

Abstract
Modulation of voltage-gated potassium (Kv) channels by protein phosphorylation plays an essential role in the regulation of the membrane properties of cells. Protein–protein binding domains, such as Src homology 3 (SH3) domains, direct ion channel modulation by coupling the channels with intracellular signaling enzymes. The conventional view is that protein kinase binding to ion channels leads to modulation by bringing the channel substrate into physical proximity to the enzyme, thereby fostering covalent modification of the channel. The SH3 domain binding-dependent functional suppression of Kv1.5 currents by Src family protein tyrosine kinases (PTKs) is considered a canonical example of this type of mechanism. In the present study we address whether the SH3-dependent binding of Src family PTKs toShaker family Kvs mediates modulatory events that are independent of and/or dependent on Src-catalyzed tyrosine phosphorylation of the channel. We find that Src binding and tyrosine phosphorylation are each able to modulate Kv1 family macroscopic channel currents independently. SH3-dependent binding of Src leads to the suppression of both Kv1.5 and Kv1.4 (modified to contain proline-rich SH3 domain binding sites) macroscopic currents even in the absence of Src-catalyzed tyrosine phosphorylation, whereas binding-independent tyrosine phosphorylation by Src leads to the suppression of Kv1.5 macroscopic currents and the modulation of Kv1.4 inactivation kinetics.
Keywords: Kv1.4, Kv1.5, potassium channel, protein–protein interaction, tyrosine phosphorylation, Shaker, Src, protein engineering
Voltage-gated potassium (Kv) channels regulate the membrane properties of neurons, including action potential dynamics and the maintenance of resting potential. Kv channels are important functional targets for covalent modification by protein tyrosine kinases (PTKs). Tyrosine phosphorylation of Kv channels modulates voltage-evoked currents (Huang et al., 1993; Holmes et al., 1996a,b; Szabo et al., 1996; Bowlby et al., 1997; Fadool et al., 1997; Fadool and Levitan, 1998; Cayabyab et al., 2000; Nitabach et al., 2001; Cook and Fadool, 2002). Ion channels also can be modulated by direct protein–protein interactions between channels and signaling molecules. For example, G-protein binding to inward rectifier potassium (Kir) channels modulates channel function without catalyzing covalent modification of the channel (Logothetis et al., 1987; He et al., 2002).
Many PTKs associate with their substrates via protein–protein binding domains, including the Src homology 2 (SH2) and 3 (SH3) domains first described for Src family PTKs. Ion channel subunits from diverse protein families possess canonical proline-rich SH3 ligand sequences in their cytoplasmic N or C termini that interact directly with SH3 domains (Holmes et al., 1996a; Kanemitsu et al., 1997; Santoro et al., 1997; Maximov et al., 1999; Nitabach et al., 2001). Kv1.5 associatesin vivo with Src family PTKs in heart, hippocampus, and Schwann cells (Holmes et al., 1996a; Sobko et al., 1998; Nitabach et al., 2001). Kv1.5 biophysical modulation depends on the binding of the Kv1.5 proline-rich SH3 domain ligand sequence to the SH3 domain of Src family PTKs Src and Fyn (Holmes et al., 1996a, 1998; Sobko et al., 1998; Nitabach et al., 2001). In addition, ion channel subunits can act as adaptor proteins in the context of heteromultimers: channel subunits with SH3 domain binding sites allow Src family PTKs to phosphorylate coassembled subunits that lack SH3 domain ligand sequences and biophysically modulate the heteromultimeric channel (Nitabach et al., 2001).
The conventional view has been that the binding of signaling enzymes to ion channels leads to modulation by bringing the channel substrate into physical proximity to the enzyme, thereby fostering covalent modification of the channel (Chung et al., 1991). Although the functional importance of SH3 domain binding-dependent covalent modification of ion channels by signaling enzymes is clear, whether SH3 domain-dependent binding and covalent modification might mediate distinct modes of modulation has not been examined. In the present study we ask whether the SH3 domain-dependent binding of Src family PTKs to Shaker family Kvs mediates modulatory events that are independent of Src-catalyzed tyrosine phosphorylation. We also examine whether modified Src PTKs that are incapable of binding Kv channels are competent to modulate channels via their tyrosine kinase catalytic activity. We find that Src binding and tyrosine phosphorylation are each able to modulate independently both Kv1.4 and Kv1.5 macroscopic channel currents. SH3 domain-dependent binding of Src leads to the suppression of Kv1.5 and Kv1.4 (modified to contain proline-rich SH3 domain binding sites; Kv1.4-Pro) macroscopic currents even in the absence of Src-catalyzed tyrosine phosphorylation, whereas binding-independent tyrosine phosphorylation by Src causes the suppression of Kv1.5 macroscopic currents and the modulation of Kv1.4 inactivation kinetics.
MATERIALS AND METHODS
cDNA constructs and cRNA in vitrotranscription. All cDNA constructs for the transfection of human embryonic kidney (HEK) 293 cells were under the control of the mammalian cytomegalovirus CMV promoter. Rat Kv1.4 was provided by M. Sheng (Massachusetts General Hospital, Boston, MA), human Kv1.5 tagged with the human insulin C-terminal peptide epitope (CP) was provided by L. Philipson (University of Chicago, Chicago, IL), and v-Src was provided by R. Huganir (Johns Hopkins University, Baltimore, MD). Kinase-dead v-Src (SrcKD) was generated by introducing the K295A point mutation (Snyder et al., 1985; Kamps and Sefton, 1986; Ebina et al., 1987), and catalytically impaired v-Src (SrcCI) was generated by introducing the R385G point mutation (Nitabach et al., 2001), in both cases into v-Src cDNA [Stratagene's Quickchange method (La Jolla, CA) was used for the construction of all mutant cDNAs used herein]. SrcCISH3KO was generated by introducing the D99N mutation into SrcCI. A synthetic gene fragment encoding proline-rich amino acids 64–82 of Kv1.5 with HindIII cohesive ends was inserted into aHindIII site introduced between the codons encoding amino acids 649 and 650 of Kv1.4, resulting in Kv1.4-C-Pro. Kv1.4-N-Pro was generated by insertion of the same synthetic gene fragment into aHindIII site introduced between the codons encoding amino acids 170 and 171. The position of the proline-rich insertions was chosen by sequence alignments of Kv1.4 with related Kv channels that possess endogenous proline-rich SH3 ligand sequences via the program CLUSTAL W. Kv1.5-Δ-Pro was generated by deleting amino acids 64–82 of Kv1.5. The c-Src SH3–GST fusion construct was generated by subcloning cDNA encoding amino acids 81–141 of Gallus gallus c-Src into pGEX-2T (New England Biolabs, Beverly, MA), which expresses an N-terminal glutathione S-transferase (GST) fusion protein under the control of the tac-inducible promoter (Holmes et al., 1996a). The D99N mutation was introduced into the SH3–GST construct.
cDNA constructs used as templates for the in vitrotranscription of cRNA for injection into Xenopus laevisoocytes were subcloned into the pCS2+vector (provided by D. Turner, Fred Hutchinson Cancer Center, Seattle, WA), which contains (in order from 5′ to 3′) an SP6 phage RNA polymerase promotor, Xenopus β-globin 5′-untranslated leader sequence, multiple cloning site, SV40 late polyadenylation signal, and inverted T7 phage RNA polymerase promoter. Templates for SP6 in vitro transcription reactions were generated by PCR with Pfu Turbo (Stratagene) thermostable polymerase, pCS2+-based cDNA templates, and SP6 and T7 primers. Then 5 μl of the completed PCR was used without further purification as template for SP6 in vitrotranscription (mMessage mMachine, Ambion, Austin, TX). cRNA purified by phenol/chloroform/isoamyl alcohol extraction, chloroform/isoamyl alcohol extraction, and isopropanol precipitation, followed by resuspension in water, was analyzed by formaldehyde-agarose gel electrophoresis to assess integrity and concentration.
HEK 293 cell culture, transfection, pervanadate treatment, and cell lysis. HEK 293 cells were maintained as described previously (Holmes et al., 1996b, 1997). Cells were cultured in EMEM with l-glutamine and Earle's salts supplemented with 10% fetal bovine serum (Mediatech, Washington, DC). Each 60 mm dish of ∼80% confluent cells was transfected with 5 μg of plasmid DNA by using the Fugene 6 reagent (Roche, Hertforshire, UK) according to the manufacturer's instructions. Sodium pervanadate was prepared by mixing 100 mm sodium orthovanadate with 1/10 volume of hydrogen peroxide and incubating for 5 min at room temperature. Then this stock was diluted to 250 μmpervanadate in cell culture medium and applied to the cells for 10 min before lysis (Holmes et al., 1996b). Cells were pervanadate treated and lysed ∼48 hr after transfection in 1 ml/dish modified RIPA lysis buffer [containing (in mm) 25 Tris, pH 7.5, 150 NaCl, 100 NaF, 5 EDTA, and 1 Na3VO4plus 1% Triton X-100] containing protease inhibitors (1 mm PMSF, 2 μg/ml aprotinin, 1 μg/ml pepstatin A, 0.5 μg/ml leupeptin). Cell lysate samples for Western blotting were mixed 1:1 with 2× Laemmli sample buffer containing 2% SDS and 2% β-mercaptoethanol.
Immunoprecipitation and SH3–GST precipitation. α-Kv1.4 rabbit serum [α-Kv1.4 polyclonal antibody (pAb)] was raised against a maltose binding protein fusion protein containing amino acids 581–655 of rat Kv1.4. Preparation of antiserum was performed in accordance with National Institutes of Health (NIH) and New York University (NYU) institutional guidelines and in accordance with theGuide for Care and Use of Laboratory Animals of the National Academy of Sciences. α-Kv1.4 pAb (5 μl), α-CP pAb (5 μl; Linco Research, St. Charles, MO), or c-Src SH3–GST fusion protein (5 μg) was added to 1 ml of HEK 293 cell lysate and incubated overnight at 4°C with shaking. Then 25 μl of either protein A/G beads (Pierce, Rockford, IL) or glutathione-conjugated beads (New England Biolabs) was added to α-Kv1.4–, α-CP–, or SH3–GST-treated lysates, as appropriate, and incubated for 2 hr with shaking. Beads were pelleted with a 20 sec spin at maximum speed in a microcentrifuge and were washed three times with the cell lysis buffer described above, modified to contain only 0.1% Triton X-100. Washed pellets were resuspended in 25 μl of 2× Laemmli sample buffer.
Western blotting. Samples were separated on 10% acrylamide discontinuous Laemmli SDS-PAGE gels. After separation the gels were electroblotted onto nitrocellulose membranes and blocked overnight in TBS-Tw (100 mm Tris, pH 7.5, 150 mm NaCl, 0.05% Tween 20) with 10% nonfat dry milk. Membranes were incubated for 2 hr in 0.1 μg/ml α-Kv1.4 monoclonal antibody (α-Kv1.4 mAb; clone K13/31; Upstate Biotechnology, Lake Placid, NY), 0.05 μg/ml α-phosphotyrosine (α-pY) mAb (clone 4G10; Upstate Biotechnology), 0.3 μg/ml α-CP mAb (BiosPacific, Emeryville, CA), or 0.1 μg/ml α-GST mAb (New England Biolabs) in TBS-Tw, followed by extensive washing in TBS-Tw. Then the membranes were incubated in 1:3000 α-mouse IgG coupled to horseradish peroxidase in TBS-Tw (Amersham Biosciences, Arlington Heights, IL) for 1 hr, followed by extensive washing in TBS-Tw. Membranes were immersed in SuperSignal (Pierce) enhanced chemiluminescence reagent and exposed to X-Omat (Kodak, Rochester, NY) x-ray film.
Exposed x-ray films were digitized by using an AcerScan 620PT flat-bed scanner in transparency mode with Adobe Photoshop software, and band densities were quantified with Scion Image software. Within each experiment, for each experimental condition, the α-pY band density was divided by the α-Kv1.4 band density to yield the normalized α-pY staining density. This normalization provides a measure of the number of phosphorylated tyrosines per quantity of Kv1.4 protein. Within each experiment the normalized α-pY staining density for each experimental condition was divided by the normalized α-pY staining density for Kv1.4-wt to convert the normalized staining densities into relative density units. This controls for any variation between experiments in the relative staining efficiencies of the α-pY and α-Kv1.4 Western blots. Mean relative normalized α-pY staining was analyzed by one-way ANOVA and Bonferroni's multiple comparison test.
Preparation of X. laevis oocytes and electrophysiological recordings. Maintenance of frogs and oocyte harvest, injection, and storage procedures are standard (Goldin, 1992), in accordance with NIH and NYU institutional guidelines and in accordance with the Guide for Care and Use of Laboratory Animals of the National Academy of Sciences. Ovary lobes were removed surgically from anesthetized female frogs through a small abdominal incision, cut into pieces containing ∼20 oocytes, and incubated at room temperature with gentle rocking for ∼1.5 hr in 1 mg/ml collagenase type IA (C-9891; Sigma, St. Louis, MO) in ND96 [containing (in mm) 96 NaCl, 2 KC1, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.5], but without calcium, to separate oocytes from follicular membranes. After extensive washing in ND96, healthy-appearing stage V and VI oocytes were selected and stored in oocyte incubation medium (OIM; 50% Liebovitz's L-15 cell culture medium, 50 μg/ml gentamycin, 100 U/ml nystatin, and 10 mm HEPES, pH 7.5).
Approximately 24 hr after harvesting the oocytes were removed from OIM into ND96, injected with 50 nl of cRNA (∼5 ng) encoding the appropriate Kv subunit, and replaced in OIM. Each oocyte was injected a second time, ∼48 hr after injection of the Kv subunit cRNA, with 50 nl of water or one of the various Src cRNAs. Src cRNAs were of identical concentration (∼0.3 mg/ml), as assessed on an ethidium bromide-stained formaldehyde-agarose gel. Voltage-evoked currents were recorded 9–12 hr after the reinjection with a two-electrode voltage-clamp amplifier (model OC-725C; Warner Instruments, Grand Haven, MI), Digidata 1200 analog/digital hardware, and Clampex 8.0 data collection software (both from Axon Instruments, Foster City, CA). Electrodes were filled with 3 m KCl and exhibited resistances of 0.5–2.5 MΩ, while the bath solution was ND96 with the NaCl replaced with Na-glutamate to reduce the magnitude of endogenous oocyte outward chloride currents. Inactivation time constants were calculated for each Kv1.4-expressing oocyte after reinjection by fitting a first-order exponential function to the 125 msec region of the voltage-evoked current beginning 25 msec after the voltage step. Peak currents and inactivation time constants were averaged for each experimental condition and analyzed by one-way ANOVA, with further paired comparison analysis via Bonferroni's test.
RESULTS
Src PTK modulates Kv1.5 subunits via multiple mechanisms
To address the role in channel modulation of the SH3-dependent binding of Src PTKs to Shaker family Kvs and consequent increased phosphorylation of the channel, we coexpressed wild-type and various mutant forms of Kv1.5 and v-Src in Xenopus oocytes and then measured macroscopic voltage-evoked currents. Oocytes were injected with channel cRNAs and then reinjected with Src cRNA ∼48 hr later. Voltage-evoked channel currents were recorded from each reinjected oocyte 9–12 hr after the Src cRNA injection. This protocol ensures that channel currents have reached nearly peak levels before the expression of exogenous Src (data not shown). Because cRNA-expressed Kv subunits are potentially in great molar excess to endogenous oocyte Src family PTKs, coexpression of Src cRNA allows for a closer molar equivalence of Src and Kv molecules and minimizes the potential confounding variable of kinase substrate stoichiometry. Unlike in HEK 293 cells, the extended pervanadate treatment ofXenopus oocytes leads to little detectable increase in the tyrosine phosphorylation of cellular proteins, suggesting a relative paucity of endogenous PTK activity (data not shown).
We initially intended the use of the kinase-dead v-Src point mutant K295A (v-SrcKD) as a negative control for any possible nonspecific phosphorylation-independent effects of the reinjection of kinase-encoding cRNAs into channel-expressing oocytes. However, we found that v-SrcKD coexpression markedly suppresses voltage-evoked Kv1.5 macroscopic currents. As shown in Figures 1A (left column) and 2, both v-SrcKD and highly active wild-type v-SrcWT significantly suppress Kv1.5 macroscopic voltage-evoked currents. This indicates that expression of a catalytically inactive Src kinase is sufficient for Kv1.5 current suppression.
Fig. 1.
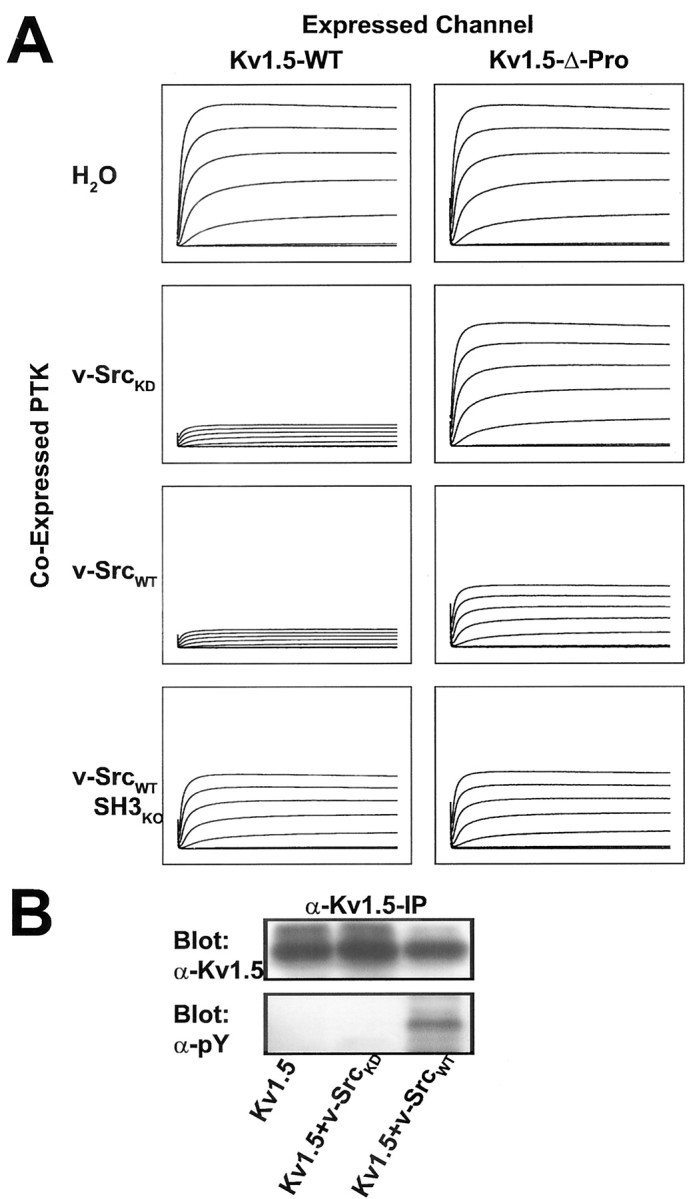
Phosphorylation-dependent and phosphorylation-independent modes of modulation of voltage-evoked Kv1.5 peak currents by Src family protein tyrosine kinase are distinguished by SH3 domain binding dependence. A, Currents evoked in Kv subunit cRNA-injected X. laevis oocytes by 200 msec steps up to +80 mV in 20 mV increments from a holding potential of –80 mV were measured 9–12 hr after a second injection with H2O, kinase-dead v-Src (v-SrcKD) cRNA, wild-type v-Src (v-SrcWT) cRNA, or cRNA encoding v-SrcWT with an inactivated SH3 domain (v-SrcWTSH3ko). The traces that are depicted are averages of the currents measured after the second injection (n > 10 oocytes for each experimental condition). Kv1.5-wt currents are modulated by v-SrcKD, v-SrcWT, and v-SrcWTSH3ko, whereas Kv1.5-Δ-Pro currents are modulated only by v-SrcWT and v-SrcWTSH3ko. All traces for each channel type are normalized to the peak average current for the water-injected group: Kv1.5-wt = 9.5 μA and Kv1.5-Δ-Pro = 4.5 μA.B, Coexpression of v-SrcKD with Kv1.5 does not increase the tyrosine phosphorylation of Kv1.5, as shown by Western blot, with specific anti-phosphotyrosine (α-pY) antibodies of α-Kv1.5 immunoprecipitates of HEK 293 cells transfected with the indicated cDNAs. Kv1.5 protein levels and the proportion of higher-molecular-weight forms of Kv1.5 do not diminish when Kv1.5 is coexpressed with kinase-dead v-SrcKD. Higher-molecular-weight forms of Kv channel subunits indicate channel maturation and plasma membrane targeting.
We next sought to determine whether current suppression of Kv1.5 by v-SrcKD was a nonspecific effect of the reinjection protocol or a specific phosphorylation-independent/binding-dependent action of Src on the channel. We examined the effects of reciprocal channel and kinase mutations that abolish SH3-dependent channel kinase binding. v-SrcKD-induced Kv1.5 current suppression is abolished completely when the Kv1.5 proline-rich SH3 domain ligand is deleted (Figs. 1A, right column, 2). Thus Kv1.5 voltage-evoked current suppression by v-SrcKD is attributable to channel/kinase binding mediated by an SH3 domain interaction. This binding-dependent suppression of Kv1.5 peak current is not attributable to increased phosphorylation of Kv1.5 mediated by coexpressed v-SrcKD (Fig. 1B). However, in the case of v-SrcWT there is a significant component of SH3 binding-independent/phosphorylation-dependent current suppression, as shown by the suppression of Kv1.5 voltage-evoked current by v-SrcWTSH3KO(Figs. 1A, right column, 2), a catalytically active v-Src mutant (D99N) that exhibits a 40- to 50-fold reduction of SH3 domain binding (Feng et al., 1995). The binding-independent component of Kv1.5 modulation is attributable to v-SrcWT catalytic activity, as shown by the equal suppression of Kv1.5 and Kv1.5-Δ-Pro by v-SrcWTSH3KO (Figs. 1, 2). Binding-dependent and phosphorylation-dependent suppression of Kv1.5 currents does not appear to be attributable to decreased Kv1.5 protein expression (Fig. 1B), consistent with previous studies of PTK modulation of mammalian Shaker-like channels (Huang et al., 1993; Holmes et al., 1996a,b; Fadool et al., 1997; Nitabach et al., 2001). Our results show that modulation of Kv1.5 by Src kinase exhibits two distinct modes: SH3 binding-dependent, phosphorylation-independent suppression and phosphorylation-dependent, SH3 binding-independent suppression.
Fig. 2.

Kv1.5 subunits are suppressed functionally by Src family protein tyrosine kinase via both a tyrosine phosphorylation-independent/SH3 domain binding-dependent mechanism and by a tyrosine phosphorylation-dependent/SH3 domain binding-independent mechanism. The bars depict normalized mean ± SEM peak currents for each oocyte that was recorded after reinjection with kinase cRNA; p < 0.001 for overall effect by ANOVA (n > 10 oocytes for each experimental condition). Paired comparisons were performed by using Bonferroni's multiple comparison test with experiment-wise p < 0.05. Significant differences for Kv1.5-wt: a versusb, a versus c,a versus d, b versusd, c versus d; significant differences for Kv1.5-Δ-Pro: e versusg, e versus h,f versus g, f versush.
Stable association of Src family PTK with Kv1.5 depends on direct interaction of the Src SH3 domain and the Kv1.5 proline-rich SH3 domain ligand sequence
In light of these findings regarding Src modulation of Kv1.5, we sought to determine biochemically whether the stable association of Kv1.5 with Src family PTKs depends on direct interaction between the Kv1.5 proline-rich SH3 domain ligand sequence and the Src SH3 domain. Src SH3–GST fusion protein was added to lysates of transfected cells, precipitated with glutathione-conjugated beads, and analyzed by Western blot to detect bound Kv1.5 proteins. As seen in Figure3, Src SH3 domain–GST fusion protein coprecipitates with Kv1.5 subunits expressed in HEK 293 cells. This association is abolished when the Kv1.5 proline-rich SH3 ligand sequence is deleted or when the Src SH3 domain is subject to the D99N point mutation (Fig. 3), thus confirming that the stable association between Kv1.5 and Src is mediated by an interaction between the Kv1.5 proline-rich sequence and the Src SH3 domain.
Fig. 3.

Kv1.5 subunits with intact Src family SH3 domain ligand sequence precipitate from lysates of transfected HEK 293 cells by purified bacterially expressed Src SH3 domains. Shown are Western blots of lysates (top panel) and wild-type Src SH3 domain (wt) and D99N point mutant Src SH3 domain (ko) precipitates (middle andbottom panels) from HEK 293 cells transfected with no channel (control), Kv1.5-wt, or Kv1.5-Δ-Pro. Starting lysate samples contained uniform amounts of Kv1.5 protein (top panel). Identical amounts of Src SH3 fusion protein were added to, and precipitated from, each precipitation reaction (bottom panel). Blots are representative of four experiments.
Kv1.4 subunits modified to contain the Kv1.5 proline-rich SH3 domain ligand sequence are expressed and post-translationally modified in HEK 293 cells
To determine whether the multimodal modulation we observed with Kv1.5 generalizes to other Shaker family Kv subunits, we examined the relative roles of SH3 domain-dependent Src binding and tyrosine phosphorylation in the modulation of rapidly inactivating Kv1.4 currents. Unlike Kv1.5, Kv1.4 contains no endogenous proline-rich SH3 domain ligand sequences. We created chimeric Kv1.4 subunits that contain the Kv1.5 proline-rich SH3 ligand sequence placed in either the N- or C-terminal cytoplasmic domain. The placement of the proline-rich SH3 domain binding sequences in Kv1.4 was guided by analysis of Kv channel sequence alignment, which shows that proline-rich sequences tend to be found in either of those domains (see Fig. 11). Although such chimeric subunits do not exist in nature, Kv1.4/Kv1.5 heteromultimers are found both in rat pituitary cells and in rabbit hippocampus, with the Kv1.4 subunits of such heteromultimers subject totrans-phosphorylation by stably associated Src family PTKs (Takimoto and Levitan, 1996; Nitabach et al., 2001). We use proline-rich sequence-inserted chimeras here as an experimental proxy for Kv1.4/Kv1.5 heteromultimers that allows us to dissociate the effects of Kv1.4 phosphorylation from the effects of Kv1.5 phosphorylation while still allowing for SH3 domain-dependent Src binding.
Fig. 11.

SH3 domain ligand proline-rich sequences and putative SH3 domain ligand proline-rich sequences cluster in the distal N- and C-terminal modulatory domain sequences of Kv channel subunits (shown in black). Modulatory domains also can contain phosphorylation sites, sequences mediating N-type inactivation, and sequences that inhibit N-type inactivation. Proline-rich sequences in Kv channel subunits were determined by MotifFinder, by CLUSTAL W alignments of Kv channel subunit protein sequences, and by visual inspection of Kv channel subunit protein sequences. In contrast, putative SH3 ligand proline-rich sequences are not found in other Kv channel subunit functional domains, including the ion conduction domain (S5–S6), the voltage sensor (S2–S4), or the channel assembly domain (T1–S1).
Kv1.4-N-Pro and Kv1.4-C-Pro proteins were generated by inserting amino acids 64–82 of Kv1.5 into the N or C terminus of Kv1.4. This proline-rich region of Kv1.5 contains two copies of the canonical Src family SH3 domain ligand RPLPPLP (Rickles et al., 1994; Yu et al., 1994). The Kv1.4-N-Pro insertion is located between the “ball-and-chain” N-terminal inactivation domain and the T1 tetramerization domain, and the Kv1.4-C-Pro insertion is located at the distal C terminus. Western blot analysis of α-Kv1.4 immunoprecipitations (IPs) of transfected HEK 293 cell lysates reveals that Kv1.4-N-Pro and Kv1.4-C-Pro, like wild-type Kv1.4 (Kv1.4-wt), exhibit two major immunoreactive bands (Figs.4A, top panel, 5A, top panel, 6A,top panel). Kv1.4-wt bands migrate with apparent molecular weights of 97 and 87 kDa. The corresponding bands of Kv1.4-N-Pro migrate slightly slower, and those of Kv1.4-C-Pro migrate slower still. The 23 amino acid insertion into Kv1.4-N-Pro and Kv1.4-C-Pro accounts for their slower migration than Kv1.4-wt. The slight difference in mobility between Kv1.4-N-Pro and Kv1.4-C-Pro could be attributable to a difference in SDS-resistant secondary structure, because the two chimeric proteins have identical molecular weights and polypeptide chain lengths. The slower and faster migrating Kv1.4 bands represent fully post-translationally processed highly glycosylated Kv1.4 polypeptide and immature unglycosylated Kv1.4 polypeptide, respectively (Li et al., 2000). Kv1.4-C-Pro always exhibits a greater ratio of the immature to the mature form compared with either Kv1.4-wt or Kv1.4-N-Pro. The Kv1.4-C-Pro insertion is not expected to disrupt the Kv1.4 C-terminal plasma membrane targeting signal (Li et al., 2000).
Fig. 4.

Kv1.4 subunits with inserted Src family SH3 domain ligand sequence (Kv1.4-N-Pro and Kv1.4-C-Pro) precipitate from lysates of transfected HEK 293 cells by purified bacterially expressed Src SH3 domains. A, α-Kv1.4 Western blots of lysates and Src SH3 domain precipitates from HEK 293 cells transfected with Kv1.4-wt, Kv1.4-N-Pro, or Kv1.4-C-Pro. Blots are representative of three experiments. B, α-Kv1.4 Western blot of wild-type Src SH3 domain (wt) and D99N point mutant Src SH3 domain (ko) precipitates from HEK 293 cells transfected with Kv1.4-wt, Kv1.4-N-Pro, or Kv1.4-C-Pro. Both short and long exposures are shown. Identical amounts of Src SH3 fusion protein were added to, and precipitated from, each precipitation reaction (data not shown). The blot is representative of four experiments.
Fig. 5.

Kv1.4 subunits with inserted Src family SH3 domain ligand sequence (Kv1.4-N-Pro and Kv1.4-C-Pro) stably associate with native c-Src and c-Fyn protein tyrosine kinases by a phosphorylation-independent mechanism in transfected HEK 293 cells. Shown are α-Kv1.4, α-Src, and α-Fyn Western blots of α-Kv1.4 immunoprecipitates from cells transfected with Kv1.4-wt, Kv1.4-N-Pro, or Kv1.4-C-Pro in the presence (+) or absence (−) of a 10 min treatment with 200 μm sodium pervanadate (PerV). c-Src (60 kDa) and c-Fyn (59 kDa) exhibit electrophoretic mobilities just greater than that of the immunoglobulin (IgG) used for immunoprecipitation. Blots are representative of five experiments.
Fig. 6.

Kv1.4 subunits with inserted Src family SH3 domain ligand sequence (Kv1.4-N-Pro and Kv1.4-C-Pro) exhibit greater basal and pervanadate-induced (PerV) tyrosine phosphorylation than wild-type Kv1.4 (Kv1.4-wt) in transfected HEK 293 cells. A, α-Kv1.4 and anti-phosphotyrosine (α-pY) Western blots of α-Kv1.4 immunoprecipitates from cells transfected with Kv1.4-wt, Kv1.4-N-Pro, or Kv1.4-C-Pro in the presence (+) or absence (−) of a 10 min treatment with 200 μm PerV. Both short and long exposures of the α-pY blot are shown. Long exposures permit the visualization of basal nonpervanadate-induced phosphorylation.B, α-pY staining density of α-Kv1.4 immunoprecipitates from PerV-treated cells as in A, normalized to α-Kv1.4 staining density for each sample. Short exposure blots were digitized. Normalized α-pY staining density of Kv1.4-wt in each experiment is defined as one relative density unit;n = 5 experiments. **p < 0.001 for overall effect by ANOVA; experiment-wise p < 0.05 for differences between Kv1.4-wt and each of Kv1.4-N-Pro and Kv1.4-C-Pro by Bonferroni's multiple comparison test; no significant difference between Kv1.4-N-Pro and Kv1.4-C-Pro.
Kv1.4 subunits modified to contain a proline-rich SH3 domain ligand sequence associate stably with bacterially expressed Src family PTK SH3 domain fusion proteins
To determine whether chimeric insertion of the Kv1.5 proline-rich Src SH3 ligand sequence into Kv1.4-N-Pro or Kv1.4-C-Pro confers interaction with the Src family PTK SH3 domain, we used bacterially expressed c-Src SH3 domain–GST fusion protein to precipitate associated proteins from lysates of transfected HEK 293 cells. Kv1.4-wt, which lacks native proline-rich sequences, does not exhibit any detectable association with the Src SH3 domain (Fig.4A, bottom panel). Kv1.4-N-Pro and Kv1.4-C-Pro, unlike Kv1.4-wt, robustly coprecipitate with Src SH3–GST (Fig. 4A, bottom panel). Thus the stable association of the chimeric subunits with the c-Src SH3 domain depends on the presence of the Kv1.5 proline-rich SH3 domain ligand sequence. This difference in coprecipitation between Kv1.4-wt and the chimeric subunits is not attributable to differences in the quantity of subunits present in the transfected cell lysates (Fig.4A, top panel).
To confirm the specificity of the SH3 domain-dependent interaction between Src SH3–GST and the chimeric Kv1.4 subunits, we introduced the D99N point mutation into the SH3 domain that decreases binding to proline-rich ligand sequences by 40- to 50-fold (Feng et al., 1995). This point mutation (SH3ko) dramatically reduces the coprecipitation of Kv1.4-N-Pro and Kv1.4-C-Pro subunits with GST–SH3ko (Fig. 4B). Although long exposure reveals the expected residual binding of SH3ko–GST to the chimeric subunits, there is no detectable association of either SH3–GST or SH3ko–GST to Kv1.4-wt (Fig.4B, bottom panel). Thus the specific interaction of the c-Src SH3 domain ligand binding site with the proline-rich ligand in the modified Kv1.4 subunits is the structural basis for protein–protein association.
Kv1.4 subunits modified to contain a proline-rich SH3 domain ligand sequence associate stably with native Src family PTKs
To determine whether chimeric insertion of the SH3 ligand sequence into Kv1.4-N-Pro and Kv1.4-C-Pro confers binding to native Src family PTKs, we analyzed α-Kv1.4 IPs from transfected HEK 293 cell lysates for the presence of coassociated native Src and Fyn. v-Src, c-Src, and c-Fyn SH3 domains exhibit identical or nearly identical binding specificity for proline-rich ligands (Rickles et al., 1994). Native HEK 293 cell Src family PTKs c-Src and c-Fyn are each detected in Western blots of α-Kv1.4 IPs from cells transfected with Kv1.4-N-Pro or Kv1.4-C-Pro, but not with Kv1.4-wt (Fig.5). The thick bands migrating slightly faster than c-Src and c-Fyn are the anti-Src or anti-Fyn rabbit IgGs used for immunoprecipitation. This band is visible, although mouse monoclonal antibodies were used for the Western blots because of the large relative quantity of immunoprecipitating IgG compared with the quantity of immunoprecipitated PTK. This shows that the Kv1.5 proline-rich Src family SH3 domain ligand sequence functions heterologously in chimeric Kv1.4 subunits to mediate stable Src family PTK association. c-Src and c-Fyn associate with chimeric Kv1.4 subunits either with or without a 10 min treatment with 250 μmsodium pervanadate [a cell-permeant specific protein tyrosine phosphatase (PTP) inhibitor] (Fig. 5), indicating that phosphotyrosine-dependent binding is not necessary for channel/Src family PTK association under these conditions.
Kv1.4 subunits modified to contain a proline-rich SH3 domain ligand sequence exhibit increased protein tyrosine phosphorylation by endogenous PTKs
We tested whether modular insertion of the proline-rich Src SH3 domain ligand sequence of Kv1.5 mediates increased tyrosine phosphorylation of SH3 ligand-modified Kv1.4 subunits by native PTKs. Cells were transfected to express Kv1.4-wt, Kv1.4-N-Pro, or Kv1.4-C-Pro. At 2 d after transfection the cells were treated for 10 min before lysis with 250 μm sodium pervanadate. Sodium pervanadate inhibits cellular PTPs, thus unmasking the relative activity of PTKs toward their substrates. α-Kv1.4 IPs from cell lysates were analyzed by Western blot with α-phosphotyrosine (α-pY) antibodies. The inhibition of HEK 293 cell native PTPs for 10 min leads to much greater accumulation of phosphotyrosine in Kv1.4-N-Pro and Kv1.4-C-Pro relative to Kv1.4-wt (Fig.6A, middle panel, B). The higher-molecular-weight fully processed forms of chimeric Kv1.4 are phosphorylated much more efficiently than the lower-molecular-weight forms (Fig. 6A), suggesting that c-Src- and c-Fyn-mediated phosphorylation occurs in channel/PTK complexes that form at the plasma membrane.
The increased tyrosine phosphorylation of Kv1.4-N-Pro and Kv1.4-C-Pro is not attributable to differences in the level of expression or efficiency of precipitation of these modified forms of Kv1.4 (Fig.6A, top panel). This shows that native HEK 293 cell PTKs phosphorylate Kv1.4-N-Pro and Kv1.4-C-Pro to a far greater extent than Kv1.4-wt. A ceiling of tyrosine phosphorylation is observed when the cells are treated with sodium pervanadate for 30 min before lysis. The accumulated phosphotyrosine ceiling levels are the same for Kv1.4-wt, Kv1.4-N-Pro, and Kv1.4-C-Pro (data not shown). Thus the differences in accumulated phosphotyrosine between Kv1.4-wt and the modified forms do not appear to be attributable to a greater capacity for maximal phosphotyrosine accumulation in the modified forms. Basal steady-state phosphotyrosine levels in the absence of sodium pervanadate treatment are greater for Kv1.4-N-Pro and Kv1.4-C-Pro than for Kv1.4-wt (Fig. 6A, bottom panel). This provides further support for the conclusion that native PTKs more rapidly phosphorylate chimeric Kv1.4-N-Pro and Kv1.4-C-Pro than they do Kv1.4-wt.
Src PTK physiologically modulates channels composed of Kv1.4 subunits modified to contain a proline-rich SH3 domain ligand sequence via multiple mechanisms
We examined the binding-dependent physiological regulation of Kv1.4-N-Pro and Kv1.4-C-Pro by Src family PTKs by using two-electrode voltage clamp of X. laevis oocytes expressing both a Kv1.4 subunit and a Src family PTK. We introduced the SH3 domain-inactivating D99N mutation into SrcCI, a catalytically impaired but still active point mutant (R385G) of v-Src (Nitabach et al., 2001). This allowed us to examine the dependence of physiological regulation on SH3 domain binding to the Kv1.5 proline-rich ligand sequence. An electrostatic interaction between the Src autophosphorylation site Y416 and R385 underlies the positive regulation of Src activity (Fig.7A) (Johnson et al., 1996;Hubbard, 1999). This activating interaction is disrupted by the R385G mutation of SrcCI, resulting in a form of v-Src that exhibits greatly decreased binding-independent substrate phosphorylation (Fig. 7B). In contrast, wild-type v-Src promiscuously phosphorylates target proteins in a binding-independent manner, as shown by the high levels of phosphorylation of cellular proteins catalyzed by v-SrcWT in comparison to the binding-deficient v-SrcWTSH2KO and v-SrcWTSH3KO point mutants. However, SrcCI retains sufficient catalytic activity for both autophosphorylation and the efficient phosphorylation of stably associated target substrates (Fig. 7B) (Nitabach et al., 2001). We developed SrcCI for the introduction of the D99N SH3-inactivating mutation, rather than using c-Src or c-Fyn, because disabling mutations of the c-Src (or c-Fyn) SH3 domain constitutively release the kinase catalytic domain from allosteric inhibition, thus allowing for nonphysiological promiscuous phosphorylation of cellular proteins (Seidel-Dugan et al., 1992; Okada et al., 1993; Xu et al., 1999). v-Src, c-Src, and c-Fyn have identical or nearly identical SH3 domain binding affinity for proline-rich ligands (Rickles et al., 1994) and strongly overlapping relative inherent catalytic specificity toward different tyrosine-containing substrate sequences (Seidel-Dugan et al., 1992; Stein et al., 1994).
Fig. 7.
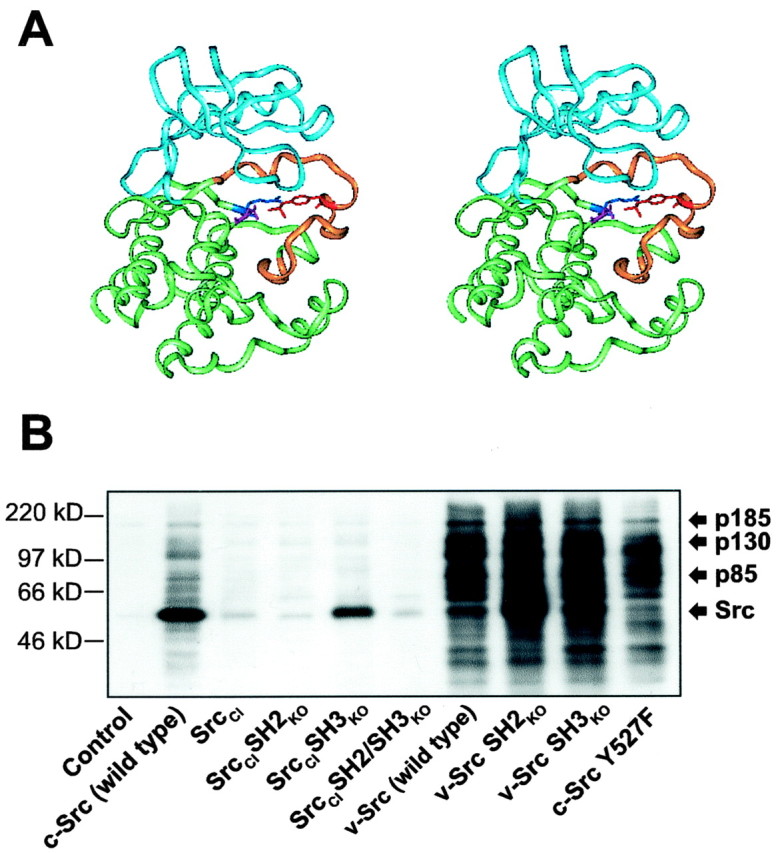
Structural basis for the design of SrcCI and tyrosine phosphorylation of cellular substrates by Src isoforms. A, Structural basis for the R385G mutation of SrcCI depicted in the rendering of the crystal structure of the Src PTK catalytic domain (Xu et al., 1999). The N- and C-terminal lobes are colored blue andgreen, respectively, and the activation segment isyellow. Electrostatic coordination of positively charged R385 (dark blue) of the catalytic loop with negatively charged autophosphorylated Y416 (red) of the activation segment orients the catalytic residue D386 (magenta) for efficient nucleophilic attack on the tyrosine-containing substrate (data not shown). The R385G mutation disrupts the coordination of the activation segment and the catalytic loop. Additional rotational flexibility of the D386 catalytic residue by the substitution of glycine for the adjacent arginine could lower kinase activity further (Xu et al., 1999). B, α-pY Western blot of lysates from HEK 293 cells transfected with various wild-type and mutated isoforms of c-Src, v-Src, and SrcCI. SrcCIexhibits greatly reduced promiscuous binding-independent phosphorylation of cellular targets compared with both c-Src and v-Src while still detectably phosphorylating itself and the physiological Src substrates p85, p130, and p185. The Y527F mutation, which disables the intramolecular interaction of the Src SH2 and SH3 domains with the Src C-terminal domain, increases the promiscuous phosphorylation of cellular targets by c-Src. In contrast, SrcCISH3KO (SH3-disabled SrcCI) exhibits no increase in promiscuous phosphorylation of cellular targets in comparison to unmodified SH3-intact SrcCI, whereas autophosphorylation increases, possibly because of further relaxation of inhibitory intramolecular interactions.
Kv1.4-wt voltage-evoked peak currents are unaffected by v-SrcKD or SrcCI (Figs. 8, left column,9). However, introduction of the Kv1.5 proline-rich SH3 ligand sequence into either the N or C terminus of Kv1.4 confers sensitivity to Src-mediated physiological suppression (Figs. 8, middle and right columns, 9). A similar degree of suppression of voltage-evoked peak currents is observed with either v-SrcKD or SrcCI, indicating that this modulation can be independent of Src-mediated tyrosine phosphorylation of the Kv1.4 subunits. Physiological suppression of chimeric Kv1.4 voltage-evoked currents is abolished when SrcCI is replaced with SrcCISH3KO (Figs. 8,middle and right columns, 9). Thus, as we observed with Kv1.5, there is a mode of modulation of Kv1.4 that depends only on the SH3 domain-dependent binding of Src to the channel, and that is independent of Src-catalyzed tyrosine phosphorylation.
Fig. 8.

Phosphorylation-independent and phosphorylation-dependent modes of modulation of voltage-evoked Kv1.4 currents by Src family protein tyrosine kinase are distinguished by SH3 binding dependence. Peak voltage-gated currents evoked in Kv subunit cRNA-injected X. laevis oocytes by a 1 sec step to +80 mV from a holding potential of –80 mV were measured 9–12 hr after a second injection with H2O, kinase-dead v-Src (v-SrcKD) cRNA, catalytically impaired SrcCI cRNA, or cRNA encoding SrcCI with an inactivated SH3 domain (SrcCISH3ko). The traces that are depicted are averages of the currents measured after the second injection (n > 11 oocytes for each experimental condition). Kv1.4 currents are not modulated significantly by either v-SrcKD or SrcCI, whereas Kv1.4-N-Pro and Kv1.4-C-Pro currents are modulated significantly by both v-SrcKD and SrcCI (Fig. 9). All traces for each channel type are normalized to the peak average current for the water-injected group: Kv1.4-wt = 19.6 μA, Kv1.4-N-Pro = 9.3 μA, and Kv1.4-C-Pro = 13.9 μA.
Fig. 9.

Voltage-gated peak currents evoked from oocytes expressing Kv1.4 subunits with the inserted Src family SH3 domain ligand sequence (Kv1.4-N-Pro and Kv1.4-C-Pro) are suppressed by Src family protein tyrosine kinase via a tyrosine phosphorylation-independent mechanism, whereas Kv1.4-wt is not modulated. The bars depict normalized mean ± SEM peak currents for each oocyte that was recorded after reinjection with kinase cRNA.p < 0.001 for overall effect by ANOVA (n > 11 oocytes for each experimental condition). Paired comparisons were performed with Bonferroni's multiple comparison test with experiment-wise p < 0.05. Significant differences for Kv1.4-wt: none; significant differences for Kv1.4-N-Pro: d versus e, dversus f, e versus g,f versus g; significant differences for Kv1.4-C-Pro: h versus i, hversus j, i versus k,j versus k.
The suppression of Kv1.4 macroscopic currents depends on SH3-mediated Src binding to the chimeric channel subunit, and not on the catalytic activity of the bound Src. However, modulation of Kv1.4 inactivation kinetics does depend on the catalytic activity of the bound Src. As seen in Figure 8 (middle and right columns) and quantified in Figure 10, it is only the binding of the catalytically impaired, but still active, SrcCI that leads to substantial increases in the time constant of rapid N-type inactivation. These results show that SH3-dependent Src-mediated physiological modulation can be conferred on an otherwise insensitive Kv channel subunit via chimeric introduction of a proline-rich Src SH3 domain ligand sequence. In addition, they reveal two distinct binding-dependent mechanisms for Src modulation: the pho-sphorylation-independent suppression of channel current and the phosphorylation-dependent increase in the time constant of inactivation.
Fig. 10.

Inactivation kinetics of Kv1.4 subunits with inserted Src family SH3 domain ligand sequence (Kv1.4-N-Pro and Kv1.4-C-Pro) are slowed by Src-mediated tyrosine phosphorylation. Inactivation time constants derived from fits to a first-order exponential decay function for each oocyte that was recorded after reinjection are depicted as bars (mean ± SEM).p < 0.001 for overall effect by ANOVA (n > 11 oocytes for each experimental condition). Paired comparisons were performed with Bonferroni's multiple comparison test with experiment-wise p < 0.05. Significant differences for Kv1.4-wt: none; significant differences for Kv1.4-N-Pro: d versus f, eversus f, g versus f; significant differences for Kv1.4-C-Pro: h versusj, i versus j,k versus j.
DISCUSSION
Our results demonstrate that channels composed of theShaker family subunits Kv1.4 and Kv1.5 are subject to at least two distinct modes of modulation by Src family PTKs. Surprisingly, SH3 domain-dependent binding of the Src kinase in the absence of catalytic activity is sufficient in itself to suppress the Kv1.4 and Kv1.5 voltage-evoked currents substantially by a phosphorylation-independent mechanism. In turn, Src catalytic activity in the absence of SH3 domain-dependent binding is sufficient for the suppression of Kv1.5 currents by a phosphorylation-dependent mechanism. Furthermore, in the context of SH3 domain-dependent binding, Src catalytic activity induces an increase in the time constant of inactivation of Kv1.4 currents. These results point to a departure from the conventional view that ion channel modulation by protein kinases is mediated wholly by the covalent modification of the channel subunits by the catalytic activity of the enzyme. It is now apparent that Src family PTKs can suppress greatly the macroscopic Shakerfamily Kv currents solely via SH3 domain-dependent binding to the channel.
Although the mechanism for this modulation remains to be elucidated, there are at least two main possibilities. First, binding-dependent/phosphorylation-independent modulation might be mediated by alterations in intracellular processing and/or trafficking of the Kv subunits, as has been demonstrated for the tyrosine kinase-dependent modulation of inward rectifier K+ channels (Tong et al., 2001). Because the dramatic current suppression we observe is caused by SH3 domain-dependent binding of Src that first is expressed when channel currents in control oocytes already have reached near-peak levels, it is possible that the suppression is attributable to increased degradation and/or internalization of already-expressed channels rather than to the suppression of assembly or membrane insertion. However, we observe no apparent decrease in Kv1.5 protein levels when Kv1.5 is coexpressed with kinase-dead v-SrcKD. Second, modulation might be mediated by direct allosteric modification of channel biophysical properties, as has been suggested by previous work on the Src modulation of Kv channels (Fadool et al., 1997). Assessment of the relative roles of these two modulatory mechanisms will require further cell biological analysis and single-channel recordings.
We demonstrate here a direct, noncatalytic role of protein kinase channel binding in ion channel modulation. A noncatalytic modulatory interaction between a PTK and a cyclic nucleotide-gated channel has been observed, although the involvement of direct binding of the PTK to the channel is still unclear (Molokanova et al., 1999). A direct, noncatalytic, protein–protein interaction-based role for G-protein binding to ion channels has been known for quite some time (Holz et al., 1986; Logothetis et al., 1987; Ma et al., 1997; Jing et al., 1999) (for review, see Zamponi and Snutch, 1998; Mark and Herlitze, 2000). Although G-protein βγ heterodimers activate an inward rectifier potassium channel via direct binding to the cytoplasmic N and C termini, G-protein α-subunits are thought to terminate that effect (Mark and Herlitze, 2000). Similarly, it is believed that G-protein βγ subunits inhibit some classes of neuronal voltage-gated calcium channels via direct binding to the cytoplasmic linker between domains I and II of the pore-forming α-subunit and that the bound G-protein βγ allosterically hinders the channel from opening (Zamponi and Snutch, 1998). In the case of voltage-gated sodium channels, it has been observed that direct binding of G-protein βγ to the C-terminal domain stabilizes a gating mode that underlies a persistent current (Ma et al., 1997). By analogy to these results, it is possible that SH3-mediated Src binding to the N- or C-terminal cytoplasmic domains ofShaker family Kv subunits allosterically constrains the channel in a stable closed or inactive state.
There is at least some degree of positional independence within a Kv subunit of the modulatory functions of a Src SH3 domain ligand sequence. We observe a similar degree of Src-induced physiological modulation (either phosphorylation-independent current suppression or phosphorylation-dependent slowing of inactivation) regardless whether the 2xRPLPPLP sequence is introduced in the N- or C-terminal cytoplasmic domain of Kv1.4. This positional independence for Kv1.4 current modulation indicates that there are not likely to be essential steric constraints on the site of Src PTK binding in comparison to the sites of tyrosine phosphorylation or allosteric effects necessary for channel modulation. Because the crystallographic structure of the distal N- or C-terminal domains of a Kv channel have yet to be obtained, one can only speculate about the reasons for this. One possibility is that the N and C termini are flexible and thus allow the bound Src kinase relatively free movement to catalyze phosphorylation of tyrosines or otherwise interact with amino acid residues regardless of their locations in the subunit. Sequence alignment indicates that proline-rich SH3 domain ligand sequences do tend to cluster in the distal N and C termini of Kv channels (Fig.11), suggesting that SH3 domain binding-dependent mechanisms may be conserved.
In addition to the phosphorylation-independent current suppression we observe, Src also modulates Kv currents via the phosphorylation of the channel subunits, causing current suppression of Kv1.5 and slowed inactivation of Kv1.4. This difference in the biophysical effects of phosphorylation on Kv1.4 and Kv1.5 is potentially attributable to structural differences between the channels; it is only Kv1.4 that contains an N-terminal ball-and-chain domain and thus only Kv1.4 that undergoes modulation of N-type inactivation. Indeed, previous studies have addressed the effects of serine/threonine phosphorylation of the N-terminal domain on Kv1.4 physiological properties. Calcium/calmodulin-dependent protein kinase has been shown to slow the inactivation of Kv1.4 currents by phosphorylating S123 in the cytoplasmic N-terminal (Roeper et al., 1997). Treatment of Kv1.4-expressing Xenopus oocytes with phorbol 12-myristate 13-acetate, a protein kinase C activator, has been shown to lead to a biphasic change in the magnitude of peak current: an initial increase in peak current was followed by a later reduction (Murray et al., 1994). Our preliminary results indicate that the increase in the time constant of inactivation caused by Src does not depend on the phosphorylation of S123 (M. N. Nitabach and T. C. Holmes, unpublished results). The possible functional interaction between serine/threonine and tyrosine phosphorylation of Kv1.4 is an interesting question but remains to be examined further, as do the tyrosine phosphorylation sites in Kv1.4 that underlie the modulation of inactivation kinetics. The multiple modes of modulation of ion channels by a single signaling enzyme that we describe here could underlie differential modulation of native channels contingent on the presence or absence of coassembled subunits containing proline-rich SH3 domain binding sequences.
Footnotes
This research was supported by the New York University Whitehead Fellowship for Junior Faculty in Biomedical or Biological Sciences, the New York University Research Challenge Fund, and the National Science Foundation (IBN-0092753). M.N.N. was supported in part by a National Institutes of Health National Research Service Award. We thank M. Sheng, L. Philipson, R. Huganir, and D. Turner for the provision of reagents and S. Broyde and S. Roy for assistance with the rendering of the Src catalytic domain.
Correspondence should be addressed to Todd C. Holmes, Department of Biology, New York University, 1009 Main Building, 100 Washington Square East, New York, NY 10003. E-mail: todd.holmes@nyu.edu.
REFERENCES
- 1.Bowlby MR, Fadool DA, Holmes TC, Levitan IB. Modulation of the Kv1.3 potassium channel by receptor tyrosine kinases. J Gen Physiol. 1997;110:601–610. doi: 10.1085/jgp.110.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cayabyab FS, Khanna R, Jones OT, Schlichter LC. Suppression of the rat microglia Kv1.3 current by Src family tyrosine kinases and oxygen/glucose deprivation. Eur J Neurosci. 2000;12:1949–1960. doi: 10.1046/j.1460-9568.2000.00083.x. [DOI] [PubMed] [Google Scholar]
- 3.Chung SK, Reinhart PH, Martin BL, Brautigan D, Levitan IB. Protein kinase activity closely associated with a reconstituted calcium-activated potassium channel. Science. 1991;253:560–562. doi: 10.1126/science.1857986. [DOI] [PubMed] [Google Scholar]
- 4.Cook KK, Fadool DA. Two adaptor proteins differentially modulate the phosphorylation and biophysics of Kv1.3 ion channel by Src kinase. J Biol Chem. 2002;277:13268–13280. doi: 10.1074/jbc.M108898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebina Y, Araki E, Taira M, Shimada F, Mori M, Craik CS, Sidd K, Pierce SB, Roth RA, Rutter WJ. Replacement of lysine residue 1030 in the putative ATP-binding region of the insulin receptor abolishes insulin- and antibody-stimulated glucose uptake and receptor kinase activity. Proc Natl Acad Sci USA. 1987;84:704–708. doi: 10.1073/pnas.84.3.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fadool DA, Levitan IB. Modulation of olfactory bulb neuron potassium current by tyrosine phosphorylation. J Neurosci. 1998;18:6126–6137. doi: 10.1523/JNEUROSCI.18-16-06126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fadool DA, Holmes TC, Berman K, Dagan D, Levitan IB. Tyrosine phosphorylation modulates current amplitude and kinetics of a neuronal voltage-gated potassium channel. J Neurophysiol. 1997;78:1563–1573. doi: 10.1152/jn.1997.78.3.1563. [DOI] [PubMed] [Google Scholar]
- 8.Feng S, Kasahara C, Rickles RJ, Schreiber SL. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc Natl Acad Sci USA. 1995;92:12408–12415. doi: 10.1073/pnas.92.26.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldin AL. Maintenance of Xenopus laevis and oocyte injection. Methods Enzymol. 1992;207:266–279. doi: 10.1016/0076-6879(92)07017-i. [DOI] [PubMed] [Google Scholar]
- 10.He C, Yan X, Zhang H, Mirshahi T, Jin T, Huang A, Logothetis DE. Identification of critical residues controlling G-protein-gated inwardly rectifying K+ channel activity through interactions with the βγ subunits of G-proteins. J Biol Chem. 2002;277:6088–6096. doi: 10.1074/jbc.M104851200. [DOI] [PubMed] [Google Scholar]
- 11.Holmes TC, Fadool DA, Ren R, Levitan IB. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science. 1996a;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- 12.Holmes TC, Fadool DA, Levitan IB. Tyrosine phosphorylation of the Kv1.3 potassium channel. J Neurosci. 1996b;16:1581–1590. doi: 10.1523/JNEUROSCI.16-05-01581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmes TC, Berman K, Swartz JS, Dagan D, Levitan IB. Expression of voltage-gated potassium channels decreases cellular protein tyrosine phosphorylation. J Neurosci. 1997;17:8964–8974. doi: 10.1523/JNEUROSCI.17-23-08964.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmes TC, Marquez J, Levitan IB. Fyn tyrosine kinase associates with human Kv1.5 potassium channel by an SH3 domain interaction. Soc Neurosci Abstr. 1998;24:1332. [Google Scholar]
- 15.Holz GG, Rane SG, Dunlap K. GTP-binding proteins mediate transmitter inhibition of voltage-dependent calcium channels. Nature. 1986;319:670–672. doi: 10.1038/319670a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang XY, Morielli AD, Peralta EG. Tyrosine kinase-dependent suppression of a potassium channel by the G-protein-coupled m1 muscarinic acetylcholine receptor. Cell. 1993;75:1145–1156. doi: 10.1016/0092-8674(93)90324-j. [DOI] [PubMed] [Google Scholar]
- 17.Hubbard SR. Src autoinhibition: let us count the ways. Nat Struct Biol. 1999;6:711–714. doi: 10.1038/11468. [DOI] [PubMed] [Google Scholar]
- 18.Jing J, Chikvashvilli D, Singer-Lahat D, Thornhill WB, Reuveny E, Lotan I. Fast inactivation of a brain K+ channel composed of Kv1.1 and Kvβ1.1 subunits modulated by G-protein βγ subunits. EMBO J. 1999;18:1245–1256. doi: 10.1093/emboj/18.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 20.Kamps MP, Sefton BM. Neither arginine nor histidine can carry out the function of lysine-295 in the ATP-binding site of p60src. Mol Cell Biol. 1986;6:751–757. doi: 10.1128/mcb.6.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanemitsu MY, Loo LW, Simon S, Lau AF, Eckhart W. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J Biol Chem. 1997;272:22824–22831. doi: 10.1074/jbc.272.36.22824. [DOI] [PubMed] [Google Scholar]
- 22.Li D, Takimoto K, Levitan ES. Surface expression of Kv1 channels is governed by a C-terminal motif. J Biol Chem. 2000;275:11597–11602. doi: 10.1074/jbc.275.16.11597. [DOI] [PubMed] [Google Scholar]
- 23.Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 24.Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G-protein βγ subunits. Neuron. 1997;19:443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 25.Mark MD, Herlitze S. G-protein-mediated gating of inward-rectifier K+ channels. Eur J Biochem. 2000;267:5830–5836. doi: 10.1046/j.1432-1327.2000.01670.x. [DOI] [PubMed] [Google Scholar]
- 26.Maximov A, Sudhof TC, Bezprozvanny I. Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem. 1999;274:24453–24456. doi: 10.1074/jbc.274.35.24453. [DOI] [PubMed] [Google Scholar]
- 27.Molokanova E, Savchenko A, Kramer RH. Noncatalytic inhibition of cyclic nucleotide-gated channels by tyrosine kinase induced by genistein. J Gen Physiol. 1999;113:45–56. doi: 10.1085/jgp.113.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray KT, Fahrig SA, Deal KK, Po SS, Hu NN, Snyders DJ, Tamkun MM, Bennett PB. Modulation of an inactivating human cardiac K+ channel by protein kinase C. Circ Res. 1994;75:999–1005. doi: 10.1161/01.res.75.6.999. [DOI] [PubMed] [Google Scholar]
- 29.Nitabach MN, Llamas DA, Araneda RC, Intile JL, Thompson IJ, Zhou YI, Holmes TC. A mechanism for combinatorial regulation of electrical activity: potassium channel subunits capable of functioning as SH3-dependent adaptors. Proc Natl Acad Sci USA. 2001;98:705–710. doi: 10.1073/pnas.031446198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada M, Howell BW, Broome MA, Cooper JA. Deletion of the SH3 domain of Src interferes with regulation by the phosphorylated carboxyl-terminal tyrosine. J Biol Chem. 1993;268:18070–18075. [PubMed] [Google Scholar]
- 31.Rickles RJ, Botfield MC, Weng Z, Taylor JA, Green OM, Brugge JS, Zoller MJ. Identification of Src, Fyn, Lyn, PI3K, and Abl SH3 domain ligands using phage display libraries. EMBO J. 1994;13:5598–5604. doi: 10.1002/j.1460-2075.1994.tb06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roeper J, Christoph L, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel Kv1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santoro B, Grant SGN, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-Src identifies a new brain-specific ion channel protein, with homology to Eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 1997;94:14815–14820. doi: 10.1073/pnas.94.26.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidel-Dugan C, Meyer BE, Thomas SM, Brugge JS. Effects of SH2 and SH3 deletions on the functional activities of wild-type and transforming variants of c-Src. Mol Cell Biol. 1992;12:1835–1845. doi: 10.1128/mcb.12.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snyder MA, Bishop JM, McGrath JP, Levinson AD. A mutation at the ATP-binding site of pp60v-Src abolishes kinase activity, transformation, and tumorigenicity. Mol Cell Biol. 1985;5:1772–1779. doi: 10.1128/mcb.5.7.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sobko A, Peretz A, Attali B. Constitutive activation of delayed-rectifier potassium channels by a Src family tyrosine kinase in Schwann cells. EMBO J. 1998;17:4723–4734. doi: 10.1093/emboj/17.16.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stein PL, Vogel H, Soriano P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994;8:1999–2007. doi: 10.1101/gad.8.17.1999. [DOI] [PubMed] [Google Scholar]
- 38.Szabo I, Gulbins E, Apfel H, Zhang X, Barth P, Busch AE, Schlottmann K, Pongs O, Lang F. Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J Biol Chem. 1996;271:20465–20469. doi: 10.1074/jbc.271.34.20465. [DOI] [PubMed] [Google Scholar]
- 39.Takimoto K, Levitan ES. Altered K+ channel subunit composition following hormone induction of Kv1.5 gene expression. Biochemistry. 1996;35:14149–14156. doi: 10.1021/bi961290s. [DOI] [PubMed] [Google Scholar]
- 40.Tong Y, Brandt GS, Li M, Shapovalov G, Slimko E, Karschin A, Dougherty DA, Lester HA. Tyrosine decaging leads to substantial membrane trafficking during modulation of an inward rectifier potassium channel. J Gen Physiol. 2001;117:103–118. doi: 10.1085/jgp.117.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 42.Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 43.Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G-proteins. Curr Opin Neurobiol. 1998;8:351–356. doi: 10.1016/s0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]