrathecal injections of vehicle (aCSF; n = 10) or drug (methysergide, emetine, or cycloheximide; n = 6 each). In addition, measurements were made in rats during 2 hr of isocapnic hyperoxia (time controls) in rats with intrathecal injections of vehicle or drug (n = 3 each).
In a series of preliminary experiments, a limited dose–response curve was performed for each drug. Our goal was to find a dose range that selectively blocked phrenic but not hypoglossal LTF. Thus, for each drug we found drug dose ranges that (1) did not affect phrenic or hypoglossal LTF, (2) selectively blocked phrenic LTF, and (3) blocked phrenic and hypoglossal LTF. With the exception of the high-dose emetine series (see below), only experiments using the drug dose that selectively blocked phrenic LTF are reported. Our interpretation of these preliminary drug–dose experiments is as follows: higher doses that blocked phrenic and XII LTF crossed the blood–brain barrier and reached the brainstem at effective concentrations via the circulation; lower doses that did not block phrenic or XII LTF did not produce effective concentrations in either the brainstem or spinal cord, whereas intermediate concentrations that blocked phrenic but not XII LTF produced effective concentrations at the spinal cord only.
To investigate the role of serotonin receptors in LTF, we used a broad-spectrum serotonin receptor antagonist, methysergide maleate (250 μg/kg, 20 mm; Sandoz, Hanover, NJ). Intrathecal injections of more specific serotonin receptor antagonists, such as ketanserin tartrate, ritanserin, mesulergine HCl, lisuride hydrogen maleate (Research Biochemicals, Natick, MA), and Ly-53857 (Sigma, St. Louis, MO) were attempted, but these drugs did not dissolve at a high enough concentration in aCSF, pH 7.4. Even when these drugs were initially dissolved in saline or DMSO, a precipitate formed on injection into CSF (as visualized under a microscope).
To investigate the role of spinal protein synthesis in LTF, we used two different protein synthesis inhibitors: emetine (1 μg/kg, 70 μm; Sigma; Grollman, 1966) and cycloheximide (250 μg/kg, 35 mm; Sigma; Grollman, 1966). We also report preliminary dose–response experiments using emetine at varying doses (20–250 μg/kg; n = 6; average dose, 112 ± 38 μg/kg).
Blood samples (0.3 ml in a heparinized syringe) were drawn before, during the first hypoxic episode, and 15, 30, and 60 min after the last hypoxic episode to ensure that blood gases met the criteria outlined above. Phrenic and XII measurements corresponding to these time points were used in the analysis. At the conclusion of all experiments, rats were killed via urethane overdose.
Statistical analysis. Peak amplitudes and frequency (bursts per minute) of phrenic and XII nerve activity were averaged in 1 min bins at each recorded data point (baseline, during the first hypoxia, and 15, 30, and 60 min after hypoxia). Changes in amplitude were normalized as a percentage of the baseline value. Burst frequency (bursts per minute) is reported as an absolute change from baseline. We compared phrenic and hypoglossal responses of rats receiving intrathecal aCSF injections (vehicle control) versus drug injections (methysergide, emetine, or cycloheximide). Statistical analyses were conducted using a two-way ANOVA with a repeated measures design, and individual comparisons were made using the Student–Neuman–Keulspost hoc test. Differences were considered significant atp < 0.05. All values are expressed as mean ± SE.
RESULTS
Intrathecal artificial CSF (control)
There were no changes in baseline phrenic or XII burst amplitude or frequency after intrathecal aCSF injections (data not shown). Increases in integrated phrenic and XII burst amplitude or frequency during hypoxia in control rats were consistent with other reports from our laboratory on rats without an intrathecal catheter (Table1) (Bach and Mitchell, 1996; Fuller et al., 2000). Shortly after intermittent hypoxia, integrated phrenic and hypoglossal burst amplitude declined toward baseline levels (Fig.1; XII not shown). However, over the course of the next hour, phrenic and XII burst amplitude progressively increased, with significant increases in phrenic and XII amplitude above baseline at 15, 30, and 60 min after hypoxia, indicating the development of phrenic and XII LTF (Fig.2). At 60 min after hypoxia, integrated phrenic burst amplitude had increased 78 ± 15%, whereas integrated XII burst amplitude increased 44 ± 10% above baseline values (both p < 0.05). Changes in phrenic and XII burst amplitudes from baseline at 60 min were significantly greater than values 15 min after hypoxia (33 ± 10 and 16 ± 10% above baseline, respectively; p < 0.05). Similarly, the change in phrenic burst amplitude from baseline at 60 min was significantly greater than values obtained at 30 min after hypoxia (49 ± 11% above baseline; p < 0.05). These data indicate that LTF develops progressively for at least 1 hr after intermittent hypoxia.
Table 1.
Hypoxic ventilatory responses in rats pretreated with artificial CSF, methysergide, emetine (1 μg/kg), or cycloheximide
| Amplitude change in hypoxia (% above baseline) | Burst frequency (bursts/min) | |||
|---|---|---|---|---|
| ∫Phr | ∫XII | Baseline | Hypoxia | |
| Artificial CSF | 111 ± 161-a | 137 ± 281-a | 36.2 ± 1.3 | 44.5 ± 1.31-a |
| Methysergide | 88 ± 101-a | 119 ± 181-a | 44.3 ± 1.61-b | 48 ± 2 |
| Emetine | 172 ± 491-a | 155 ± 141-a | 40.2 ± 1.7 | 45.8 ± 1.81-a |
| Cycloheximide | 89 ± 131-a | 199 ± 451-a | 40.7 ± 2.4 | 49.4 ± 2.71-a |
All rat groups significantly increased integrated phrenic (∫Phr) and hypoglossal (∫XII) amplitude (percentage above baseline) similarly during hypoxia (p > 0.05). Rats pretreated with artificial CSF, emetine, or cycloheximide had a significant increase in burst frequency in hypoxia (p < 0.05). Rats pretreated with methysergide had a significantly elevated baseline frequency (p < 0.05) but did not have a significant increase in burst frequency during hypoxia (p > 0.05).
Significantly different from baseline.
Significantly different from artificial CSF control.
Fig. 1.

Representative phrenic neurogram taken before, during, and 1 hr after intermittent hypoxia in rats pretreated with artificial CSF (A), methysergide (B; 250 μg/kg), or emetine (C; 1 μg/kg). In all rats, the integrated phrenic amplitude returned close to baseline levels immediately after intermittent hypoxia. Only rats pretreated with artificial CSF had a subsequent progressive increase in phrenic amplitude over the course of the next hour, indicating phrenic long-term facilitation.
Fig. 2.

Spinal serotonin receptors are required for phrenic long-term facilitation. The mean percentage change in integrated phrenic (Phr; A) and hypoglossal (XII; B) discharge from baseline at 15, 30, and 60 min after intermittent hypoxia is shown. Intermittent hypoxia elicits phrenic and hypoglossal long-term facilitation in rats pretreated with intrathecal artificial CSF (●). Intrathecal methysergide (○; 250 μg/kg) abolished phrenic but not hypoglossal long-term facilitation. *Significantly increased from baseline (p < 0.05).#Significantly different from artificial CSF controls (p < 0.05).
A small but significant burst frequency LTF was also observed in rats injected with aCSF (36.2 ± 1.3 bursts/min during baseline and 42.5 ± 1.5 bursts/min at 60 min after hypoxia) (Fig.3; p < 0.05). The change in frequency from baseline (6.2 ± 1 bursts/min) was only 17% (vs 78% for amplitude).
Fig. 3.

Spinal serotonin receptor activation is required for burst frequency long-term facilitation. The change in burst frequency from baseline 15, 30, and 60 min after intermittent hypoxia is shown. Intermittent hypoxia elicits long-term facilitation of burst frequency in rats intrathecally injected with artificial CSF (●) but not in rats intrathecally injected with methysergide (○; 250 μg/kg). #Significantly different from artificial CSF controls (p < 0.05). *Significantly increased from baseline (p < 0.05).
Intrathecal methysergide
Intrathecal methysergide significantly increased baseline phrenic burst amplitude (28.1 ± 13.8%; p < 0.05), consistent with a previous report using intravenous drug administration (Bach and Mitchell, 1996). Baseline phrenic and XII measurements were taken ∼10 min after phrenic activity had reached this new stable level. Hypoglossal burst amplitude and burst frequency were not affected by intrathecal methysergide (change of 2.9 ± 5.6% and 1.5 ± 0.9 bursts/min, respectively; p > 0.05), indicating a spinal site of action. However, despite similar baseline PaCO2 levels (47.5 mmHg), rats treated with intrathecal methysergide (250 μg/kg, 20 mm) had a significantly higher baseline burst frequency (44.3 ± 1.6 bursts/min) than rats injected with aCSF (36.2 ± 1.3 bursts/min; Table 1; p < 0.05).
Integrated phrenic and XII burst amplitude increased during hypoxia in rats with intrathecal methysergide (88 ± 10 and 119 ± 18%; respectively; Table 1; p < 0.05), and neither response was significantly different from that in aCSF controls (111 ± 16 and 137 ± 28%, respectively; p > 0.05). Although burst frequency did not increase significantly during hypoxia in rats with intrathecal methysergide (baseline, 44.3 ± 1.6 bursts/min; hypoxia, 48 ± 2 bursts/min; Table 1;p > 0.05), the hypoxic burst frequency was not significantly different between control and methysergide-treated rats (control hypoxia, 44.5 ± 1.3 bursts/min; p > 0.05).
Immediately after intermittent hypoxia, integrated phrenic and XII burst amplitude returned toward baseline values in a manner similar to aCSF controls (Fig. 1). However, in contrast to aCSF controls, phrenic burst amplitude remained near baseline levels for the duration of the protocol in rats pretreated with intrathecal methysergide, whereas XII burst amplitude steadily increased in a manner similar to that of control rats. At 60 min after hypoxia, phrenic burst amplitude was not significantly above baseline (20 ± 4% above baseline;p > 0.05 in overall ANOVA) and was significantly less than in control rats at all time points (control, 78 ± 15%, 60 min after hypoxia) (Fig. 2; p < 0.05). In contrast, intrathecal methysergide had no effect on XII LTF at any time point (Fig. 2), suggesting that methysergide was restricted at an effective dose to the spinal cord. The increase in XII burst amplitude at 60 min after hypoxia in rats with intrathecal methysergide was 40 ± 5% above baseline (p < 0.05), a response similar to that in control rats (44 ± 10% above baseline). Thus, intrathecal methysergide attenuated phrenic, but not XII LTF.
Intrathecal methysergide also attenuated burst frequency LTF (Fig. 3). At 60 min after intermittent hypoxia, burst frequency was 45.2 ± 1.5 bursts/min, a nonsignificant increase of 0.9 ± 0.6 bursts/min from baseline (44.3 ± 1.6 bursts/min; p > 0.05).
Protein synthesis inhibition
Intrathecal emetine (dose–response)
In preliminary experiments, we tested a series of high emetine doses (20–250 μg/kg) that apparently resulted in unintended drug distribution. At these high emetine doses, a complete block of phrenic and XII LTF was observed at all time points (Fig.4; p < 0.05). At 60 min after intermittent hypoxia, the change in phrenic burst amplitude was –4 ± 11%, and XII was 7 ± 4% from baseline (bothp > 0.05). Likewise, burst frequency LTF was abolished after high emetine doses (Fig. 5) (1.4 ± 1.2 bursts/min above baseline, 60 min after hypoxia; p > 0.05). These data contrast with those of control rats, which show significant LTF of burst amplitude and frequency as early as 15 min after intermittent hypoxia (see above). Thus, LTFs of burst amplitude and frequency are associated with rapid protein synthesis. Because XII LTF was also abolished, we concluded that the doses of emetine used were sufficiently high to reach the brainstem, either diffusing through the CSF or (more likely) crossing into and distributing via the circulation. Thus, we progressively lowered the emetine dose until differential effects on phrenic and XII burst discharge were observed (1 μg/kg, 70 μm).
Fig. 4.

Spinal protein synthesis is required for phrenic long-term facilitation. The mean percentage change in integrated phrenic (Phr; A) and hypoglossal (XII; B) discharge from baseline at 15, 30, and 60 min after intermittent hypoxia is shown in rats intrathecally injected with emetine (1 or 20–250 μg/kg), cycloheximide (250 μg/kg), or artificial CSF. Intermittent hypoxia elicits phrenic and hypoglossal long-term facilitation in rats injected with intrathecal artificial CSF (●). Intrathecal emetine (■; 1 μg/kg) and cycloheximide (○; 250 μg/kg) abolished phrenic, but not hypoglossal long-term facilitation. Rats pretreated with high doses of emetine (▪; 20–250 μg/kg) had no significant phrenic or hypoglossal long-term facilitation. *Significantly increased from baseline (p < 0.05).#Significantly different from artificial CSF controls (p < 0.05).
Fig. 5.

Spinal protein synthesis is not required for burst frequency long-term facilitation. The change in burst frequency 15, 30, and 60 min after intermittent hypoxia is shown. Intermittent hypoxia elicits burst frequency long-term facilitation in rats intrathecally injected with artificial CSF (●), emetine (■; 1 μg/kg), or cycloheximide (○; 250 μg/kg). High doses of emetine that were likely not restricted to the spinal cord (▪; 20–250 μg/kg) blocked burst frequency LTF (p > 0.05). *Significantly increased from baseline (p < 0.05).
Intrathecal emetine (low dose)
Similar to aCSF control rats, rats injected with intrathecal emetine (1 μg/kg) increased respiratory burst amplitude and frequency during hypoxia (Table 1; p < 0.05). Immediately after intermittent hypoxia, integrated phrenic and XII burst amplitude returned toward baseline in rats treated with intrathecal emetine (Fig.1). However, in contrast to aCSF control rats, phrenic burst amplitude remained near baseline for the duration of the protocol. At 60 min after intermittent hypoxia, the change in integrated phrenic burst amplitude from baseline was 0.2 ± 11% in rats injected with intrathecal emetine, a response significantly lower than in rats injected with aCSF (78 ± 15% from baseline) (Fig. 4;p < 0.05). In contrast, XII burst amplitude progressively increased after intermittent hypoxia, reaching a value 35 ± 9% above baseline at 60 min after intermittent hypoxia (p < 0.05). This response was similar to that in control rats injected with aCSF (44 ± 10% above baseline) (Fig. 4; p > 0.05), suggesting that emetine was restricted at an effective dose to the spinal cord.
Rats injected with low doses of emetine (1 μg/kg) exhibited small but statistically significant burst frequency LTF (Fig. 5). In rats receiving emetine, burst frequency increased from a baseline of 40.2 ± 1.7 to 44.1 ± 1.4 bursts/min at 60 min after intermittent hypoxia (i.e., 3.9 ± 1.1 bursts/min above baseline;p < 0.05); this response was not significantly different from that in aCSF control rats (6.2 ± 1 bursts/min above baseline; p > 0.05), although there was a trend toward attenuation (Fig. 5).
Intrathecal cycloheximide
Rats injected with intrathecal cycloheximide (250 μg/kg, 35 mm) continued to show significant increases in respiratory burst amplitude and frequency during hypoxia (p< 0.05), and these responses were not significantly different from those in aCSF control rats (Table 1; p > 0.05). Intrathecal cycloheximide attenuated phrenic LTF (Fig. 4). At 60 min after intermittent hypoxia, the change in integrated phrenic burst amplitude in rats pretreated with cycloheximide was 20 ± 2% above baseline (p > 0.05 in overall ANOVA), a response significantly less than in aCSF control rats (78 ± 15% baseline; p < 0.05). In contrast, XII LTF was not affected by intrathecal cycloheximide (57 ± 29% above baseline, 60 min after intermittent hypoxia; p < 0.05), a response similar to that in control rats (44 ± 10% baseline;p > 0.05).
Similar to low doses of emetine, burst frequency LTF was not significantly affected by intrathecal cycloheximide (Fig. 5). Sixty minutes after intermittent hypoxia, burst frequency increased from a baseline value of 40.7 ± 2.4 to 44.2 ± 2.4 bursts/min (3.5 ± 0.8 bursts/min above baseline; p < 0.05). This response was not significantly different from that in control aCSF rats (6.2 ± 1 bursts/min; p > 0.05), although there was a trend toward attenuation.
Time controls
Rats intrathecally injected with aCSF, methysergide, cycloheximide or emetine, but without hypoxia (time controls), exhibited no time-dependent changes in phrenic or XII nerve activity from 30 min to 2 hr after injection (data not shown). Thus, phrenic and XII activity were stable in the time frame of our experiments.
DISCUSSION
These studies indicate that phrenic amplitude LTF after intermittent hypoxia requires spinal serotonin receptor activation and protein synthesis, although we do not rule out additional brainstem mechanisms. To investigate spinal mechanisms in phrenic LTF, a serotonin receptor antagonist or protein synthesis inhibitors were injected intrathecally in the cervical spinal cord. As an internal control, XII LTF was assessed to determine whether unintended drug distribution affected LTF in cranial respiratory motoneurons.
Respiratory rhythm is postulated to originate within a small group of neurons in the caudal medulla (Smith et al., 1991; Feldman and McCrimmon, 1999). Once the basic timing is established, respiratory premotoneurons in the brainstem modify the burst pattern (amplitude and duration) and transmit this modified respiratory drive to motoneurons in the spinal cord and brainstem. Although phrenic and XII motoneurons do not receive respiratory synaptic inputs from the same premotoneurons in rats (Peever et al., 2001), their respective premotoneurons have similar rostrocaudal distributions within the medulla, although the phrenic premotoneurons tend to be more ventral (Dobbins and Feldman, 1994, 1995). Thus, dissociation between phrenic and XII LTF should be possible only when drug distribution is restricted to the respective motor nuclei, surrounding interneurons or synaptic inputs. For each drug, a dose was defined for differential effects on phrenic and XII LTF. At lower doses, LTF was not blocked in either nerve, whereas LTF could be blocked in both nerves at higher doses. High drug doses do not allow differentiation between actions within motor nuclei (phrenic and XII) versus brainstem premotoneurons. At intermediate drug doses blocking phrenic but not XII LTF, we suggest that drugs were restricted at effective concentrations to the spinal cord.
Role of spinal serotonin receptor activation in phrenic LTF
Phrenic LTF requires spinal serotonin receptor activation, because intrathecal methysergide abolished burst amplitude LTF in phrenic (but not XII) motor output. Although these experiments do not allow firm conclusions regarding the receptor subtype or their precise spinal location, the known effects of systemic ketanserin on phrenic LTF (Kinkead and Mitchell, 1999) suggest an involvement of 5-HT2A receptors. Ketanserin is nearly two orders of magnitude more selective for 5-HT2A versus 5-HT2C receptors (Barnes and Sharp, 1999), and 5-HT2A receptors have been localized on phrenic motoneurons (Basura et al., 2001). Because 5-HT2Areceptors are primarily somatodendritic (Cornea-Hebert et al., 1999), phrenic LTF is unlikely the result of 5-HT2Areceptor activation on axon terminals that synapse on phrenic motoneurons. However, an involvement of 5-HT2Areceptors on spinal interneurons cannot be ruled out. Systemic ketanserin also blocks XII LTF (Fuller et al., 2001b), and 5-HT2A receptors are abundant in the XII motor nucleus (Fay and Kubin, 2000); thus we suggest that similar, although translocated, mechanisms underlie phrenic and XII LTF.
Role of spinal protein synthesis in phrenic LTF
Phrenic burst amplitude LTF requires spinal protein synthesis, because intrathecal emetine and cycloheximide blocked phrenic (but not XII) LTF. The protein synthesis dependence was rapid; phrenic motor output differed between control rats and rats treated with protein synthesis inhibitors as early as 15 min after hypoxia. Thus, respiratory LTF is similar to other models of neural plasticity in its protein synthesis dependence (Bailey et al., 1996; Steward and Schuman, 2001).
We hypothesize that intermittent (but not continuous) spinal 5-HT2A receptor activation initiates synthesis of the relevant proteins. Similarly, intermittent but not continuous serotonin receptor activation increases protein synthesis 2 hr after stimulation in Aplysia pleural ganglia (Yanow et al., 1998). It is uncertain whether the protein synthesis required for phrenic LTF results from translation of existing mRNA versus increased gene transcription. However, given the time frame of phrenic LTF and the location of serotonergic terminals on distal dendrites of phrenic motoneurons (Pilowsky et al., 1990), increased translation of existing dendritic mRNA is most likely necessary for LTF maintenance. Dendritic protein synthesis occurs rapidly in neurons after activation of certain postsynaptic receptors, such as tyrosine kinase B receptors (Crino and Eberwine, 1996; Kang and Schuman, 1996; Smith et al., 1999), metabotropic glutamate receptors (Huber et al., 2000; Kacharmina et al., 2000), and NMDA receptors (Scheetz et al., 2000). Furthermore, protein synthesis machinery is found in neuronal dendrites in the ventral cervical spinal cord (Gardiol et al., 1999). Thus, we hypothesize that phrenic motoneuron dendrites have the capacity to synthesize new proteins in response to intermittent serotonin receptor activation.
Burst frequency LTF
Most studies on anesthetized rats demonstrate LTF in burst amplitude but not frequency (Hayashi et al., 1993; Bach and Mitchell, 1996; Kinkead et al., 1998; Kinkead and Mitchell, 1999; Baker and Mitchell, 2000; Fuller et al., 2001a,b; Zabka et al., 2001a,b). Because frequency LTF after intermittent hypoxia is not commonly observed in anesthetized rats, we are reluctant to draw firm conclusions regarding its mechanisms. A retrospective meta-analysis of frequency LTF (n = 86) (Bach and Mitchell, 1996; Baker and Mitchell, 2000; Fuller et al., 2000, 2001a,b; Zabka et al., 2001b) reveals that rats with lower baseline burst frequencies are more likely to exhibit frequency LTF (our unpublished data). Frequency LTF is generally observed only when baseline burst frequency is <41 bursts/min; higher baseline frequencies exhibit little or no frequency LTF. In newborn rat brainstem–spinal cords, serotonin effects on respiratory frequency depend similarly on baseline burst frequency (Onimaru et al., 1998). Thus, rats treated with intrathecal methysergide may not show frequency LTF because of higher baseline burst frequencies in that group (Table1). Alternatively, intrathecal methysergide may block frequency LTF via indirect effects on respiratory rhythm generation. Electrical stimulation of phrenic afferent fibers increases respiratory frequency (Marlot et al., 1987; Road et al., 1987), likely via spinal projections to brainstem respiratory neurons. Serotonergic neurons synapse profusely in the dorsal horn (Skagerberg and Bjorklund, 1985; Leger et al., 2001), and serotonin receptor activation at this site facilitates synaptic strength (Zhuo, 2000). Thus, serotonin receptor activation during intermittent hypoxia may enhance synaptic transmission from phrenic afferent neurons, thereby increasing phrenic burst frequency (i.e., frequency LTF). Finally, it remains possible that frequency LTF was attenuated by unintended methysergide distribution to the brainstem, although we do not favor this hypothesis (see above). Regardless, when present, frequency LTF must involve direct or indirect effects on neurons involved with respiratory rhythm generation. Serotonin effects on brainstem rhythm-generating neurons are likely; indeed, serotonin receptor activation elicits a persistent respiratory frequency increase in adult turtle brainstems (Johnson et al., 2001), an effect similar to frequency LTF.
Because high emetine doses (which may have reached the brainstem) blocked frequency LTF, whereas lower spinal doses did not, protein synthesis in brainstem neurons generating respiratory rhythm may be required for frequency LTF. Thus, mechanisms of amplitude and frequency LTF may be distinct, with phrenic amplitude LTF originating primarily within the spinal cord and frequency LTF primarily the result of effects on brainstem respiratory neurons. More detailed analysis of frequency LTF is necessary before firm conclusions can be made regarding its significance or mechanisms.
Working model of LTF
Our working model of phrenic LTF and, by extension, LTF in other respiratory motoneurons has been discussed in recent reviews (Baker et al., 2001; Mitchell et al., 2001). In brief, we propose that increased caudal raphe neuron activity during hypoxia (Erickson and Millhorn, 1994; Teppema et al., 1997) and the subsequent release of serotonin activate dendritic 5-HT2A receptors on respiratory motoneurons, thereby giving rise to LTF (Fig.6). Serotonin receptor activation is required during but not after intermittent hypoxia (Fuller et al., 2001b), suggesting that 5-HT2 receptors activate a signaling cascade that maintains LTF. We hypothesize that LTF maintenance involves new protein synthesis within phrenic motoneurons via translational regulation of existing mRNA. These newly translated proteins may act presynaptically, postsynaptically, or both between bulbospinal respiratory premotoneurons and respiratory motoneurons, thereby giving rise to a facilitated respiratory motor output. The required proteins are currently unknown, although we suspect an involvement of brain-derived neurotrophic factor (Mitchell et al., 2001).
Fig. 6.
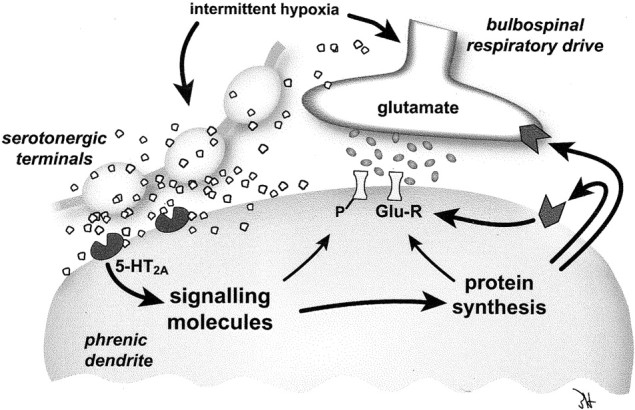
Proposed mechanism of phrenic long-term facilitation. Intermittent hypoxia increases the release of serotonin in the vicinity of the synapse between descending bulbospinal respiratory neurons and phrenic dendrites. The activation of 5-HT2A receptors initiates a signal transduction cascade that leads to new protein synthesis. These newly synthesized proteins may act both presynaptically and postsynaptically to increase synaptic efficacy between bulbospinal respiratory neurons and phrenic motoneurons and hence may give rise to a facilitated phrenic motor output.
Significance of LTF
There are other examples of spinal serotonin-dependent plasticity in mammals. Facilitation of nociceptive synaptic transmission (Calejesan et al., 1998; Zhuo, 2000) results from 5-HT2 receptor activation in the spinal dorsal horn (Hori et al., 1996). Serotonin receptor activation converts preexisting but functionally ineffective synapses in the spinal dorsal horn into functional synapses through PKC-mediated recruitment of AMPA receptors (Li and Zhuo, 1998; Li et al., 1999). Serotonin also promotes recovery of phrenic burst activity ipsilateral to a high cervical hemisection, possibly by converting ineffective crossed spinal pathways to effective pathways (Ling et al., 1994; Zhou and Goshgarian, 2000).
Phrenic LTF is a unique model of spinal plasticity initiated by descending modulatory pathways. Although we do not yet know the physiological significance of respiratory LTF, it may be important in offsetting mechanisms of respiratory inhibition during or after hypoxia or hypercapnia (Bisgard and Neubauer, 1995; Powell et al., 1998;Mitchell et al., 2001). Respiratory LTF implies an ability to differentially modulate select respiratory motoneuron pools. Thus, respiratory LTF may be a useful mechanism, strengthening motor output to specific respiratory muscles under altered physiological or pathophysiological conditions such as weight gain or loss, pregnancy, injury, and certain respiratory disorders such as emphysema and sleep-disordered breathing. We have reported recently that male (but not female) rats have reduced phrenic and XII LTF with increasing age (Zabka et al., 2001a,b), suggesting an age–gender interaction in respiratory LTF. These patterns bear a striking similarity to the prevalence of obstructive sleep apnea in humans (Bixler et al., 2001). Mechanisms such as respiratory LTF may augment upper airway muscle tone during vulnerable periods (such as during sleep), thereby maintaining upper airway patency. Diminished LTF with age may therefore predispose an individual to sleep-disordered breathing.
Regardless of its physiological role, phrenic LTF after intermittent hypoxia is a useful model for studying mechanisms of serotonin-dependent (spinal) plasticity. Intermittent hypoxia is physiologically relevant and reproducible, and meaningful, quantitative assessment of plasticity and its manifestations can be obtained (i.e., breathing). Investigations of the mechanisms giving rise to LTF may yield fundamental insights into general principles of plasticity in the CNS.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants HL 53319 and HL 65383. T.L.B.-H. was supported by NIH Training Grant HL 07654.
Correspondence should be addressed to Dr. Tracy L. Baker-Herman, Department of Comparative Biosciences, University of Wisconsin, 2015 Linden Drive, Madison, WI 53706. E-mail: BakerT@svm.vetmed.wisc.edu.
REFERENCES
- 1.Abel T, Kandel E. Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res Brain Res Rev. 1998;26:360–378. doi: 10.1016/s0165-0173(97)00050-7. [DOI] [PubMed] [Google Scholar]
- 2.Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- 3.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol (Lond) 2000;15:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respir Physiol. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- 6.Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 7.Basura GJ, Zhou SY, Walker PD, Goshgarian HG. Distribution of serotonin 2A and 2C receptor mRNA expression in the cervical ventral horn and phrenic motoneurons following spinal cord hemisection. Exp Neurol. 2001;169:255–263. doi: 10.1006/exnr.2001.7682. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi AL, Denavit-Saubie M, Champagnat J. Central control of breathing in mammals: neuronal circuitry, membrane properties and neurotransmitters. Physiol Rev. 1995;75:1–45. doi: 10.1152/physrev.1995.75.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Bisgard GE, Neubauer JA. Peripheral and central effects of hypoxia. In: Dempsey JA, Pack AI, editors. Regulation of breathing. Dekker; New York: 1995. pp. 151–218. [Google Scholar]
- 10.Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Rein J, Vela-Bueno A, Kales A. Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med. 2001;163:608–613. doi: 10.1164/ajrccm.163.3.9911064. [DOI] [PubMed] [Google Scholar]
- 11.Bonham AC. Neurotransmitters in the CNS control of breathing. Respir Physiol. 1995;101:219–230. doi: 10.1016/0034-5687(95)00045-f. [DOI] [PubMed] [Google Scholar]
- 12.Calejesan AA, Ch'ang MH, Zhuo M. Spinal serotonergic receptors mediate facilitation of a nociceptive reflex by subcutaneous formalin injection into the hindpaw in rats. Brain Res. 1998;798:46–54. doi: 10.1016/s0006-8993(98)00394-1. [DOI] [PubMed] [Google Scholar]
- 13.Carew TJ. Molecular enhancement of memory formation. Neuron. 1996;16:5–8. doi: 10.1016/s0896-6273(00)80016-1. [DOI] [PubMed] [Google Scholar]
- 14.Cornea-Hebert V, Riad M, Wu C, Singh SK, Descarries L. Cellular and subcellular distribution of the serotonin 5-HT2A receptor in the central nervous system of adult rat. J Comp Neurol. 1999;409:187–209. doi: 10.1002/(sici)1096-9861(19990628)409:2<187::aid-cne2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 15.Crino PB, Eberwine J. Molecular characterization of the dendritic growth cone: regulated mRNA transport and local protein synthesis. Neuron. 1996;17:1173–1187. doi: 10.1016/s0896-6273(00)80248-2. [DOI] [PubMed] [Google Scholar]
- 16.Dobbins EG, Feldman JL. Brainstem network controlling descending drive to phrenic motoneurons in rat. J Comp Neurol. 1994;347:64–86. doi: 10.1002/cne.903470106. [DOI] [PubMed] [Google Scholar]
- 17.Dobbins EG, Feldman JL. Differential innervation of protruder and retractor muscles of the tongue in rat. J Comp Neurol. 1995;357:376–394. doi: 10.1002/cne.903570305. [DOI] [PubMed] [Google Scholar]
- 18.Edagawa Y, Saito H, Abe K. Endogenous serotonin contributes to a developmental decrease in long-term potentiation in the rat visual cortex. J Neurosci. 2001;21:1532–1537. doi: 10.1523/JNEUROSCI.21-05-01532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erickson JT, Millhorn DE. Hypoxia and electrical stimulation of the carotid sinus nerve induce Fos-like immunoreactivity within catecholaminergic and serotonergic neurons of the rat brainstem. J Comp Neurol. 1994;348:161–182. doi: 10.1002/cne.903480202. [DOI] [PubMed] [Google Scholar]
- 20.Fay R, Kubin L. Pontomedullary distribution of 5-HT2A receptor-like protein in the rat. J Comp Neurol. 2000;418:323–345. [PubMed] [Google Scholar]
- 21.Feldman JL, McCrimmon DR. Neural control of breathing. In: Zigmond MJ, Bloom FE, Landis SC, Roberts JL, Squire LR, editors. Fundamental neuroscience. Academic; San Diego: 1999. pp. 1063–1090. [Google Scholar]
- 22.Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic motor output. Respir Physiol. 2000;121:135–146. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- 23.Fuller D, Baker T, Behan M, Mitchell G. Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol Genomics. 2001a;4:175–181. doi: 10.1152/physiolgenomics.2001.4.3.175. [DOI] [PubMed] [Google Scholar]
- 24.Fuller DD, Zabka AG, Baker TL, Mitchell GS. Selected contribution: phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001b;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- 25.Gardiol A, Racca C, Triller A. Dendritic and postsynaptic protein synthetic machinery. J Neurosci. 1999;19:168–179. doi: 10.1523/JNEUROSCI.19-01-00168.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grollman AP. Structural basis for inhibition of protein synthesis by emetine and cycloheximide based on an analogy between ipecac alkaloids and glutarimide antibiotics. Proc Natl Acad Sci USA. 1966;56:1867–1874. doi: 10.1073/pnas.56.6.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayashi F, Coles SK, Bach KB, Mitchell GS, McCrimmon DR. Time-dependent phrenic nerve responses to carotid afferent activation: intact vs. decerebellate rats. Am J Physiol. 1993;265:R811–R819. doi: 10.1152/ajpregu.1993.265.4.R811. [DOI] [PubMed] [Google Scholar]
- 28.Hori Y, Endo K, Takahashi T. Long-lasting synaptic facilitation induced by serotonin in superficial dorsal horn neurones of the rat spinal cord. J Physiol (Lond) 1996;492:867–876. doi: 10.1113/jphysiol.1996.sp021352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 30.Johnson SM, Wilkerson JE, Henderson DR, Wenninger MR, Mitchell GS. Serotonin elicits long-lasting enhancement of rhythmic respiratory activity in turtle brain stems in vitro. J Appl Physiol. 2001;91:2703–2712. doi: 10.1152/jappl.2001.91.6.2703. [DOI] [PubMed] [Google Scholar]
- 31.Kacharmina JE, Job C, Crino P, Eberwine J. Stimulation of glutamate receptor protein synthesis and membrane insertion within isolated neuronal dendrites. Proc Natl Acad Sci USA. 2000;97:11545–11550. doi: 10.1073/pnas.97.21.11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 33.Kinkead R, Mitchell GS. Time-dependent hypoxic ventilatory responses in rats: effects of ketanserin and 5-carboxyamidotryptamine. Am J Physiol. 1999;277:R658–R666. doi: 10.1152/ajpregu.1999.277.3.R658. [DOI] [PubMed] [Google Scholar]
- 34.Kinkead R, Zhan WZ, Prakash YS, Bach KB, Sieck GC, Mitchell GS. Cervical dorsal rhizotomy enhances serotonergic innervation of phrenic motoneurons and serotonin-dependent long-term facilitation of respiratory motor output in rats. J Neurosci. 1998;18:8436–8443. doi: 10.1523/JNEUROSCI.18-20-08436.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kojic L, Gu Q, Douglas RM, Cynader MS. Serotonin facilitates synaptic plasticity in kitten visual cortex: an in vitro study. Brain Res Dev Brain Res. 1997;101:299–304. doi: 10.1016/s0165-3806(97)00083-7. [DOI] [PubMed] [Google Scholar]
- 36.Leger L, Charnay Y, Hof PR, Bouras C, Cespuglio R. Anatomical distribution of serotonin-containing neurons and axons in the central nervous system of the cat. J Comp Neurol. 2001;433:157–182. [PubMed] [Google Scholar]
- 37.Li P, Zhuo M. Silent glutamatergic synapses and nociception in mammalian spinal cord. Nature. 1998;393:695–698. doi: 10.1038/31496. [DOI] [PubMed] [Google Scholar]
- 38.Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M. AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci. 1999;2:972–977. doi: 10.1038/14771. [DOI] [PubMed] [Google Scholar]
- 39.Ling L, Bach KB, Mitchell GS. Serotonin reveals ineffective spinal pathways to contralateral phrenic motoneurons in spinally hemisected rats. Exp Brain Res. 1994;101:35–43. doi: 10.1007/BF00243214. [DOI] [PubMed] [Google Scholar]
- 40.Marlot D, Macron JM, Duron B. Inhibitory and excitatory effects on respiration by phrenic nerve afferent stimulation in cats. Respir Physiol. 1987;69:321–333. doi: 10.1016/0034-5687(87)90086-7. [DOI] [PubMed] [Google Scholar]
- 41.McCrimmon DR, Dekin MS, Mitchell GS. Glutamate, GABA and serotonin in ventilatory control. In: Dempsey JA, Pack AI, editors. Regulation of breathing: central nervous system. Dekker; New York: 1995. pp. 151–218. [Google Scholar]
- 42.Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by endogenous central serotonin. Respir Physiol. 1980;41:171–188. doi: 10.1016/0034-5687(80)90113-9. [DOI] [PubMed] [Google Scholar]
- 43.Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- 44.Mongeau R, Blier P, de Montigny C. The serotonergic and noradrenergic systems of the hippocampus: their interactions and the effects of antidepressant treatments. Brain Res Brain Res Rev. 1997;23:145–195. doi: 10.1016/s0165-0173(96)00017-3. [DOI] [PubMed] [Google Scholar]
- 45.Onimaru H, Shamoto A, Homma I. Modulation of respiratory rhythm by 5-HT in the brainstem-spinal cord preparation from newborn rat. Pflügers Arch. 1998;435:485–494. doi: 10.1007/s004240050543. [DOI] [PubMed] [Google Scholar]
- 46.Peever JH, Mateika JH, Duffin J. Respiratory control of hypoglossal motoneurones in the rat. Pflügers Arch. 2001;442:78–86. doi: 10.1007/s004240000502. [DOI] [PubMed] [Google Scholar]
- 47.Pilowsky PM, de Castro D, Llewellyn-Smith I, Lipski J, Voss MD. Serotonin immunoreactive boutons makes synapses with feline phrenic motoneurons. J Neurosci. 1990;10:1091–1098. doi: 10.1523/JNEUROSCI.10-04-01091.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- 49.Road JD, West NH, Van Vliet BN. Ventilatory effects of stimulation of phrenic afferents. J Appl Physiol. 1987;63:1063–1069. doi: 10.1152/jappl.1987.63.3.1063. [DOI] [PubMed] [Google Scholar]
- 50.Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1A) receptors. Proc Natl Acad Sci USA. 2000;97:14731–14736. doi: 10.1073/pnas.97.26.14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheetz AJ, Nairn AC, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci. 2000;3:211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- 52.Skagerberg G, Bjorklund A. Topographic principles in the spinal projections of serotonergic and non-serotonergic brainstem neurons in the rat. Neuroscience. 1985;15:445–480. doi: 10.1016/0306-4522(85)90225-8. [DOI] [PubMed] [Google Scholar]
- 53.Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith WB, Aakalu GN, Tsung MI, Reis GF, Schuman EM. Neurotrophin-induced increase in local protein synthesis in cultured hippocampal neurons. Soc Neurosci Abstr. 1999;25:467. [Google Scholar]
- 55.Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001;24:299–325. doi: 10.1146/annurev.neuro.24.1.299. [DOI] [PubMed] [Google Scholar]
- 56.Tecott LH, Logue SF, Wehner JM, Kauer JA. Perturbed dentate gyrus function in serotonin 5-HT2C receptor mutant mice. Proc Natl Acad Sci USA. 1998;95:15026–15031. doi: 10.1073/pnas.95.25.15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teppema LJ, Veening JG, Kranenburg A, Dahan A, Berkenbosch A, Olievier C. Expression of c-fos in the rat brainstem after exposure to hypoxia and to normoxic and hyperoxic hypercapnia. J Comp Neurol. 1997;388:169–190. doi: 10.1002/(sici)1096-9861(19971117)388:2<169::aid-cne1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 58.Voss MD, de Castro D, Lipski J, Pilowsky PM, Jiang C. Serotonergic immunoreactive boutons form close appositions with respiratory neurons of the DRG in the cat. J Comp Neurol. 1990;295:1132–1135. doi: 10.1002/cne.902950205. [DOI] [PubMed] [Google Scholar]
- 59.Yaksh TL, Rudy TA. Chronic catheterization of the subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 60.Yanow SK, Manseau F, Hislop J, Castellucci VF, Sossin WS. Biochemical pathways by which serotonin regulates translation in the nervous system of Aplysia. J Neurochem. 1998;70:572–583. doi: 10.1046/j.1471-4159.1998.70020572.x. [DOI] [PubMed] [Google Scholar]
- 61.Zabka AG, Behan M, Mitchell GS. Selected contribution: Time-dependent hypoxic respiratory responses in female rats are influenced by age and by the estrus cycle. J Appl Physiol. 2001a;91:2831–2838. doi: 10.1152/jappl.2001.91.6.2831. [DOI] [PubMed] [Google Scholar]
- 62.Zabka AG, Behan M, Mitchell GS. Long term facilitation of respiratory motor output decreases with age in male rats. J Physiol (Lond) 2001b;531:509–514. doi: 10.1111/j.1469-7793.2001.0509i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou SY, Goshgarian HG. 5-Hydroxytryptophan-induced respiratory recovery after cervical spinal cord hemisection in rats. J Appl Physiol. 2000;89:1528–1536. doi: 10.1152/jappl.2000.89.4.1528. [DOI] [PubMed] [Google Scholar]
- 64.Zhuo M. Silent glutamatergic synapses and long-term facilitation in spinal dorsal horn neurons. Prog Brain Res. 2000;129:101–113. doi: 10.1016/S0079-6123(00)29008-0. [DOI] [PubMed] [Google Scholar]