

Abstract
The cholinergic system is thought to play an important role in hippocampal-dependent learning and memory. However, the mechanism of action of the cholinergic system in these actions in not well understood. Here we examined the effect of muscarinic receptor stimulation in hippocampal CA1 pyramidal neurons using whole-cell recordings in acute brain slices coupled with high-speed imaging of intracellular calcium. Activation of muscarinic acetylcholine receptors by synaptic stimulation of cholinergic afferents or application of muscarinic agonist in CA1 pyramidal neurons evoked a focal rise in free calcium in the apical dendrite that propagated as a wave into the soma and invaded the nucleus. The calcium rise to a single action potential was reduced during muscarinic stimulation. Conversely, the calcium rise during trains of action potentials was enhanced during muscarinic stimulation. The enhancement of free intracellular calcium was most pronounced in the soma and nuclear regions. In many cases, the calcium rise was distinguished by a clear inflection in the rising phase of the calcium transient, indicative of a regenerative response. Both calcium waves and the amplification of action potential-induced calcium transients were blocked the emptying of intracellular calcium stores or by antagonism of inositol 1,4,5-trisphosphate receptors with heparin or caffeine. Ryanodine receptors were not essential for the calcium waves or enhancement of calcium responses. Because rises in nuclear calcium are known to initiate the transcription of novel genes, we suggest that these actions of cholinergic stimulation may underlie its effects on learning and memory.
Keywords: gene, learning, memory, nucleus, IP3, acetylcholine, ryanodine
Acetylcholine is an important neuromodulatory transmitter affecting hippocampal-dependent learning and memory (Hagan and Morris, 1988). Loss of cholinergic function has been hypothesized to underlie age-related learning impairments and memory loss that accompanies Alzheimer's disease (Bartus et al., 1982;Kasa et al., 1997), and activation of the cholinergic system has been shown to modulate processing in sensory and visual cortex (Furey et al., 2000; Shulz et al., 2000). However, the mechanisms by which the cholinergic system affects neuronal processing are not understood. It is well known that acetylcholine, acting via muscarinic receptors, modulates a number of potassium conductances in hippocampal pyramidal neurons, including those that mediate spike frequency adaptation (Nicoll et al., 1989). These actions of acetylcholine have been suggested to underlie the mechanism of action of acetylcholine on learning and memory (Hasselmo and Bower, 1993; Disterhoft et al., 1999).
The long-term changes that underlie the final mechanisms in learning and memory involve changes in gene transcription (Kandel and Pittenger, 1999). These changes are in large part initiated by rises in cytosolic calcium (Ghosh and Greenberg, 1995; Finkbeiner and Greenberg, 1998), which activates a number of signaling pathways. Under resting conditions, cytosolic free calcium [Ca2+]i is maintained at 50–100 nm and can rise either as a result of influx from the extracellular space or by release from intracellular stores (Tsien and Tsien, 1990; Berridge, 1998). The pathways by which calcium can enter neurons from the extracellular space are well characterized and include voltage-gated calcium channels and receptor-operated calcium channels, such as NMDA receptors. Much is understood about the activation of these pathways, their modulation, and the downstream consequences of the calcium rises that follow (Ghosh and Greenberg, 1995; Berridge, 1998). Thus, rises in cytosolic calcium attributable to influx via either L-type voltage-dependent calcium channels or NMDA receptors has been shown to cause translocation of calmodulin to the nucleus and initiation of gene transcription by phosphorylation of cAMP response element-binding protein (CREB) (Deisseroth et al., 1996, 1998). Calcium is also sequestered within the smooth endoplasmic reticulum (Henzi and MacDermott, 1991) and can be released by activation of either inositol 1,4,5-trisphosphate (InsP3) or ryanodine receptors (RyRs) (Tsien and Tsien, 1990; Berridge, 1998), both of which are present in abundance in central neurons (Sharp et al., 1993). Release of calcium from intracellular stores and the consequent rise in nuclear calcium levels has been shown to be effective in initiating gene transcription (Dolmetsch et al., 1998; Li et al., 1998; Hardingham et al., 2001). Notably, in hippocampal neurons, a rise in nuclear calcium without translocation of CaM is sufficient to activate the CREB transcription pathway (Hardingham et al., 2001).
In addition to its actions on potassium currents, acetylcholine, acting at muscarinic receptors, also stimulates the formation of InsP3 and diacylglycerol (Fowler and Tiger, 1991;Fisher et al., 1992). In non-neuronal cells, receptor-mediated generation of InsP3 results in release of calcium from intracellular stores, often in an oscillatory manner (Henzi and MacDermott, 1991; Berridge, 1998). Here we show that activation of muscarinic acetylcholine receptors on hippocampal pyramidal neurons leads to rises of cytosolic calcium that are initiated in the apical dendrite and propagate as a wave to the soma in which they invade the nucleus. These calcium rises are attributable to release of calcium from InsP3-sensitive intracellular calcium stores. Because of the calcium dependence of the InsP3 receptor, this mechanism also acts to amplify nuclear calcium rises in response to trains of action potentials. Thus, this mechanism functions as a nuclear detector of cholinergic activation and may explain how memory formation is enhanced during cholinergic activation.
MATERIALS AND METHODS
All experiments were done on transverse hippocampal slices prepared from 17- to 28-d-old rats using standard methods (Sah and Isaacson, 1995). For recording, slices were transferred to the stage of a BX50 microscope (Olympus Optical, Tokyo, Japan) and superfused with artificial CSF (aCSF) containing (in mm): 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.0 Na2PO4, 26.2 NaHCO3, and 11 glucose (equilibrated with 95% CO2–5% O2 and heated to 33°C). Whole-cell recordings were made from the soma of pyramidal neurons in area CA1 using infrared differential interference videomicroscopy. Patch pipettes (2–5 MΩ) were fabricated from borosilicate glass and filled with an internal solution containing 135 mm KMeSO4, 8 mm NaCl, 10 mm HEPES, 2 mm Mg2-ATP, 0.3 mm Na3-GTP, pH 7.3 with KOH (osmolarity 280–290 mOsm), and 50 μm Oregon green BAPTA-1 (Molecular Probes, Eugene, OR). Cells were selected close to the surface of the slice. We estimate that the depth of the cell body and proximal dendrite were within 50 μm from the surface. Electrophysiological signals were recorded with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA), digitized at 20 kHz with an ITC-16 board (InstruTech, Port Washington, NY), and controlled using custom software running under Igor Pro (WaveMetrics, Lake Oswego, OR) or Axograph (Axon Instruments). Electrophysiological data were analyzed using Axograph.
Whole-field fluorescence measurements were made using a monochromator-based imaging system (Polychrome II; T.I.L.L. Photonics, Martinsried, Germany). Neurons were visualized using a 60× water immersion objective (0.9 numerical aperture; Olympus Optical) and illuminated with 488 nm light. Images were acquired with an in-line transfer, cooled CCD camera (T.I.L.L. Photonics) in which the scan lines were binned by two in both horizontal and vertical directions, giving a spatial resolution of 0.33 μm per pixel. During collection of image sequences, the exposure time was 10 msec per frame to improve temporal resolution and minimize photobleaching of the dye. When examining calcium transients in response to action potentials, frames were collected at either 16 or 33 Hz. During bath application of agonists, frames were collected at 1 or 2 Hz. Single action potentials were evoked by a 10 msec current pulse (200–700 pA), and trains of action potentials were evoked by either a 300 msec current pulse or a train of 10 msec current pulses. Images were analyzed offline using Vision (T.I.L.L Photonics). Small rectangular regions (∼10 × 10 pixels) were selected over the nucleus, extranuclear soma, and the proximal dendrite, and the fluorescence over this region was averaged. Confocal fluorescence images were obtained using a Zeiss (Oberkochen, Germany) Axioskop 2FS, with a 510 laser scanning head and equipped with an argon laser. Confocal fluorescence images were acquired in line scan mode in which the selected line encompassed nuclear, somatic, and proximal apical dendritic regions at a resolution of 10–20 pixels/μm. The laser power was reduced to 1% to prevent bleaching. The detector pinhole aperture was set to give a vertical resolution of <2 μm. Lines were acquired at 20 Hz. Small segments (∼3 μm) were selected over the nucleus, extranuclear soma, and the proximal dendrite, and the fluorescence over this region was averaged.
Kinetic sequences were then constructed over time for each of the selected regions. Calcium signals were measured as the relative change in fluorescence over baseline fluorescence (ΔF/F). Additional experiments were performed using fura-2 and Fluo4 (Molecular Probes). Fura-2 calcium signals were quantified as background subtracted ratio of fluorescence signals using excitation wavelengths of 340 and 380 nm (F340/F380). Most recordings concentrated on the soma and proximal 25–50 μm of the apical dendrite.
Muscarine or carbachol (Sigma, St. Louis, MO) were either bath applied (10–20 μm) or applied by focal pressure application (2–100 μm in aCSF) through a patch pipette. Focal pressure was applied either by manually applying pressure through via a 5 ml syringe or via a Picospritzer (30 psi, 50–350 msec; Parker Hannifin, Fairfield, NJ). In some experiments, 500 μg/ml low molecular weight heparin (Sigma), 20 μm ruthenium red (Sigma), and/or 50 μm caged myo-inositol 1,4,5-trisphosphate (Molecular Probes) were added to the internal solution. The lamp used for the photolysis of caged InsP3 was a pulsed xenon arc lamp (T.I.L.L. Photonics), which illuminated the entire field of view and discharged ∼80 J in 2 msec. Light from both the monochromator and the flash were delivered to the BX50 microscope via a quartz light guide and a custom epifluorescence attachment provided by T.I.L.L. Photonics and then to the cells through the objective. Atropine, ryanodine, dantrolene, and cyclopiazonic acid (CPA) were purchased from Sigma. Thapsigargin, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 2-amino-5-phosphono- valeric acid (APV), and α-methyl-4-carboxyphenylglycine [(+)-MCPG] were purchased from Research Biochemicals (Natick, MA). Tetrodotoxin (TTX) was purchased from Alomone Labs (Jerusalem, Israel).
Statistical comparisons were made using a paired Student'st test or as otherwise indicated. All data are presented as mean ± SEM.
RESULTS
Whole-cell recordings were obtained from CA1 hippocampal pyramidal neurons. All cells had resting potentials more negative than −55 mV. The nucleus could be seen as an area of higher fluorescence within the soma (Fig. 1, 3). The higher fluorescence is attributable to the lack of organelles within the nucleus, resulting in a greater ratio of free intracellular space to total volume compared with the soma (O'Malley, 1994) and not attributable to differences in resting calcium. In confirmation of this, ratiometric imaging with fura-2 AM did not show a difference in the 340/380 ratio between the nucleus and the soma (Fig.1e). Activation of muscarinic acetylcholine receptors by bath application of muscarine (10–20 μm) caused a transient rise in cytosolic calcium, as indicated by an increase in Oregon green fluorescence. The rise in calcium began as a focal increase in the proximal apical dendrite (30–50 μm from the soma) and propagated as a wave toward the soma and into the nucleus (Fig. 1). Backpropagation of the calcium wave was observed on occasion; however, the extent of the backpropagation was usually limited to a few micrometers. On some occasions, calcium waves were also propagated from basal dendrites (n = 6). The average speed of wave propagation was 26 ± 2 μm/sec (n= 35). Entry of calcium into the nucleus was associated with a slight delay (Fig. 1d,e). The movement of free calcium from the dendrite to the soma cannot be attributable to simple diffusion because the amplitude of the calcium transient was not decremental (Fig. 1c). The time course of the calcium rise was fastest in the dendrite with a half-width of 1.18 ± 0.14 sec and became broader as it propagated to the soma; the half-width of the calcium transient at the soma was 2.67 ± 0.40 and 3.48 ± 0.57 sec at the nucleus (n = 10). In many cases (18 of 42), repetitive rises in calcium were observed in the continued presence of agonist, occurring at a frequency of ∼0.03 Hz (data not shown). Each cycle of the oscillation generally took the form of a wave that originated in the dendrite and propagated to the soma and nucleus. Membrane depolarization was not required for the rise in cytosolic calcium because muscarine was equally effective in raising calcium when cells were voltage clamped between −60 and −70 mV (28 of 34) as when cells were held in current clamp mode (19 of 22). The calcium rises were rarely associated changes in whole-cell current, suggesting that the calcium was released from intracellular calcium stores (Fig.1e). Bath application of cholinergic agonists has been reported to produce oscillatory network activity in hippocampal slices (Fellous and Sejnowski, 2000). Calcium waves were not initiated by network activity because calcium waves were evoked when recurrent synaptic activity was blocked with tetrodotoxin (500 nm; two of three) (Fig.2a). Furthermore, brief focal pressure application of muscarine onto the soma and proximal apical dendrite of neurons reliably and repeatedly evoked calcium waves (16 of 18), even when ionotropic and metabotropic glutamate and GABA receptors were blocked (two of two) (Fig. 2b) or when voltage-gated calcium channels were blocked with 5 mmNiSO4 (two of two) (Fig. 2c). The onset of the calcium rise occurred 828 ± 134 msec after Picospritzer application of agonist muscarine. The half-width of the calcium transients evoked by focal application was similar to that of bath-applied agonist (dendrite, 0.37 ± 0.02 sec; soma, 1.67 ± 0.15 sec; nucleus, 2.33 ± 0.16 sec). Focal application of muscarine to the distal dendrites (>150 μm from the soma) did not evoke a calcium rise (n = 2) in either the distal dendrites or the proximal dendrites and soma.
Fig. 1.

Muscarinic stimulation evokes calcium waves in hippocampal pyramidal neurons. A fluorescent image of a cell loaded with 50 μm Oregon Green BAPTA-1 is shown ina. The boxes indicate the regions in which fluorescence measurements were taken. The nucleus can be seen as a bright area in the soma (area 1). b, Selected pseudocolor frames are baseline-subtracted images (ΔF) of the cell shown in aduring bath application of 20 μm muscarine. The first detectable increase in fluorescence is labeled as time 0, and subsequent frames at the indicated times are shown below.c, Rises in calcium, plotted as ΔF/F, measured over the regions indicated in a are shown over time. The calcium transient had the fastest time course in the dendrite and became slower as it propagated to the soma and nucleus. d, Different cell loaded with the ratiometric indicator fura-2 AM (100 μm). The panel above shows image recorded using the isobestic 360 nm excitation wavelength. Theboxes indicate the regions in which fluorescence measurements were made. The selected pseudocolor frame below shows the calcium wave propagating into the soma before entry into the nucleus.e, Rises in calcium plotted as the ratioF340/F360over time for the three regions (dendrite, soma, and nucleus) indicated in the frames above. Note that [Ca2+]iis similar in the soma and nuclease, and there is a clear delay between the calcium rises in the extranuclear soma surrounding the nuclear region and the calcium rise in the nucleus itself.
Fig. 2.
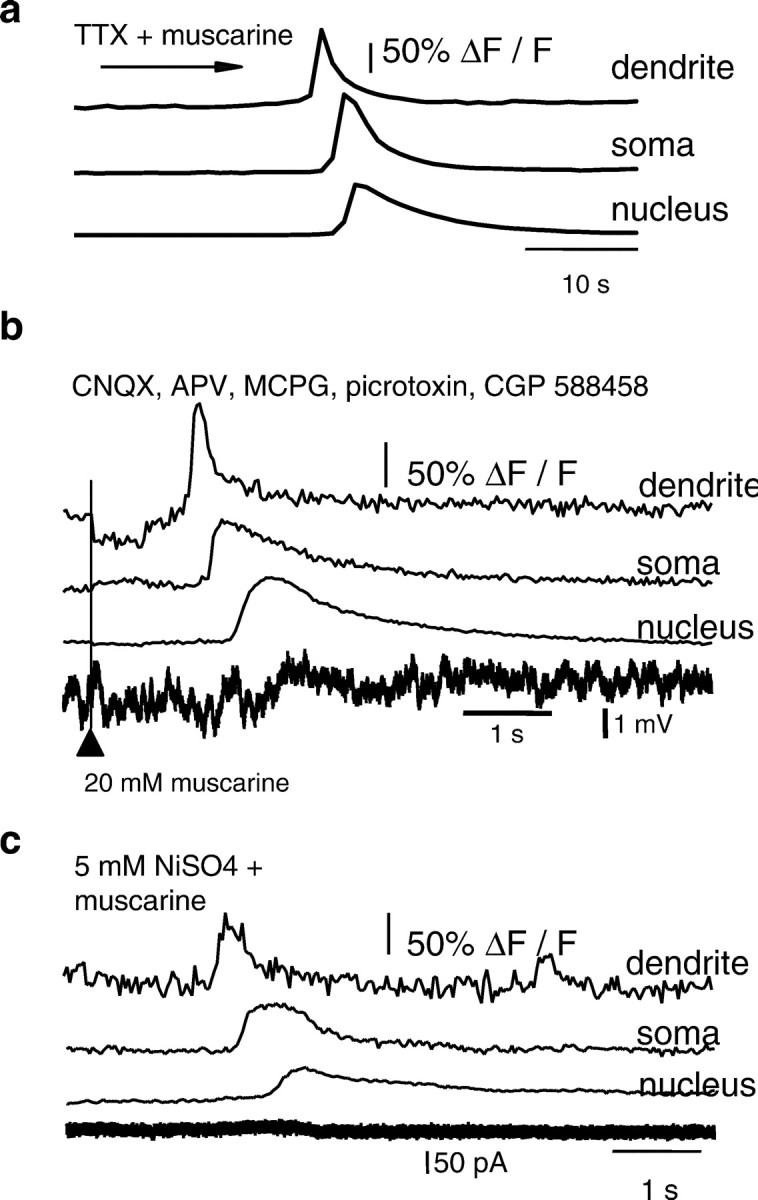
Calcium waves are not the result of enhanced network activity. a, Rises in calcium, plotted as ΔF/F, to bath application of muscarine in the presence of TTX (500 nm) are shown ina. b, Focal application of muscarine onto the soma and proximal dendrite readily evoked calcium waves, even when glutamatergic and GABAergic synaptic activity was blocked by bath application of APV, CNQX, MCPG, picrotoxin, and CGP 588458. Rises in calcium are plotted as ΔF/F and are shown at the top. This neuron was recorded in current-clamp mode. The simultaneously recorded voltage is shown below. The timing of the Picospritzer activation is indicated by thearrowhead and the vertical line. Note the delay between application of muscarine and the onset of the calcium wave. c, Calcium waves do not depend on extracellular calcium because a calcium wave could be evoked in 5 mmNiSO4. Rises in calcium are plotted as ΔF/F and are shown for the dendrite, soma, and nucleus, along with the simultaneously recorded whole-cell current.
Although the kinetics of nuclear calcium rises measured with whole-field fluorescence imaging are consistent with calcium rises occurring in the nucleus itself, it is possible that the nuclear fluorescence signal is contaminated by somatic signals above and below the nucleus. To address this possibility, muscarinic calcium waves were measured with a confocal imaging system (Fig.3). The confocal line scan data clearly show the delayed invasion of the calcium wave into the nucleus. In addition, the slow kinetics of the nuclear calcium rise observed with confocal imaging are in complete agreement with the whole-field fluorescence data. The half-width of the calcium rise was 0.66 ± 0.03 sec at the apical dendrite, 2.19 ± 0.20 sec at the soma, and 3.02 ± 0.05 sec at the nucleus (n = 3).
Fig. 3.
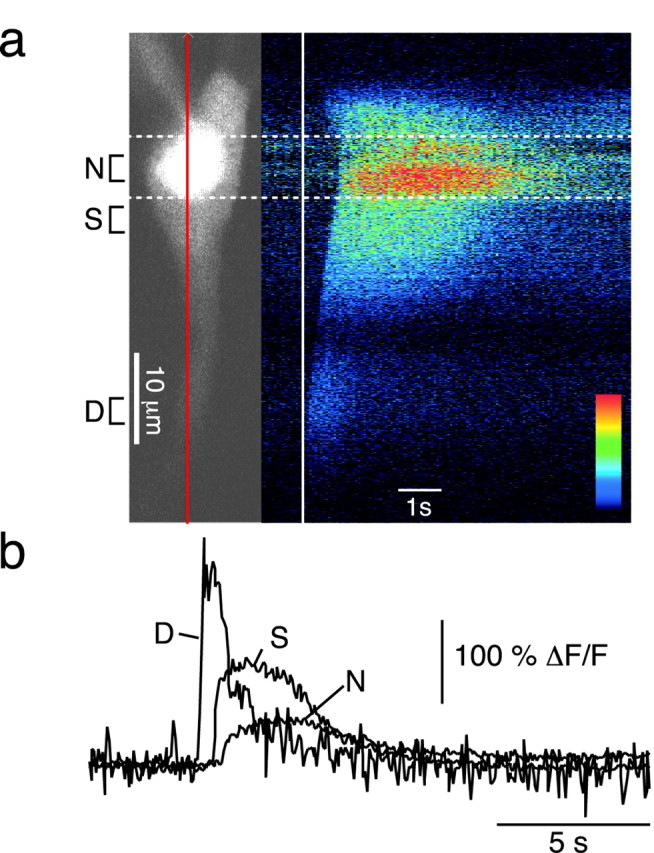
Calcium waves invade the nucleus. A confocal fluorescent image of a cell loaded with 50 μm Oregon Green BAPTA-1 is shown on the left of a. The nucleus can be seen as a bright area in the soma. Thevertical red line through the cell indicates the line of interest used in subsequent line scan time series. The baseline-subtracted time series (ΔF) displayed in pseudocolor after focal application of carbachol (20 μm) is shown on the right. Thevertical white line indicates the time of the first detectable rise in calcium. Dashed white lines mark the nucleus. b, Rises in calcium, plotted as ΔF/F, measured over the dendritic (D), somatic (S), and nuclear (N) regions indicated in aare shown over time. Note the latency between the initiation of the calcium wave and its subsequent nuclear invasion.
Action potentials in hippocampal pyramidal neurons initiated rises in both somatic and nuclear calcium. As shown previously (Jaffe et al., 1992; Markram et al., 1995; Sah and Clements, 1999), [Ca2+]i peaked at a higher level and decayed more rapidly in the dendrites than in the soma. Compared with extranuclear somatic calcium, however, the rise in nuclear calcium was smaller in amplitude (Schiller et al., 1995) and had a slower time course than that of the extranuclear soma (Fig.4). This is likely to be attributable to the diffusional barrier presented by the nuclear pores in the nuclear membrane (Rogue and Malviya, 1999). Because voltage-activated calcium currents are modulated by muscarinic receptors (Gahwiler and Brown, 1987), we next tested whether action potential-induced calcium rises are affected during muscarinic stimulation. Calcium transients in one cell recorded from the soma and the apical dendrite in response to a single action potential (Fig.5a) and an action potential train (Fig. 5b) are shown in Figure 5. After muscarinic stimulation, the peak [Ca2+]i in response to single action potentials was typically smaller (Fig.5a) than the control response. The median peak ΔF/F in the presence of agonist was reduced by 26% in the nucleus (p < 0.001;n = 38), 27% in the soma (p < 0.002; n = 38), and 19% in the proximal dendrite (p < 0.001; n = 36; Wilcoxon signed rank test). In contrast, during action potential trains, there was a large amplification of the calcium response; both the amplitude and duration of the calcium response were larger (Fig. 5b). Average peak ΔF/F increased by 145 ± 39% in the nucleus, 78 ± 25% in the soma, and 39 ± 17% proximal dendrite (n = 10). To test whether the enhancement of the action potential-induced calcium rise in the presence of muscarine was simply attributable to the reduction of spike frequency adaptation and corresponding increase in the number of action potentials (Müller et al., 1988; Tsubokawa and Ross, 1997), we examined the effect of trains of action potentials evoked by four 10 msec current pulses given at 20 Hz. Both the amplitude and duration of the calcium response evoked by four spike trains of action potentials were larger during muscarinic stimulation (Fig. 5c). Average peak ΔF/F increased by 56 ± 21% in the nucleus (p < 0.05; n = 28), 61 ± 23% at the soma (p < 0.01;n = 28), and 10 ± 7% in the proximal dendrite. In addition to the increase in peak amplitude, there was a dramatic prolongation of the calcium transient. Under control conditions, [Ca2+]i began to decline immediately after the last action potential. However, during muscarinic stimulation, the calcium transient showed a pronounced plateau phase and greatly outlasted the action potential train (Fig.5b,c). During muscarinic stimulation, the latency from the last action potential to the peak of the calcium transient increased from 18 ± 8 to 304 ± 58 msec in the nucleus (p < 0.001; n = 28) and from 4 ± 7 to 122 ± 29 msec over the soma (p < 0.001; n = 28). The integrated area of the calcium transient evoked by four action potentials increased by 147 ± 47% at the nucleus (p < 0.005), 190 ± 59% at the soma (p < 0.004), and 42 ± 18% in the apical dendrite (p < 0.02; n = 26) during muscarinic stimulation. In many cases, a clear inflection could be seen in the rising phase of the calcium transient, which is reminiscent of a regenerative response (Fig. 5c). This inflection was never observed under control conditions, even with longer trains of action potentials. The muscarinic amplification of the calcium response during action potential trains was unlikely to result from blockade of the afterhyperpolarization (AHP) because isoprenaline (10 μm), which also blocked the AHP, did not cause any regenerative rises in calcium in response to spike trains (n = 4; data not shown) (Sah and Clements, 1999). All effects of muscarinic agonists were blocked by atropine and fully reversible upon washout (n = 5).
Fig. 4.

Action potential-evoked calcium rises in the nucleus are smaller and have a slower time course than transients recorded in extranuclear soma. a, Cell loaded with fura-2 AM. The squares show the regions selected for measurement of calcium transients. b, Calcium transients evoked by a single action potential are shown recorded from the proximal dendrite, extranuclear soma, and nuclear soma. Thetraces have been normalized and superimposed on theright. c, Calcium rises in the nucleus are smaller in amplitude, have a slower time-to-peak, and have a slower decay to baseline. Average data are shown in the histograms below. *p < 0.05.
Fig. 5.

Action potential-evoked regenerative rises in free calcium in response to muscarinic stimulation. Action potentials were evoked by a single or train of current injections as shown in the traces at theleft. Calcium transients, plotted as ΔF/F, recorded over the nucleus, extranuclear soma, and dendrite are shown to the right.a, the calcium transient in response to a single action potential (thin traces) was reduced in the presence of muscarine (thick traces). b, In response to a 200 msec current injection, which evokes a train of action potentials, the calcium response showed a regenerative amplification in the nucleus, extranuclear soma, and the proximal dendrite during muscarinic stimulation. c, The amplification is not attributable to increased number of action potentials evoked in muscarine because a similar amplification is seen when only four action potentials are evoked at a frequency of 20 Hz. The responses in control Ringer's solution are shown as thin traces, and the response in the presence of muscarine is shown as a thick trace. Note that, in the presence of muscarine, the rising phase of the calcium transients greatly outlast the action potential train, indicated by the solid bar below thetraces.
Synaptic activation of muscarinic receptors
The hippocampal formation receives a dense innervation by cholinergic fibers from the septal nucleus (Lewis and Shute, 1967). Activation of cholinergic fibers with a brief tetanus (33 Hz; 300–600 msec) in stratum oriens evokes a slow depolarizing synaptic potential and blockade of the AHP by activation of muscarinic receptors (Cole and Nicoll, 1984). In neurons in which synaptic stimulation produced a reduction of the current underlying the slow AHP, calcium waves were also observed (n = 14) (Fig.6a). Similar to bath-applied carbachol and muscarine, the calcium rise from synaptic stimulation produced a focal rise in calcium in the proximal segment of the apical dendrite, which began 803 ± 207 msec after the first stimulus and propagated toward the soma at 32 ± 4 μm/sec (n= 8). Similar to results obtained with agonist application, backpropagation of the calcium wave was spatially limited (Fig.6b,c). After reaching the soma, as with bath application of agonists, the calcium wave invaded the nucleus. The half-width of the calcium transient was 0.69 ± 0.21 sec in the proximal dendrite, 1.11 ± 0.17 sec in the soma, and 1.68 ± 0.18 sec in the nucleus (n = 4).
Fig. 6.

Synaptic stimulation of cholinergic afferents evokes calcium waves that invade the nucleus.a, A fluorescent image of a cell loaded with 50 μm Oregon Green BAPTA-1. b, The selected pseudocolor frames are baseline-subtracted images (ΔF) taken at 300 msec intervals after synaptic stimulation (10 pulses at 33 Hz). c, Rises in calcium, plotted as ΔF/F, measured at the soma (s), nucleus (n), and different distances from the soma (in micrometers), are shown in thetop traces. The bottom trace shows the whole-cell current over the same time period. A 30 mV hyperpolarizing step was applied during the tetanus to ensure that voltage-gated channels were not activated during the tetanus. d, In neurons in which calcium waves evoked by brief (10–20 pulses at 33 Hz) synaptic stimulation did not propagate to the nucleus, the propagation distance could be increased by increasing the number of stimuli (data not shown) or application of eserine. e, Synaptic stimulation of cholinergic afferents amplifies of action potential-induced calcium transients. The nuclear calcium rise to a train of four action potentials, plotted as ΔF/F, was greater when action potentials immediately followed synaptic stimulation (33 Hz, 1 sec;thick traces) than without previous synaptic stimulation (thin traces). Sequential application of eserine, MCPG, and atropine in the presence of CNQX and APV demonstrate that the amplification was attributable to activation of cholinergic synapses.
Synaptically evoked calcium waves were not blocked by the addition of CNQX and APV (eight of nine cells) or the metabotropic glutamate receptor antagonist MCPG (1 mm; n = 2) but were blocked by the muscarinic antagonist atropine (1 μm; five of six). Consistent with the waves being evoked by acetylcholine, rises in response to synaptic stimulation were enhanced by increasing the number of stimuli or by addition of the acetylcholinesterase blocker eserine (1–2 μm;n = 6) (Fig. 6d).
In cells in which cholinergic stimulation reduced the AHP, an amplification of action potential-induced calcium rise was also observed. The amplitude of the calcium transient to a train of action potentials (4–10 spikes 20 Hz) after synaptic activation was 22 ± 3% greater in the nucleus, 30 ± 14% greater in the soma, and 17 ± 14% greater in the proximal dendrite than the additive response of the unpaired tetanus and action potential train (n = 3) (Fig. 6e). These effects of cholinergic stimulation were unaffected by glutamate receptor antagonists, potentiated by eserine, but fully blocked by atropine (n = 3) (Fig. 6e), showing that they are attributable to activation of muscarinic receptors.
Involvement of intracellular calcium stores
All five types of muscarinic receptor (m1 to m5) are expressed in hippocampal pyramidal neurons (Levey et al., 1991, 1995). Because m1 and m3 receptors are coupled to phospholipase C and lead to generation of InsP3 (Caulfield, 1993), we tested whether the changes in calcium dynamics resulting from muscarinic stimulation are attributable to activation of InsP3-sensitive calcium stores. The uptake of calcium into InsP3-sensitive and ryanodine-sensitive stores is mediated by a Ca2+-ATPase (Henzi and MacDermott, 1991), which can be blocked by CPA and thapsigargin, thus emptying these stores. Application of muscarine in the presence thapsigargin (100 nm; n = 3) (Fig.7a,b) or CPA (30 μm; n = 5) (Fig.7c,d) led to a depolarization and blockade of the AHP and reduction in spike frequency adaptation. However, calcium waves (Fig. 7a,c) and amplification of action potential-evoked calcium rises (Fig. 7b,d) were blocked after application of thapsigargin or CPA. To confirm that CPA had indeed emptied the InsP3-sensitive stores, neurons were loaded with 20–50 μm caged InsP3. Photolytic uncaging of InsP3 readily evoked a large calcium rise in all cells. This response to InsP3 was blocked by CPA (Fig. 7e), confirming that InsP3-sensitive stores had been depleted. It should be noted that intracellular calcium stores were not depleted after muscarinic stimulation because the calcium response in response to uncaging of InsP3 after muscarinic stimulation was not blocked (Fig. 7e).
Fig. 7.

Intracellular calcium stores are required for generation of calcium waves and the amplification of action potential-evoked calcium rises. a, In the presence of thapsigargin (100 nm), application of muscarine reduced spike frequency adaptation and blocked the slow afterhyperpolarization (inset). Action potentials were evoked using a 200 msec, 300 pA depolarizing current injection. b, Oregon Green fluorescence recorded in response to a train of action potentials (shown in a) from the soma and proximal dendrite before (thin traces) and after (thick traces) application of muscarine. No regenerative rises in calcium were seen after the application of muscarine. c, Calcium rise in response to muscarinic stimulation is completely blocked in the presence of the calcium ATPase blocker cyclopiazonic acid.d, the amplification of action potential-induced calcium rises in the presence of muscarine are also blocked by cyclopiazonic acid. In each case, the control response is shown as the thin line, and the response in muscarine is shown as thethick trace. e, Photolysis of caged InsP3 causes a rapid rise in cytosolic calcium that is only slightly affected after muscarinic stimulation (thick trace) but blocked by application of CPA, confirming that InsP3-sensitive stores have been emptied in CPA.
Caffeine at high concentrations reversibly antagonizes the actions of InsP3 (Parker and Ivorra, 1991). Application of muscarine in the presence of caffeine (10 mm) blocked the AHP. However, no calcium waves or regenerative calcium rises in response to action potential trains were seen (Fig.8a,b). After washout of caffeine, a second application of muscarine led to regenerative calcium rises (Fig. 8). Finally, cells were loaded with the InsP3-receptor antagonist heparin (Ghosh et al., 1988) by passive diffusion from the patch pipette. The response to uncaging InsP3 was completely blocked by heparin (Fig. 8d), showing that InsP3receptors had been inhibited. Calcium waves were never observed when muscarine was applied in the presence of heparin (n = 7) (Fig. 8c). The electrophysiological properties of cells loaded with heparin were indistinguishable from control neurons; application of muscarine to these cells caused a membrane depolarization, blockade of the AHP (Fig. 8e), and reduced spike frequency adaptation, showing that heparin had not disrupted coupling of muscarinic receptors to G-protein-mediated second-messenger systems. However, the regenerative changes in intracellular calcium evoked by trains of action potentials were fully blocked in heparin-loaded cells (Fig. 8c). In heparin-loaded cells, muscarine reduced the average peak calcium rise to a train of action potentials by 27 ± 5% in the nucleus and 29 ± 10% in the soma. The integrated area of the calcium rise to a train of action potentials was reduced by 16 ± 17% in the nucleus and 12 ± 18% in the soma. Together, these results indicate that calcium waves and amplification of action potential-induced calcium rises is attributable to regenerative calcium release from InsP3-sensitive stores.
Fig. 8.

Inositol trisphosphate receptor activation is required for generation of calcium waves and the amplification of action potential-evoked calcium rises. a, Muscarine-evoked calcium waves were reversibly blocked by 10 mm caffeine. Rises in calcium are plotted as ΔF/F in arbitrary units (A.U.). Nuc, Nucleus;Dend, dendrite. b, Superimposed somatic calcium transients, plotted as ΔF/F, in response to a train of action potentials are shown in the presence of caffeine (thin trace) and after addition of muscarine (thick trace). After washout of caffeine (traces onright), reapplication of muscarine to the same cell now evokes a large regenerative calcium rise. c, When heparin (500 μg/ml) was included in the internal solution, calcium waves (top traces) and action potential-induced amplification (bottom traces) were not observed after application of muscarine. d, In heparin-loaded cells, InsP3 receptors are blocked, as shown by the lack of response to uncaging of InsP3. e, Muscarine application still blocked the AHP evoked by a train of action potentials in heparin-loaded cells, showing that heparin did not disrupt the activation of second-messenger systems.
Because RyRs are also present in hippocampal pyramidal neurons (Sharp et al., 1993), we tested whether the RyR-sensitive calcium stores were also involved in the regenerative calcium response. Application of muscarine in the presence of ryanodine (10 μm) failed to produce a calcium wave or amplification of the action potential-induced calcium rises (n = 5) (Fig.9a,b). In contrast, waves were readily observable when muscarine was applied in the presence of the RyR antagonists ruthenium red (three of four) or dantrolene (two of four). The calcium amplification of the action potential train-induced calcium rise was also not occluded by ruthenium red or dantrolene (Fig. 9a,b). Ryanodine at low concentrations depletes RyR-sensitive calcium stores by locking the RyRs in the open state (Rousseau et al., 1987). To test whether the ryanodine blockade of the muscarinic calcium amplification may be the result of depletion of the InsP3 store, we tested the status of the InsP3 stores by uncaging InsP3. The response to uncaged InsP3 was blocked by ryanodine but not by ruthenium red or dantrolene (Fig. 9c), indicating that ryanodine and InsP3 receptors in CA1 neurons share a common calcium pool. These results indicate that blockade of muscarinic calcium response by ryanodine is not attributable to the block of ryanodine receptors.
Fig. 9.

Ryanodine receptors share the same receptor store as InsP3 receptors but are not required for calcium waves or amplification of action potential-induced calcium transients. Application of ryanodine (10 μm) blocks muscarine-induced calcium waves (a) and amplification of action potential-induced calcium transients (b). In contrast, neither calcium waves (top) nor amplification (bottom traces) are blocked by dantrolene (100 μm). The control response is shown by the thin lines, and the response in the presence of ryanodine and dantrolene are shown as the thick traces.c, Photolytic uncaging of InsP3 leads to release of calcium from an intracellular store (baseline response) that is abolished by application of ryanodine but is unaffected in the presence of the ryanodine receptor antagonist dantrolene (trace on right). A.U., Arbitrary units.
DISCUSSION
These results show that activation of muscarinic receptors on hippocampal pyramidal neurons causes a rise of cytosolic calcium that begins in the apical dendrite, propagates toward the soma as a wave, and invades the nucleus. Pairing trains of action potentials with muscarinic stimulation leads to an amplification of cytosolic and nuclear calcium by its synergistic release from intracellular stores. Potentiation of the calcium response to trains of action potentials in the presence of muscarinic agonists has been reported previously in CA1 pyramidal neurons (Müller and Connor, 1991; Tsubokawa and Ross, 1997; Beier and Barish, 2000). However, this effect was suggested to be attributable to a combination of reduced spike frequency adaptation and enhancement of backpropagation of action potentials into the dendritic tree (Müller and Connor, 1991; Tsubokawa and Ross, 1997) or attributable to alterations in Ca2+clearance from the cytosol (Beier and Barish, 2000). We showed that both calcium waves and action potential-evoked release are inhibited by antagonists of InsP3 receptors and by emptying intracellular calcium stores with CPA or thapsigargin, indicating that they involve the release of calcium from InsP3-sensitive intracellular stores.
An InsP3-mediated dendritic calcium rise has been shown recently in response to metabotropic glutamate receptors in CA1 pyramidal (Nakamura et al., 1999, 2000) and cerebellar Purkinje (Finch and Augustine, 1998; Takechi et al., 1998) neurons. However, calcium waves evoked by metabotropic glutamate stimulation are confined to the dendritic tree. In CA1 pyramidal neurons, calcium waves evoked by metabotropic glutamate stimulation are also initiated in the proximal apical dendrite, ∼30–50 μm from the soma. Cholinergic stimulation of distal dendrites did not evoke a calcium rise. Interestingly, cholinergic stimulation of the proximal apical dendrite is necessary for cholinergic blockade of the AHP (Egorov et al., 1999). This initiation site may reflect a differential distribution of metabotropic receptors and/or InsP3 receptors. Type I metabotropic glutamate receptors, which generate InsP3, are located on the dendrites (Lujan et al., 1996). However, neither muscarinic receptors (Levey et al., 1995) nor InsP3 receptors (Sharp et al., 1993) show any obvious somatodendritic distribution gradient. The dendritic origin of the calcium wave may instead be attributable to a faster rise in the concentration of InsP3 attributable to the smaller volume of the dendritic compartment. Interestingly, dendritic spines, which contain IP3 receptors (Sala et al., 2001) and are thought to compartmentalize calcium (Sabatini et al., 2001), first appear 30–50 μm from the soma.
Because InsP3 and calcium act as coagonists at InsP3 receptors (Bezprozvanny et al., 1991; Finch et al., 1991; Mak et al., 1999), propagation of the calcium wave into the soma and nucleus may require both calcium and generation of InsP3 in the soma by somatic cholinergic receptors. Ryanodine receptors are not required for calcium waves or the amplification of action potential-evoked calcium transients, although they share a common intracellular calcium pool. Similarly, in cerebellar Purkinje neurons, ryanodine and InsP3-sensitive calcium stores share a common calcium pool (Khodakhah and Armstrong, 1997). Although not necessary for the regenerative calcium response, it is possible that calcium-induced calcium release from ryanodine receptor activation also occurs in concert with calcium release from InsP3receptors.
Action potentials lead to a rise in free calcium by opening voltage-dependent calcium channels (Markram et al., 1995). In the presence of muscarinic agonists, there was a reduction in the calcium transient in response to single action potentials. Activation of muscarinic receptors has been shown previously to reduce the amplitude of voltage-activated calcium currents in CA3 pyramidal neurons (Gahwiler and Brown, 1987). The smaller peak amplitude of the calcium transient in the presence of muscarinic agonists likely reflects this reduction of the calcium current. In contrast to single spikes, trains of action potentials evoked regenerative, prolonged rises in calcium in the presence of muscarinic agonists. Type I and type II InsP3 receptors show a bell-shaped dose–response curve to free calcium (Bezprozvanny et al., 1991; Finch et al., 1991) so that, in the presence of InsP3, increases in free calcium cause a positive feedback and amplification of calcium release (Mak et al., 1999). Thus, the regenerative component of the calcium response is most likely attributable to calcium-induced calcium release via InsP3 receptors (Taylor and Marshall, 1992; Nakamura et al., 1999). The observation that the regenerative increases is seen after action potential trains but not after single action potentials (Yamamoto et al., 2000) is consistent with this idea.
The cholinergic system has been implicated in a variety of cognitive tasks involving learning and memory. Its role in the pathophysiology of memory loss such as in Alzheimer's disease is well recognized (Bartus et al., 1982). Extracellular ACh concentrations rise in the hippocampus by as much as fourfold during a variety of hippocampal-dependent learning tasks (Stancampiano et al., 1999; Nail-Boucherie et al., 2000). It is now clear that long-term synaptic changes associated with memory and learning require new gene expression (Ghosh and Greenberg, 1995; Berridge, 1998), and rises in nuclear calcium levels form an integral part of the mechanisms responsible for activation of cascades, which lead to new gene transcription (Dolmetsch et al., 1998; Li et al., 1998; Hardingham et al., 2001). Our finding that cholinergic stimulation leads to rises in nuclear calcium and amplification of action potential-induced calcium transients in the nucleus may explain the role of cholinergic system in regulating memory. The nuclear envelope is well known to be continuous with the smooth endoplasmic reticulum and has been shown to have both calcium stores and InsP3 receptors (Malviya et al., 1990; Nicotera et al., 1990). Thus, it is possible that the rise in nuclear calcium reported here may be attributable to calcium release from these stores in the nuclear envelope. These results suggest that the cholinergic stimulation amplifies and may improve signal-to-noise ratio for stimulus transcription coupling dependent on nuclear calcium signals.
Footnotes
Correspondence should be addressed to Pankaj Sah, Division of Neuroscience, John Curtin School of Medical Research, GPO Box 334, Canberra, ACT 2601, Australia. E-mail: pankaj.sah@anu.edu.au.
REFERENCES
- 1.Bartus RT, Dean RL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–417. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 2.Beier SM, Barish ME. Cholinergic stimulation enhances cytosolic calcium ion accumulation in mouse hippocampal CA1 pyramidal neurones during short action potential trains. J Physiol (Lond) 2000;526:129–142. doi: 10.1111/j.1469-7793.2000.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berridge MJ. Neuronal calcium signalling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 4.Bezprozvanny I, Watras J, Ehrilich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 5.Caulfield MP. Muscarinic receptors-characterisation, coupling and function. Pharmacol Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- 6.Cole AE, Nicoll RA. Characterization of a slow cholinergic postsynaptic potential recorded in vitro from rat hippocampal cells. J Physiol (Lond) 1984;352:173–188. doi: 10.1113/jphysiol.1984.sp015285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 8.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 9.Disterhoft JF, Kronforst-Collins M, Oh MM, Power JM, Preston AR, Weiss C. Cholinergic facilitation of trace eyeblink conditioning in aging rabbits. Life Sci. 1999;64:541–548. doi: 10.1016/s0024-3205(98)00599-2. [DOI] [PubMed] [Google Scholar]
- 10.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 11.Egorov AV, Gloveli T, Müller W. Muscarinic control of dendritic excitability and Ca2+ signaling in CA1 pyramidal neurons in rat hippocampal slice. J Neurophysiol. 1999;82:1909–1915. doi: 10.1152/jn.1999.82.4.1909. [DOI] [PubMed] [Google Scholar]
- 12.Fellous JM, Sejnowski TJ. Cholinergic induction of oscillations in the hippocampal slice in the slow (0.5–2 Hz), theta (5–12 Hz), and gamma (35–70 Hz) bands. Hippocampus. 2000;10:187–197. doi: 10.1002/(SICI)1098-1063(2000)10:2<187::AID-HIPO8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 13.Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- 14.Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- 15.Finkbeiner S, Greenberg ME. Ca2+ channel-regulated neuronal gene expression. J Neurobiol. 1998;37:171–189. [PubMed] [Google Scholar]
- 16.Fisher SK, Heacock AM, Agranoff BW. Inositol lipids and signal transduction in the nervous system: an update. J Neurochem. 1992;58:18–38. doi: 10.1111/j.1471-4159.1992.tb09273.x. [DOI] [PubMed] [Google Scholar]
- 17.Fowler CJ, Tiger G. Modulation of receptor-mediated inositol phospholipid breakdown in the brain. Neurochem Int. 1991;19:171–206. [Google Scholar]
- 18.Furey ML, Pietrini P, Haxby JV. Cholinergic enhancement and increased selectivity of perceptual processing during working memory. Science. 2000;290:2315–2319. doi: 10.1126/science.290.5500.2315. [DOI] [PubMed] [Google Scholar]
- 19.Gahwiler BH, Brown DA. Muscarine affects calcium currents in rat hippocampal pyramidal cells in vitro. Neurosci Lett. 1987;76:301–306. doi: 10.1016/0304-3940(87)90419-8. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh A, Greenberg ME. Calcium signalling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–246. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. J Biol Chem. 1988;15:11075–11079. [PubMed] [Google Scholar]
- 22.Hagan JJ, Morris RGM. The cholinergic hypothesis of memory: a review of animal experiments. In: Iversen LL, Iversen SD, Snyder SH, editors. Handbook of psychopharmacology. Plenum; New York: 1988. pp. 237–323. [Google Scholar]
- 23.Hardingham GE, Arnold FJL, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 24.Hasselmo ME, Bower JM. Acetylcholine and memory. Trends Neurosci. 1993;16:218–222. doi: 10.1016/0166-2236(93)90159-j. [DOI] [PubMed] [Google Scholar]
- 25.Henzi V, MacDermott AB. Characteristics and function of Ca2+- and inositol 1,4,5-trisphosphate-releasable stores of Ca2+ in neurons. Neuroscience. 1991;46:251–273. doi: 10.1016/0306-4522(92)90049-8. [DOI] [PubMed] [Google Scholar]
- 26.Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- 27.Kandel ER, Pittenger C. The past, the future and the biology of memory storage. Philos Trans R Soc B Biol Sci. 1999;354:2027–2052. doi: 10.1098/rstb.1999.0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasa P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer's disease. Prog Neurobiol. 1997;52:511–535. doi: 10.1016/s0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- 29.Khodakhah K, Armstrong CM. Inositol trisphosphate and ryanodine receptors share a common functional Ca2+ pool in cerebellar purkinje neurons. Biophys J. 1997;73:3349–3357. doi: 10.1016/S0006-3495(97)78359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of m1–m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis PR, Shute CC. The cholinergic limbic system: projections to hippocampal formation, medial cortex, nuclei of the ascending cholinergic reticular system, and the subfornical organ and supra-optic crest. Brain. 1967;90:521–540. doi: 10.1093/brain/90.3.521. [DOI] [PubMed] [Google Scholar]
- 33.Li W-H, Llopis J, Whitney M, Zlokarnik G, Tsien RY. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimise gene expression. Nature. 1998;392:936–941. doi: 10.1038/31965. [DOI] [PubMed] [Google Scholar]
- 34.Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 35.Mak D-OD, McBride S, Foskett JK. Inositol 1,4,5-tris-phosphate activation of inositol tris-phosphate receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc Natl Acad Sci USA. 1999;95:15821–15825. doi: 10.1073/pnas.95.26.15821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malviya A, Rogue P, Vincendon G. Stereospecific inositol 1,4,5-[32P]trisphosphate binding to isolated rat liver nuclei: evidence for inositol trisphosphate receptor-mediated calcium release from the nucleus. Proc Natl Acad Sci USA. 1990;87:9270–9274. doi: 10.1073/pnas.87.23.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. J Physiol (Lond) 1995;485:1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Müller W, Connor JA. Cholinergic input uncouples Ca2+ changes from K+ conductance activation and amplifies intradendritic Ca2+ changes in hippocampal neurons. Neuron. 1991;6:901–905. doi: 10.1016/0896-6273(91)90230-w. [DOI] [PubMed] [Google Scholar]
- 39.Müller W, Misgeld U, Heinemann U. Carbachol effects on hippocampal neurons in vitro: dependence on the rate of use of carbachol tissue concentration. Exp Brain Res. 1988;77:1–12. doi: 10.1007/BF00250251. [DOI] [PubMed] [Google Scholar]
- 40.Nail-Boucherie K, Dourmap N, Jaffard R, Costentin J. Contextual fear conditioning is associated with an increase of acetylcholine release in the hippocampus of rat. Brain Res Cogn Brain Res. 2000;9:193–197. doi: 10.1016/s0926-6410(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura T, Barbar J-G, Nakamra K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura T, Nakamura K, Lasser-Ross N, Barbara J-G, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in the mammalian central nervous system. Physiol Rev. 1989;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- 44.Nicotera P, Orrenius S, Nilsson T, Berggren P. An inositol 1,4,5-trisphosphate-sensitive Ca2+ pool in liver nuclei. Proc Natl Acad Sci USA. 1990;87:6858–6862. doi: 10.1073/pnas.87.17.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Malley DM. Calcium permeability of the neuronal nuclear envelope: evaluation using confocal volumes and intracellular perfusion. J Neurosci. 1994;14:5741–5758. doi: 10.1523/JNEUROSCI.14-10-05741.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parker I, Ivorra I. Caffeine inhibits inositol tisphosphate-mediated liberation of intracellular calcium in Xenopus oocytes. J Physiol (Lond) 1991;422:229–240. doi: 10.1113/jphysiol.1991.sp018423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogue PJ, Malviya AN. Calcium signals in the cell nucleus. EMBO J. 1999;18:5147–5152. doi: 10.1093/emboj/18.19.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rousseau E, Smith JS, Meissner G. Ryanodne modifies conductance and gating behaviour of single Ca2+ release channel. Am J Physiol. 1987;253:C364–C368. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- 49.Sabatini BL, Maravall M, Svoboda K. Ca2+ signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- 50.Sah P, Clements JD. Photolytic manipulation of [Ca2+]i reveals slow kinetics of potassium channels underlying the afterhyperpolaristion in hippocampal pyramidal neurons. J Neurosci. 1999;19:3657–3664. doi: 10.1523/JNEUROSCI.19-10-03657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sah P, Isaacson JS. Channels underlying the slow afterhyperpolarization in hippocampal pyramidal neurons: neurotransmitters modulate the open probability. Neuron. 1995;15:435–441. doi: 10.1016/0896-6273(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 52.Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31:115–130. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- 53.Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol (Lond) 1995;487:583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH. Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in the rat brain. J Neurosci. 1993;13:3051–3063. doi: 10.1523/JNEUROSCI.13-07-03051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shulz DE, Sosnik R, Ego V, Ahissar E. A neuronal analogue of state-dependent learning. Nature. 2000;403:549–553. doi: 10.1038/35000586. [DOI] [PubMed] [Google Scholar]
- 56.Stancampiano R, Cocco S, Cugusi C, Sarais L, Fadda F. Serotonin and acetylcholine release response in the rat hippocampus during a spatial memory task. Neuroscience. 1999;89:1135–1143. doi: 10.1016/s0306-4522(98)00397-2. [DOI] [PubMed] [Google Scholar]
- 57.Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- 58.Taylor CW, Marshall CB. Calcium and inositol 1,4,5-trisphosphate receptors: a complex relationship. Trends Biochem Sci. 1992;17:403–407. doi: 10.1016/0968-0004(92)90009-x. [DOI] [PubMed] [Google Scholar]
- 59.Tsien RW, Tsien RY. Calcium channels, stores, and oscillations. Annu Rev Cell Biol. 1990;6:715–760. doi: 10.1146/annurev.cb.06.110190.003435. [DOI] [PubMed] [Google Scholar]
- 60.Tsubokawa H, Ross WN. Muscarinic modulation of spike backpropagation in the apical dendrites of hippocampal CA1 pyramidal neurons. J Neurosci. 1997;17:5782–5791. doi: 10.1523/JNEUROSCI.17-15-05782.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamamoto K, Hashimoto K, Isomura Y, Shimohama S, Kato N. An IP3-assisted form of Ca2+-induced Ca2+ release in neocortical neurons. NeuroReport. 2000;11:535–539. doi: 10.1097/00001756-200002280-00022. [DOI] [PubMed] [Google Scholar]