

Abstract
μ-Opioid receptor (MOR) desensitization and endocytosis have been implicated in tolerance and dependence to opioids. The efficiency of each process is known to be agonist dependent; however, it is not known what determines the relative efficiency of various agonists at either process. In the present study, homologous MOR desensitization in locus ceruleus (LC) neurons and MOR internalization in HEK293 cells were examined using a series of agonists. The results show that the rank order of this series of agonists was different when comparing the magnitude of hyperpolarization and the ability to cause desensitization in LC neurons. Endocytosis of MOR was also examined in HEK293 cells using the same agonists. The relative ability to cause endocytosis in HEK293 cells correlated with the degree of desensitization in LC cells. This strong correlation suggests that the two processes are closely linked. The results also suggest that agonist efficacy is not necessarily a predictor of the ability to cause MOR desensitization or endocytosis. Identification and characterization of the biophysical properties of agonists that favor desensitization and internalization of receptors will lead to a better understanding of opioid signaling.
Keywords: opioids, locus ceruleus, homologous desensitization, endocytosis, electrophysiology, imaging
Desensitization and internalization of μ-opioid receptor (MOR) are two regulatory mechanisms thought to contribute to the development of tolerance to opioids (Whistler et al., 1999; Williams et al., 2001). Morphine, the most widely used analgesic, is well known to produce tolerance, although it promotes neither rapid desensitization nor receptor endocytosis when studied in heterologous expression systems or native preparations in several regions of brain and spinal cord (Keith et al., 1996; Sternini et al., 1996; Trafton et al., 2000). In contrast, endogenous opioid peptides and certain nonpeptide agonists (such as etorphine and methadone) promote both rapid desensitization and induce internalization. These observations suggest that there are important differences in the effects of specific agonists on the physiological regulation of opioid receptors in neurons.
Like other G-protein-coupled receptors, activation of MOR by an agonist can result in receptor phosphorylation mediated by G-protein receptor kinases (GRKs). β-Arrestins bind to the phosphorylated receptor, and this complex is unable to couple to G-proteins and to activate downstream effectors, resulting in receptor desensitization (Ferguson, 2001). This receptor complex is recruited to the clathrin-coated pit and then removed from the plasma membrane by a multistep process known as endocytosis. Although endocytosis has been thought to contribute directly to functional desensitization of opioid signaling by rapidly reducing the number of receptors present at the cell surface (Law et al., 2000), it has been proposed that endocytosis also mediates receptor “resensitization.” This process involves delivering receptors to an endosome-associated phosphatase and then returning the dephosphorylated receptors to the plasma membrane via a rapid recycling pathway (Koch et al., 1998) (for review, see Lefkowitz, 1998). Indeed, cloned μ-opioid receptors (MOR1 and MOR1B) predominantly recycle to the plasma membrane of transfected cells after endocytosis and traffic to lysosomes at a much slower rate (Koch et al., 1998; Law et al., 2000; Sternini et al., 2000; Finn and Whistler, 2001).
Opioid agonists differ in coupling efficacy to various effectors i.e., G-protein-coupled inwardly rectified potassium channels (GIRK), voltage-gated calcium channels, adenylyl cyclase, as well as GRKs, and these differences appear to depend on the preparation studied. Efficacy was first defined by the ability of an agonist to activate these primary effectors. Desensitization and receptor internalization mediated by GRKs was considered secondary or a follow-on consequence of receptor activation. It has become clear that these regulatory processes may also depend on agonist properties (Kenakin, 2002). The question that arises is whether the activation of effectors and the regulation of receptors (desensitization and endocytosis) are related or are independent responses.
In the present study, a series of opioid agonists were examined for their ability to activate potassium currents, cause homologous desensitization, and induce receptor internalization. The results showed that the degree of desensitization of MOR signaling observed in locus ceruleus (LC) neurons correlates well with that of agonist-induced MOR endocytosis assessed in HEK293 cells.
MATERIALS AND METHODS
Electrophysiological recordings. Horizontal slices (225–250 μm) containing the LC were prepared from 6- to 8-week-old male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) as described previously (Ishimatsu and Williams, 1996). Extracellular solution contained (in mm):126 NaCl, 2.5 KCl, 2.4 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 glucose (equilibrated with 95%O2–5%CO2 at 35°C). Whole-cell recordings were made using Nomarski optics and infrared illumination. Recordings were made with an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA) in current-clamp mode with pipettes (2–3 MΩ) filled with internal solution containing (in mm): 115 MES potassium salt, 20 KCl, 1.5 MgCl2, 1 BAPTA, 5 HEPES, 4 Mg-ATP, and 0.4 Na-GTP, pH 7.3. For intracellular recordings, pipettes (30–50 MΩ) were filled with 2 m KCl, and recordings were made with an Axoclamp 2A (Axon Instruments). Data was acquired at 100 Hz (Chart version 4.0, MacLab System; DInstruments, Castle Hill, Australia) and analyzed in Axograph (version 4.5; Axon Instruments) when necessary.
All of the experiments were performed at 35°C, and, unless otherwise stated, pharmacological agents were applied by bath perfusion. All experiments with [Met]5enkephalin (ME) were done in the presence of the peptidase inhibitors bestatin (10 μm) and thiorphan (1 μm). In some cases, fluorescent agonists were applied at high concentrations using pressure ejection from a patch pipette placed in the slice within 50 μm from the recording site.
Desensitization protocols. Three different protocols were used to measure MOR desensitization of the hyperpolarization response in LC neurons (Fig. 1). One method compared the amplitude of the hyperpolarization produced by an EC50 concentration of ME (300 nm) before and after exposure to a high desensitizing concentration of ME (Harris and Williams, 1991) (Fig.1A). The amplitude of the opioid response was calculated as the average value of the membrane potential during the opioid perfusion minus the average resting membrane potential before the application. This protocol was particularly good for studying desensitization induced by ME because it cleared rapidly from the preparation after washout so that an EC50concentration of ME could be tested within 2–3 min. To examine desensitization of other opioid agonists, including dermorphin-Bodipy Texas Red (DERM-BTR), a new protocol that did not require rapid agonist washout was used (Fig. 1B). Previous work has shown that, after MOR desensitization, the maximal amplitude of the opioid response was depressed (Osborne and Williams, 1995). This protocol takes advantage of that observation and measures the maximal response to ME (10 μm) at various times during a prolonged application of other agonists applied at submaximal concentrations. The application of ME (10 μm) was limited to 1.5 min with a 5 min washout, to reduce the induction of desensitization by the ME applications alone. Decreases in the amplitude of the maximal ME response produced by a prolonged application (25 min) of the test agonist were taken as a measure of desensitization. At the end of the agonist treatment, naloxone (1 μm) was used to reverse the opioid-induced hyperpolarization. Once the potential returned to baseline, a maximal concentration of the α2-adrenoreceptor agonist UK14304 (3 μm) was tested to control for both rundown of the signal pathway during the experiment and heterologous desensitization. This method was used for measuring desensitization induced by agonists that did not wash from the preparation. A third method measured the decline in the amplitude of the hyperpolarization induced by a high agonist concentration (Fig. 1C). This decline was taken as a sign of desensitization. At the end of each experiment, a maximal concentration of the α2-adrenoreceptor agonist UK14304 (3 μm) was applied. The hyperpolarization induced by UK14304 was reversed by superfusion with the α2-adenoceptor antagonist yohimbine (1 μm).
Fig. 1.

Protocols used in recordings from LC neurons to measure desensitization. A, An EC50concentration of ME (300 nm) was used as a test before (1) and after (2) treating the tissue with a maximal concentration of agonist. The decrease in amplitude of the hyperpolarization immediately after washout was taken as a measure of desensitization. Over time, the amplitude of the hyperpolarization in response to the EC50 test returned toward control values. B, The hyperpolarization induced by a maximal concentration of ME (10 μm) was tested before (1) and during (2) the continuous superfusion with an EC50 concentration of opioid agonists. The decline in the peak hyperpolarization (3) was taken as a sign of desensitization. The hyperpolarization induced by the EC50 concentration of agonist was reversed with naloxone (1 μm). The hyperpolarization induced by a maximal concentration of UK14304 (3 μm) was determined for each experiment. C, A maximal concentration of opioid agonist was applied, and the difference between the peak hyperpolarization (1) and the amplitude of the hyperpolarization after 15 min (2) was taken as a sign of desensitization. In each experiment, naloxone was used to reverse the opioid-induced hyperpolarization, and the hyperpolarization induced by UK14304 was determined.
MOR endocytosis. MOR endocytosis was determined by feeding experiments as described previously (Finn and Whistler, 2001). Human embryonic kidney 293 (HEK293) cells stably expressing flag-tagged MOR (flag-MOR) were grown in DMEM supplemented with 10% fetal bovine serum and propagated in the presence of 700 μg/ml G418 (all tissue culture reagents were purchased from Invitrogen, Gaithersburg, MD). For the experiments, cells were grown on poly-lysine-coated coverslips (Fisher Scientific) to ∼60% confluence. They were incubated with 1:500 dilution of M1 mouse anti-FLAG antibody (Sigma, St. Louis, MO) for 5 min at 37°C and then with agonists. At the end of incubation period, the antibody was stripped off by a brief 1 min rinse at 37°C with a hypertonic acid solution (DMEM adjusted to pH 4 with acetic acid plus 0.5 m NaCl plus 5 mm EDTA). This treatment disrupted the extracellular calcium-sensitive M1–epitope interaction and the residual membrane-bound agonist. Cells were fixed in 4% paraformaldehyde in PBS, permeabilized in 0.1% Triton X-100, and stained with AlexaFlour 488 goat anti-mouse antibody (Molecular Probes, Eugene, OR) or Cy5 goat anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA). Coverslips were mounted in Mowiol (Sigma).
Image acquisition and analyses. Images were acquired under a confocal microscope equipped with a krypton–argon laser coupled with a Bio-Rad (Hercules, CA) MRC-1000 and an Optiphot II Nikon (Tokyo, Japan) microscope. Cells were visualized under a Plan Apo 60× objective lens (1.4 numerical aperture, oil). The filters used for scanning Alexa488 were 488 nm for excitation and 522 nm for emission and, for Cy5, were 647 nm for excitation and 680 nm for emission. Pictures were taken from four to five fields from each coverslip and analyzed by Scion Image for Windows software (version Beta 4.0.2; Scion, Frederick, MD). The fluorescence intensity of ∼50 individual cells (10 cells per field) was determined for each coverslip, and one mean fluorescence intensity value was obtained. For each condition, a duplicate in one experiment and three to five separate experiments were performed.
Statistics and curve fitting. Statistical difference was determined by unpaired t test unless specified, and the two-tailed p values are presented in the text or figure legends. Statistical analysis, curve fits, and correlations were performed with GraphPad Prism (GraphPad Software, San Diego, CA).
Materials. [Met]5enkephalin, dermorphin, bestatin, and yohimbine were obtained from Sigma. Naloxone and UK14304 were obtained from Research Biochemicals (Natick, MA). Thiorphan was from Bachem (Torrance, CA). Dermorphin-BTR and dermorphin-A488 were prepared as described by Arttamangkul et al., 2000. Morphine, methadone, normorphine, and etorphine were obtained from the National Institute on Drug Abuse.
RESULTS
MOR desensitization
The response to an EC50 concentration of ME (300 nm) was significantly reduced after treatment with a high concentration of ME (30 μm) (Figs.1A, for protocol, 2A, top trace). The amplitude of the EC50 response measured 10 min after washout of the high concentration of ME (MEpost) was approximately half of the initial EC50 response value (MEpost/MEpre = 0.46 ± 0.05; n = 4) (Fig. 2). The amplitude of the MEpost response recovered over a period of 40 min in accordance with previous reports (Harris and Williams, 1991). The same amount of desensitization and similar kinetics of recovery were obtained with both intracellular and whole-cell recording configurations. Thus, the recording conditions did not apparently interfere with the rapid desensitization process.
Fig. 2.

Induction of and recovery from desensitization. In this and other figures, the recordings are of membrane potential. In some recordings, the presence of spontaneous oscillations in membrane potential resulted in “noise” in the trace. The oscillations and thus the noise was abolished in the presence of opioid agonists.A, An EC50 concentration of ME (300 nm) was tested every 5 min before and after the application of a maximal concentration of ME (30 μm; top trace) and DERM-A488 (1 μm; bottom trace; applied by pressure ejection). The amplitude of the EC50 MOR response was significantly decreased after the desensitizing treatment with ME (top trace) but only slightly reduced after DERM-A488 (bottom trace). Desensitization recovered over 30–40 min. B, Summarized data from desensitization experiments done with whole-cell (left) and intracellular (right) recordings. The decrease in amplitude of the hyperpolarization after desensitization is presented as a fraction of the amplitude of the initial response to ME (300 nm). Black barsare the ratio obtained after 10 min application, and gray bars are the ratio after 40 min.
This protocol was also used to examine the desensitization of two other agonists that washed quickly from the tissue, an active metabolite of morphine, morphine 6-glucuronide (M6G) (10 μm), and a fluorescent analog of dermorphin (DERM-A488) (1 μm, applied by pressure ejection). The effect of ME (300 nm) was determined 10 and 40 min after washout of the high concentration of these compounds. The amount of desensitization induced by DERM-A488 (1 μm; n = 6) was present but significantly smaller than that induced by ME, and there was little or no desensitization caused by M6G (10 μm;n = 6) (Fig. 2B).
Desensitization by DERM-A488 and DERM-BTR
When the same experiment was used to examine the desensitization induced by another dermorphin analog, DERM-BTR, the washout was too slow to permit measurements of desensitization. In fact, this protocol was not suitable for testing many agonists. Compounds that washed slowly from the tissue were tested with a protocol that used submaximal concentrations, and desensitization was measured using maximal ME concentration applied at various intervals during the prolonged treatment (Fig. 1B). Using this protocol, DERM-BTR but not DERM-A488 caused significant desensitization (Fig.3). There was a significant decline in the maximal hyperpolarization induced by ME induced by DERM-BTR (30 nm; 50 ± 7% after 20 min). Neither DERM-A488 (30 nm; 20 min) nor dermorphin (30 nm; 20 min) changed the amplitude of maximal ME hyperpolarization (p > 0.05; one-way ANOVA), indicating that there was no desensitization when applied at this low concentration. Although the EC50 concentration of DERM-A488 did not cause desensitization, a maximal concentration did (Fig. 2), indicating that the desensitization was dependent on the concentration applied. It appears that concentration was not the only factor in causing desensitization, because DERM-BTR (30 nm) induced desensitization, although it caused a hyperpolarization that was smaller than that caused by both DERM-A488 (30 nm) and dermorphin (30 nm) (Fig. 3C). Thus, DERM-BTR was more potent at causing desensitization.
Fig. 3.

DERM-BTR induced desensitization in LC.A, Examples of experiments done with the low-dose protocol using DERM-A488 (30 nm; top) and DERM-BTR (30 nm; bottom). The maximum hyperpolarization induced by ME (10 μm) was not changed during treatment with DERM-A488, whereas the maximum hyperpolarization was reduced in the presence of DERM-BTR. B, Summarized data showing the amplitude of the ME-induced hyperpolarization during the treatment of slices with dermorphin, DERM-A488, and DERM-BTR (n = 5 for each experiment). The amplitude is plotted as a fraction of that observed during the first application of ME in the presence of the low concentration of each dermorphin analog.C, A summary of the acute effects of dermorphin, DERM-A488, and DERM-BTR, all applied at 30 nm(left). Right side indicates that the maximal hyperpolarization induced by DERM-A488 (1 μm; applied by pressure ejection) and DERM-BTR (1 μm; applied by pressure ejection) is the same as that induced by ME (10 μm).
MOR endocytosis
Previous work with DERM-A488 and DERM-BTR showed that these agonists differ substantially in their internalization properties, although these agonists had similar binding affinity (Ki ∼2.5 nm) and biological activity (EC50 ∼30 nm) (Arttamangkul et al., 2000). Although the only distinction between these analogs is the fluorescent dye attached to the C terminus, only DERM-BTR was internalized by CHO cells expressing MOR. Given the current observation that these agonists also desensitized MOR responses differently, one prediction was that the ability of the two fluorescent agonists to cause MOR internalization would be different. The ability of the dermorphin analogs to stimulate receptor internalization was examined directly by measuring the endocytosis of flag-MOR stably expressed in HEK293 cells (Fig.4). These cells express functional receptors, and receptor endocytosis was a reliable indicator of arrestin-dependent regulation of downstream signaling (Whistler and von Zastrow, 1998; Zhang et al., 1998).
Fig. 4.

Endocytosis of flag-tagged MOR in HEK293 cells by opioid peptide agonists. A, Examples of experiments examining the endocytosis induced by DERM-A488 (top) and DERM-BTR (bottom). The far left images(a, e) show total receptor binding using anti-flag antibodies. In b–d and f–h, the cells were incubated with DERM-A488 and DERM-BTR for the period indicated. At each time point, the anti-flag antibody remaining on the plasma membrane was stripped off so that only internalized label remained. The results show more internalization during treatment with DERM-BTR. B, Summarized data showing the time course of internalization induced by different concentrations of DERM-A488 and DERM-BTR. C, Examples of the maximal internalization caused by DERM-BTR, dermorphin, and DERM-A488 all applied at 30 nm for 30 min. D, Summary of many experiments with the dermorphin analogs and ME after a 30 min incubation period at 37°C. Each of the opioid peptides caused internalization that was dependent on the concentration. Also shown is constitutive internalization, which is the amount of receptor that was internalized in 30 min in the absence of any opioid agonist (No Agonist). ∗p < 0.05; ∗∗p < 0.001; one-way ANOVA; Tukey's multiple comparison test.
In the absence of opioid agonists, these cells showed constitutive endocytosis of ∼34 ± 3% of the total membrane receptors over a 30 min incubation period at 37°C (Fig.4B,D). Both dermorphin analogs caused receptor internalization, but the concentrations of agonist required for internalization were very different. Whereas DERM-A488 at an EC50 concentration (30 nm) did not cause endocytosis above constitutive levels (45 ± 3%), a very low concentration of DERM-BTR (3 nm) caused a constant increase of receptor internalization that reached maximal levels over a 60 min incubation (80 ± 8%) (Fig. 4B). Higher concentrations of both fluorescent agonists produced rapid and maximal receptor internalization with a similar time constant (DERM-A488, 1 μm, τ of 3.1 ± 1 min, maximum of 78 ± 6%; DERM-BTR, 30 nm, τ of 3.5 ± 1 min, maximum of 91 ± 6%). Maximal agonist-induced endocytosis reached ∼80% of the total membrane receptors, and it was not further increased with longer incubation times, presumably attributable to receptor recycling.
These results indicate that DERM-BTR itself was not only internalized, as it has been shown previously (Arttamangkul et al., 2000), but it also caused efficient receptor endocytosis. In fact, DERM-BTR was the most potent agonist studied for promoting receptor endocytosis, more potent than dermorphin itself or dermorphin plus the cysteine linker (DERM-Cys) (Fig. 4D). The endocytosis induced by DERM-A488 was similar to dermorphin and DERM-Cys in that it caused receptor endocytosis only when applied at maximal concentrations (Fig.4). ME-induced internalization was also apparent only when incubated at a high concentration (3 μm). The results of the two experiments indicate that DERM-A488 at high concentration can both desensitize and internalize MORs, whereas DERM-BTR does each at low concentration.
The internalization of a number of well characterized alkaloid agonists was examined under the same experimental conditions to put the results obtained with the two fluorescent dermorphin analogs into perspective (Fig. 5). In agreement with previous reports, morphine and M6G failed to promote MOR internalization even at very high concentrations. Normorphine at 1 μm did not induce receptor internalization, but, when the concentration was increased to 30 μm, 65 ± 6% of the total membrane receptors were internalized. Methadone and etorphine both caused internalization that was dependent on the concentration of agonist used. No etorphine-induced endocytosis was observed at concentrations up to 100 nm, whereas at 300 nm, a maximum effect was observed (Fig. 5B). Methadone at 1 μm caused some endocytosis, and a maximal response was observed at 30 μm. Thus, the results are completely consistent with the work of others and suggest that the difference in endocytosis induced by DERM-A488 and DERM-BTR results from an agonist-specific property conferred by chemical modification of this opioid peptide.
Fig. 5.

Methadone but not morphine caused endocytosis of flag-tagged MOR in HEK293 cells. A, Representative images from endocytosis experiments. Fluorescence stains internalized receptor after 30 min exposure to 30 μm methadone (left), morphine (middle), and M6G (right). B, Summarized data from several endocytosis experiments testing exposure to alkaloid opioids for 30 min at 37°C. Normorphine, methadone, and etorphine caused internalization that was dependent on the concentration. Morphine and M6G did not cause MOR endocytosis above the constitutive level (dotted lines), even at high concentrations. ∗∗p < 0.05; one-way ANOVA; Tukey's multiple comparison test.
Desensitization by alkaloid opioid agonists
To correlate the degree of desensitization with the ability of agonists to induced endocytosis, the desensitization measured in LC neurons was determined using the same opioid agonists. Given that the washout of most agonists was slow, a combination of the low-dose (Fig. 1B) and high-dose (Fig. 1C) protocols was used. With the low-dose protocol, morphine (1 μm) and M6G (1 μm) failed to induce changes in the maximal ME response (Fig.6). The results with M6G are consistent with those obtained with the high-dose protocol (Fig.2B). These results confirmed that neither morphine nor M6G were capable of inducing desensitization.
Fig. 6.

Methadone-induced desensitization using the low-dose protocol. A, Examples of experiments using the low-dose protocol with morphine and methadone. The amplitude of the ME-induced hyperpolarization was not changed during treatment with morphine (3 μm; top trace) but decreased progressively during superfusion with methadone (3 μm;bottom trace). B, Summarized data showing the decline in the ME-induced hyperpolarization in the presence of methadone and the lack of any change in the presence of morphine (3 μm), M6G (3 μm), and UK14304 (100 nm).
Methadone caused both receptor internalization and desensitization. Perfusion with methadone (1 μm) reduced the amplitude of maximal ME response by 50 ± 5% (n = 5) after 20 min (Fig. 6). There was no difference between the mean amplitude of the first ME response (before exposure to methadone) (30 ± 1.5 mV) and the amplitude of first pulse of ME response (32 ± 2 mV) immediately (3 min) after the onset of superfusion with methadone. However, in the continued presence of methadone, a reduction was detected as early as 10 min and declined continuously over 20 min (Fig.6).
Desensitization was also measured using the decline in the hyperpolarization during a prolonged exposure to a high agonist concentration (Fig. 1C). This protocol can be used for any agonist, regardless of the washout kinetics. The decline in the hyperpolarization was measured at the end of a 15 min application of morphine (30 μm; n = 4), methadone (10 μm; n = 4), etorphine (1 μm; n = 4), and ME (10 μm; n = 5) (Fig.7). Morphine was the only agonist that did not cause desensitization. The decline in response during the 15 min test was the same for each of the other agonists (∼10 mV). Naloxone (1 μm) was used to reverse the effect, and a maximal concentration of the α2-adenoceptor agonist UK14304 (3 μm) was applied to control for rundown or heterologous desensitization (Fig. 7). In one set of experiments, UK14304 (3 μm) was tested before application of any opioid agonist. The amplitude of the UK14304-induced hyperpolarization in these experiments was the same as that observed after desensitization to opioids (Fig. 7C) (n = 4 for each experiment).
Fig. 7.
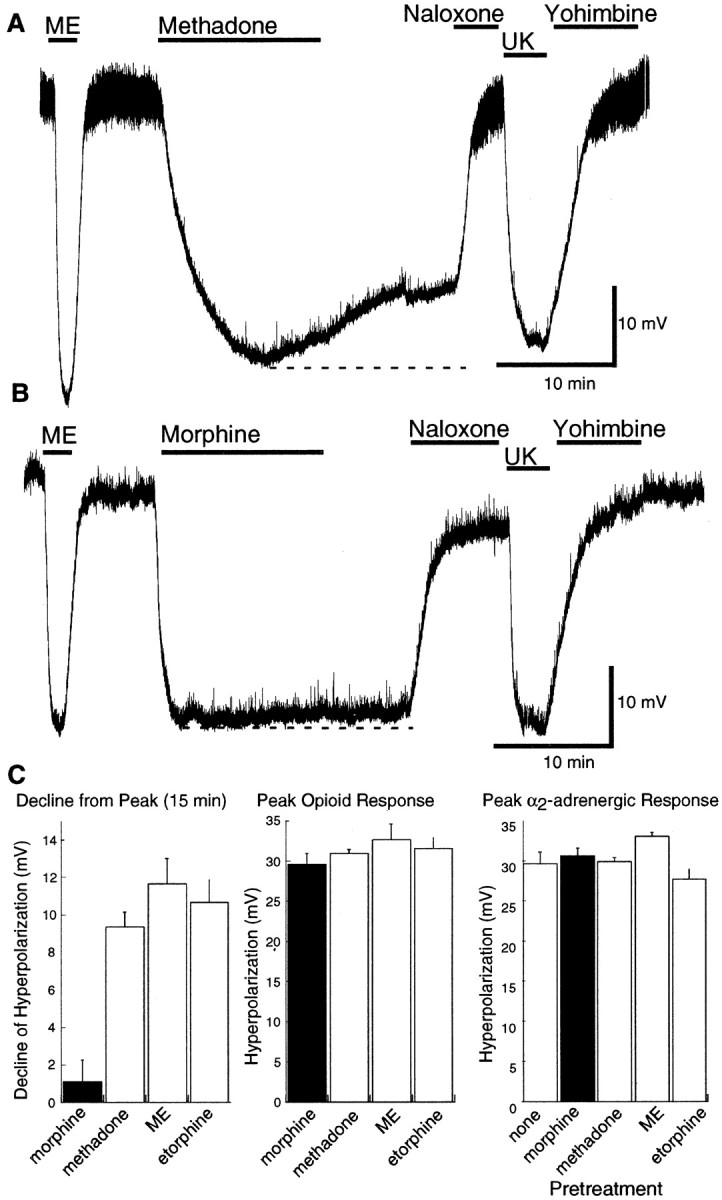
Methadone-induced desensitization using the high-dose protocol. A, An example of the desensitization caused by a high concentration of methadone (30 μm). The peak hyperpolarization caused by methadone was approximately the same as the hyperpolarization induced by ME (1 μm) and UK14304 (3 μm). The decline in the hyperpolarization during the 15 min application was taken as a sign of desensitization.B, An example of the same experiment using a high concentration of morphine (30 μm). There is little or no decline in the hyperpolarization induced by morphine. C,Left, Summarized data showing the amplitude of the decline in hyperpolarization during a 15 min application of several agonists. This decline was ∼10 mV for methadone, ME, and etorphine but <2 mV for morphine. Middle shows the peak amplitude of the hyperpolarization caused by each agonist. Rightshows the amplitude of the hyperpolarization induced by UK14304 (3 μm) before (none) and after treatment with each of the indicated opioid agonists. There is no sign of heterologous desensitization induced by any of the opioid agonists.
Correlation between endocytosis and desensitization
A summary of the results obtained with LC neurons in brain slices and HEK293 cells is illustrated in Figure8. Desensitization measured in locus ceruleus neurons showed a positive correlation with agonist-induced receptor endocytosis in HEK293 cells. Independent of the assay used to measure desensitization, whether it was a change in EC50 or a change in maximal hyperpolarization, agonists that failed to induce desensitization also failed to produce endocytosis. Furthermore, desensitization was only observed at agonist concentrations that also caused internalization of at least 70% of total membrane receptors.
Fig. 8.

A comparison of MOR endocytosis and desensitization. Summarized data from all endocytosis and desensitization experiments. The percentage of the total MOR internalized by a given concentration of agonist (taken from Figs. 4,5) was plotted as a function of amount of desensitization achieved when tested in LC neurons (expressed as percentage of the maximal opioid hyperpolarization). A significant correlation was found (nonparametric Spearman's test; r = 0.77; two-tailedp = 0.01269). The lowest possible value on they-axis (35%) is the level of constitutive endocytosis.
DISCUSSION
GIRK activation and desensitization
Striking differences were found among the opioid agonists with respect to the relative ability to activate GIRK-mediated hyperpolarization and to trigger desensitization. The rank of order for desensitization of MOR activation of potassium current in LC neurons was as follows: DERM-BTR > etorphine > methadone ≈ ME > dermorphin ≈ DERM-A488 > morphine ≈ M6G. The desensitization was dependent on the concentration of agonist applied. At approximately EC50 concentrations, DERM-A488 (30 nm) and etorphine (1 nm; data not shown) did not result in detectable desensitization, whereas at maximal concentrations, both induced significant desensitization. DERM-BTR, however, caused both desensitization and internalization at a low concentration (30 nm).
Based on both our experiments and previous literature, the rank of order for receptor activation is as follows: etorphine ≈ dermorphin > DERM-A488 ≈ DERM-BTR > ME ≈ methadone > morphine. These differences in the rank of order is particularly relevant because it has been suggested by studies in heterologous expression systems that the rate at which μ-opioid agonists induce rapid desensitization is dependent on agonist efficacy (Kovoor et al., 1998). Other studies, however, found that differences in the ability of certain agonists to induce MOR internalization in transfected fibroblasts were not positively correlated with either potency (Keith et al., 1996, 1998) or apparent efficacy (Whistler et al., 1999). Here we examined the relationship between efficacy and desensitization in neurons expressing endogenous μ-opioid receptors. Hence, the relationship between efficacy and desensitization were studied under conditions in which expression levels of receptors and signaling molecules were not manipulated.
The fluorescent opioid peptides were powerful tools for addressing the question of the relationship between agonist efficacy and desensitization and/or internalization of receptors. Created from the same opioid agonist, dermophin, DERM-BTR and DERM-A488 share several characteristics, such as EC50 and binding affinity (Kd of 2.3 and 2.5 nm for DERM-A488 and DERM-BTR, respectively) (Arttamangkul et al., 2000) but were very different in the ability to desensitize and internalize the receptor. DERM-BTR had a much greater ability to induce MOR desensitization and internalization. This observation questions the idea that receptor activation alone is required for rapid receptor desensitization.
Conjugation of BTR to dermorphin resulted in a very hydrophobic peptide. Given that this is the primary difference between DERM-BTR and DERM-A488, it is reasonable to propose that the unique desensitization properties of DERM-BTR could be related to its hydrophobicity. At least three possible explanations could account for the results. First, DERM-BTR could accumulate in the proximity of the plasma membrane and create very high local increases in agonist concentration that could saturate receptors, causing desensitization and endocytosis. This explanation, however, should result in the maximal activation of a physiological response. Second, the hydrophobic properties of DERM-BTR could enhance clustering and/or dimerization of receptor, which could facilitate desensitization and endocytosis. Third, DERM-BTR could slow recycling of internalized receptors such that an apparent increase in endocytosis and desensitization could result from failure to recycle.
In both the brain slice and HEK293 cell experiments, the onset and recovery from application of DERM-BTR was slower than DERM-A488. Although this observation may be related simply to differences in hydrophobicity, it may also suggest that the receptor binding kinetics are different for the two ligands. A slower dissociation rate might imply that DERM-BTR remains bound to receptors, even after endocytosis, thereby impairing receptor recycling. This notion is supported by the observation that a low concentration of DERM-BTR induced a progressive accumulation of MOR in the intracellular compartment (Fig.4B). The slow but constant rate of receptor accumulation might suggest a reduced rate of receptor recycling.
Desensitization and internalization
Desensitization was tested using different protocols that included exposure to maximal or submaximal agonist concentrations, all yielding consistent results. In agreement with previous studies, signaling through α2-adrenergic receptors was not affected by the exposure to opioid agonists, indicating a homologous mechanism for MOR desensitization (Harris and Williams, 1991; Osborne and Williams, 1995, Fiorillo and Williams, 1996). A strong correlation was found between desensitization observed in LC neurons and internalization observed in HEK293 cells. Ideally, both internalization and desensitization should be studied in the same neurons; however, to date, imaging experiments in brain slices have been limited technically. Although the correlation between cell types does not definitively demonstrate a link between these processes, it does not disprove a connection.
The opioid agonists normorphine, methadone, etorphine, dermorphin, DERM-Cys, and DERM-A488 all induced MOR desensitization and internalization in a concentration-dependent manner. Interestingly, supramaximal concentrations of these agonists were often required to cause significant desensitization and endocytosis. An important example is etorphine, which is both potent and efficacious at activating a number of different effectors but required relatively high concentrations to induce both desensitization and endocytosis. This observation suggests that higher receptor occupancy may be required for internalization and desensitization than for activation of other effectors. The exception to this statement is DERM-BTR.
Significance
Acute desensitization and receptor endocytosis have been proposed to be protective mechanisms that come into play during prolonged agonist exposure (Finn and Whistler, 2001; He et al., 2002). According to this hypothesis, when endocytosis is not triggered, adaptive responses in the cell and/or animal are activated that oppose the acute action of opioids to restore equilibrium. The prediction is that opioids with differing ability to induce tolerance and dependence can be distinguished based on the ratio of receptor activation divided by receptor endocytosis (RA/VE) (Whistler et al., 1999).
A recent study compared the effect of chronic treatment with morphine (high RAVE value) and etorphine (low RAVE value) on analgesic tolerance and receptor regulation in vivo (Stafford et al., 2001). The results showed that the analgesic potency of morphine declined linearly over the 7 d treatment, and there was no change in opioid receptor number during this period, as it has been previously reported. A very different pattern was observed in animals treated with etorphine. With low doses of etorphine (that were equipotent to the morphine treatment), no analgesic tolerance developed. As the dose was increased, however, there was a sharp increase in tolerance, which was characterized by receptor downregulation. High doses of etorphine, thus, reduced analgesic potency by a decline in receptor signaling through a decrease in receptor number. Other work has suggested that the decline in receptor number induced by etorphine is mediated by a pertussis toxin-insensitive pathway (Gomes et al., 2002). The present paper does not address the G-proteins involved but shows that high concentrations of etorphine triggered receptor endocytosis very efficiently. Chronic treatment with etorphine might then cause receptor downregulation by increased endocytosis and degradation.
In summary, acute homologous desensitization and MOR endocytosis were examined using several opioid agonists. A close link between desensitization and endocytosis was found, and both processes were dependent on the individual characteristics and concentration of the agonists used. This study demonstrated that dermorphin, a compound with a relatively high RAVE value was transformed to a compound with a lower RAVE value by the addition of a hydrophobic dye molecule. This observation may lead to a better understanding of the chemical properties of agonists that govern receptor activation, desensitization, and endocytosis.
V.A.A. and S.A. contributed equally to this work.
This work was supported by National Institutes of Health Grants DA08163 (J.T.W.), DA07262 (S.A.), DA12864 (M.v.Z.), and DA08562 and DA10703 (D.K.G.). We thank Drs. Amara and Watt for the use of the confocal microscope and Drs. Torrecilla and Morikawa for comments on this work.
Correspondence should be addressed to John T. Williams, Vollum Institute, L474 Oregon Health and Sciences University, 3181 S.W. Sam Jackson Park Road, Portland, OR 97201. E-mail williamj@ohsu.edu.
V. A. Alvarez's present address: Department of Neurobiology, Harvard Medical School, Boston, MA 02115.
REFERENCES
- 1.Arttamangkul S, Alvarez-Maubecin V, Thomas G, Williams JT, Grandy DK. Binding and internalization of fluorescent opioid peptide conjugates in living cells. Mol Pharmacol. 2000;58:1570–1580. doi: 10.1124/mol.58.6.1570. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 3.Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 4.Fiorillo CD, Williams JT. Opioid desensitization: interactions with G-protein-coupled receptors in the locus coeruleus. J Neurosci. 1996;16:1479–1485. doi: 10.1523/JNEUROSCI.16-04-01479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes BA, Shen J, Stafford K, Patel M, Yoburn BC. μ-Opioid receptor down-regulation and tolerance are not equally dependent upon G-protein signaling. Pharmacol Biochem Behav. 2002;72:273–278. doi: 10.1016/s0091-3057(01)00757-2. [DOI] [PubMed] [Google Scholar]
- 6.Harris GC, Williams JT. Transient homologous mu-opioid receptor desensitization in rat locus coeruleus neurons. J Neurosci. 1991;11:2574–2581. doi: 10.1523/JNEUROSCI.11-08-02574.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He L, Fong J, von Zastrow M, Whistler JL. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108:271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- 8.Ishimatsu M, Williams JT. Synchronous activity in locus coeruleus results from dendritic interactions in pericoerulear regions. J Neurosci. 1996;16:5196–5204. doi: 10.1523/JNEUROSCI.16-16-05196.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- 10.Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von Zastrow M. mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377–384. [PubMed] [Google Scholar]
- 11.Kenakin T. Efficacy and G-protein-coupled receptors. Nature Rev. 2002;1:103–110. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- 12.Koch T, Schulz S, Schroder H, Wolf R, Raulf E, Hollt V. Carboxyl-terminal splicing of the rat mu opioid receptor modulates agonist-mediated internalization and receptor resensitization. J Biol Chem. 1998;273:13652–13657. doi: 10.1074/jbc.273.22.13652. [DOI] [PubMed] [Google Scholar]
- 13.Kovoor A, Celver JP, Wu A, Chavkin C. Agonist induced homologous desensitization of mu-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol. 1998;54:704–711. [PubMed] [Google Scholar]
- 14.Law PY, Erickson LJ, El-Kouhen R, Dicker L, Solberg J, Wang W, Miller E, Burd AL, Loh HH. Receptor density and recycling affect the rate of agonist-induced desensitization of mu-opioid receptor. Mol Pharmacol. 2000;58:388–398. doi: 10.1124/mol.58.2.388. [DOI] [PubMed] [Google Scholar]
- 15.Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 16.Osborne PB, Williams JT. Characterization of acute homologous desensitization of mu-opioid receptor-induced currents in locus coeruleus neurones. Br J Pharmacol. 1995;115:925–932. doi: 10.1111/j.1476-5381.1995.tb15899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stafford K, Gomes AB, Shen J, Yoburn BC. mu-Opioid receptor downregulation contributes to opioid tolerance in vivo. Pharmacol Biochem Behav. 2001;69:233–237. doi: 10.1016/s0091-3057(01)00525-1. [DOI] [PubMed] [Google Scholar]
- 18.Sternini C, Spann M, Anton B, Keith DE, Jr, Bunnett NW, von Zastrow M, Evans C, Brecha NC. Agonist-selective endocytosis of mu opioid receptor by neurons in vivo. Proc Natl Acad Sci USA. 1996;93:9241–9246. doi: 10.1073/pnas.93.17.9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sternini C, Brecha NC, Minnis J, D'Agostino G, Balestra B, Fiori E, Tonini M. Role of agonist-dependent receptor internalization in the regulation of mu opioid receptors. Neuroscience. 2000;98:233–241. doi: 10.1016/s0306-4522(00)00118-4. [DOI] [PubMed] [Google Scholar]
- 20.Trafton JA, Abbadie C, Marek K, Basbaum AI. Postsynaptic signaling via the μ-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. J Neurosci. 2000;20:8578–8584. doi: 10.1523/JNEUROSCI.20-23-08578.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by beta-arrestin. Proc Natl Acad Sci USA. 1998;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- 23.Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci USA. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]