

Abstract
Syntaxin 1A/HPC-1 is a key component of the exocytotic molecular machinery, namely, the solubleN-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor mechanism. Although >10 syntaxin-binding proteins have been identified, they cannot completely explain the regulation of exocytosis. Thus, novel proteins may interact with syntaxin. Because exocytosis requires both Ca2+ and ATP, we searched for Ca2+/ATP-dependent syntaxin-binding proteins from the rat brain and discovered Ca2+/calmodulin-activated protein kinase II (CaMKII)-α. At Ca2+ concentrations of >10−6m, only autophosphorylated CaMKII bound to syntaxin. Bound CaMKII was released from syntaxin by EGTA or by phosphatase, indicating that the binding is reversible. CaMKII bound to the linker domain of syntaxin, unlike any other known syntaxin-binding proteins. CaMKII–syntaxin complexes were also detected in synaptosomes by immunoprecipitation, and when reconstitutedin vitro, they recruited larger amounts of synaptotagmin and SNAP-25 than syntaxin alone. The microinjected CaMKII-binding domain of syntaxin specifically affected exocytosis in chromaffin cells and in neurons. These results indicate that the Ca2+/ATP-dependent binding of CaMKII to syntaxin is an important process in the regulation of exocytosis.
Keywords: syntaxin 1A/HPC-1, CaMKII, SNARE mechanism, exocytosis, linker domain, Munc-18
Studies of yeast and mammals have identified the protein components involved in the molecular machinery controlling exocytosis and have clarified this process (Jahn and Südhof, 1999; Brunger, 2000; Misura et al., 2000a). The solubleN-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) mechanism has become the standard explanation of the molecular processes of exocytosis (Jahn and Südhof, 1999;Brunger, 2000). Among the identified protein components, syntaxin homologs are some of the most important molecules in vesicular trafficking (Bennett et al., 1993). Several lines of biochemical and electrophysiological evidence indicate that syntaxin 1A/HPC-1 is a key component of the SNARE mechanism (Inoue et al., 1992; Marsal et al., 1997; O'Connor et al., 1997). Although syntaxin and another synaptic protein interact (Jahn and Südhof, 1999), the SNARE-dependent regulation of exocytosis is not yet completely understood. In particular, most of the known interactions between syntaxin and other proteins are independent of Ca2+ or ATP, both of which are essential to exocytosis, and the manner in which Ca2+/ATP regulates the protein association and dissociation involving syntaxin during exocytosis remains unclear. We believe that the missing link in the molecular explanation of exocytosis is an unknown protein–protein interaction involving syntaxin that is regulated by Ca2+/ATP and is essential to the molecular regulation of exocytosis.
We searched for such a protein in the rat brain using a glutathione S-transferase (GST)-syntaxin pull-down study and screened brain syntaxin-binding proteins in the presence of Ca2+/ATP. We found a 50 kDa protein that specifically binds to syntaxin in a Ca2+/ATP-dependent manner, which we called Ca2+/calmodulin-activated protein kinase II (CaMKII)-α. At Ca2+ concentrations of >10−6m, only the autophosphorylated form of CaMKIIα bound to syntaxin. We then demonstrated that the binding site for CaMKII resides in the linker domain of syntaxin, where no other known syntaxin-binding proteins bind. Immunoprecipitation showed that the CaMKII–syntaxin complex formed in the presynaptic terminal recruited larger amounts of synaptotagmin and SNAP-25 than syntaxin alone. Finally, we showed that the microinjected CaMKII-binding domain of syntaxin specifically decreased the frequency of exocytosis in chromaffin cells and in neurons, probably because of its interference with the endogenous CaMKII–syntaxin complex. These results indicate that the binding of CaMKII to syntaxin is an important step in the regulation of exocytosis.
MATERIALS AND METHODS
Recombinant proteins. The cDNA fragments corresponding to the various regions of syntaxin 1A and those encoding syntaxin-binding proteins were amplified by PCR and inserted into pGEX-6P-1 (Amersham-Pharmacia Biotech, Uppsala, Sweden) in frame with GST. Site-directed mutagenesis proceeded as described byImai et al. (1991). The cDNAs encoding synaptotagmin, vesicle-associated membrane protein (VAMP), SNAP-25, and the glutamate receptor NR2A (Ikeda et al., 1992) were provided by Drs. T. Abe and K. Sakimura (Brain Research Institute, Niigata University, Niigata, Japan). In vitro translation was performed using the TNT SP6-coupled reticulocyte lysate system (Promega, Madison, WI), and the translated protein was labeled using biotinylated tRNA (Promega). The cDNA encoding rat CaMKIIα or T286A-CaMKIIα (provided by Dr. H. Schulman, Stanford University, Stanford, CA) was added to the above system, and the proteins were expressed after a 1.5 hr incubation at 30°C. The binding of the labeled proteins was detected as described previously (Ohyama et al., 2001).
Protein-binding studies. The rat brain S2 fraction was prepared as described previously (Fujita et al., 1998). GST alone or GST-fusion proteins produced byEscherichia coli bound to glutathione Sepharose were incubated with the S2 fraction from the adult rat brain at 4°C for 1 hr with or without 1 mmCaCl2 and/or 0.5 mm ATP, and then digested with PreScission protease (Amersham-Pharmacia Biotech). After cleavage, the supernatant was incubated with glutathione Sepharose to remove the protease. Purified CaMKIIα from the rat brain was also autophosphorylated (see below) and incubated with GST–syntaxin as described above. Recombinant CaMKIIα and CaMKIIβ, obtained using the baculovirus-Sf21 cell system (Kolb et al., 1998), were provided by Dr. Neal Waxham (University of Texas, Houston, TX). CaMKIIα lacking the association domain [1–325] (i.e., monomeric CaMKII) was purchased from Calbiochem (La Jolla, CA). Anti-CaMKII polyclonal antibody (pAb) was a generous gift from Prof. E. Miyamoto (Kumamoto University School of Medicine, Kumamoto, Japan). Anti-syntaxin 1A monoclonal antibody (mAb) was provided by Dr. M. Takahashi (Mitsubishi Institute of Life Science, Machida, Tokyo, Japan).
Determination of the partial primary structure of the 50 kDa syntaxin-binding protein. The 50 kDa syntaxin-binding protein was excised and digested by trypsin, and its partial structure was then analyzed by mass spectrometry using the nanospray method. Sequence tags from the two fragment ions confirmed that this protein was rat CaMKIIα (FTEEYQLFEELGK [9–21] and ITQYLDAGGIPR [434–445]).
Autophosphorylation and dephosphorylation of CaMKII. After dephosphorylation by protein phosphatase 1 (PP1; Calbiochem) at 30°C for 30 min, the purified CaMKIIα was re-autophosphorylated again for 5 min (Katoh and Fujisawa, 1991) and incubated with GST–syntaxin. In some experiments, the autophosphorylated CaMKII was bound to immobilized GST–syntaxin and then incubated with the EGTA treatment buffer (CaCl2 in the binding buffer was replaced with 10 mm EGTA) or with PP1 for 30 min. The remaining bound CaMKII was visualized by immunoblotting.
Activity of CaMKII in the presence of syntaxin. We examined whether or not the autophosphorylation of CaMKII is altered by syntaxin binding. Autophosphorylation proceeded in the absence or presence of the CaMKII-binding fragment of syntaxin ([145–184]; 20 μm) as described by Katoh and Fujisawa (1991). Ca2+/CaM-dependent synaptosomal protein was phosphorylated by the method of Popoli et al. (1995). Syntaxin and synaptotagmin I were phosphorylated by CaMKII in vitro by the method of Hilfiker et al. (1999). The32P-labeled proteins were resolved by electrophoresis and quantified using a BAS 2000 system (Fuji Film Co., Tokyo, Japan).
Affinity between syntaxin and CaMKII. The binding of syntaxin 1A [1–262], [145–184], and [1–262; R151G] to recombinant CaMKIIα or CaMKIIβ was monitored using a BIAcore system (BIAcore Ltd., Uppsala, Sweden) by the method of Suetsugu et al. (2001). The BIAcore system was operated at 25°C and at a constant flow of 5 ml/min of running buffer containing 10 mm HEPES-NaOH, pH 7.5, 150 mm NaCl, 3 mm EDTA, and 0.005% Tween 20. CaMKII was fixed on the CM5 sensor chip, and recombinant syntaxin fragments were then individually diluted to ∼4 mg/ml running buffer and injected over the chip. The efficiency of immobilization was confirmed by an increase in resonance units to ∼1000. After washing with high-salt buffer (running buffer plus 1m MgCl2), the binding of recombinant syntaxin at various concentrations loaded onto the sensor chip was monitored. Specific binding was calculated by subtracting the resonance units found in controls (background binding) from those detected in samples. The apparent association (Ka) and dissociation (Kd) rate constants were estimated using linear transformation of the biosensor curves. The primary data were analyzed using BIA evaluation software (BIAcore, Inc.). Because the model assumes that the kinetics of the syntaxin–CaMKII interaction are pseudo-first order, the binding rate equation is dR/dt =KaCRmax − (KaC +Kd)R, where R is the signal response, Rmax is maximum response level, and C is the molar concentration of CaMKII.
Immunoprecipitation. Synaptosomes were prepared from the adult rat brain as described previously (Pevsner et al., 1994; Igarashi et al., 1997, 2000). The buffer composition for immunoprecipitation was 20 mm HCl, pH 7.6, 150 mmNaCl, 1% Nonidet P-40, 1 mg/ml BSA, 1 mmCaCl2, 2 mmMgCl2, 0.5 mm ATPγS, and protease inhibitor mixtures. Complexes were immunoprecipitated as described previously (Pevsner et al., 1994; Igarashi et al., 1997), except that the washing buffer contained (in mm): 1 CaCl2, 2 MgCl2, and 0.5 ATPγS. Glycerol gradient experiments were performed as described byPevsner et al. (1994).
In vitro reconstitution of syntaxin-CaMKII complex. Purified CaMKII complexed with GST–syntaxin 1A [1–262] was immunoprecipitated using anti-CaMKII antibody, incubated with SNAP-25 and synaptotagmin, resolved by SDS-PAGE, and immunoblotted. The amount of immunoprecipitated GST–syntaxin was 0.2 μg. Control GST–syntaxin alone was immobilized and incubated with SNAP-25 and synaptotagmin.
Amperometry of chromaffin cells. Exocytotic events in single cells were measured by amperometry as follows: chromaffin cells were cultured on collagen-coated coverslips. Recordings were taken using carbon fiber electrodes (Dagan Corp., Minneapolis, MN), and the reference electrodes were silver wires coated with AgCl. The cytosol of 50–150 6-d-old cultured cells was microinjected for 0.2 sec at 10 kPa of pressure using an Eppendorf injection system. The estimated cytosolic concentration of the injected fragments was 60–120 μg/ml. Release of Ca from single cells induced by a pressure ejection of 0.1 mm nicotine was determined using an amperometer 30 min after microinjection.
Electrophysiology of cultured superior cervical ganglion neurons after microinjection of recombinant proteins. Conventional intracellular recordings were taken from pairs of neighboring superior cervical ganglion (SCG) neurons cultured for 4–5 weeks. Postsynaptic responses were recorded from one neuron when action potentials were generated in the others by passing a current through an intracellular recording electrode once every 5 sec (0.2 Hz). The recombinant proteins dissolved in the solution (in mm: 150 K-acetate, 5 Mg2+-ATP, and 10 HEPES, pH 7.4) in the glass suction pipette were introduced into the presynaptic cell body by diffusion from the pipette as described previously (Mochida et al., 1998). Data were analyzed using software written by L. Tauc (Centre National de la Recherche Scientifique, Gif sur Yvette, France). The resultant values were smoothed by an eight point moving-average algorithm and plotted against recording time, with t = 0 indicating the start of presynaptic injection. The EPSPs of one representative experiment, in which values were recorded 5 min before injection and 50 and 90 min thereafter, are illustrated (see Fig.10A). EPSPs were recorded once every 5 sec, at 0.2 Hz. Normalized average EPSPs were obtained by plotting the results of five experiments using syntaxin-derived fragments.
Fig. 10.

Activity-dependent inhibition of synaptic transmission by the CaMKII-binding domain of syntaxin-1A in cultured SCG neurons. A, Syntaxin [145–184] was introduced into presynaptic neurons by diffusion from a pipette from the start of membrane disruption by suction at t = 0.B, EPSPs of neurons microinjected with the CaMKII-binding fragment were significantly lower than those of neurons microinjected with deletion mutant that was unable to bind CaMKII. Normalized average EPSPs were plotted from five experiments with syntaxin [145–184] (CaMKII-binding fragment;triangles) and with syntaxin [152–184] (fragment unable to bind CaMKII; Fig. 3A; circles). CaMKII-binding fragments of NR2A ([1389–1461];diamonds) and NR2B ([1290–1309];squares) glutamate receptors were also microinjected (Strack et al., 2000; Gardoni et al., 2001). EPSP amplitudes at 80 min after the start of injection with syntaxin [145–184] and syntaxin [152–184] were −39 ± 7.0% (n = 5; mean ± SEM) and −14 ± 5.8% (n = 5), respectively. psps, Postsynaptic potentials.
RESULTS
CaMKII binds to syntaxin only when autophosphorylated
To search for a protein that might be the missing link in the molecular explanation of exocytosis in the rat brain, we used a GST–syntaxin pull-down study and found a 50 kDa protein that specifically binds to syntaxin in a Ca2+/ATP-dependent manner (Fig.1A). This protein was identified as CaMKIIα by mass spectrometry (see Materials and Methods). Immunoblotting showed that CaMKII specifically binds to syntaxin (Fig. 1B) only in the presence of Ca2+ and ATP (Fig. 1B). In the same study, CASK, a presynaptic terminal protein homologous with the catalytic domain of CaMKII, did not bind to syntaxin (data not shown). Binding experiments using ATP analogs revealed that ATP and ATPγS, both of which are available to protein kinases (Takahashi et al., 1999), are effective at binding CaMKII and syntaxin (Fig.1C). CaMKII–syntaxin binding was completely Ca2+-dependent, whereas tomosyn or mammalian unc (Munc)-18 syntaxin complexes required Ca2+ concentrations of at least 10−6m for binding (Fig. 1D).
Fig. 1.

CaMKII binds syntaxin in a Ca2+/ATP-dependent manner. A, A 50 kDa protein, which bound syntaxin 1A specifically in a Ca2+/ATP-dependent manner, was found using a GST pull-down study. GST or GST–syntaxin 1A (4 nmol each) was immobilized to glutathione Sepharose for 1 hr at 4°C and incubated with (or without) 1 ml of brain S2 fraction (protein concentration was 10 mg/ml) for 1 hr at 4°C in the presence or absence of Ca2+/Mg-ATP. Next, GST fusion protein was cleaved by incubation with PreScission protease (10 U) for 1 hr at 4°C. After centrifugation, the supernatant was again incubated with glutathione Sepharose for 1 hr at 4°C and centrifuged to remove the protease. The supernatant was mixed with an equal volume of 2× SDS sample buffer and loaded onto 7.5% SDS-PAGE. The silver staining of the gel is shown.Lane 1, GST alone; lane 2, GST plus brain S2 fraction with Ca2+/Mg-ATP;lane 3, GST–syntaxin alone; lane 4, GST–syntaxin plus S2 fraction with Ca2+/Mg-ATP; lane 5, GST–syntaxin plus S2 fraction without Ca2+/Mg-ATP. The Ca2+/Mg-ATP conditions consisted of an incubation with (in mm): 1 CaCl2, 0.5 ATP, and 2 MgCl2. To reproduce these conditions in the absence of Ca2+/Mg-ATP, 1 mm EGTA and 2 mm EDTA were added instead of CaCl2 and MgCl2, and ATP was omitted. Molecular mass (in kilodaltons) is shown on the left, and the 50 kDa protein enriched in lane 4 is shown as anarrow. B, CaMKIIα in brain S2 fraction specifically binds syntaxin in the presence of Ca2+ and ATP. C, Nucleotide requirements for binding. Instead of ATPγS, 0.5 mm of each nucleotide was added to the incubation mixture. D, Ca2+ requirement for CaMKIIα binding to syntaxin. Various final concentrations of Ca2+ were added to the incubation mixture AMP-PNP, 5′-Adenylylimidodiphosphate. The syntaxin-binding protein fraction, eluted by PreScission protease (Amersham-Pharmacia Biotech), was resolved by 10% SDS-PAGE and immunoblotted using anti-CaMKII pAb (B–D) and anti-tomosyn (Fujita et al., 1998) or anti-Munc-18 pAb (D). Blots in Band C were stained with anti-CaMKII pAb.
We considered that an activation process involving CaMKII autophosphorylation (Braun and Schulman, 1995; Soderling et al., 2001) is necessary for binding. Autophosphorylated CaMKII bound syntaxin, whereas dephosphorylated CaMKIIα purified from the rat brain did not (Fig. 2A). T286A–CaMKIIα, a mutant lacking an autophosphorylation site, did not bind to syntaxin even in the presence of Ca2+/ATP (Fig. 2B). The CaMKIIα produced using in vitro translation also bound to syntaxin in a Ca2+-dependent manner (Fig.2C). CaMKII bound to syntaxin in a dose-dependent manner, suggesting that the binding is stoichiometric (Fig.2D). Among the three domains of CaMKII (i.e., the catalytic, regulatory, or association domains), the association domain is required for oligomerization (Kanaseki et al., 1991). When the association domain was deleted, CaMKII could not bind syntaxin, even after autophosphorylation (Fig. 2E). This indicates that CaMKII binding to syntaxin requires the oligomerization of CaMKII, as well as its autophosphorylation. After binding to syntaxin, EGTA (Fig. 3A) or the dephosphorylation of CaMKII (Fig. 3B) caused the release of CaMKII from the complex. These results suggest that binding between CaMKII and syntaxin is reversible and regulated by both [Ca2+] and the autophosphorylation–dephosphorylation cycle.
Fig. 2.

CaMKII binds to syntaxin only when autophosphorylated. A, Purified CaMKIIα also binds syntaxin after autophosphorylation. The fraction unbound to GST–syntaxin after re-autophosphorylation was not recognized by anti-autophosphorylated CaMKII antibody. CaMKIIα (5 μg) purified from rat brain was autophosphorylated (Auto-P) in buffer containing 50 mm HEPES-NaOH, pH 8.0, 8 mmMg(CH3COO)2, 0.25 mmCaCl2, 2 μm CaM, and 0.5 mm ATP for 5 min at 30°C, and then incubated with immobilized GST or GST–syntaxin (4 nmol) for 1 hr at 4°C. After centrifugation, the supernatant was collected as the unbound fraction. The PreScission protease fraction was collected as the bound fraction. Samples were resolved by 10% SDS-PAGE and immunoblotted using anti-CaMKIIα mAb or anti-autophosphorylated CaMKII mAb.B, T286A-CaMKIIα could not bind syntaxin.Top, Both wild-type CaMKIIα (wt) and T286A-CaMKIIα (T286A) were produced by in vitro translation. Bottom, Only wt CaMKIIα bound to syntaxin in the pull-down study in presence of Ca2+/ATP. The cDNA encoding rat CaMKIIα or T286A-CaMKIIα (provided by Dr. H. Schulman) was added to thein vitro translation kit (Promega). Proteins were expressed by incubating kit components for 1.5 hr at 30°C and then adding immobilized GST–syntaxin as above. After elution with SDS sample buffer, bound proteins were blotted and detected using streptavidin-conjugated alkaline phosphatase. C, CaMKIIα produced using in vitro translation also shows Ca2+ sensitivity for binding to syntaxin after autophosphorylation. Translation in vitro proceeded as described above, and CaMKIIα was autophosphorylated in buffer containing Tris-HCl, pH 7.6, 0.5 mmCaCl2, 2 μm CaM, 2 mmMgCl2, and 0.5 mm ATP at 30°C for 15 min. The autophosphorylated CaMKIIα was incubated with immobilized GST–syntaxin (4 nmol) at 4°C for 1 hr and then with binding buffer containing various concentrations of Ca2+ for an additional 1 hr. D, Dose-dependent binding of CaMKII to syntaxin. GST–syntaxin (4 nmol) was incubated with recombinant CaMKIIα at various concentrations. E, CaMKIIα lacking the association domain [1–325] (i.e., monomeric CaMKII) does not bind to syntaxin, even when autophosphorylated. The monomeric CaMKIIα was autophosphorylated and incubated with the immobilized GST–syntaxin as described above (A).
Fig. 3.

Dissociation of the autophosphorylated CaMKII from syntaxin 1A is regulated by decrease of [Ca2+] (A) and its dephosphorylation (B). In both A andB, anti-CaMKIIα (top) and anti-autophosphorylated CaMKIIα (bottom) antibodies were used in immunoblots. Purified rat brain CaMKIIα was autophosphorylated and incubated with immobilized GST–syntaxin as described above (A). Protein complex was additionally incubated with EGTA-treated buffer (see Materials and Methods) for 1 hr at 4°C (A) or with PP1 for 30 min at 25°C (B). Bound CaMKIIα was then eluted with PreScission protease and analyzed using anti-CaMKIIα mAb. EGTA (A) or CaMKII dephosphorylated by PP1 (B) after binding causes CaMKIIα release from syntaxin. Under these conditions, retained CaMKII accounted for only 18.7 ± 1.5% (A) and 14.3 ± 2.3% (B) of the control, respectively (four independent experiments each). In A, Munc-18 and tomosyn were compared with CaMKII under the same experimental conditions, and their binding to syntaxin was completely independent of Ca2+.
CaMKII-binding site is the linker domain of syntaxin
We determined which domain of syntaxin was responsible for binding. The core complex region [191–261] of syntaxin (Dulubova et al., 1999; Brunger, 2000; Misura et al., 2000b), where most syntaxin-binding proteins bind, was not the CaMKII-binding site. We found that the linker domain [145–184] of syntaxin was the site of CaMKII binding (Fig.4A). Until now, the minimal binding site was thought to be region 145–172 of syntaxin. The linker domain is believed to connect the core complex region to three N-terminus helical domains, but proteins that specifically bind to this portion have not been found (Dulubova et al., 1999; Brunger, 2000;Misura et al., 2000a) (Fig. 4B). With respect to the requirement of CaMKII autophosphorylation for binding, we found three basic amino acids (K146, R148, and R151) within the linker domain. Deletion of all three of these amino acids or the substitution of R151 alone resulted in a total loss of CaMKII binding (Fig. 4A). Within cytoplasmic syntaxin [1–262], CaMKII binding was selectively lost with R151G substitution, but the binding of other known syntaxin-binding proteins was unaffected (Fig. 4B). CaMKII was displaced from syntaxin [1–262] after addition of 10 μm syntaxin [145–184] (Fig. 4C). We tested other CaMKII-binding fragments derived from NR2A [1349–1461] (Gardoni et al., 2001) or NR2B [1290–1309] (Strack et al., 2000), both of which bind to CaMKII but do not inhibit syntaxin–CaMKII binding. These fragments did not interfere with CaMKII–syntaxin interaction (Fig. 4C), indicating that the CaMKII-binding fragment of syntaxin specifically inhibits CaMKII–syntaxin binding.
Fig. 4.

Determination of CaMKIIα-binding site in syntaxin. A, Binding visualized using anti-CaMKII antibody. Each recombinant fragment derived from syntaxin 1A as a GST-fusion protein (4 nmol) was immobilized to glutathione Sepharose, incubated with autophosphorylated purified CaMKIIα (5 μg), and then cleaved by PreScission protease like the brain S2 fraction (see legend to Fig. 1). Eluted fractions were resolved by 10% SDS-PAGE and immunoblotted against anti-CaMKIIα mAb. B, R151G is critical to binding of CaMKII to syntaxin, but mutation in this amino acid did not affect interactions between syntaxin and other syntaxin-binding proteins. Each GST-fusion protein was immobilized as well as those described in A and incubated with each recombinant protein (1 nmol) or with CaMKIIα (5 μg). After PreScission protease digestion, eluted proteins were resolved by 15% SDS-PAGE and immunoblotted. C, Dissociation of CaMKII from syntaxin by CaMKII-binding fragments derived from syntaxin 1A ([145–184]; squares) and glutamate receptors of NR2A ([1349–1461]; circles) and NR2B ([1290–1309];triangles). GST–syntaxin 1A [1–262] (4 nmol) was immobilized with glutathione Sepharose and then incubated with purified autophosphorylated CaMKII (5 μg). After rinsing with PBS, immobilized syntaxin–CaMKII complex was incubated with syntaxin [145–184] for 1 hr at 4°C and centrifuged. In some experiments, various concentrations of NR2A or NR2B fragments were incubated with immobilized syntaxin–CaMKII complex as described above. The supernatant containing released CaMKII was resolved by 10% SDS-PAGE and immunoblotted against anti-CaMKIIα mAb. The amount of the bound CaMKII before incubation with syntaxin fragments [145–184] or derived from glutamate receptors is designated as 100%.
We confirmed the reversible and specific binding of CaMKII and syntaxin using the BIAcore system (data not shown). The dissociation constant (Kd) between the autophosphorylated CaMKIIα and syntaxin [1–262] was 4.3 × 10−7m (Table1). In contrast, the affinity of nonphosphorylated CaMKII to syntaxin was ∼250-fold lower than that of phosphorylated CaMKII (Table 1). We also found that CaMKIIβ, another abundant isoform of CaMKII in neurons, binds syntaxin (Table 1). The dissociation constants of CaMKII to the linker domain of syntaxin and to syntaxin were similar [1–262] (Table 1).
Table 1.
Dissociation constants of CaMKII–syntaxin interactions determined by surface plasmon resonance
| Ligand (CaMKII) | Analyte (Syntaxin) | Kd (μm) |
|---|---|---|
| Auto-P α | 1–262 | 0.43 |
| Auto-P α | 145–184 | 1.6 |
| Auto-P α | 145–184 (T161A) | 0.99 |
| Auto-P β | 1–262 | 1.52 |
| Non-Auto-P α | 1–262 | 110 |
Binding was monitored using the BIAcore system. Ligand (CaMKII) was fixed on the CM5 sensor chip. For Kdcalculation, four or five concentrations of recombinant syntaxin fragments were injected. Each Kd was calculated by binding using the mass transfer program. Results are means of four independent experiments. Auto-P, Autophosphorylated; Non-Auto-P, non-autophosphorylated.
The binding of syntaxin to CaMKII does not affect the catalytic activity of CaMKII and is independent of syntaxin phosphorylation by CaMKII
Fragment [145–184] of syntaxin had little effect on the autophosphorylation of CaMKIIα, indicating that binding to this fragment does not primarily affect CaMKIIα activity (Fig.5A). Syntaxin itself was not a good substrate for CaMKIIα (∼0.05 mol of phosphate per mole of syntaxin 1A), although slight phosphorylation was detected (Fig.5A) (Risinger and Bennett, 1999). The autophosphorylation of CaMKII was not affected by syntaxin [145–184] (Fig. 5B). The immunoprecipitation of synaptosomal proteins by anti-syntaxin mAb after Ca2+-dependent phosphorylation revealed that the major and minor phosphorylated proteins were CaMKIIα and CaMKIIβ and synaptotagmin I and syntaxin, respectively. The phosphorylation of all of these proteins was KN-93-sensitive. Among the syntaxin-binding proteins, synaptotagmin I is a substrate of CaMKIIin vitro (Hilfiker et al., 1999). The phosphorylation of synaptotagmin I was 103.5 ± 5.8% in the presence of 12.5 μm syntaxin [145–184] and 104.7 ± 6.5% in the presence of 12.5 μm syntaxin ([145–184]; R151G), respectively (p > 0.05 using t test; four independent experiments; the amount of32P incorporated into phosphorylated synaptotagmin I, 1 μm, was designated as 100%). Thus, the phosphorylation of synaptotagmin I by CaMKII was not altered in the presence of syntaxin [145–184] or ([145–184]; R151G). In addition, T112A–syntaptotagmin I (Hilfiker et al., 1999), a mutant that could not undergo the phosphorylation by CaMKII, was still bound to syntaxin 1A as well as the wild type (Fig. 5C). We located a consensus sequence of CaMKII substrates (RxxS/T) in the CaMKII-binding site of syntaxin 1A at 158–161 (RTTT). The substitution mutant T161A was not phosphorylated by CaMKII, but the syntaxin fragment ([145–184]; T161A) still bound CaMKII (Fig. 5D), and the dissociation constant of this fragment was similar to that of the wild type (Table 1). These results indicate that CaMKII–syntaxin 1A binding is independent of the phosphorylation of syntaxin 1A by CaMKII.
Fig. 5.

Binding of syntaxin to CaMKII does not affect the catalytic activity of CaMKII. A, Ca2+/CaM-dependent protein phosphorylation in synaptosomes is not altered by microinjection of a syntaxin-derived CaMKII-binding fragment ([145–184]; 20 μm). Molecular masses are shown on the left (in kilodaltons). The synaptosomal proteins (10 μg) were phosphorylated as described byPopoli et al. (1995). Samples were resolved by 15% SDS-PAGE and the gel dried on filter paper was then analyzed using a BAS 2000 system.B, Autophosphorylation of CaMKII is not altered in the absence (squares) or presence (circles) of the CaMKII-binding fragment of syntaxin ([145–184], 20 μm). Autophosphorylation proceeded as described by Katoh and Fujisawa (1991). C, T112A–synaptotagmin I was dephosphorylated by CaMKII, but it normally binds to syntaxin.Top, Autoradiogram visualized using the BAS 2000 system. Synaptotagmin I was phosphorylated by CaMKII, but the T112A mutant was not phosphorylated. Bottom, Binding visualized by immunoblotting against anti-synaptotagmin I antibody. The amount of bound T112A mutant was 94.1 ± 7.0% of the bound wild type (four independent experiments), with no statistically significant difference. Phosphorylation proceeded according to Hilfiker et al. (1999).D, Syntaxin ([145–184]; T161A) is not phosphorylated, but binding to CaMKII is similar to that of the wild type.Left, Autoradiogram visualized using the BAS 2000 system. Syntaxin [145–184] was phosphorylated faintly, but the T161A mutant was not phosphorylated at all. Right, Binding visualized by immunoblotting using anti-CaMKII antibody. The amount of CaMKII that bound to syntaxin ([145–184]; T161A) was 104 ± 8% of that bound to wild-type syntaxin [145–184] (four independent experiments), with no statistically significant difference. Phosphorylation proceeded according to Risinger and Bennett (1999). The binding experiment and the following analysis proceeded under standard conditions (see legend to Fig. 4). Incorporation of 32P by CaMKII-dependent phosphorylation was analyzed using the BAS 2000 system as above.
CaMKII interacts with syntaxin in the presynaptic terminal
We examined whether or not CaMKIIα binding to syntaxin regulates formation of the SNARE complex. Using synaptosomal proteins solubilized with Triton X-100 (Pevsner et al., 1994), immunoprecipitation in the presence of Ca2+ and ATP confirmed that CaMKIIα and syntaxin form a complex in vivo as wellin vitro (Fig.6A). We surmised that the CaMKII–syntaxin complex recruits other proteins that regulate the SNARE mechanism. Among solubilized synaptic proteins separated in a glycerol gradient, SNAP-25, synaptotagmin, and tomosyn colocalized with syntaxin (Fig. 6B). Anti-CaMKIIα antibody coimmunoprecipitated synaptotagmin and SNAP-25 with syntaxin but not tomosyn or VAMP (Fig. 6C). A reconstitution study showed that the CaMKIIα–syntaxin complex bound more synaptotagmin and SNAP-25 than syntaxin alone (Fig.7A). Synaptotagmin and SNAP-25 did not bind to CaMKIIα itself (data not shown). In contrast, VAMP, which is not associated with the CaMKII–syntaxin complex in vivo (Fig. 6C), bound to CaMKII–syntaxin in vitro, but the amount of VAMP bound to this complex was similar to that bound to syntaxin alone (Fig. 7A). The complex composed of four proteins did not show SDS resistance at 65°C as the SNARE complex does (data not shown). The CaMKII-binding fragment derived from NR2A did not interfere with the formation of this complex (Fig.7A). The syntaxin-binding activity of T112A–synaptotagmin I, a mutant without a CaMKII phosphorylation site (Hilfiker et al., 1999), was increased to a level similar to that of the wild type in the presence of CaMKII (Fig. 7B). In addition, the activity of T161A–syntaxin lacking a putative phosphorylation site caused by CaMKII was similar to that of wild-type syntaxin (Fig. 7B). Thus, this effect of CaMKII is probably not based on the phosphorylation of either synaptotagmin I or syntaxin by CaMKII.
Fig. 6.
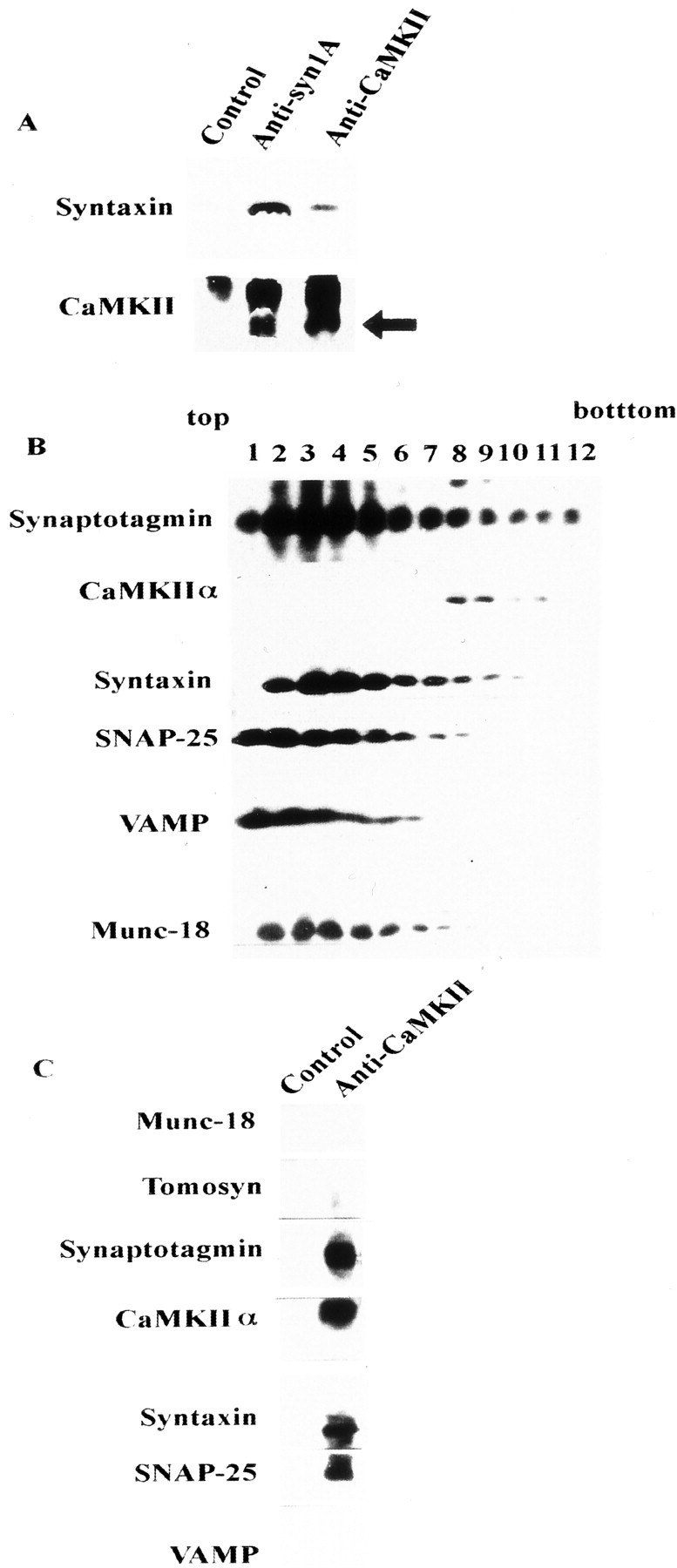
Relationship between the CaMKII–syntaxin complex and other syntaxin-binding proteins. A, Immunoprecipitation of Triton X-100-solubilized synaptosomal proteins using anti-CaMKII or anti-syntaxin antibodies. Complexes were immunostained using anti-syntaxin 1A (top) and anti-CaMKIIα (bottom) mAbs. The arrowindicates the position of CaMKII (the top bandrepresents the IgG heavy chain). Normal mouse IgG was used as the control. Each antibody was incubated with protein G-Sepharose at 4°C for 2 hr, and 10 mg of solubilized synaptosomal proteins (Pevsner et al., 1994; Igarashi et al., 1997) was then added to each antibody–protein G–Sepharose complex at 4°C for an additional 2 hr. Protein–antibody complex eluted with SDS sample buffer was loaded onto 15% SDS-PAGE and immunoblotted against these antibodies. B, Fractionation of synaptosomal proteins solubilized by 1% Triton X-100 and separated on a 10–35% glycerol gradient, as described previously (Igarashi et al., 1997). Fractions (1 ml) were collected from the top. One milliliter of solubilized synaptosomal proteins (protein content was ∼1 mg) was loaded onto 10 ml of a 10–35% continuous glycerol gradient and centrifuged at 41,000 rpm for 17 hr. Fractions (100 μl each) were resolved on 15% SDS-PAGE, and the distribution of each protein was analyzed by immunoblotting. C, Immunoprecipitation of solubilized proteins in fraction 8 of B, which contained both CaMKII and syntaxin, proceeded as described previously (Igarashi et al., 1997, 2000).
Fig. 7.

In vitro reconstitution of the syntaxin 1A–CaMKII complex with other presynaptic proteins.A, Syntaxin–CaMKII complexes formed in vitro recruited more synaptotagmin and SNAP-25 than syntaxin alone. After the formation of CaMKII–syntaxin complex with synaptotagmin I and SNAP-25, NR2A [1389–1464] (10−5m) was added in some experiments to a complex composed of those four proteins for 1 hr.B, Relative binding of synaptotagmin I to syntaxin binding in the absence or presence of CaMKII. Combinations were as follows: wt/wt, Wild-type syntaxin/wild-type synaptotagmin I; wt/T112A, wild-type syntaxin/T112A–synaptotagmin I; T161A/wt, T161A–syntaxin/wild-type synaptotagmin I; andT161A/T112A, T161A–syntaxin/T112A–synaptotagmin I. T112A–synaptotagmin I, a mutant without a CaMKII-dependent phosphorylation site, binds to syntaxin 1A in a manner similar to wild type in the presence of CaMKII. Binding was quantified by densitometry of immunoblots. The ratio was calculated as amount of syntaxin-bound synaptotagmin I in the presence of CaMKII compared with that in the absence of CaMKII.
CaMKII and Munc-18 alternatively binds to syntaxin
CaMKII did not bind syntaxin in the presence of Munc-18 (Fig.8A), and CaMKII and Munc-18 alternatively bind syntaxin in a dose-dependent manner (Fig.8B). However, CaMKII bound to syntaxin 1A ([1–262]; L165A and E166A) (Fig. 8C), a mutant with a forced open configuration (Dulubova et al., 1999) to which Munc-18 could not bind. Rather, more CaMKII bound to the forced open form than to the wild type (130 ± 5.5%; p < 0.05; four independent experiments). These results indicated that the open but not the closed form of syntaxin could bind CaMKII.
Fig. 8.

CaMKII and Munc-18 bind to syntaxin alternatively.A, CaMKII is not bound to Munc-18–syntaxin complex. After recombinant Munc-18 and syntaxin were incubated, the protein complex was immunoprecipitated by anti-syntaxin antibody, and purified CaMKII was added to this complex. Total complex bound to syntaxin was released from GST as described in Materials and Methods.B, CaMKII and Munc-18 alternatively bind to syntaxin in a dose-dependent manner. Both proteins were incubated with the same concentration of immobilized GST–syntaxin 1A at 4°C for 1 hr and eluted with SDS sample buffer. Samples were resolved by 10% SDS-PAGE and immunoblotted. C, CaMKII can bind mutant syntaxin 1A ([1–262]; L165A and E166A; mutant), a forced open form, to which Munc-18 never binds (Dulubova et al., 1999). Control represents normal syntaxin 1A [1–262]. The amount of CaMKII bound to open-form syntaxin was 130.1 ± 4.3% of that bound to wild type, which was significantly increased (n = 4;p < 0.05, t test).
Effects of CaMKII-binding fragment in chromaffin cells and in SCG neurons
We examined the physiological significance of CaMKII–syntaxin interactions in exocytosis by electrophysiological means using chromaffin cells and SCG neurons into which the CaMKII-binding fragment derived from syntaxin was microinjected. We confirmed that syntaxin 1A binds CaMKII of the chromaffin cell lysate (Fig.9A). Microinjection of the CaMKII-binding fragment of syntaxin [145–184] decreased the frequency of exocytosis, as determined by amperometry of catecholamine release (Fig. 9B). In contrast, microinjection of the mutant R151G fragment, which cannot bind CaMKIIα, had no effect (Fig.9B,C). We tested another CaMKII-binding fragment derived from NR2A [1349–1461] (Gardoni et al., 2001) that binds CaMKII (Fig.9A) but does not inhibit syntaxin–CaMKII binding (Fig.4C). The fragment did not affect the exocytotic frequency of the chromaffin cells, unlike syntaxin [145–184] (Fig.9C).
Fig. 9.
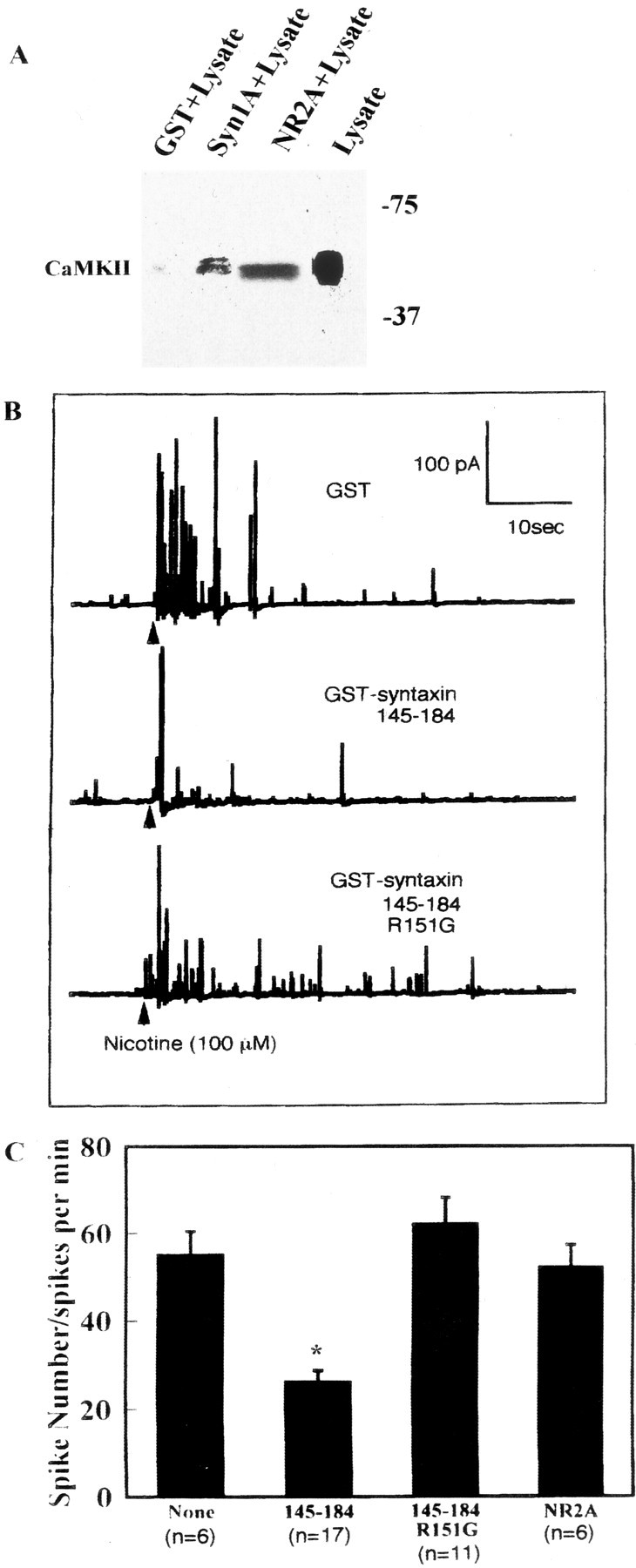
Effects of a CaMKII-binding fragment [145–184] derived from syntaxin in adrenal chromaffin cells. A, CaMKII specifically bound syntaxin in chromaffin cell lysate. The molecular mass is shown on theright (in kilodaltons). This isoform was thought to be CaMKIIδ. Chromaffin cells were separated from adrenal medulla, and cell lysate was made by solubilization with buffer containing 20 mm HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1 mg/ml BSA, 1 mm CaCl2, 2 mm MgCl2, 0.5 mm ATPγS, and protease inhibitor mixtures for 2 hr at 4°C. Lysate (protein concentration 10 mg/ml) was incubated with GST, GST–syntaxin 1A [1–262], or GST-NR2A [1349–1461], and binding proteins were eluted with PreScission protease as described above (see legend to Fig. 1A). Samples were resolved by 10% SDS-PAGE and immunoblotted against anti-CaMKII pAb. B, C, Effects of microinjected syntaxin [145–184] and syntaxin ([145–184]; R151G) on nicotine-evoked catecholamine release from single chromaffin cells detected by single-cell amperometry.B, Typical catecholamine release responses of nicotine-stimulated chromaffin cells into which GST, GST–syntaxin [145–184], or GST–syntaxin ([145–184]; R151G) was microinjected. Both recombinant syntaxin fragments (15 μm) were microinjected into cytosol of chromaffin cells. C, Summary of experiments using chromaffin cells. Frequency (spike numbers) of catecholamine release was significantly lower within the CaMKII-binding fragment of syntaxin [145–184] than in cells injected with control or syntaxin fragment ([145–184]; R151G). The NR2A fragment interacted with CaMKII (A) in chromaffin cells but did not inhibit exocytosis (C). Data are means ± SEM of indicated determinations. Data are typical of several cell preparations. *p < 0.05 versus R151G. R151G and NR2A were not significantly different from controls (no addition).
In addition, EPSPs decreased in SCG neurons microinjected with the CaMKII-binding fragment of syntaxin [145–184] compared with those microinjected with a mutated fragment having a deletion that prevents CaMKIIα binding (Fig.10A,B). Fragments derived from NR2A and NR2B (Strack et al., 2000), as well as those from chromaffin cells, did not affect the EPSPs in SCG neurons (Fig. 10B). These results suggest that the CaMKII-binding fragment decreases the frequency of exocytosis, probably through competitive inhibition of the CaMKII–syntaxin interaction (Fig. 4C). Thus, we concluded that the Ca2+/ATP-dependent binding of CaMKII to syntaxin is physiologically important in the regulation of exocytosis.
DISCUSSION
The present study found that autophosphorylated CaMKII specifically binds to syntaxin at Ca2+concentrations of >10−6mand that bound CaMKII is released from syntaxin when the Ca2+ concentration decreases or the CaMKII is dephosphorylated (Fig. 3A,B). Thus, the binding is thought to be reversible and regulated by Ca2+/ATP. This protein complex was also detected in solubilized synaptosomes, indicating that the CaMKII–syntaxin complex is formed in vivo (Fig.7A). The microinjected CaMKII-binding domain of syntaxin, namely the linker domain where no known syntaxin-binding proteins bind, specifically decreased the frequency of exocytosis in chromaffin cells and in SCG neurons (Figs. 9, 10), probably because the endogenous complex was perturbed by the injected fragments (Fig. 4C). These findings indicated that the binding of CaMKII to syntaxin plays a physiologically important role in the regulation of exocytosis. We originally supposed that the catalytic activity of CaMKII is affected by its binding to syntaxin. However, neither the in vitronor in vivo results confirmed this supposition (Fig. 5). Together, these results indicate that neither the catalytic activity of CaMKII–syntaxin binding nor the phosphorylation of other syntaxin-binding proteins by CaMKII is significantly affected by CaMKII–syntaxin interactions. Moreover, the phosphorylation of syntaxin by CaMKII is not involved in CaMKII binding to syntaxin (Fig.5D). Thus, it is unlikely that the physiological role of this binding is CaMKII accessibility to the membrane via syntaxin for CaMKII to phosphorylate syntaxin-binding proteins. This binding has considerable physiological significance. Syntaxin-bound CaMKII might act as a Ca2+ sensor by trapping CaM (Meyer et al., 1994), thereby stimulating vesicular fusion (Peters and Meyer, 1998), or CaM trapping might help the translocation of CaMKII to the plasma membrane. However, based on the function of the linker domain where CaMKII binds, we believe that the latter possibility is more likely. The linker domain might play a role in structural conversion of the closed form (unable to bind other SNAREs) to the open form (able to bind other SNAREs) (Dulubova et al., 1999; Richmond et al., 2001). This hypothesis assumes that Munc-18 is a protein that binds the closed form. Our results showed that CaMKII and Munc-18 alternatively bind syntaxin (Figs. 6B,C,8A). Thus, these two proteins are thought to bind two distinct forms of free syntaxin (Dulubova et al., 1999). The present results indicate that CaMKII initially assumes the open form that alternatively binds syntaxin. This in turn opens syntaxin, a notion that is supported by the length of its oligomers (8–12 mer, the estimated molecular mass of which is > 400 kDa) (Lin et al., 1987; Morris and Török, 2001). In this state, the core complex region is released. This allows syntaxin to recruit SNAP-25 or synaptotagmin on the vesicle [CaMKII may be also on the vesicle (Benfenati et al., 1992)], which serves as a priming protein complex composed of the target-SNARE complex plus a putative Ca2+ sensor. After [Ca2+] elevation and additional stimulation, vesicles having this complex are more likely to be released than those in which this priming complex is absent. Although the affinity of CaMKII to syntaxin was lower than that of Munc-18 or tomosyn (Table 1) (Fujita et al., 1998), Ca2+ and ATP induced a rapid increase in CaMKII binding (Figs. 1C,D, 2C). Because CaMKII is abundant in the synaptic area, a largeBmax must cover the lowerKd value. The value 10−6m represents a physiological increase of Ca2+ when [Ca2+] is elevated in the presynaptic terminal.
Our evidence indicated that VAMP does not coimmunoprecipitate with CaMKII and syntaxin (Fig. 6C) and that the recruitment of VAMP to the syntaxin–CaMKII complex in vitro proceeds in a different manner from that of synaptotagmin I or SNAP-25 (i.e., an increase in recruitment was not detected compared with syntaxin alone) (Fig. 7A). We therefore concluded that VAMP is not a core member of the CaMKII–syntaxin complex and considered that this large complex of four proteins is an intermediate at a transition state before the complete SNARE complex is formed.
The level of exocytosis was not significantly altered in transfected PC12 cells overexpressing syntaxin [145–184] (M. Igarashi, unpublished observation), although transmitter release from chromaffin cells and SCG neurons was affected by this fragment, as shown in Figures 9 and 10. This is probably a result of the much lower amount of CaMKII in PC12 cells than in neurons (Massé and Kelly, 1997) and an ineffective CaMKII-dependent machinery (Chen et al., 1999). These data indicate that the CaMKII–syntaxin complex aids efficient, continual exocytosis, although it may not be the minimal exocytotic requirement.
CaMKII has α, β, γ, and δ isoforms (Soderling et al., 2001). The α and β isoforms are dominant in neurons, whereas the γ and δ isoforms are distributed among various tissues (Braun and Schulman, 1995). Both the α and β isoforms of CaMKII bind syntaxin in vitro (Table 1). In chromaffin cells, CaMKIIδ is localized (Tashima et al., 1996) and binds syntaxin (Fig.9A). Thus, all of the isoforms are thought to have syntaxin 1A-binding ability. Even if one CaMKII isoform is lacking, others can compensate. CaMKIIα-null mice do not have significantly defective exocytosis (Silva et al., 1992), probably because of functional compensation by the β isoform, which, like CaMKIIα, is abundant in the brain. To reconfirm the present results, it will be necessary to examine the dynamics of exocytosis using electrophysiological techniques in gene-targeted mice. We have started to generate knock-in mice with a point mutation modifying CaMKII–syntaxin interaction.
Finally, our results indicate that in addition to the postsynaptic machinery (Soderling et al., 2001), the autophosphorylation of CaMKII also affects the presynaptic mechanism of synaptic transmission through regulation of SNARE complex formation. This interaction might provide new insight into the molecular mechanism of presynaptic synaptic plasticity (Giese et al., 1998).
Footnotes
This work was supported in part by grants for Scientific Research on Priority Areas (A)-Neural Circuit Project and (C)-Advanced Brain Science Projects from the Ministry of Education, Sciences, Culture, and Sports of Japan to M.I. and Y.K., as well as from the Life Science Foundation, the Brain Science Foundation, the Yujin Memorial Foundation, and the Ichiro Kanehara Memorial Medical Science Foundation to M.I. We thank T. Abe, E. Miyamoto, K. Sakimura, H. Schulman, T. C. Südhof, M. Takahashi, and Y. Takai for cDNAs and antibodies. We also thank E. Akaishi, K. Nomura, and K. Honda for technical assistance and S. Suetsugu for help with the BIAcore measurements. We are grateful to T. Abe, F. Gotoh, M. Itakura, H. Kasai, T. Manabe, H. Shirataki, M. Takahashi, T. Takahashi, M. Watanabe, and H. Yamamoto for discussion and advice and to M. Neal Waxham for recombinant CaMKII and for critical comments regarding this manuscript.
Correspondence should be addressed to Dr. Michihiro Igarashi, Division of Molecular and Cellular Biology, Department of Signal Transduction Research, Niigata University, Graduate School of Medical and Dental Sciences, 1-757 Asahi-machi, Niigata, Niigata 951-8510, Japan. E-mail:tarokaja@med.niigata-u.ac.jp.
REFERENCES
- 1.Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- 2.Bennett MK, García-Arrarás JE, Elferink LA, Peterson K, Fleming AM, Hazuka CD, Scheller RH. The syntaxin family of vesicular transport receptors. Cell. 1993;74:863–873. doi: 10.1016/0092-8674(93)90466-4. [DOI] [PubMed] [Google Scholar]
- 3.Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- 4.Brunger AT. Structural insights into the molecular mechanisms of Ca2+-dependent exocytosis. Curr Opin Neurobiol. 2000;10:293–302. doi: 10.1016/s0959-4388(00)00098-2. [DOI] [PubMed] [Google Scholar]
- 5.Chen YA, Duvuuri V, Schulman H, Scheller RH. Calmodulin and protein kinase C increase Ca2+-stimulated secretion by modulating membrane-attached exocytic machinery. J Biol Chem. 1999;274:26469–26476. doi: 10.1074/jbc.274.37.26469. [DOI] [PubMed] [Google Scholar]
- 6.Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, Rizo J. A conformational switch in syntaxin during exocytosis: role of munc 18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujita Y, Shirataki H, Sakisaka T, Asakura T, Ohya T, Kotani H, Yokoyama M, Mizoguchi A, Scheller RH, Takai Y. Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron. 1998;20:905–915. doi: 10.1016/s0896-6273(00)80472-9. [DOI] [PubMed] [Google Scholar]
- 8.Gardoni F, Schrama LH, Kamal A, Gispen WH, Cattabeni F, Di Luca M. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J Neurosci. 2001;21:1501–1509. doi: 10.1523/JNEUROSCI.21-05-01501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the α calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 10.Hilfiker S, Pieribone VA, Nordstedt C, Greengard P, Czernik AJ. Regulation of synaptotagmin I phosphorylation by multiple protein kinases. J Neurochem. 1999;73:921–932. doi: 10.1046/j.1471-4159.1999.0730921.x. [DOI] [PubMed] [Google Scholar]
- 11.Igarashi M, Tagaya M, Komiya Y. The SNARE mechanisms in growth cones: molecular aspects of the axon terminal development. J Neurosci. 1997;17:1460–1470. doi: 10.1523/JNEUROSCI.17-04-01460.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Igarashi M, Ohyama A, Ohbayashi K, Kozaki S, Komiya Y. The mechanism of the neurotransmitter release in growth cones. J Neurosci Res. 2000;60:743–753. doi: 10.1002/1097-4547(20000615)60:6<743::AID-JNR6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda K, Nagasawa M, Mori H, Araki K, Sakimura K, Watanabe M, Inoue Y, Mishina M. Cloning and expression of the epsilon 4 subunit of the NMDA receptor channel. FEBS Lett. 1992;313:34–38. doi: 10.1016/0014-5793(92)81178-o. [DOI] [PubMed] [Google Scholar]
- 14.Imai Y, Matsushima Y, Sugimura T, Terada M. A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res. 1991;19:2785. doi: 10.1093/nar/19.10.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue A, Obata K, Akagawa K. Cloning and sequence analysis of cDNA for a neuronal cell membrane antigen, HPC-1. J Biol Chem. 1992;267:10613–10619. [PubMed] [Google Scholar]
- 16.Jahn R, Südhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- 17.Kanaseki T, Ikeuchi Y, Sugiura H, Yamauchi T. Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy. J Cell Biol. 1991;115:1049–1060. doi: 10.1083/jcb.115.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katoh T, Fujisawa H. Autoactivation of calmodulin-dependent protein kinase II by autophosphorylation. J Biol Chem. 1991;266:3039–3044. [PubMed] [Google Scholar]
- 19.Kolb SJ, Hudmon A, Ginsberg TR, Waxham MN. Identification of domains essential for the assembly of calcium/calmodulin-dependent protein kinase II holoenzymes. J Biol Chem. 1998;273:31555–31564. doi: 10.1074/jbc.273.47.31555. [DOI] [PubMed] [Google Scholar]
- 20.Lin CR, Kapiloff MS, Durgerian S, Tatemoto K, Russo AF, Hansson P, Schulman H, Rosenfeld MG. Molecular cloning of a brain-specific calcium/calmodulin-dependent protein kinase. Proc Natl Acad Sci USA. 1987;84:5962–5966. doi: 10.1073/pnas.84.16.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsal J, Ruiz-Motasell B, Blasi J, Moreira JE, Contreras D, Sugimori M, Llinás R. Block of transmitter release by botulinum C1 action on syntaxin at the squid giant synapse. Proc Natl Acad Sci USA. 1997;94:14871–14876. doi: 10.1073/pnas.94.26.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massé T, Kelly PT. Overexpression of Ca2+/calmodulin-dependent protein kinase II in PC12 cells alters cell growth, morphology, and nerve growth factor-induced differentiation. J Neurosci. 1997;17:924–931. doi: 10.1523/JNEUROSCI.17-03-00924.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer T, Hanson PI, Stryer L, Schulman H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 1994;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 24.Misura KMS, May AP, Weis WI. Protein-protein interactions in intracellular membrane fusion. Curr Opin Struct Biol. 2000a;10:662–671. doi: 10.1016/s0959-440x(00)00151-2. [DOI] [PubMed] [Google Scholar]
- 25.Misura KMS, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000b;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 26.Mochida S, Yokoyama CT, Kim DK, Itoh K, Caterall WA. Evidence for a voltage-dependent enhancement of neurotransmitter release mediated via the synaptic protein interaction site of N-type Ca2+ channels. Proc Natl Acad Sci USA. 1998;95:14523–14528. doi: 10.1073/pnas.95.24.14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris EP, Török K. Oligomeric structure of α-calmodulin-dependent protein kinase II. J Mol Biol. 2001;308:1–8. doi: 10.1006/jmbi.2001.4584. [DOI] [PubMed] [Google Scholar]
- 28.O'Connor V, Heuss C, De Bello WM, Dresbach T, Charlton MP, Hunt JH, Pellegrini LL, Hodel A, Burger MM, Betz H, Augustine GJ, Schäfer T. Disruption of syntaxin-mediated protein interactions blocks neurotransmitter secretion. Proc Natl Acad Sci USA. 1997;94:12186–12191. doi: 10.1073/pnas.94.22.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohyama A, Komiya Y, Igarashi M. Globular tail of myosin V binds to VAMP/synaptobrevin. Biochem Biophys Res Commun. 2001;280:888–891. doi: 10.1006/bbrc.2001.4236. [DOI] [PubMed] [Google Scholar]
- 30.Peters C, Meyer A. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature. 1998;396:575–580. doi: 10.1038/25133. [DOI] [PubMed] [Google Scholar]
- 31.Pevsner J, Hsu SC, Braun JE, Calakos N, Ting AE, Bennett MK, Scheller RH. Specificity and regulation of a synaptic vesicle fusion complex docking complex. Neuron. 1994;13:353–364. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 32.Popoli M, Vocaturo-Perez CJ, Smeraldi E, Racagni G. Presynaptic Ca2+/calmodulin-dependent protein kinase II: autophosphorylation and activity increase in the hippocampus after long-term blockade of serotonin reuptake. Mol Pharmacol. 1995;48:623–629. [PubMed] [Google Scholar]
- 33.Richmond JE, Weimer RM, Jorgensen EM. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature. 2001;412:338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Risinger C, Bennett MK. Differential phosphorylation of syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) isoforms. J Neurochem. 1999;72:614–624. doi: 10.1046/j.1471-4159.1999.0720614.x. [DOI] [PubMed] [Google Scholar]
- 35.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 36.Soderling TR, Chang B, Brickey D. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2001;276:3719–3722. doi: 10.1074/jbc.R000013200. [DOI] [PubMed] [Google Scholar]
- 37.Strack S, McNeil RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- 38.Suetsugu S, Miki H, Takenawa T. Identification of another actin-related protein (Arp) 2/3 complex binding site in neural Wiskott-Aldrich syndrome protein (N-WASP) that complements actin polymerization induced by the Arp2/3 complex activating (VCA) domain of N-WASP. J Biol Chem. 2001;276:33175–33180. doi: 10.1074/jbc.M102866200. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi N, Kadowaki T, Yazaki Y, Ellis-Davies GCR, Miyashita Y, Kasai H. Post-priming actions of ATP on Ca2+-dependent exocytosis in pancreatic beta cells. Proc Natl Acad Sci USA. 1999;96:760–765. doi: 10.1073/pnas.96.2.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tashima K, Yamamoto H, Setoyama C, Ono T, Miyamoto E. Overexpression of Ca2+/calmodulin-dependent protein kinase II inhibits neurite outgrowth of PC12 cells. J Neurochem. 1996;66:57–64. doi: 10.1046/j.1471-4159.1996.66010057.x. [DOI] [PubMed] [Google Scholar]