

Abstract
Tolerance to the analgesic effects of an opioid occurs after its chronic administration, a pharmacological phenomenon that has been associated with the development of abnormal pain sensitivity such as hyperalgesia. In the present study, we examined the role of spinal glutamate transporters (GTs) in the development of both morphine tolerance and associated thermal hyperalgesia. Chronic morphine administered through either intrathecal boluses or continuous infusion induced a dose-dependent downregulation of GTs (EAAC1 and GLAST) in the rat's superficial spinal cord dorsal horn. This GT downregulation was mediated through opioid receptors because naloxone blocked such GT changes. Morphine-induced GT downregulation reduced the ability to maintain in vivo glutamate homeostasis at the spinal level, because the hyperalgesic response to exogenous glutamate was enhanced, including an increased magnitude and a prolonged time course, in morphine-treated rats with reduced spinal GTs. Moreover, the downregulation of spinal GTs exhibited a temporal correlation with the development of morphine tolerance and thermal hyperalgesia. Consistently, the GT inhibitorl-trans-pyrrolidine-2-4-dicarboxylate (PDC) potentiated, whereas the positive GT regulator riluzole reduced, the development of both morphine tolerance and thermal hyperalgesia. The effects from regulating spinal GT activity by PDC were at least in part mediated through activation of the NMDA receptor (NMDAR), because the noncompetitive NMDAR antagonist MK-801 blocked both morphine tolerance and thermal hyperalgesia that were potentiated by PDC. These results indicate that spinal GTs may contribute to the neural mechanisms of morphine tolerance and associated abnormal pain sensitivity by means of regulating regional glutamate homeostasis.
Keywords: tolerance, opioid, glutamate transporter, NMDA, hyperalgesia, riluzole, PDC
Opioids are a class of the most effective analgesics for treating many forms of acute and chronic pain. Besides the known side effects, the clinical utility of opioid analgesics is often hampered by the development of analgesic tolerance that necessitates dose escalation regardless of the disease progression. The development of opioid tolerance also has been associated with enhanced pain sensitivity such as hyperalgesia (exacerbated pain in response to noxious stimulation) in both laboratory and clinical settings (Sjogren et al., 1993; Mao et al., 1994; Ossipov et al., 1995; Devulder, 1997; Wegert et al., 1997;Vanderah et al., 2000; Celerier et al., 2001). Several lines of recent research have shed light on the neurobiology of opioid tolerance. For instance, β-arrestin, a regulatory protein, has been shown to play an important role in the development of opioid tolerance (Bohn et al., 1999, 2000; Whistler and von Zastrow, 1999). More recently, the activity of μ-opioid receptor oligomerization and endocytosis has been suggested to be critical to the prevention of morphine tolerance (Finn and Whistler, 2001; He et al., 2002). As such, an opioid agonist that would facilitate the μ-opioid receptor endocytosis has been shown to reduce the development of morphine tolerance (He et al., 2002). Another interesting development is that activation of excitatory amino acid receptors such as the NMDA receptor (NMDAR) has been implicated in the mechanisms of opioid tolerance, particularly μ-opioid tolerance, and associated abnormal pain sensitivity (Marek et al. 1991a,b; Trujillo and Akil, 1991; Tiseo and Inturrisi, 1993;Elliott et al., 1994a,b; Mao et al., 1994, 1996; Manning et al., 1996). There is also emerging evidence suggesting that opioid tolerance and abnormal pain sensitivity may share common cellular mechanisms mediated in part through NMDARs (Mao et al., 1994, 1995b).
Despite a large number of studies over a decade that indicate the involvement of NMDARs in the development of μ-opioid tolerance and associated abnormal pain sensitivity, it remains unclear how activation of NMDARs could be initiated in response to opioids that are known to have overwhelmingly inhibitory effects. Opioids such as morphine do not have detectable binding affinity with NMDARs (Mao, 1999), making it unlikely that opioids directly interact with NMDARs at the receptor level. On the other hand, neither acute opioid analgesic effects nor the expression of opioid tolerance is affected by an NMDAR antagonist (Trujillo and Akil, 1991; Mao et al., 1994), indicating that the NMDAR itself does not directly modulate opioid receptor functions.
The homeostasis of the extracellular glutamate level, a primary endogenous ligand for the NMDAR, is actively and tightly regulated by the glutamate transporter (GT) system (Robinson and Dowd, 1997; Semba and Wakuta, 1998; Mennerick et al., 1999; 9Jabaudon et al., 2000;Danbolt, 2001). There are at least five identified Na+-dependent GT proteins, which are differentially expressed in specific cell types with EAAC1 primarily being in neurons and GLAST and GLT-1 in glial cells (Robinson and Dowd, 1997; Danbolt, 2001). A number of studies have shown that GTs play a critical role in the prevention of glutamate neurotoxicity under both physiological and pathological conditions (Mennerick et al., 1999; Lievens et al., 2000; Vorwerk et al., 2000;Trotti et al., 2001). In brain regions, changes in GLT-1 mRNAs have been observed after naloxone-precipitated morphine withdrawal (Ozawa et al., 2001), and morphine tolerance decreases after subcutaneous injection of a proposed glial GT activator, MS-135 (Nakagawa et al., 2001). Conceivably, activation of NMDARs would become possible, despite the overwhelmingly inhibitory opioid effects, if the GT function were reduced after chronic opioid administration with a resultant increase in synaptic glutamate availability. In this series of experiments, we examined the possibilities that chronic morphine administration downregulates both neuronal and glial GTs in the spinal cord, which in turn contributes to the neural mechanisms of morphine tolerance and abnormal pain sensitivity in part through the activation of NMDARs.
MATERIALS AND METHODS
Experimental animals and drugs
Adult male Sprague Dawley rats weighing 300–350 gm were used. Rats were housed in individual cages with water and food pellets available ad libitum. The animal room was artificially illuminated from 7:00 A.M. to 7:00 P.M.. The protocols were approved by our Institutional Animal Care and Use Committee. The following drugs were purchased from Sigma: MK-801, morphine, riluzole, glutamate, naloxone, andl-trans-pyrrolidine-2-4-dicarboxylate (PDC).
Intrathecal catheter and osmotic pump implantation
A polyethylene (PE)-10 intrathecal catheter was implanted in each rat (Yaksh and Rudy, 1976). Those animals that exhibited neurological deficits after intrathecal catheter implantation were excluded from the experiments. Drugs were delivered via an intrathecal catheter in a total volume of 10 μl followed by a saline flush. For continuous intrathecal infusion, osmotic minipumps (Alza, Mountain View, CA) were implanted as described previously (Granados-Soto et al., 2000; Vanderah et al., 2000). An osmotic pump was connected to an intrathecal catheter via a piece of PE-60 catheter. The filled minipumps were soaked in normal saline for 4 hr before the insertion to ensure an immediate drug delivery. The integrity of the pump delivery system was reexamined when the spinal cords were harvested for the immunocytochemical or Western blot analysis. The experiments including behavioral testing were conducted so that the experimenters were blinded to treatment conditions.
Induction of morphine tolerance
Tolerance to the antinociceptive effect of morphine was induced using two intrathecal treatment regimens: repeated boluses and continuous infusion. Morphine was given twice daily for 7 d in the repeated bolus regimen, whereas continuous morphine infusion was given for 7 d via an implanted osmotic pump system. Because the osmotic pump infusion began on day 1, day 8 was the last day of a full 7 d delivery using an osmotic pump. Differences in morphine antinociception among treatment groups were assessed on the test day using the tail-flick test at 30 min after a probe dose containing either 10 μg of morphine (intrathecal) for repeated bolus groups or 5 mg/kg of morphine (intraperitoneal) for continuous infusion groups.
Behavioral tests
The routine tail-flick test was made with baseline latencies of 4–6 sec and a cutoff time of 10 sec to assess the antinociceptive effects of morphine (D'Amour, Smith, 1941; Akil and Mayer, 1972). The percentage of maximal possible antinociceptive effect (%MPAE) was calculated by comparing the test latency before [baseline (BL)] and after a drug injection (TL) using the equation: %MPAE = [(TL − BL)/(cutoff − BL)] × 100. In dose–response experiments, the generation of cumulative dose–response curves, as described in the literature (Paronis and Holtzman, 1991; Elliott et al., 1994a,b; Mao et al. 1995a), was used to reduce the total number of rats used in experiments. To examine changes in baseline nociceptive responses before and after chronic administration of morphine or a GT regulator, the paw-withdrawal test with baseline latencies of 9–11 sec and a cutoff time of 22 sec was used as described previously (Hargreaves et al., 1988). The paw-withdrawal test was used because this test has been shown to be sensitive in detecting subtle changes of a baseline nociceptive response because of its slow-rising temperature change during the test (Mao et al., 1994). In this study, the development of morphine tolerance was assessed by the tail-flick test, whereas the development of thermal hyperalgesia was evaluated by the paw-withdrawal test.
Statistics for behavioral data
Data obtained from the tail-flick test were first calculated to yield mean %MPAE as shown previously (Mao et al., 1994). The data for both tail-flick and paw-withdrawal tests were then analyzed by using two-way ANOVA to detect overall differences among treatment groups. When significant main effects were observed, the Waller–Duncan K-ratiot tests were performed to determine sources of differences. For the dose–response data analysis, AD50 values and 95% confidence intervals (CIs) were computed using a computerized Litchfield and Wilcoxon calculation.
Immunocytochemistry and Western blotting
The routine immunostaining procedure was followed. Briefly, rats were perfused through the ascending aorta with saline followed by 4% paraformaldehyde. Spinal cord lumbar segments in which the intrathecal catheter was implanted were removed, postfixed for 2 hr, and kept overnight in 15% sucrose. Spinal cord samples from both experimental and control groups were mounted on the same block, and 10 μm transverse sections were cut together on a cryostat. These sections were then treated under the same condition during the immunocytochemical procedure to minimize the between-group variability. Sections were blocked with 1% goat serum in 0.3% Triton X-100 for 1 hr at room temperature and incubated overnight at 4°C with a primary antibody (EAAC1 or GLAST, 1:2000; Chemicon). The sections were then incubated for 1 hr at room temperature with the corresponding CY3-conjugated secondary antibody (1:300; Chemicon).
For Western blotting, rats were killed rapidly (<1 min) in a CO2 chamber. The dorsal horn from the lumbar spinal cord, corresponding to the site where samples for immunocytochemistry were collected, was removed and homogenized in SDS sample buffer containing a mixture of proteinase inhibitors (Sigma). The spinal cord dorsal horn was sampled because the immunocytochemical staining showed that EAAC1 and GLAST, as well as their changes after morphine treatment, were presented primarily in this spinal region. Protein samples were separated on SDS-PAGE gel (4–15% gradient gel; Bio-Rad) and transferred to polyvinylidene difluoride filters (Millipore). The filters were blocked with 3% milk and incubated overnight at 4°C with a primary antibody (EAAC1, 1:2000; GLAST, 1:2000) and for 1 hr at room temperature with HRP-conjugated secondary antibody (Amersham; 1:10,000). The blots were then visualized in ECL solution (NEN) for 1 min and exposed onto hyperfilms (Amersham) for 1–10 min.
Image analysis
For the immunostaining analysis, six spinal sections were randomly selected and scanned using a Nikon fluorescence microscope. Images were then captured with a CCD Spot camera (Diagnostic Instruments, Inc.), and image densities were analyzed (Adobe PhotoShop) according to the division of spinal cord dorsal horn regions (Molander et al., 1984; Mao et al., 1992, 1993). Relative density of images was determined by subtracting the background density in each image. The percentage change of staining density in morphine-treated groups from the corresponding saline group was calculated by the following equation: (density of the saline group − density of the morphine group)/(density of the saline group) × 100. For Western blotting, the developed films were scanned, and the density of immunoreactive bands was measured and normalized with internal control bands (ERK2 as loading control). For both immunostaining and Western blotting, differences in image density were compared using the Student'st test (two groups) or ANOVA (multiple groups) followed by the Waller–Duncan K-ratio t tests.
Experimental design
Experiment 1: changes in spinal GTs after chronic morphine. A total of 10 groups of rats were used in this experiment. To investigate whether repeated exposure to morphine boluses would result in changes in spinal GTs, three groups of rats (n = 5) were given intrathecal 10 or 20 μg of morphine or saline twice daily for 7 d. In addition, three more groups of rats (n = 5) were infused, via an intrathecal osmotic pump for 7 d, with 10 or 20 nm · μl−1 · hr−1morphine or saline. The continuous infusion regimen was included because it has been suggested that the cellular mechanisms of opioid tolerance might differ between repeated boluses and continuous administration (Ibuki et al., 1997; Dunbar and Pulai, 1998). In either case, morphine doses were chosen on the basis of previous studies that showed the reliable development of morphine tolerance using these doses (Mao et al., 1994; Ibuki et al., 1997). Additional two groups of rats (n = 4) were included to examine whether the EAAC1 or GLAST protein content (Western blot) would be changed after a 7 d continuous intrathecal infusion with either saline or 20 nm · μl−1 · hr−1morphine.
To examine whether blockade of opioid receptors would prevent morphine-induced changes in spinal GTs, morphine (20 μg) was coadministered intrathecally (twice daily) with naloxone (10 μg, a generic opioid receptor antagonist) for 7 d (n = 5). As a control, naloxone (10 μg) also was given alone twice daily for 7 d (n = 5). In all groups, spinal cords were harvested after the final behavioral test on day 8 (see above for the time line for pump groups), and samples were prepared for either immunocytochemical or Western blot analysis.
Experiment 2: effect of spinal GT changes on the response to exogenous glutamate. To examine whether changes in spinal GTs would alter the baseline latency to noxious thermal stimulation (i.e., the development of thermal hyperalgesia) and the response to exogenous glutamate, baseline paw-withdrawal latencies were compared between day 0 and day 8 in four groups of rats (n = 5), each receiving intrathecal infusion of saline, 10 nm · μl−1 · hr−1morphine, or 20 nm · μl−1 · hr−1morphine (two groups). On day 8 after determining the baseline latency, glutamate (5 nm) was given intrathecally to the saline and morphine (20 nm · μl−1 · hr−1) groups. Paw-withdrawal latencies to noxious thermal stimulation were again measured at 30, 60, 120, and 240 min after the glutamate administration. In the second 20 nm · μl−1 · hr−1morphine group, 20 μg of riluzole (a positive GT regulator) was given intrathecally at 30 min before the injection of 5 nm glutamate to determine whether increasing GT activity with acute riluzole treatment would affect either the response magnitude or the time course of exogenous glutamate. A control group of rats (n = 4) received a single injection of 20 μg of riluzole on day 8 to examine the effect of riluzole alone on the baseline nociceptive response.
Experiment 3: time course between GT changes, morphine tolerance, and thermal hyperalgesia. To examine the temporal relationship between changes in spinal GTs and the development of morphine tolerance and thermal hyperalgesia, eight groups (n = 8–9) of rats were used, each receiving 20 μg of either morphine or saline (twice daily), and their spinal cords were harvested for either immunocytochemistry or Western blotting on days 2, 4, 6, or 8 after the behavioral tests. The behavioral tests included the paw-withdrawal test to examine thermal hyperalgesia and the tail-flick test (cumulative dose–response) to examine the antinociceptive effect of intrathecal morphine.
Experiment 4: effect of regulating spinal GT activity and blocking NMDARs on morphine tolerance and thermal hyperalgesia. To investigate the role of spinal GTs in the development of morphine tolerance and associated thermal hyperalgesia, the GT inhibitor PDC (Lievens et al., 2000; Matthews et al., 2000) and the positive GT regulator riluzole (Azbill et al., 2000) were used to determine whether inhibiting and activating spinal GT activity would enhance and attenuate, respectively, morphine tolerance and thermal hyperalgesia. Ten groups of rats (n = 5) were used as follows: groups 1–3 (10 μg of morphine + 5, 10, or 20 μg of riluzole), group 4 (20 μg of riluzole alone), groups 5–7 (10 μg of morphine + 5, 10, or 20 μg of PDC), group 8 (20 μg of PDC alone), group 9 (10 μg of morphine + vehicle), and group 10 (vehicle alone). The drug combinations were given intrathecally twice daily for 7 d. The equivalent doses of PDC and riluzole have been demonstrated to be effective in regulating the extracellular glutamate concentration and NMDAR-mediated activities under both in vivo and in vitro experimental conditions (Semba and Wakuta, 1998; ; Azbill et al., 2000; Jabaudon et al., 2000; Lievens et al., 2000; Matthews et al., 2000). Additionally, two more groups of rats each received a 7 d intrathecal treatment (twice daily) with either 10 μg of morphine + 10 nm MK-801 + 20 μg of PDC (n = 6) or 10 nm MK-801 alone (n = 4). The MK-801 dose was selected on the basis of a previous study that showed its prevention of morphine tolerance and hyperalgesia (Mao et al., 1994). The data from these two groups were compared with the above morphine alone and morphine plus PDC group to examine whether the effect of PDC on the development of morphine tolerance and hyperalgesia is mediated through NMDARs.
RESULTS
Downregulation of spinal GTs after chronic morphine administration
In naïve rats there was a basal level of EAAC1 and GLAST immunoreactivity (ir) primarily within laminas I–II of the spinal cord dorsal horn. When examined on day 8, both EAAC1-ir and GLAST-ir in laminas I–II were significantly reduced in rats receiving a 7 d intrathecal morphine treatment given either as repeated boluses (10 or 20 μg, twice daily) or continuous infusion (10 or 20 nm · μl−1 · hr−1), as compared with the corresponding saline group (Figs.1A,B,D,E,2A,B). There were no differences in the level of EAAC1-ir and GLAST-ir between saline-treated and naïve rats, indicating a specific effect of morphine on EAAC1-ir and GLAST-ir. A much lower basal level of EAAC1-ir and GLAST-ir was observed in laminas III–VI (Fig.1A,D), as compared with that in laminas I–II, and the EAAC1 and GLAST immunostaining in laminas III–VI was not significantly changed after chronic morphine.
Fig. 1.
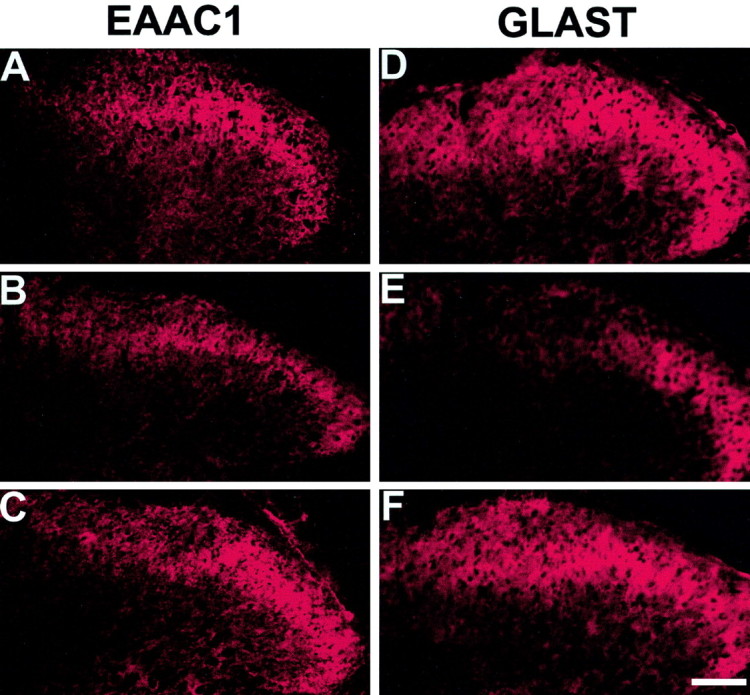
Downregulation of spinal GTs after chronic morphine. Both EAAC1-ir (B) and GLAST-ir (E) were reduced in rats receiving a 7 d intrathecal, twice daily treatment with 20 μg of morphine as compared with the corresponding saline control (A, D). Coadministration of morphine (20 μg) with naloxone (10 μg), twice daily for 7 d, blocked the reduction of both EAAC1-ir (C) and GLAST-ir (F). Scale bar, 50 μm.
Fig. 2.

Quantification of the EAAC1-ir and GLAST-ir reduction. The relative density of immunostaining in laminas I–II was measured by subtracting the background density in each image. The percentage reduction of density from the corresponding saline group was calculated as described in Materials and Methods. A,B10, B20, 10 or 20 μg of morphine boluses; B, C10, C20, 10 or 20 nm · μl−1 · hr−1morphine infusion; C, B20, 20 μg of morphine bolus alone; B20+NX, 20 μg of morphine plus 10 μg of naloxone boluses; NX, 10 μg of naloxone bolus alone. **p < 0.01 as compared with the saline group and + p < 0.01 as compared with the corresponding low morphine dose or saline groups.
Quantitatively, chronic morphine administration resulted in a 30–40% reduction of EAAC1-ir and GLAST-ir in laminas I–II from that of saline-treated rats on day 8, and such reductions were morphine dose dependent (Fig. 2A,B). In addition, the level of reduction for both EAAC1-ir and GLAST-ir was comparable between two morphine treatment regimens (Fig.2A,B). The downregulation of EAAC1 and GLAST expression also was revealed by the Western blot assay. There was a clear reduction of the EAAC1 and GLAST protein content in the corresponding Western blots after a 7 d infusion of 20 nm · μl−1 · hr−1morphine as compared with the saline control (Fig.3). Thus, spinal EAAC1 and GLAST expression was downregulated dose dependently after two independent morphine treatment regimens.
Fig. 3.

Morphine-induced EAAC1 and GLAST reduction in Western blotting. Both EAAC1 and GLAST protein contents were reduced in rats receiving a 7 d intrathecal infusion with 20 nm · μl−1 · hr−1morphine as compared with the saline control. *p < 0.05; two-tailed Student's t test. ERK2 is a loading control.
The downregulation of EAAC1 and GLAST was prevented by coadministration of morphine (20 μg) with naloxone (10 μg) twice daily for 7 d (Figs. 1C,F, 2C), indicating that changes in spinal GTs were mediated through opioid receptors. The naloxone treatment alone did not affect the GT level (Fig.2C). Naloxone (10 μg) also blocked the development of tolerance to morphine (20 μg) antinociception and thermal hyperalgesia in the same group of rats (Figs.4B,5B).
Fig. 4.

Development of morphine tolerance and its blockade by naloxone. A, Morphine antinociception was dose-dependently reduced on day 8 in rats receiving a 7 d intrathecal morphine treatment of either twice daily boluses or continuous infusion. B, Coadministration of morphine (20 μg) with naloxone (10 μg) for 7 d blocked the development of morphine tolerance (B20+NX), and a 7 d naloxone (10 μg; NX) treatment alone did not affect the morphine antinociception. See Figure 2 for the details of each group. **p < 0.01 as compared with the saline group, and + p < 0.05 as compared with the corresponding low morphine dose group.
Fig. 5.
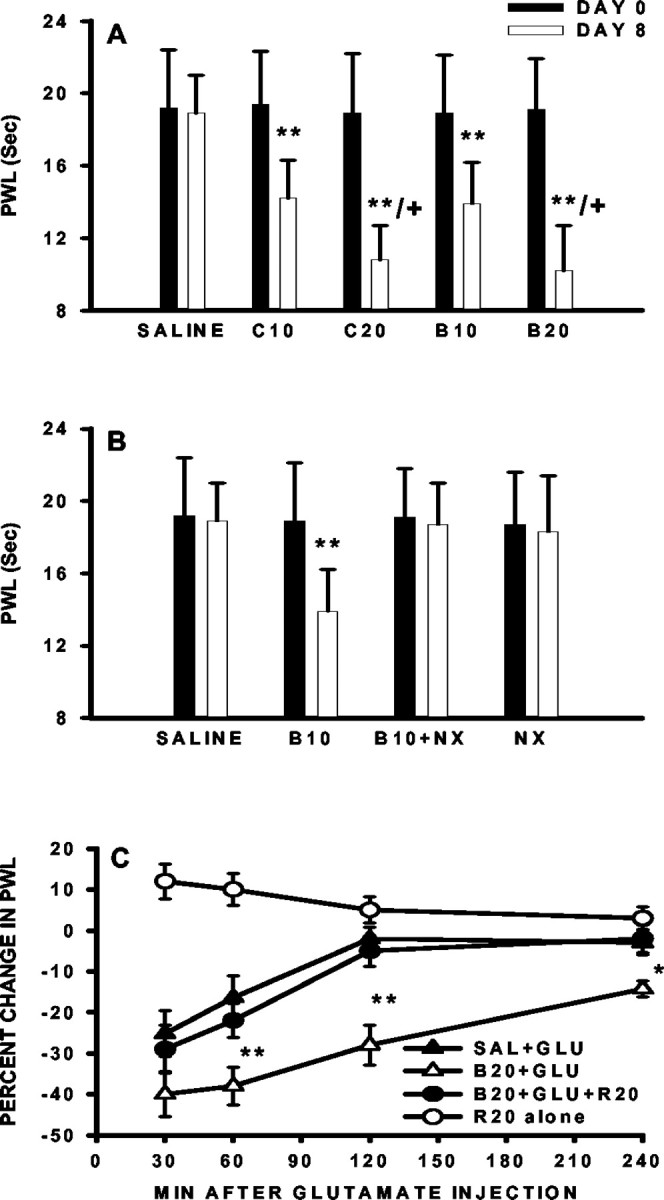
Development of thermal hyperalgesia and its reversal by riluzole. A, The paw-withdrawal latency (PWL) was reduced on day 8 in the absence of exogenous glutamate as compared with that on day 1 in rats receiving either 10 (B10) or 20 μg (B20) of morphine boluses or continuous infusion of 10 (C10) or 20 nm · μl−1 · hr−1(C20) morphine for 7 d. B, Coadministration of morphine (20 μg) with naloxone (10 μg) for 7 d blocked the development of thermal hyperalgesia (B20+NX), and a 7 d naloxone (10 μg;NX) treatment alone did not affect baseline paw-withdrawal latency. C, The response to intrathecal 5 nm glutamate was exacerbated in morphine-infused rats (B20+GLU) as compared with saline-treated rats (SAL+GLU). A single intrathecal pretreatment with 20 μg of riluzole at 30 min before the glutamate injection attenuated the hyperalgesia (B20+GLU+R20). Riluzole alone (R20 alone) transiently increased the baseline paw-withdrawal latency. The data were presented as the percentage change of the paw-withdrawal latency from that of before the glutamate treatment on day 8 in each group. *p < 0.05, **p < 0.01, as compared with the correspondingSAL+GLU group. The paw-withdrawal latency in theR20 alone group was compared before and after riluzole treatment, and its change did not reach statistical significance.
Temporal correlation between GT downregulation, morphine tolerance, and thermal hyperalgesia
Consistent with the downregulation of spinal GTs revealed by the immunocytochemical and Western blot assays, tolerance to the antinociceptive effects of morphine developed dose dependently in morphine- but not saline-treated rats (bolus or infusion) when tested on day 8 (Fig. 4A). In these same morphine-treated rats, the baseline paw-withdrawal latency to noxious radiant heat was dose-dependently reduced on day 8 as compared with the baseline latency before the morphine treatment (Fig. 5A), indicating the development of thermal hyperalgesia.
The time course between the GT downregulation and the development of morphine tolerance and thermal hyperalgesia was compared at days 2, 4, 6, and 8 of a repeated morphine (20 μg, twice daily) treatment regimen. Neither reduction of GTs (EAAC1 and GLAST) in Western blotting nor the development of morphine tolerance or thermal hyperalgesia was observed on days 2 and 4 (Fig. 6), and there was a transient increase in both EAAC1 and GLAST in Western blots at least on day 2 (Fig. 6). However, both EAAC1 and GLAST in Western blotting were clearly reduced on days 6 and 8 as compared with the saline group (Fig. 6). Consistent with the GT downregulation, the reduction of both morphine antinociception (tolerance) and baseline paw-withdrawal latency (hyperalgesia) also was present on days 6 and 8 (Fig. 7, Table1). Together, these results indicate a temporal correlation between the GT downregulation and the development of morphine tolerance and associated thermal hyperalgesia after chronic morphine.
Fig. 6.

Time course of EAAC1 and GLAST changes after chronic morphine. Both EAAC1 and GLAST protein contents were progressively reduced after twice daily intrathecal treatment with 20 μg of morphine. The M2 to M8 groups stand for rats receiving 20 μg of morphine, and their spinal cords were harvested at day 2, 4, 6, or 8 of the treatment period. *p < 0.05, **p < 0.01, as compared with the corresponding saline group. ERK2 is a loading control.
Fig. 7.

Time course of the development of morphine tolerance and thermal hyperalgesia after chronic morphine. Both morphine tolerance and thermal hyperalgesia developed on days 6 (D6) and 8 (D8) after twice daily intrathecal treatment with 20 μg of morphine. Note that the time course of behavioral changes correlated with that of EAAC1 and GLAST changes in Western blot analysis (Fig. 6). **p < 0.01, as compared with the corresponding saline group.
Table 1.
Time course of morphine tolerance
| Group | AD50 (μg) | 95% CI (μg) |
|---|---|---|
| Saline | 5.1 | 1.6–7.9 |
| M2 | 5.8 | 2.9–8.2 |
| M4 | 8.1 | 3.5–15.2 |
| M6 | 53.5 | 31.2–88.2 |
| M8 | 67.1 | 37.7–101.4 |
The M2 to M8 groups stand for rats receiving 20 μg of morphine twice daily, and their spinal cords were harvested at day 2, 4, 6, or 8 of the treatment period. Note that both M6 and M8 groups were significantly different from the remaining groups based on the AD50 value and 95% CI.
Exacerbation of the hyperalgesic response to exogenous glutamate after the GT downregulation
Thermal hyperalgesia in rats treated with morphine (20 nm · μl−1 · hr−1) was further exacerbated in response to exogenous glutamate (5 nm, i.t.), as compared with the corresponding saline group (Fig. 5C). The exacerbation of thermal hyperalgesia to exogenous glutamate was reflected by (1) an increased magnitude, i.e., a further reduction of the paw-withdrawal latency, and (2) a prolonged time course of hyperalgesia lasting at least 4 hr versus <2 hr in saline-treated rats (Fig. 5C). Moreover, riluzole (20 μg, a positive GT regulator), given at 30 min before administering 5 nm glutamate, attenuated the hyperalgesic response to exogenous glutamate (Fig. 5C). Riluzole (20 μg) alone in the absence of exogenous glutamate resulted in an increase in the baseline paw-withdrawal latency (10–12% from the baseline) in saline-treated rats (Fig. 5C). These results indicate that the downregulation of spinal GTs induced by chronic morphine has a functional impact on maintaining in vivoglutamate homeostasis within the spinal cord dorsal horn.
Potentiation of morphine tolerance and thermal hyperalgesia by the GT inhibitor PDC
The antinociceptive effect of morphine examined by the tail-flick test was significantly reduced beginning on day 4 in rats treated repeatedly with 10 μg of morphine plus 20 μg of PDC (Fig.8A). In contrast, the antinociceptive effects of morphine were not significantly reduced until day 6 in the morphine (10 μg) plus vehicle group (Fig.8A). That is, the onset for the development of morphine tolerance was shortened in rats coadministered with morphine and PDC. The potentiation of morphine tolerance by PDC was further indicated by an increased rightward shift of the antinociceptive dose–response curve in the morphine plus PDC groups when tested on day 8 as compared with the morphine alone group (Fig. 8B, Table 2). Taken together, the GT inhibitor PDC potentiated the development of morphine tolerance by shortening the onset and enhancing the level of antinociceptive tolerance.
Fig. 8.
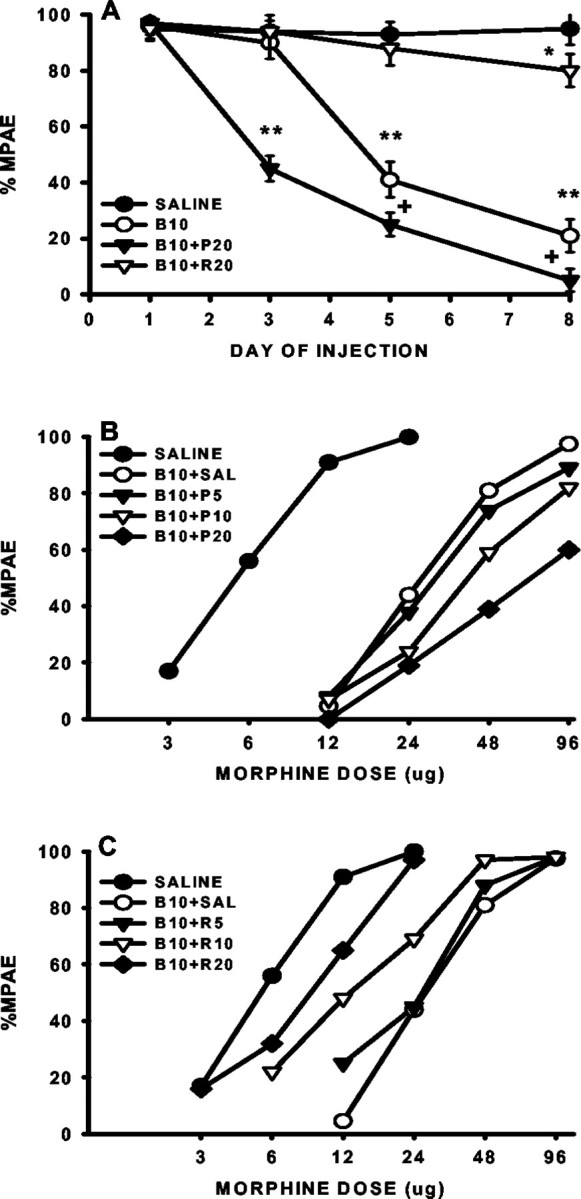
Regulation of morphine tolerance by the GT inhibitor PDC and activator riluzole. A, The onset for the development of morphine tolerance was shortened by coadministration of 10 μg of morphine (B10) with 20 μg of PDC (B10+P20) but prolonged by coadministration of 10 μg of morphine (B10) with 20 μg of riluzole (B10+R20). **p < 0.01, as compared with the saline group, and + p < 0.05, as compared with the morphine alone group. B,C, The cumulative dose–response curves were shifted dose dependently to the right in rats treated with 10 μg of morphine (B10) with 5, 10, or 20 μg of PDC (B10+P5, B10+P10,B10+P20), whereas the dose–response curves were shifted dose dependently to the left in rats treated with 10 μg of morphine with 5, 10, or 20 μg of riluzole (B10+R5,B10+R10, B10+R20), as compared with the rats receiving either saline alone or 10 μg of morphine plus saline (B10+SAL).
Table 2.
Effect of PDC and riluzole on morphine tolerance
| Group | AD50 (μg) | 95% CI (μg) |
|---|---|---|
| Saline | 4.8 | 1.3–8.2 |
| B10+SAL | 22.6 | 6.9–38.2 |
| B10+P5 | 24.4 | 3.5–45.2 |
| B10+P10 | 30.5 | 5.6–66.8 |
| B10+P20 | 68.1 | 27.7–108.5 |
| B10+R5 | 19.5 | 14.4–53.4 |
| B10+R10 | 10.5 | 4.8–25.7 |
| B10+R20 | 6.1 | 1.4–16.3 |
See the Figure 8 legend for the details for each group. Note that both B10+P20 and B10+R20 groups were significantly different from the B10+saline group based on the AD50 value and 95% CI.
Although repeated intrathecal treatment with PDC (20 μg) alone for 7 d did not produce detectable changes in the baseline tail-flick latency, this treatment reduced the baseline paw-withdrawal latency on day 8 (Fig. 9A), indicating a role of PDC in regulating GT activity. However, there was a further reduction of the paw-withdrawal latency in the morphine (10 μg) plus PDC (20 μg) group, as compared with either the PDC or morphine alone group (Fig. 9A). Thus, PDC also potentiated the development of thermal hyperalgesia associated with morphine tolerance.
Fig. 9.
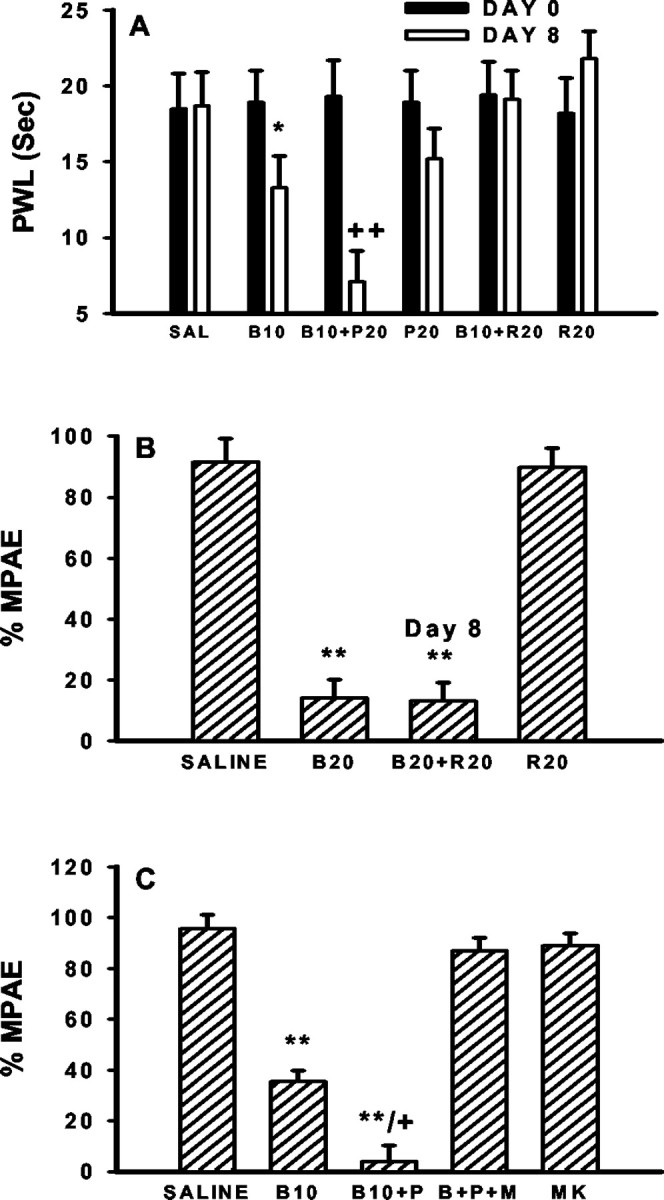
Inhibition by MK-801 of morphine tolerance and thermal hyperalgesia potentiated by PDC. A, The development of thermal hyperalgesia was potentiated in rats treated with 10 μg of morphine plus 20 μg of PDC (B10+P20) but prevented in rats receiving 10 μg of morphine plus 20 μg of riluzole (B10+R20). PDC or riluzole alone changed baseline paw-withdrawal latencies (PWL) on day 8 but did not reach the statistical significance at the current dose.B, The morphine antinociception was dose-dependently reduced on day 8 in rats receiving 7 d intrathecal 20 μg of morphine boluses (B20). The GT activator riluzole (20 μg), given intrathecally at 30 min before the morphine antinociceptive test on day 8 (B20+R20; Day 8), did not reverse the behavioral manifestation of morphine tolerance. C, The development of morphine tolerance was potentiated by intrathecal coadministration of 10 μg of morphine with 20 μg of PDC (B10+P) twice daily for 7 d. This potentiation was blocked by adding 10 nm MK-801 into this combination (B+P+M). Treatment with 10 nm MK-801 alone (MK) for 7 d did not change the antinociceptive effects of morphine. *p < 0.05, **p < 0.01, as compared with the corresponding saline group, and+ p < 0.05,++ p < 0.01, as compared with the corresponding morphine alone group.
Reduction of morphine tolerance and thermal hyperalgesia by the positive GT regulator riluzole
There was a nearly fivefold rightward shift of the morphine antinociceptive dose–response curve in rats receiving morphine (10 μg) plus vehicle twice daily for 7 d. The positive GT regulator riluzole dose-dependently reduced the rightward shift of the dose–response curve (Fig. 8C, Table 2). Consistent with this observation, reduced antinociception as demonstrated in the morphine plus vehicle group on day 6 was absent in those rats receiving intrathecal coadministration of morphine (10 μg) with riluzole (20 μg) for 5 d (Fig. 8A), indicating that riluzole also prolonged the onset of the tolerance development. Moreover, although repeated intrathecal treatment with 20 μg of riluzole alone for 7 d had a negligent effect on the baseline tail-flick latency, it did moderately raise the paw-withdrawal latency on day 8 at the current dose (Fig. 9A). Similar to the effect of PDC on thermal hyperalgesia, coadministration of morphine (10 μg) and riluzole (20 μg) for 7 d significantly reduced the development of thermal hyperalgesia as compared with either the morphine or riluzole alone group (Fig. 9A).
Riluzole, however, did not reverse the behavioral manifestation of morphine tolerance once it had developed. Thus, an acute injection of riluzole (20 μg), given at 30 min before the behavioral test of morphine antinociception on day 8, failed to restore the antinociceptive effects of morphine in rats receiving repeated morphine (20 μg) treatment for 7 d (Fig. 9B). The results indicate that changes in spinal GT activity are contributory to the development of morphine tolerance.
Inhibition by MK-801 of morphine tolerance and thermal hyperalgesia potentiated by PDC
The noncompetitive NMDAR antagonist MK-801 (10 nm), given intrathecally with morphine (10 μg) or morphine (10 μg) plus PDC (20 μg) for 7 d, effectively blocked the development of morphine tolerance (Fig. 9C). The same treatment regimen also prevented the development of thermal hyperalgesia in the same rats when tested on day 8 (paw-withdrawal latencies: saline, 18.7 ± 2.1 sec; 10 μg of morphine, 13.1 ± 1.9 sec; 10 μg of morphine/20 μg of PDC, 7.2 ± 2.1 sec; 10 μg of morphine/20 μg of PDC/10 nm MK-801, 18.4 ± 2.0 sec;p > 0.05 as compared with the saline group). As a control, repeated intrathecal treatment with MK-801 (10 nm) alone for 7 d did not alter the baseline tail-flick or paw-withdrawal latency or the acute antinociceptive effects of morphine (Fig. 9C). The results indicated that NMDARs play a role in the development of morphine tolerance and thermal hyperalgesia that were potentiated by PDC.
DISCUSSION
The present results indicate that chronic morphine induces downregulation of spinal GTs, which contributes to the development of morphine tolerance and associated thermal hyperalgesia. First, the time course of morphine-induced GT downregulation correlated with that of the behavioral manifestation of morphine tolerance and thermal hyperalgesia. Second, morphine-induced downregulation of spinal GTs reduced the ability to maintain in vivo regional glutamate homeostasis, as shown by the development of thermal hyperalgesia in morphine-tolerant rats in the absence of exogenous glutamate and the exacerbated thermal hyperalgesia in response to exogenous glutamate. Third, perturbation of spinal GT activity by PDC or riluzole modulated the development of both morphine tolerance and associated thermal hyperalgesia. Fourth, the noncompetitive NMDAR antagonist MK-801 prevented the development of morphine tolerance and thermal hyperalgesia potentiated by the GT inhibitor PDC, indicating that the role of spinal GTs in morphine tolerance and thermal hyperalgesia is mediated at least in part through NMDARs.
Overall consideration on data interpretation
This series of experiments extended previous in vitrofindings of synaptic glutamate regulation by GTs to examine the relationship between changes in spinal GT expression and activity and the development of morphine tolerance and associated thermal hyperalgesia under an in vivo experimental condition. Several lines of evidence including the exacerbated hyperalgesia to exogenous glutamate in rats with reduced GTs and its reduction by riluzole support a functional link between changes in GT expression and activity and the regulation of in vivo glutamate homeostasis. This is consistent with the role of GTs in regulating glutamate uptake at the synaptic level in previous in vitrostudies (Semba and Wakuta, 1998; Azbill et al., 2000; Jabaudon et al., 2000; Lievens et al., 2000; Matthews et al., 2000). A caveat is that although PDC has been used extensively as a GT inhibitor, riluzole as a specific GT activator was controversial (Cheramy et al., 1992; Martin et al., 1993; Doble, 1996). This agent has recently been shown to be a positive regulator of GT activity that increases glutamate uptake in synaptosomes when given under both in vitro and in vivo conditions (Azbill et al., 2000). However, neither PDC nor riluzole is selective for neuronal or glial GT activity. As such, the effects from PDC and riluzole should be considered to be on both neuronal and glial GTs, and other GTs such as GLT-1 not examined in this study may also play a role in morphine tolerance and thermal hyperalgesia.
An interesting observation in morphine-tolerant rats is the development of abnormal nociceptive sensitivity shown as thermal hyperalgesia that was regulated by spinal GT activity. Hyperalgesia is a known sign of naloxone-precipitated withdrawal indicative of physical dependence after chronic morphine treatment (Mao et al., 1994; Jhamandas et al., 1996). A group of recent studies have shown the development of thermal hyperalgesia in association with the development of morphine tolerance in the absence of naloxone-precipitated withdrawal (Mao et al., 1994;Ossipov et al., 1995; Wegert et al., 1997; Vanderah et al., 2000). Although there is a possibility that thermal hyperalgesia after opioid boluses could result from mini-withdrawals (Jhamandas et al., 1996), thermal hyperalgesia also has been observed in rats infused continuously with morphine via an osmotic pump at the time of the hyperalgesia test (Vanderah et al., 2000). This is also observed in the present study, showing a comparable degree of thermal hyperalgesia on day 8 between rats receiving either morphine boluses or continuous infusion via an implanted osmotic pump, and the pump infusion was not disconnected during the hyperalgesic test on day 8.
Of interest to note is that PDC or riluzole treatment alone for 7 d changed the baseline paw-withdrawal latency in morphine-naïve rats, albeit less profoundly as compared with that of morphine-treated rats, although changes in the tail-flick latency in these same rats were not detected. This difference is likely attributable to the fast-rising temperature in the tail-flick test as compared with that in the paw-withdrawal test (Mao et al., 1994). Thus, the data suggest that spinal GT activity, in addition to the GT expression, could contribute to the behavioral manifestation of thermal hyperalgesia in morphine-treated rats. Indeed, a single pretreatment with riluzole attenuated thermal hyperalgesia to exogenous glutamate in morphine-treated rats, indicating that enhancing the activity of existing GTs was able to compensate for, at least in part, the GT downregulation resulting from chronic morphine administration.
Relation to mechanisms of morphine tolerance and associated thermal hyperalgesia
To date, several intriguing hypotheses have been proposed concerning the cellular and molecular mechanisms of opioid tolerance, including recent findings of the role of β-arrestin, excitatory amino acid receptors including NMDARs, and μ-opioid receptor oligomerization/endocytosis (Guitart and Nestler, 1989; Marek et al. 1991a; Trujillo and Akil, 1991; Nestler, 1992; Bohn et al., 1999, 2000;Whistler and von Zastrow, 1999; Finn and Whistler, 2001; He et al., 2002; Kieffer and Evans, 2002). With regard to the role of NMDARs, previous in vitro studies have suggested that NMDARs may be primed (i.e., increased excitability) after exposure to morphine via activation of intracellular protein kinase C (PKC) (Chen and Huang, 1991; Mao et al. 1995c). PKC may directly or indirectly modulate NMDARs by removing the Mg++ blockade from the NMDAR-Ca2+ channel site (Chen and Huang, 1992) and regulating NMDAR trafficking and gating (Xiong et al., 1998;Lan et al., 2001). The present findings of morphine-induced GT downregulation and its relation to the regulation of morphine tolerance and associated hyperalgesia that were preventable by the NMDAR inhibition provides additional evidence for the NMDAR involvement in this process.
There is a basal level of extracellular glutamate (Jhamandas et al., 1996) that is actively and tightly regulated by GTs (Robinson and Dowd, 1997; Danbolt, 2001). Chronic morphine induces downregulation of spinal GTs leading to the reduced ability to maintain regional glutamate homeostasis as indicated in the present in vivo study. That is, morphine-induced GT downregulation would increase the availability of extracellular glutamate, although such changes may not necessarily be seen as a gross increase in the regional glutamate level (Jhamandas et al., 1996). Increased glutamate availability at the extracellular level would increase the probability of excitatory amino acid receptor activation including NMDARs. Conceivably, activation of NMDARs under such circumstances could make contributions to the previously proposed intracellular mechanisms of morphine tolerance that involve PKC, cAMP, and nitric oxide (Kolesnikov et al., 1993; Elliott et al., 1994a,b; Mao et al. 1995c; Mayer et al., 1995; Bilsky et al., 1996; Narita et al., 1996, 2001; Ma et al., 2001; Zeitz et al., 2002).
Several issues are noteworthy with regard to contributions of morphine-induced GT changes to the neural mechanisms of morphine tolerance and associated hyperalgesia. First, changes in regional glutamate availability caused by the GT downregulation would modulate the activity of excitatory amino acid receptors including NMDARs at both presynaptic and postsynaptic sites, considering that NMDARs as well as μ-opioid receptors are located both presynaptically and postsynaptically (Yaksh, 1986; Liu et al., 1994). This extends previous views of NMDA and μ-opioid receptor interactions that have focused on the postsynaptic site (Mao et al. 1995b). Second, besides the interaction between NMDA and μ-opioid receptors within a single cell as demonstrated by in vitro studies (Chen and Huang, 1991), such an interaction could also occur involving neural circuits (Mao et al., 1994; Zeitz et al., 2002). Changes in regional glutamate availability from the downregulation of both glial and neuronal GTs would support such mechanisms. Third, both basic and clinical research have demonstrated the association between chronic opioid treatment and the development of abnormal pain sensitivity, and NMDAR activation is contributory to such an association (Sjogren et al., 1993; Mao et al., 1994; Ossipov et al., 1995; Devulder, 1997; Wegert et al., 1997;Vanderah et al., 2000; Celerier et al., 2001). Morphine-induced GT downregulation would play an important role in the association between morphine tolerance and hyperalgesia, because both tolerance and hyperalgesia are preventable by blocking NMDARs (Mao et al. 1995b). Fourth, the cellular and molecular mechanisms of opioid tolerance are complex, and multiple mechanisms are likely to be involved depending on opioid receptor agonists, route of treatment, assay methods, and clinical relevance (Kieffer and Evans, 2002). Of significance is that morphine-induced GT downregulation and its functional role may help explain the interaction between two opioid-related clinical observations: analgesic tolerance and associated abnormal pain sensitivity. It remains to be seen how morphine-induced GT downregulation would interact with other proposed cellular mechanisms of opioid tolerance.
Potential mechanisms of morphine-induced GT downregulation
The cellular mechanisms of morphine-induced downregulation of spinal GTs remain to be investigated. There are at least two possibilities of GT regulation by chronic morphine. Spinal GT expression could be regulated by extracellular glutamate (Danbolt, 2001). This is suggested by the observations that downregulation of GLT-1 and GLAST occurs in the rat's brain regions after an impaired cortical glutamatergic connection (Ginsberg et al., 1995), and conversely, that an increase in extracellular glutamate upregulates GLT-1 in astroglial cultures (Thorlin et al., 1998). If this is the case, a decreased level of extracellular glutamate resulting from the inhibitory effect of morphine on neurotransmitter (e.g., glutamate) release (Yaksh, 1986) could lead to a simultaneous downregulation of both EAAC1 and GLAST to maintain regional glutamate homeostasis. The present data showing a time course of progressive GT downregulation after chronic morphine would lend some support to this possibility. However, this regulation would be difficult to explain a transient increase in GT expression after morphine treatment as seen in the present study.
Another possibility is that morphine could regulate GTs via opioid receptor-mediated intracellular changes such as cAMP (She et al., 2000;Wang and Sadee, 2000), because cAMP has been shown to regulate the expression of GLT-1 and GLAST in cell cultures (Swanson et al., 1997;Schlag et al., 1998). This possibility is supported by the reduced GLT-1 mRNAs in response to a δ-opioid agonist acting directly on cultured glial cells (Thorlin et al., 1998). In addition, previous studies have shown the colocalization of μ-opioid receptors and glial cells at both developmental and adult stages (Ruzicka et al., 1995;Ruzicka and Akil, 1997; Stiene-Martin et al., 1998, 2001; Thorlin et al., 1999; Tryoen-Toth et al., 2000). Nonetheless, glutamate regulation and opioid receptor-mediated intracellular changes could each play a role in morphine-induced GT changes, and both possibilities merit future investigation.
Clinical implications
The present findings indicate a functional role of spinal GTs in the development of morphine tolerance and associated thermal hyperalgesia and suggest a new strategy for preventing opioid tolerance and the associated abnormal pain sensitivity by regulating regional glutamate homeostasis using a GT regulator such as riluzole. Furthermore, the present study may provide some insights into the neural mechanisms of substance abuse. Activation of NMDARs has been shown to play a role in the neural mechanisms of many forms of substance abuse (De Montis et al., 1992; Churchill et al., 1999; Huber et al., 2001). Thus, a corollary of the present data is that the involvement of NMDARs in substance abuse could be related to changes of brain GTs after exposure to a substance of abuse. This may be particularly relevant to the mechanisms of heroin addiction, because heroin metabolites (6-monoacetylmorphine or morphine) do indeed interact with opioid receptors (Sim-Selley et al., 2000; Kreek, 2001).
Footnotes
This work was supported by Public Health Service Grant DA08835 to J.M. We thank the Neural Plasticity Research Group at the Massachusetts General Hospital for technical support.
Correspondence should be addressed to Dr. Jianren Mao, MGH Pain Center, Suite WACC 324, Massachusetts General Hospital, Harvard Medical School, 15 Parkman Street, Boston, MA 02114. E-mail:jmao@partners.org.
REFERENCES
- 1.Akil H, Mayer DJ. Antagonization of stimulation produced analgesia by p-CPA, a serotonin synthesis inhibitor. Brain Res. 1972;44:692–697. doi: 10.1016/0006-8993(72)90338-1. [DOI] [PubMed] [Google Scholar]
- 2.Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosome. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 3.Bilsky EJ, Bernstein Z, Wang Z, Sadee W, Porreca F. Effects of naloxone and d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Phe-Thr-NH2 and the protein kinase inhibitors H7 and H8 on acute morphine dependence and antinociceptive tolerance in mice. J Pharmacol Exp Ther. 1996;277:484–490. [PubMed] [Google Scholar]
- 4.Bohn LM, Lefkowitz RJ, Gainestdinov RR, Peppel K, Caron MG, Lin F. Enhanced morphine analgesia in mice lacking beta-arrestin-2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 5.Bohn LM, Gainestdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 6.Celerier E, Laulin JP, Corcuff JB, Le Moal M, Simonnet G. Progressive enhancement of delayed hyperalgesia induced by repeated heroin administration: a sensitization process. J Neurosci. 2001;21:4074–4080. doi: 10.1523/JNEUROSCI.21-11-04074.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Huang LYM. Sustained potentiation of NMDA receptor-mediated glutamate responses through activation of protein kinase C by a μ-opioid. Neuron. 1991;7:319–326. doi: 10.1016/0896-6273(91)90270-a. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Huang LYM. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 9.Cheramy A, Barbeito L, Godeheu G, Glowinski J. Riluzole inhibits the release of glutamate in the caudate nucleus of the cat in vivo. Neurosci Lett. 1992;147:209–212. doi: 10.1016/0304-3940(92)90597-z. [DOI] [PubMed] [Google Scholar]
- 10.Churchill L, Swanson CJ, Urbina M, Kalivas PW. Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J Neurochem. 1999;72:2397–2403. doi: 10.1046/j.1471-4159.1999.0722397.x. [DOI] [PubMed] [Google Scholar]
- 11.D'Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 12.Danbolt NC. Glutamate uptake. Prog Neurol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 13.De Montis MG, Devoto P, Meloni D, Ganbarana C, Giorgi G, Tangliamonte A. NMDA receptor inhibition prevents tolerance to cocaine. Pharmacol Biochem Behav. 1992;42:179–182. doi: 10.1016/0091-3057(92)90463-p. [DOI] [PubMed] [Google Scholar]
- 14.Devulder J. Hyperalgesia induced by high-dose intrathecal sufentanil in neuropathic pain. J Neurosurg Anesthesiol. 1997;9:146–148. doi: 10.1097/00008506-199704000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Doble A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:S233–S241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- 16.Dunbar SA, Pulai IJ. Repetitive opioid abstinence causes progressive hyperalgesia sensitive to N-methyl-d-aspartate receptor blockade in the rat. J Pharmacol Exp Ther. 1998;284:678–686. [PubMed] [Google Scholar]
- 17.Elliott K, Minami N, Kolesnikov YA, Pasternak GW, Inturrisi CE. The NMDA receptor antagonists, LY274614 and MK-801, and the nitric oxide synthase inhibitor, NG-nitro-l-arginine, attenuate analgesic tolerance to the mu-opioid morphine but not to kappa opioids. Pain. 1994a;56:69–75. doi: 10.1016/0304-3959(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 18.Elliott KJ, Hynansky A, Inturrisi CE. Dextromethorphan attenuates and reverses morphine tolerance. Pain. 1994b;59:361–368. doi: 10.1016/0304-3959(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 19.Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 20.Ginsberg SD, Martin LJ, Rothstein JD. Regional deafferentation downregulates subtypes of glutamate transporter protein. J Neurochem. 1995;65:2800–2803. doi: 10.1046/j.1471-4159.1995.65062800.x. [DOI] [PubMed] [Google Scholar]
- 21.Granados-Soto V, Kalcheva I, Hua X, Newton A, Yaksh TL. Spinal PKC activity and expression: role in tolerance produced by continuous spinal morphine infusion. Pain. 2000;85:395–404. doi: 10.1016/S0304-3959(99)00281-X. [DOI] [PubMed] [Google Scholar]
- 22.Guitart X, Nestler EJ. Identification of morphine- and cyclic AMP-regulated phosphoproteins (MARPPs) in the locus coeruleus and other regions of rat brain: regulation by acute and chronic morphine. J Neurosci. 1989;9:4371–4387. doi: 10.1523/JNEUROSCI.09-12-04371.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 24.He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- 25.Huber JD, Darling SF, Park K, Soliman KF. The role of NMDA receptors in neonatal cocaine-induced neurotoxicity. Pharmacol Biochem Behav. 2001;69:451–459. doi: 10.1016/s0091-3057(01)00521-4. [DOI] [PubMed] [Google Scholar]
- 26.Ibuki T, Dunbar SA, Yaksh TL. Effect of transient naloxone antagonism on tolerance development in rats receiving continuous spinal morphine infusion. Pain. 1997;70:125–132. doi: 10.1016/s0304-3959(96)03283-6. [DOI] [PubMed] [Google Scholar]
- 27.Jabaudon D, Scanziani M, Gahwiler BH, Gerber U. Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci USA. 2000;97:5610–5615. doi: 10.1073/pnas.97.10.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jhamandas KH, Marsala M, Ibuki T, Yaksh TL. Spinal amino acid release and precipitated withdrawal in rats chronically infused with spinal morphine. J Neurosci. 1996;16:2758–2766. doi: 10.1523/JNEUROSCI.16-08-02758.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kieffer BL, Evans CJ. Opioid tolerance: in search of the holy grail. Cell. 2002;108:587–590. doi: 10.1016/s0092-8674(02)00666-9. [DOI] [PubMed] [Google Scholar]
- 30.Kolesnikov YA, Pick CG, Ciszewska G, Pasternak GW. Blockade of tolerance to morphine but not to k opioids by a nitric oxide synthase inhibitor. Proc Natl Acad Sci USA. 1993;90:5162–5166. doi: 10.1073/pnas.90.11.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreek MJ. Drug addiction. Molecular and cellular endpoints. Ann NY Acad Sci. 2001;937:27–49. [PubMed] [Google Scholar]
- 32.Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MVL, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- 33.Lievens JC, Bernal F, Forni C, Mahy N, Kerkerian-Le Goff L. Characterization of striatal lesions produced by glutamate uptake alteration: cell death, reactive gliosis, and changes in GLT-1 and GADD45 mRNA expression. Glia. 2000;29:222–232. doi: 10.1002/(sici)1098-1136(20000201)29:3<222::aid-glia4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI. Evidence for presynaptic N-methyl-d-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci USA. 1994;91:8383–8387. doi: 10.1073/pnas.91.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R. Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: an in vitro and in vivo study. Eur J Neurosci. 2001;14:1091–1104. doi: 10.1046/j.0953-816x.2001.01731.x. [DOI] [PubMed] [Google Scholar]
- 36.Manning BH, Mao J, Frenk H, Price DD, Mayer DJ. Continuous co-administration of dextromethorphan or MK-801 with morphine: attenuation of morphine dependence and naloxone-reversible attenuation of morphine tolerance. Pain. 1996;67:79–88. doi: 10.1016/0304-3959(96)81972-5. [DOI] [PubMed] [Google Scholar]
- 37.Mao J. NMDA and opioid receptors: their interactions in antinociception, tolerance and neuroplasticity. Brain Res Rev. 1999;30:289–304. doi: 10.1016/s0165-0173(99)00020-x. [DOI] [PubMed] [Google Scholar]
- 38.Mao J, Price DD, Mayer DJ, Hayes RL. Pain-related increases in spinal cord membrane-bound protein kinase C following peripheral nerve injury. Brain Res. 1992;588:144–149. doi: 10.1016/0006-8993(92)91354-h. [DOI] [PubMed] [Google Scholar]
- 39.Mao J, Mayer DJ, Hayes RL, Price DD. Spatial patterns of increased spinal cord membrane-bound protein kinase C and their relation to increases in 14C-2-deoxyglucose metabolic activity in rats with painful peripheral mononeuropathy. J Neurophysiol. 1993;70:470–481. doi: 10.1152/jn.1993.70.2.470. [DOI] [PubMed] [Google Scholar]
- 40.Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mao J, Price DD, Mayer DJ. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995a;61:353–364. doi: 10.1016/0304-3959(95)00022-K. [DOI] [PubMed] [Google Scholar]
- 42.Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and opiate tolerance: a current view of their possible interactions. Pain. 1995b;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- 43.Mao J, Price DD, Phillips LL, Lu J, Mayer DJ. Increases in protein kinase C gamma immunoreactivity in the spinal cord of rats associated with tolerance to the analgesic effects of morphine. Brain Res. 1995c;677:257–267. doi: 10.1016/0006-8993(95)00161-i. [DOI] [PubMed] [Google Scholar]
- 44.Mao J, Price DD, Caruso FS, Mayer DJ. Oral administration of dextromethorphan prevents the development of morphine tolerance and dependence in rats. Pain. 1996;67:361–368. doi: 10.1016/0304-3959(96)03120-x. [DOI] [PubMed] [Google Scholar]
- 45.Marek P, Ben Eliyahu S, Gold M, Liebeskind JC. Excitatory amino acid antagonists (kynurenic acid and MK-801) attenuate the development of morphine tolerance in the rat. Brain Res. 1991a;547:77–81. doi: 10.1016/0006-8993(91)90576-h. [DOI] [PubMed] [Google Scholar]
- 46.Marek P, Ben Eliyahu S, Vaccarino AL, Liebeskind JC. Delayed application of MK-801 attenuates development of morphine tolerance in rats. Brain Res. 1991b;558:163–165. doi: 10.1016/0006-8993(91)90736-f. [DOI] [PubMed] [Google Scholar]
- 47.Martin D, Thompson MA, Nadle JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250:473–476. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- 48.Matthews CC, Zielke HR, Wollack JB, Fishman PS. Enzymatic degradation protects from glutamate excitotoxicity. J Neurochem. 2000;75:1045–1052. doi: 10.1046/j.1471-4159.2000.0751045.x. [DOI] [PubMed] [Google Scholar]
- 49.Mayer DJ, Mao J, Price DD. The development of morphine tolerance and dependence is associated with translocation of protein kinase C. Pain. 1995;61:365–374. doi: 10.1016/0304-3959(95)00023-L. [DOI] [PubMed] [Google Scholar]
- 50.Mennerick S, Shen W, Xu W, Benz A, Tanaka K, Shimamoto K, Isenb KE, Krause JE, Zorumski CF. Substrate turnover by transporters curtails synaptic glutamate transients. J Neurosci. 1999;19:9242–9251. doi: 10.1523/JNEUROSCI.19-21-09242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molander C, Xu Q, Grant G. The cytoarchitectonic organization of the spinal cord in the rat. I. The lower thoracic and lumbosacral cord. J Comp Neurol. 1984;230:133–141. doi: 10.1002/cne.902300112. [DOI] [PubMed] [Google Scholar]
- 52.Nakagawa T, Ozawa T, Shige K, Yamamoto R, Minami M, Satoh M. Inhibition of morphine tolerance and dependence by MS-153, a glutamate transporter activator. Eur J Pharmacol. 2001;419:39–45. doi: 10.1016/s0014-2999(01)00965-7. [DOI] [PubMed] [Google Scholar]
- 53.Narita M, Mizoguchi H, Kampine JP, Tseng LF. Role of protein kinase C in desensitization of spinal delta-opioid-mediated antinociception in the mouse. Br J Pharmacol. 1996;118:1829–1835. doi: 10.1111/j.1476-5381.1996.tb15610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Narita M, Mizoguchi H, Nagase H, Suzuki T, Tseng LF. Involvement of spinal protein kinase Cγ in the attenuation of opioid μ-receptor-mediated G-protein activation after chronic intrathecal administration of [d-Ala2, N-MePhe4, Gly-Ol5]enkephalin. J Neurosci. 2001;21:3715–3720. doi: 10.1523/JNEUROSCI.21-11-03715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nestler EJ. Molecular mechanisms of drug addiction. J Neurosci. 1992;12:2439–2450. doi: 10.1523/JNEUROSCI.12-07-02439.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res. 2001;905:254–258. doi: 10.1016/s0006-8993(01)02536-7. [DOI] [PubMed] [Google Scholar]
- 57.Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci Lett. 1995;199:87–90. doi: 10.1016/0304-3940(95)12022-v. [DOI] [PubMed] [Google Scholar]
- 58.Paronis CA, Holtzman SG. Increased analgesic potency of mu agonists after continuous naloxone infusion in rats. J Pharmacol Exp Ther. 1991;259:582–589. [PubMed] [Google Scholar]
- 59.Robinson MB, Dowd LA. Heterogeneity and functional properties of subtypes of sodium-dependent glutamate transporters in the mammalian central nervous system. Adv Pharmacol. 1997;37:69–115. doi: 10.1016/s1054-3589(08)60948-5. [DOI] [PubMed] [Google Scholar]
- 60.Ruzicka BB, Akil H. The interleukin-1 beta-mediated regulation of proenkephalin and opioid receptor messenger RNA in primary astrocyte-enriched cultures. Neuroscience. 1997;79:517–524. doi: 10.1016/s0306-4522(96)00669-0. [DOI] [PubMed] [Google Scholar]
- 61.Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptors mRNA. Brain Res Mol Brain Res. 1995;34:209–220. doi: 10.1016/0169-328x(95)00165-o. [DOI] [PubMed] [Google Scholar]
- 62.Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zeleania OA, Rothstein JD, Robinso MB. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998;53:355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- 63.Semba J, Wakuta MS. Regional differences in the effects of glutamate uptake inhibitor trans-pyrrolidine-2, 4-bicarboxylic acid on extracellular amino acids and dopamine in rat brain: an in vivo microdialysis study. Gen Pharmacol. 1998;31:399–404. doi: 10.1016/s0306-3623(98)00047-0. [DOI] [PubMed] [Google Scholar]
- 64.She J, Benedict Gomes A, Gallagher A, Stafford K, Yoburn BC. Role of cAMP-dependent protein kinase A (PKA) in opioid agonist-induced mu-opioid receptor downregulation and tolerance in mice. Synapse. 2000;38:322–327. doi: 10.1002/1098-2396(20001201)38:3<322::AID-SYN11>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 65.Sim-Selley LJ, Selley DE, Vogt LJ, Childers SR, Martin TJ. Chronic heroin self-administration desensitizes micro opioid receptor-activated G-proteins in specific regions of rat brain. J Neurosci. 2000;20:4555–4562. doi: 10.1523/JNEUROSCI.20-12-04555.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sjogren P, Josson T, Jesen NH, Drenck NE, Jensen TS. Hyperalgesia and myoclonus in terminal cancer patients treated with continuous intravenous morphine. Pain. 1993;55:93–97. doi: 10.1016/0304-3959(93)90188-U. [DOI] [PubMed] [Google Scholar]
- 67.Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- 68.Stiene-Martin A, Knapp PE, Martin K, Gurwell JA, Ryan S, Thornton A, Smith FL, Hauser KF. Opioid system diversity in developing neurons, astroglia, and oligodendroglia in the subventricular zone and striatum: impact on gliagenesis in vivo. Glia. 2001;36:78–88. [PMC free article] [PubMed] [Google Scholar]
- 69.Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thorlin T, Roginski RS, Choudhury K, Nilsson M, Ronnback L, Hansson E, Eriksson PS. Regulation of the glial glutamate transporter GLT-1 by glutamate and delta-opioid receptor stimulation. FEBS Lett. 1998;425:453–459. doi: 10.1016/s0014-5793(98)00288-9. [DOI] [PubMed] [Google Scholar]
- 71.Thorlin T, Persson PA, Eriksson PS, Hansson E, Ronnback L. Delta-opioid receptor immunoreactivity on astrocytes is upregulated during mitosis. Glia. 1999;25:370–378. doi: 10.1002/(sici)1098-1136(19990215)25:4<370::aid-glia6>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 72.Tiseo PJ, Inturrisi CE. Attenuation and reversal of morphine tolerance by the competitive N-methyl-d-aspartate receptor antagonist, LY274614. J Pharmacol Exp Ther. 1993;264:1090–1096. [PubMed] [Google Scholar]
- 73.Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown RHJ, Hediger MA. Amyotrophic lateral sclerosis-linked glutamate transporter mutation has impaired glutamate clearance capacity. J Biol Chem. 2001;276:576–582. doi: 10.1074/jbc.M003779200. [DOI] [PubMed] [Google Scholar]
- 74.Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- 75.Tryoen-Toth P, Gaveriaux-Ruff C, Labourdette G. Down-regulation of mu-opioid receptor expression in rat oligodendrocytes during their development in vitro. J Neurosci Res. 2000;60:10–20. doi: 10.1002/(SICI)1097-4547(20000401)60:1<10::AID-JNR2>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 76.Vanderah TW, Gardell LR, Burgess SE, Ibrahim M, Dogrul A, Zhong CM, Malan TP, Ossipov MH, Lai J, Porreca F. Dynorphin promotes abnormal pain and spinal cord opioid antinociceptive tolerance. J Neurosci. 2000;20:7074–7079. doi: 10.1523/JNEUROSCI.20-18-07074.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vorwerk CK, Naskar R, Schuettauf F, Quinto K, Zurakowski D, Gochenauer G, Robinson, Mackler SA, Dreyer EB. Depression of retinal glutamate transporter function leads to elevated intravitreal glutamate levels and ganglion cell death. Invest Ophthalmol Vis Sci. 2000;41:3615–3621. [PubMed] [Google Scholar]
- 78.Wang Z, Sadee W. Tolerance to morphine at the mu-opioid receptor differentially induced by cAMP-dependent protein kinase activation and morphine. Eur J Pharmacol. 2000;389:165–171. doi: 10.1016/s0014-2999(99)00881-x. [DOI] [PubMed] [Google Scholar]
- 79.Wegert S, Ossipov MH, Nichols ML, Bian D, Vanderah TW, Malan TP, Jr, Porreca F. Differential activities of intrathecal MK-801 or morphine to alter responses to thermal and mechanical stimuli in normal and nerve-injured rats. Pain. 1997;71:57–64. doi: 10.1016/s0304-3959(97)03337-x. [DOI] [PubMed] [Google Scholar]
- 80.Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc Natl Acad USA. 1999;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiong ZG, Raouf R, Lu WY, Wang LY, Orser BA, Dudek EM, Browning MD, MacDonald JF. Regulation of N-methyl-d-aspartate receptor function by constitutively active protein kinase C. Mol Pharmacol. 1998;54:1055–1063. [PubMed] [Google Scholar]
- 82.Yaksh TL. The effects of intrathecally administered opioid and adrenergic agents on spinal function. In: Yaksh TL, editor. Spinal afferent processing. Plenum; New York: 1986. pp. 505–539. [Google Scholar]
- 83.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 84.Zeitz KP, Malmberg AB, Gilbert H, Basbaum AI. Reduced development of tolerance to the analgesic effects of morphine and clonidine in PKCgamma mutant mice. Pain. 2002;94:245–253. doi: 10.1016/S0304-3959(01)00353-0. [DOI] [PubMed] [Google Scholar]