

Abstract
The amyloid β-peptide (Aβ) activates microglia and promotes the generation of cytokines and oxygen species, including nitric oxide (NO) and tumor necrosis factor α (TNF-α), which can be either neurotoxic or neuroprotective. We show that neuron death in cocultures of rat cortical microglia and neurons activated by lipopolysaccharide (LPS) or Aβ1–42 plus interferon γ (IFNγ) is caused by short-lived diffusible molecules and follows the generation of superoxide and/or peroxynitrite as determined by electron paramagnetic spectroscopy. Neurotoxicity induced by LPS or Aβ1–42 plus IFNγ is blocked by inhibitors of NO synthesis and by the peroxynitrite (ONOO−) decomposition catalysts FeTMPyP [5,10,15,20-tetrakis(n-methyl-4′-pyridyl)porphinato iron (III) chloride] and FeTPPS [5,10,15,20-tetrakis(4-sulfonatophenyl)prophyrinato iron (III) chloride] but not by the TNF-α inhibitor pentoxifylline. The specificity of FeTMPyP for ONOO− was confirmed by its ability to block the toxicity of a peroxynitrite donor but not of NO donors or of high levels of superoxide in a yeast mutant lacking superoxide dismutase 1. These results implicate peroxynitrite as a mediator of the toxicity of activated microglia, which may play a major role in Aβ1–42 neurotoxicity and Alzheimer's disease.
Keywords: peroxynitrite, Aβ, LPS, microglia, neurons, superoxide, nitric oxide, TNF-α
Microglial activation is implicated in neurodegenerative disorders, including Alzheimer's disease (AD), multiple sclerosis, Parkinson's disease, and stroke. AD, the most common form of senile dementia, is accompanied by a progressive loss of neurons and synapses in brain regions characterized by senile plaques and neurofibrillary tangles. The major components of senile plaques are the β-amyloid (Aβ) peptides, which in experimental models can damage neurons directly or indirectly through the activation of microglia (Yankner et al. 1990; Pike et al., 1991; Meda et al., 1995;Combs et al., 1999; Klein et al., 2001).
Activated microglia are capable of releasing neurotoxic molecules, such as proinflammatory cytokines and toxic oxygen and nitrogen species (Colton and Gilbert, 1987; Klegeris and McGeer, 1994). Accumulating evidence shows that activated microglia can damage or kill neuronsin vitro by generating nitric oxide (NO) (Boje and Arora, 1992; Chao et al., 1992; Goodwin et al., 1995; Meda et al., 1995), tumor necrosis factor-α (TNF-α) (Wood, 1995), various toxic oxygen species (Tanaka et al., 1994), l-cysteine (Yeh et al., 2000), phenolic amine (Giulian et al., 1995), and tissue plasminogen activator (Flavin et al., 2000). NO and superoxide react to form the neurotoxic peroxynitrite (Estevez et al., 1998a,b; Koppal et al., 1999), which has been implicated in AD, in part because the levels of nitrotyrosine, a product of the reaction of peroxynitrite with tyrosine, increase in AD (Smith et al., 1997). However, a role of peroxynitrite in the toxicity of Aβ-activated microglia has not been demonstrated.
Although NO can be neurotoxic, NO is also an important signaling molecule that can protect PC12 cells and primary neurons against Aβ toxicity (Troy et al., 2000; Wirtz-Brugger and Giovanni, 2000). Furthermore, the protective effect of inhibitors of NO synthase (NOS) against Aβ toxicity (Ii et al., 1996) may be attributable to the inhibition of neuronal instead of microglial inducible NOS (iNOS) (Combs et al., 2001). Therefore, the mechanisms of Aβ and microglial neurotoxicity remain unclear.
Here we identify the mediator of Aβ and lipopolysaccharide (LPS) neurotoxicity by measuring the generation of toxic oxygen and nitrogen species by microglia and by studying the role of inhibitors and decomposition catalysts of specific molecules released by activated microglia in preventing neuron death. Neurotoxicity is studied in a cocultures system in which microglia and neurons can be separated before cell death analysis.
MATERIALS AND METHODS
Reagents. LPS (Escherichia coli strain O26:B6), superoxide dismutase (SOD), catalase, sodium nitroprusside (SNP), and fluorescein diacetate are from Sigma (St. Louis, MO). Recombinant mouse interferon γ (IFNγ) is from R & D Systems (Minneapolis, MN).NG-Monomethyl-l-arginine (l-NMMA), 3-morpholinosydnonimine (SIN-1), 5,10,15,20-tetrakis(4-sulfonatophenyl)prophyrinato iron (III) chloride (FeTPPS), 5,10,15,20-tetrakis(N-methyl-4′-pyridyl)porphinato iron (III) chloride (FeTMPyP), Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP), and NOC-18 (a nitric oxide donor; also known as DETA-NONOate) are from Calbiochem (San Diego, CA); thalidomide and pentoxifylline are from Research Biochemicals (Natick, MA). All reagents were tested to be not neurotoxic at the concentrations applied in neuron cultures.
Aβ1–42 (US Peptide, Fullerton, CA) was dissolved in DMSO (Sigma) to obtain a 5 mm stock and kept at −70°C. Before treating the cultures, a 50 μmAβ1–42 solution was prepared in F12K medium as 10× solution and incubated at 37°C for 1 d to obtain aggregated Aβ. For all treatments with Aβ1–42, 10 ng/ml IFNγ was also added as a priming factor (Meda et al., 1995; Ii et al., 1996).
Cell culture. Rat primary glial cells were derived from cerebral cortices of neonatal (postnatal day 3) Fisher 344 rat (Giulian and Baker, 1986). Dispersed cells were grown in DMEM–F12 (Cellgro; Mediatech, Herndon, VA) supplemented with 10% heat-inactivated fetal bovine serum (HyClone Laboratories, Logan, UT), 50 U/ml penicillin (Sigma), and 0.05 mg/ml streptomycin (Sigma), at 37°C in a humidified 95%–5% (v/v) mixture of air and CO2. Culture media were renewed twice per week. After 14–21 d in culture, microglia were detached from monolayer by gentle shaking and replated into cell culture inserts (Costar, Cambridge, MA; Corning, Corning, NY) or 96-well (3 × 104 cells per well) cell culture plates (Falcon; Becton Dickinson, Franklin Lakes, NJ). The microglia homogeneity achieved by this procedure was >98%, as determined by immunocytochemistry for microglial marker complement receptor type 3 (CR3) using mouse anti-rat CR3 antibody OX42 (dilution 1:50; Serotec, Raleigh, NC) (Morgan et al., 1995).
Neuron cultures were derived from fetal (embryonic day 17) Fisher 344 rat cerebral cortices as detailed previously (Banker and Goslin, 1988; Rozovsky et al., 1994) and plated at 5 × 104 viable cells per well in poly-d-lysine (Sigma) -coated 24-well plates (Costar). Culture media were renewed after 1 hr and not changed until the time of experiment at 6–7 d in culture. Microglia were harvested from mixed-glia cultures, plated in 9 mm cell culture inserts (membrane pore size 0.4 μm; Costar) at 105 cells per insert, and placed into the culture wells containing neurons. The porous membrane allows free diffusion of molecules. The distance between neuron layer on the culture plate and microglia layer on the insert membrane is 1 mm, according to the description of the manufacturer. Treatment started 3–4 hr afterward. Neuron–microglia cocultures were maintained in glial medium as described above.
Neuron viability assay. After treatment, culture inserts containing microglia were removed, and neurons were stained with 10 μg/ml fluorescein diacetate (FDA) (Sigma) for 10 min. FDA is membrane permeable and freely enters intact cells, in which it is hydrolyzed by cytosolic esterase and converted to membrane-impermeable fluorescein with a green fluorescence, exhibited only by live cells. Because neuron deaths occur primarily in the region directly underneath the microglia-containing culture inserts (see Fig. 6D), for quantification, eight images at the center of each well were taken with a Nikon (Tokyo, Japan) TE300 fluorescence microscope and analyzed with the IP Lab imaging software (version 3.54; Scanalytics, Fairfax, VA). Viable neurons were quantified by the area covered by green fluorescence after the establishment of a linear relationship between the numbers of stained cells and the green fluorescent area. The total area analyzed occupied 30% of the area in which neuron death occurred.
Fig. 6.
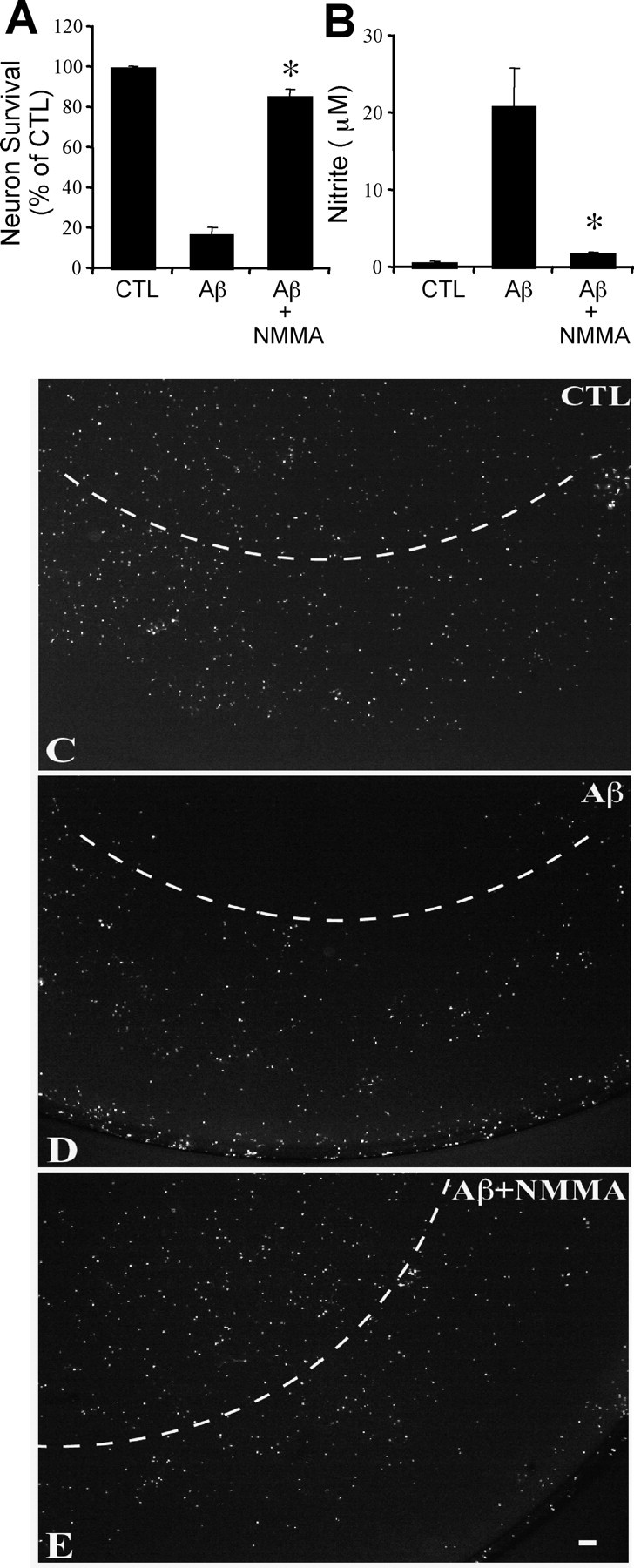
The neurotoxicity of Aβ1–42-activated microglia is nitric oxide dependent. Effect of a 48 hr treatment of microglia–neuron cocultures with 5 μm Aβ1–42 plus 10 ng/ml interferon γ on neuron survival (A) and nitrite accumulation (B) with or without 1 mm NOS inhibitor l-NMMA. Mean ± SEM from four independent experiments. *p < 0.05 compared with Aβ. Fluorescein diacetate staining of neurons untreated (C), treated with 5 μmAβ1–42 plus 10 ng/ml interferon γ (D), or treated with 5 μmAβ1–42 plus 10 ng/ml interferon γ plus 1 mm NOS inhibitor l-NMMA (E). Note the dark region in thetop portion of D, indicating dead cells primarily in the region directly underneath the microglia-containing culture inserts (see Materials and Methods), in contrast to the homogenous staining of untreated neurons shown in C.Dashed arcs delineate the projection of the microglia-containing inserts. Scale bar, 200 μm. CTL, Control.
Nitrite measurement. NO production was determined indirectly through the assay of nitrite (NO2−), a stable metabolite of NO, based on the Griess reaction (Huygen, 1970;Green et al., 1982; Ding et al., 1988). Briefly, a 50 μl aliquot of conditioned media was mixed with an equal volume of Griess reagent [0.1% N-(1-naphthyl)ethylenediamine dihydrochloride, 1% sufanilamide, and 2.5% phosphoric acid (all from Sigma)] and incubated for 10 min at 22°C, and the absorbance was read at 550 nm on a microtiter plate reader (Spectra MAX 250; Molecular Devices, Sunnyvale, CA). Nitrite concentrations were calculated from a standard curve of NaNO2 (Sigma) ranging from 0 to 100 μm. Background NO2− was subtracted from the experimental values.
Detection of superoxide–peroxynitrite by electron paramagnetic resonance. For electron paramagnetic resonance (EPR) measurements, microglia cells (100,000) with or without LPS treatment were incubated in 200 μl of culture medium containing 120 mm5,5-dimethyl-1-pyrroline-N-oxide (DMPO). After 15 min, media were removed and analyzed by EPR. EPR spectra were recorded on a Bruker ECS106 spectrometer with the following settings: receiver gain, 5 × 105; microwave power, 20 mW; microwave frequency, 9.81 GHz; modulation amplitude, 1 G; time constant, 1.3 sec; scan time, 87 sec; and scan width, 80 G. The DMPO-OH signal generated from activated microglia was quantified by comparison with 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy standard after double integration of both signals. All scans shown are an accumulation of seven scans.
TNF-α ELISA. Levels of secreted TNF-α in culture supernatants were determined by an ELISA kit following the instructions of the manufacturer (BioSource, Camarillo, CA).
Yeast sod mutants and growth assay. EG118 (sod1Δ) (DBY746 wild type withsod1:: URA3) yeast were grown in liquid media in synthetic complete media (SDC) with 2% glucose, supplemented with amino acids, adenine, and uracil, as well as a fourfold excess of the supplements tryptophan, leucine, histidine, lysine, and methionine (Longo et al., 1996). Overnight cultures were grown in selective media and inoculated with a flask volume/medium volume ratio of 5:1 at 30°C with shaking at 220 rpm. After overnight cultures in SDC medium,sod1Δ cells were diluted to an optical density at 600 nm (OD600) of 0.1 and inoculated in 3 ml of yeast complete media containing 2% ethanol (YPE) (2% ethanol) containing MnTMPyP (25 μm) or FeTMPyP (25 μm) with or without paraquat (10 μm). Cell density was determined after 48 hr by OD.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining. DNA cleavage was detected with the In Situ Cell Death Detection kit as described by the manufacturer (Roche Molecular Biochemicals, Indianapolis, IN). Briefly, cells were fixed with paraformaldehyde (4% in PBS, pH7.4) and permeabilized (0.1% Triton X-100 in 0.1% sodium citrate). After 1 hr at 37°C incubation in terminal deoxynucleotidyl transferase reaction mixture, signals were visualized under fluorescence microscope (excitation/emission wavelengths, 450–500 nm/515–565 nm). Samples were further blotted with alkaline phosphatase-conjugated anti-fluorescein antibody. After color reaction, samples were analyzed under light microscope.
Statistical analysis. Data were analyzed by one-way ANOVA, followed by post hoc tests of Newman–Keuls multiple comparison to determine whether there were significant differences between individual groups. Statistical significance was established when p < 0.05.
RESULTS
LPS-dependent neuronal death follows the generation of superoxide and/or peroxynitrite and NO–nitrite by microglia
Microglia, the resident macrophages of the CNS, during activation can produce large quantities of nitric oxide synthesized by iNOS. Activated microglia also produce abundant superoxide through the membrane-associated NADPH oxidase. In microglia– neuron cocultures, LPS induced dose-dependent nitrite generation and neuronal death after a 48 hr treatment with 0.2–100 ng/ml LPS (Fig.1A). LPS-activated microglia caused the death of 18 ± 2% of neurons by 24 hr, 50 ± 3% at 36 hr, and 78 ± 3% at 48 hr. Treatment of neurons with LPS in the absence of microglia does not cause toxicity or nitrite generation (data not shown). Cell death was observed only in neurons cultured directly under the filter insert (membrane pore size 0.4 μm; Costar) containing the microglia (1 mm distance), suggesting that neurotoxicity is mediated by short-lived diffusible molecules (see Fig. 6D). To determine whether neurotoxicity correlated with NO generation, we first investigated the time course of nitric oxide and superoxide generation. NO generation was estimated by measuring the concentration of nitrite, its stable metabolite, released into the medium (Ding et al., 1988). LPS at 100 ng/ml induced nitrite generation beginning at 6 hr (reported as micromolar generated per hour), with a peak at ∼14 hr and a gradual decline until 48 hr (Fig. 1B).
Fig. 1.
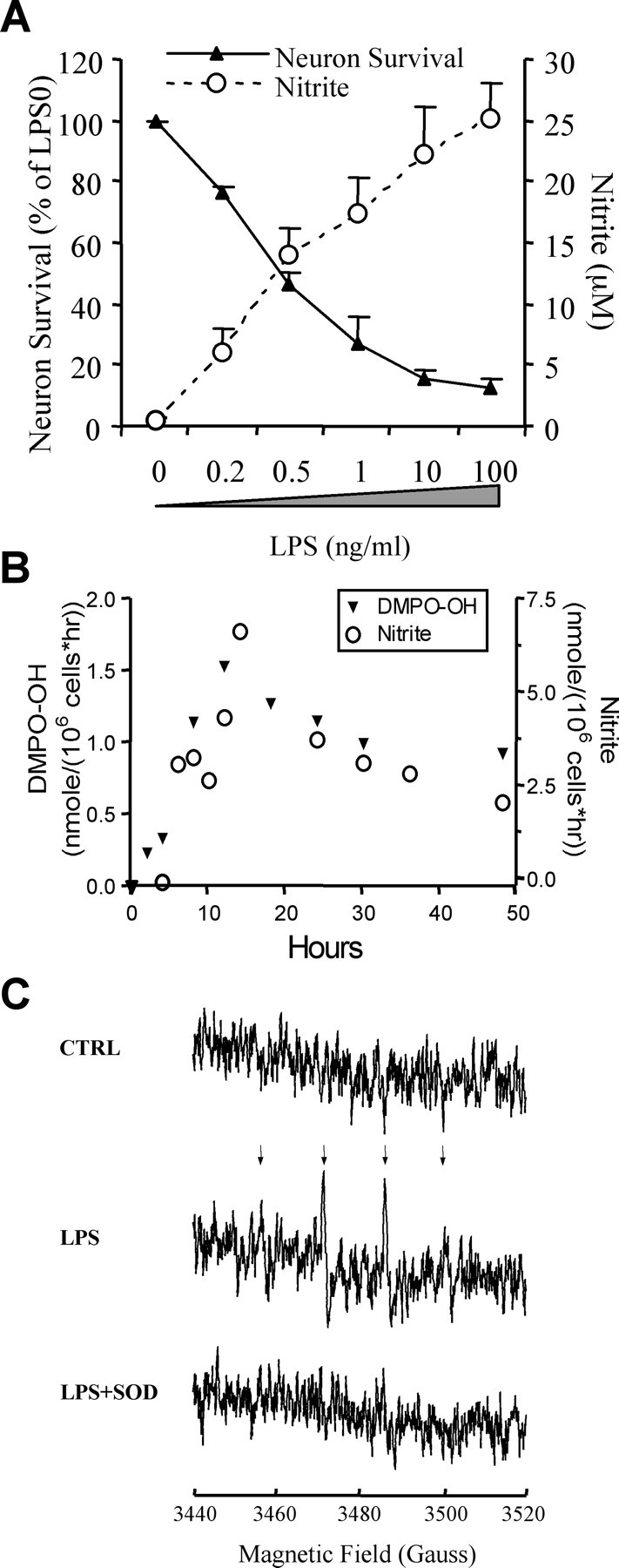
LPS-activated microglia kill neurons in a dose-dependent manner. A, Neuron survival and nitrite accumulation after a 48 hr treatment of rat microglia–neuron cocultures with 0.2–100 ng/ml LPS. Viable neurons were detected by fluorescein diacetate staining. Data represent mean ± SEM from a representative experiment. The experiment was repeated three times with similar results. B, Time course of nitrite and superoxide–peroxynitrite (DMPO-OH) generation determined by Griess reaction and EPR, respectively. Repeated three times with similar results. A representative experiment is shown. C, Detection of superoxide–peroxynitrite generation by microglia cells by EPR spectra of DMPO-OH. Arrows indicate the quartet generated by DMPO-OH. Microglia cells (100,000) with or without LPS treatment (24 hr) were incubated with 120 mm DMPO with or without 186 U/ml SOD for 15 min and analyzed by EPR. The figures shown are an accumulation of seven scans.
The generation of superoxide–peroxynitrite by activated microglia cells was monitored by EPR with the spin trap DMPO. A DMPO-OH signal (a quartet signal with signal intensity ratios of 1:2:2:1;aN =aH = 14.9) was observed in the medium of activated microglia cells but not in control cells (Fig.1C). The coincubation with exogenous SOD resulted in a complete loss of the DMPO-OH signal in LPS-treated microglia cells (Fig. 1C), indicating the EPR signal was caused by superoxide, peroxynitrite, or both but not by hydroxyl radical (although hydroxyl radical may be generated from peroxynitrite). Similar to NO, the superoxide and/or peroxynitrite generated by microglia cells peaked at 12 hr (Fig.1B,C).
It is difficult to distinguish between the EPR signal of superoxide and that of peroxynitrite because both species generate the DMPO-OH either through DMPO-OOH decomposition (superoxide) or by the peroxynitrite-dependent generation of hydroxyl radical, which subsequently reacts with DMPO (Augusto et al., 1994). Furthermore, both superoxide- and peroxynitrite-dependent quartet EPR signal can be prevented by SOD. We incubated LPS-activated microglia in the presence of both DMPO and the hydroxyl radical scavenger DMSO (200 mm). Whereas the quartet signal caused by superoxide would not be affected by DMSO, peroxynitrite is expected to generate a quartet, as well as a sextet, signal in the presence of hydroxyl radical scavengers. However, the weakness of the signal did not allow us to establish whether DMSO resulted in a sextet signal. This may be caused by the peroxynitrite-dependent decomposition of DMPO-OH (Augusto et al., 1994). Although this method cannot distinguish between superoxide and peroxynitrite, the near diffusion-limited reaction rate between nitric oxide and superoxide and the high concentrations of these two molecules in the medium would unavoidably result in the generation of peroxynitrite.
Nitric oxide is required for the neurotoxicity of activated microglia
To test whether NO or TNF-α mediate neuron-killing by activated microglia, the NO synthase inhibitor l-NMMA and the two inhibitors of TNF-α production pentoxifylline and thalidomide were added at the same time with LPS. A 48 hr treatment of LPS killed 78 ± 3% of neurons (Fig.2A).l-NMMA completely blocked LPS-induced NO synthesis and neuron death (viable neurons, 92 ± 7% of control) (Fig.2).
Fig. 2.
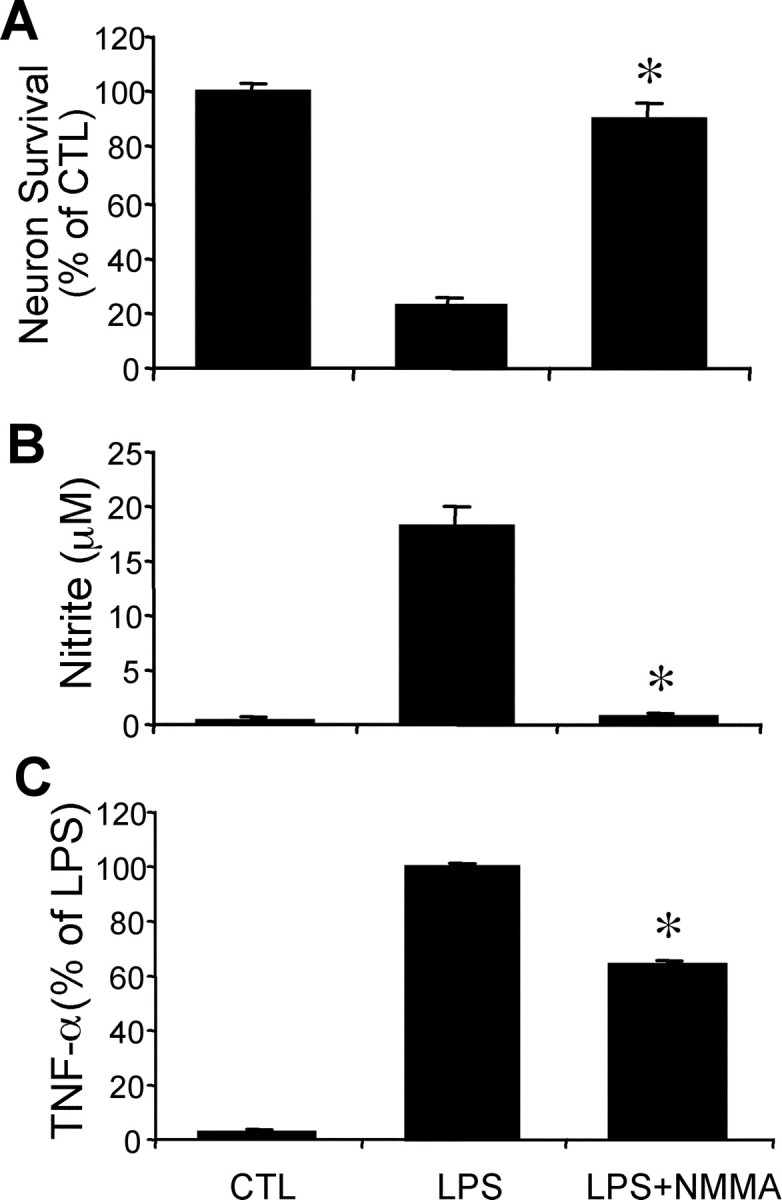
The neurotoxicity of LPS-activated microglia is nitric oxide dependent. Neuron survival (A), nitrite accumulation (B), and TNF-α secretion (C) in microglia–neuron cocultures treated with 100 ng/ml LPS with or without 1 mm NOS inhibitorl-NMMA for 48 hr. Nitrite, a metabolite of nitric oxide, and TNF-α are quantified from the same cultures used to measure neuron survival. Mean ± SEM from five independent experiments. *p < 0.05 compared with LPS alone.CTL, Control.
Pentoxifylline is a nonselective phosphodiesterase inhibitor that blocks the release of TNF-α from microglia (Chao et al., 1992). Pentoxifylline did not affect nitrite accumulation and provided only minor protection against activated microglia (Fig.3). In contrast, thalidomide, another TNF-α inhibitor that also inhibits NO production, completely blocked neuron death (Fig. 3). These results suggest that inhibition of NO synthesis is sufficient to completely block the neurotoxicity of activated microglia.
Fig. 3.
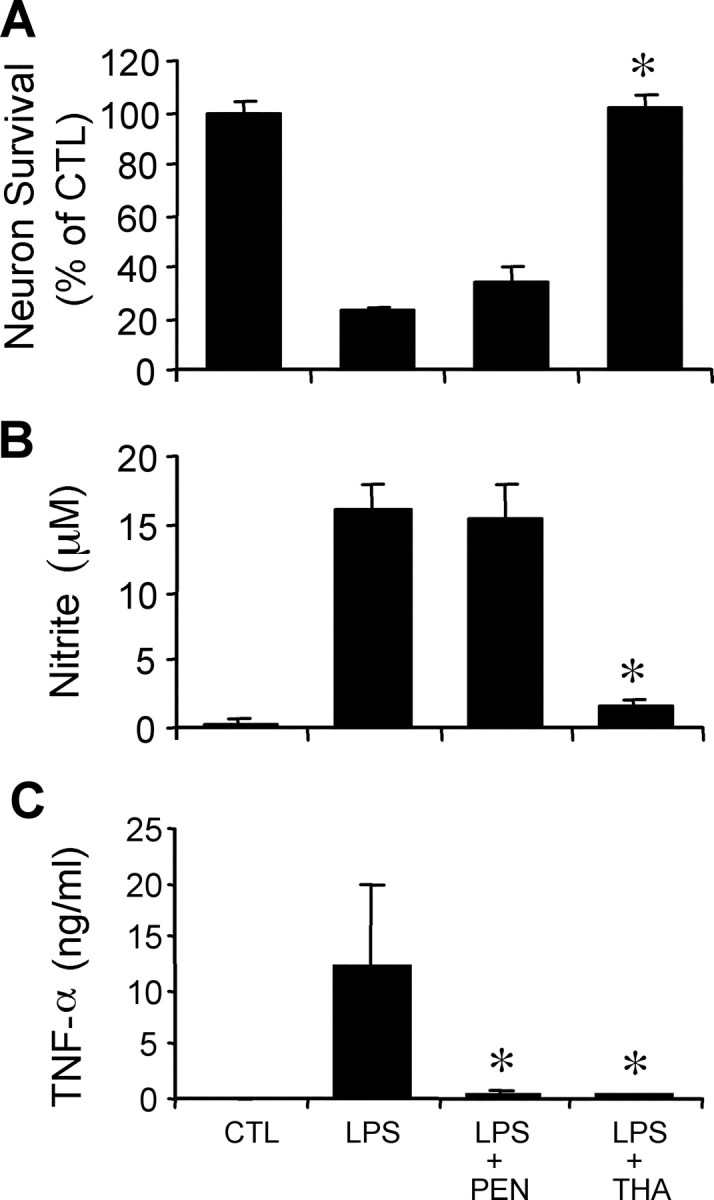
TNF-α released from activated microglia has a minor neurotoxic effect. Neuron survival (A), nitrite accumulation (B), and TNF-α secretion (C) in microglia–neuron cocultures treated with 100 ng/ml LPS for 48 hr with or without the two TNF-α inhibitors pentoxifylline (PEN; 500 μm) and thalidomide (THA; 200 μm) and 100 ng/ml LPS. Pentoxifylline inhibits TNF-α secretion but has little effect on nitrite accumulation and neuron killing. Thalidomide inhibits both TNF-α secretion and NO production. Mean ± SEM from five to eight independent experiments *p < 0.05 compared with LPS alone. CTL, Control.
Peroxynitrite mediates the neurotoxicity of microglia activated by LPS
Nitric oxide reacts with superoxide (O2·−) at a near diffusion-limited rate with a constant of 6.7 × 109m/sec, producing the highly reactive and toxic peroxynitrite (ONOO−) (Ischiropoulos et al., 1992). Furthermore, unlike superoxide, ONOO− is membrane permeable (Marla et al., 1997) and is able to reach and damage intracellular neuronal targets (Estevez et al., 1998b). Because activated microglia generate both NO and superoxide, peroxynitrite is hypothesized to be a major mediator in microglia-induced neuronal injury (Van Dyke, 1997; Combs et al., 2001).
To test the role of superoxide and peroxynitrite in the toxicity of activated microglia, we treated neurons–microglia cocultures with the membrane-permeable iron porphyrin peroxynitrite decomposition catalysts FeTMPyP and FeTPPS or the superoxide dismutase mimetic MnTMPyP (Misko et al., 1998). FeTMPyP and FeTPPS at 2 μm blocked LPS-induced microglial neurotoxicity without decreasing nitrite production (Fig.4A,B). The protective action of FeTMPyP is dose-dependent with an optimal concentration of 2 μm (Fig.4C,D). Because the superoxide anion is required to form peroxynitrite, its decrease should also attenuate neuron death. We tested the protective effect of the manganese porphyrin SOD mimetic MnTMPyP (membrane-permeable) in the coculture treated with LPS. MnTMPyP attenuated LPS-induced microglial neurotoxicity without compromising nitrite production (Fig.4A,B). In contrast, treatment with SOD plus catalase, or each scavenger alone (data not shown), did not protect against microglial neurotoxicity (Fig.4A,B), suggesting that peroxynitrite may be generated inside microglia, where superoxide cannot be reached by the nonmembrane-permeable SOD. Peroxynitrite may be also formed extracellularly as a result of the inability of SOD to efficiently compete with NO for superoxide because of the very fast rate of reaction between NO and superoxide. The inability of catalase to block neuronal death suggests that hydrogen peroxide is not a major mediator of microglial neurotoxicity. Only the neurons cultured directly under the insert containing the microglia died (data not shown) (see Fig. 6D). This is consistent with peroxynitrite toxicity, because its half-life of only 1.9 sec at pH 7.4 would prevent high levels of ONOO− to reach distant neuronal targets.
Fig. 4.
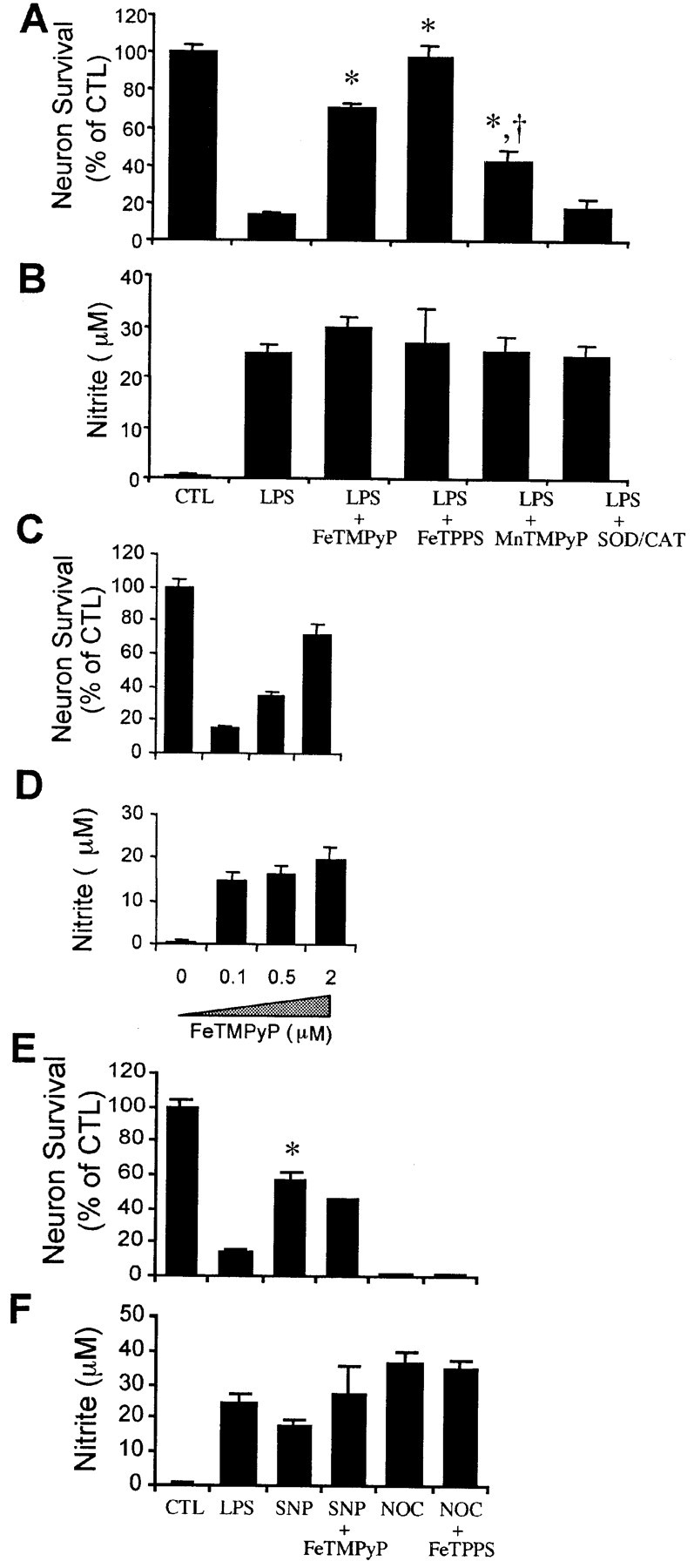
Peroxynitrite mediates the neurotoxicity of LPS-activated microglia. Neuron survival (A) and nitrite accumulation (B) after a 48 hr treatment of microglia–neuron cocultures with 100 ng/ml LPS with or without the membrane-permeable peroxynitrite decomposition catalysts FeTMPyP (2 μm) or FeTPPS (10 μm), the SOD mimetic MnTMPyP (5 μm), or 50 U/ml SOD plus 100 U/ml catalase (SOD/CAT). Values in A and B show mean ± SEM of five independent experiments. Dose-dependent effect of the peroxynitrite decomposition catalyst FeTMPyP on LPS-induced microglial neurotoxicity (C) and nitrite accumulation (D) in microglia–neuron cocultures. Effect of the NO donors SNP and NOC-18 on neuronal survival (E) and nitrite generation (F) in the presence or absence of the peroxynitrite decomposition catalysts FeTMPyP or FeTPPS. Cocultures are treated with LPS (100 ng/ml), the nitric oxide donor SNP (300 μm), or 100 μm NOC-18 plus FeTMPyP (2 μm) or FeTPPS (10 μm) for 48 hr. Mean ± SEM from five independent experiments. *p < 0.05 compared with LPS. CTL, Control. †p < 0.01 compared with LPS plus FeTMPyP or LPS plus FePPS.
To test whether peroxynitrite is formed in neurons or microglia from the reaction of exogenous nitric oxide with neuronal superoxide, we treated cells with the NO donor SNP (300 μm) at a concentration that generates nitrite–NO at levels similar to those generated by LPS-activated microglia (Fig. 4F). At 300 μm, SNP killed <50% of the neurons, significantly below the 85% cell death caused by LPS-induced activation of microglia (Fig. 4E) (p < 0.05). These results suggest that the superoxide and peroxynitrite that mediates the neurotoxicity of microglia activated by LPS are generated by the microglia. However, we have not ruled out that a portion of the superoxide that reacts to form peroxynitrite may be generated within the neurons (Longo et al., 2000).
The inability of FeTMPyP to block NO-induced neuron death (Fig.4E) also confirms that this decomposition catalyst is specific for peroxynitrite. To test further the specificity of the iron porphyrins for peroxynitrite, we treated neurons with the NO donor NOC-18, which releases NO· during decomposition (Maragos et al., 1991). NOC-18 at 100 μm resulted in the generation of high levels of nitrite and caused the death of 99% of the neurons (Fig. 4E,F). FeTPPS, which completely blocked the toxicity of peroxynitrite generated by activated microglia (Fig. 4A), did not block the toxic effect of NOC-18.
FeTMPyP is an efficient decomposition catalyst of peroxynitrite but not superoxide
To validate further the specificity and effectiveness of the peroxynitrite decomposition catalyst FeTMPyP, we tested it in neuron cultures treated with SIN-1, an exogenous NO and superoxide donor that, consequently, generates peroxynitrite under physiological conditions (Hogg et al., 1992). FeTMPyP effectively protected neurons against SIN-1-induced peroxynitrite toxicity (Fig.5A).
Fig. 5.
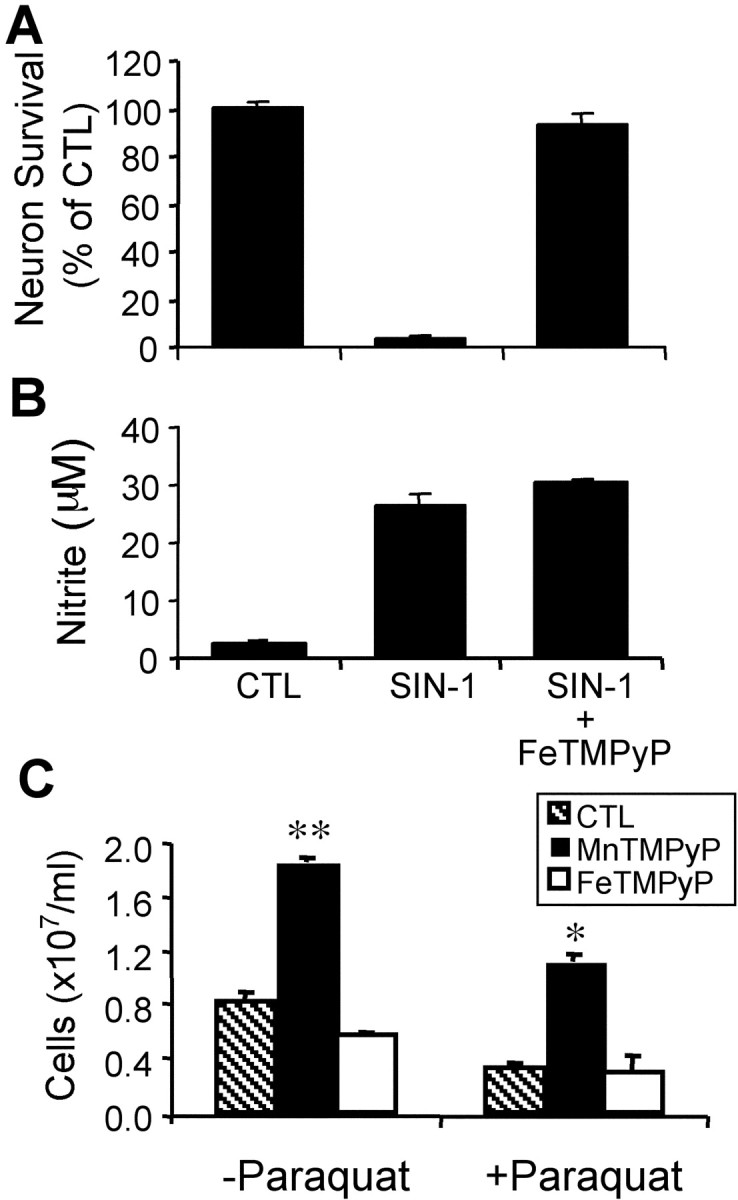
FeTMPyP and MnTMPyP are efficient and specific inhibitors of peroxynitrite and superoxide, respectively. Effect of FeTMPyP on the neurotoxicity of the NO–O2−donor SIN-1. Primary cortical neurons are treated with 50 μm SIN-1 or 50 μm SIN-1 plus 2 μm FeTMPyP for 48 hr. A, Neuron survival;B, nitrite accumulation. Data represent mean ± SEM from three independent experiments. C, Effect of FeTMPyP or MnTMPyP on the growth defects (ethanol) ofSaccharomyces cerevisiae cells lacking cytosolic superoxide dismutase (sod1Δ). S. cerevisiae cells are inoculated in SDC medium and diluted in YPE (ethanol) medium (3 ml) containing MnTMPyP (25 μm) or FeTMPyP (25 μm) with or without the intracellular superoxide generator paraquat (10 μm). Cell density is measured after 48 hr. Average ± SEM of two independent experiments with duplicate samples. *p < 0.05 compared with control plus paraquat; **p < 0.05 compared with control. CTL, Control.
To determine whether FeTMPyP may be protecting neurons by also scavenging superoxide, we studied its effect on yeast mutants lacking cytosolic SOD (sod1Δ) (Gralla and Valentine, 1991). This is a valuable system to test the specificity of these agents (Chiang et al., 2000) because the primary cause of the defects of yeastsod1Δ mutants is superoxide toxicity. In contrast, molecules such as paraquat and menadione generate other toxic oxygen species in addition to superoxide. FeTMPyP did not reverse the growth defects of sod1Δ mutants in either the absence or presence of paraquat (Fig. 5C). In contrast, the superoxide scavenger MnTMPyP blocked superoxide toxicity and reversed the growth defects of sod1Δ mutants whether or not paraquat was present. These results confirm that FeTMPyP is acting as a specific decomposition catalyst of peroxynitrite, whereas MnTMPyP functions as a permeable superoxide dismutase.
Peroxynitrite mediates the neurotoxicity of Aβ in neurons–microglia cocultures
To test whether peroxynitrite also mediates the toxicity of Aβ, implicated in AD pathology, we tested the role of agents that decompose or scavenge specific nitrogen and oxygen species in protecting neurons against fibrillar Aβ1–42. Aβ and IFNγ act synergistically in stimulating the production of nitric oxide from microglia and in causing microglial neurotoxicity (Meda et al., 1995). Treatment of microglia–neuron cocultures with 5 μmAβ1–42 plus 10 ng/ml IFNγ for 48 hr caused the death of >80% of neurons. Neuronal death required the generation of nitric oxide by microglia (Fig.6A,B), as shown by Ii et al. (1996). Treatment of neurons with 5 μm Aβ1–42 or IFNγ alone did not cause neuronal death in the absence of microglia (data not shown). Cell death was confined to the region directly underneath the inserts containing microglia, in an area slightly larger (15%) than that of the insert, as shown in Figure 6D. The spatial proximity of dead neurons to activated microglia (1 mm) is consistent with the role of a short-lived molecule, peroxynitrite, in the mediation of Aβ1–42 neurotoxicity. The peroxynitrite decomposition catalyst FeTMPyP blocked Aβ-induced microglial neurotoxicity without affecting nitrite production (Fig.7). The SOD mimetic MnTMPyP had a similar but attenuated effect (Fig. 7). These results suggest that peroxynitrite is a major mediator of the toxicity of microglia activated by Aβ.
Fig. 7.

Peroxynitrite mediates the neurotoxicity of Aβ1–42-activated microglia. Neuron survival (A) and nitrite accumulation (B) after a 48 hr treatment of microglia–neuron cocultures with 5 μm Aβ1–42 plus 10 ng/ml interferon γ, with or without the peroxynitrite decomposition catalyst FeTMPyP (2 μm) or the SOD mimetic MnTMPyP (5 μm). Mean ± SEM from four independent experiments. *p < 0.05 compared with Aβ; †p < 0.05 compared with Aβ plus FeTMPyP.CTL, Control.
Aβ1–42 causes peroxynitrite-dependent DNA fragmentation preceding neuron death
Whereas high concentrations of peroxynitrite induce necrotic cell death in neurons, low levels of peroxynitrite can induce apoptosis (Bonfoco et al., 1995; Estevez et al., 1998b). DNA fragmentation was detected in vivo by the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL). DNA strand breaks were observed at 24 hr in Aβ-treated neurons in the area below the microglial inserts (Fig.8A,B), when <30% of the cells are dead (Fig. 8C). As shown in Figure 8B, the majority of the neurons treated with Aβ 1–42 were TUNEL labeled at 24 hr, suggesting that peroxynitrite induces DNA damage. Although TUNEL assay can label both apoptotic and necrotic neurons (Adamec et al., 2001), most of the TUNEL-labeled cells at 24 hr were not necrotic, as determined by the fluorescein diacetate staining (Fig. 8C). These results are consistent with the demonstrated role of peroxynitrite in inducing macromolecular damage and apoptosis in motor neurons (Estevez et al., 1998b).
Fig. 8.
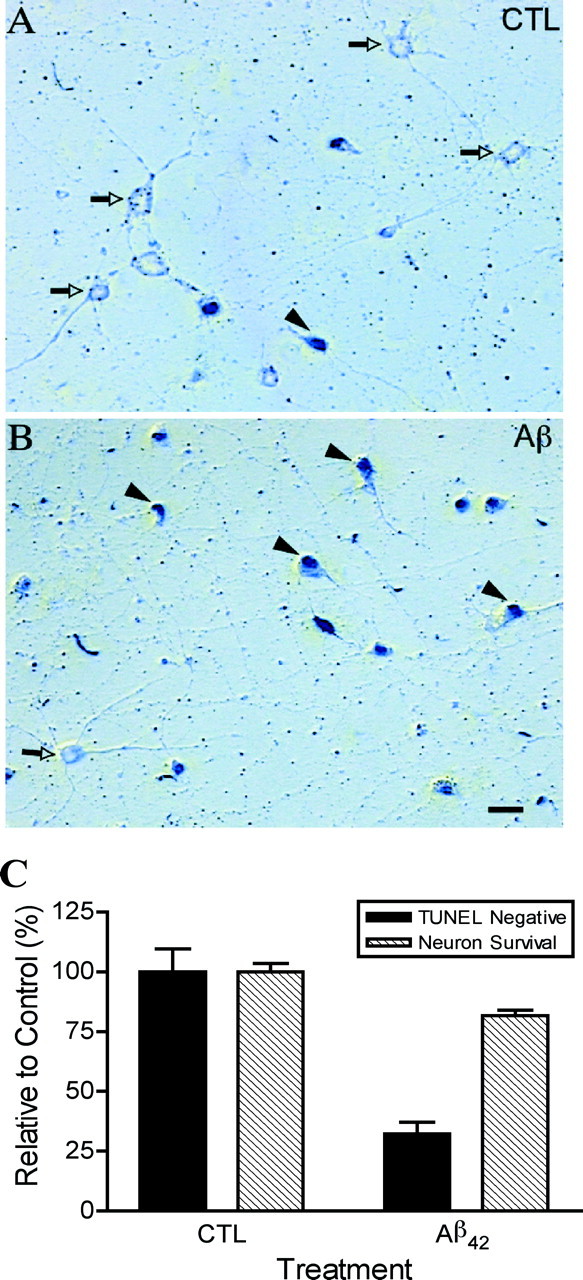
Aβ1–42-activated microglia induce TUNEL staining preceding loss of viability. TUNEL in neurons removed from microglia–neuron cocultures treated with 5 μmAβ1–42 plus 10 ng/ml interferon γ for 24 hr. Whereas neurons exposed to inactive microglia are scarcely stained (A; white arrowheads), neurons exposed to Aβ1–42-activated microglia (B) are predominantly positively stained (black arrowheads). Scale bar, 20 μm. C, Percentage of TUNEL-negative neurons and surviving neurons after 24 hr treatment of microglia–neuron cocultures with 5 μmAβ1–42 plus 10 ng/ml interferon γ.
DISCUSSION
Cell death in cocultures of cortical microglia and neurons treated with LPS or Aβ1–42 was preceded by the generation of superoxide and/or peroxynitrite and nitrite and prevented by inhibitors of NO synthesis or decomposition catalysts of peroxynitrite. Together, these results suggest that the short-lived peroxynitrite molecule generated by activated microglia is the major mediator of neurotoxicity during treatment of microglia and neurons with Aβ1–42 or LPS.
AD brains show widespread oxidative damage (Mattson, 1997; Smith et al., 2000). Both hydrogen peroxide and superoxide have been implicated in the direct toxicity of Aβ to neurons (Behl et al., 1994; Behl, 1997; Keller et al., 1998; Longo et al., 2000), whereas nitric oxide has been implicated in the neurotoxicity of activated microglia (Boje and Arora, 1992; Chao et al., 1992; Goodwin et al., 1995; Meda et al., 1995). Aβ also stimulates superoxide production in microglia (Klegeris and McGeer, 1997; McDonald et al., 1997; Colton et al., 2000), possibly by activating a NADPH oxidase (Bianca et al., 1999). Thus, the generation of peroxynitrite by microglia activated by Aβ demonstrated in our study is consistent with previous studies showing that microglia can generate both nitric oxide and superoxide. Furthermore, nitrotyrosines, the product of the reaction of peroxynitrite with tyrosine residues, and NADPH oxidase activity are elevated in AD brains (Hensley et al., 1998; Shimohama et al., 2000). Together with ours, these results suggest that peroxynitrite may play a major role in Aβ toxicity and Alzheimer's disease.
The role of peroxynitrite as the major mediator of the toxicity of microglia activated by either Aβ or LPS is supported by several results: (1) in microglia exposed to LPS, the generation of NO and superoxide–peroxynitrite reaches its peak after 10–20 hr and continues for over 50 hr; (2) the neurotoxicity of Aβ is confined to the area directly under microglia; (3) the NO synthase inhibitor NMMA blocks toxicity as reported previously (Ii et al., 1996); (4) FeTMPyP and FeTPPS decompose peroxynitrite, but not NO or superoxide, and block the toxicity of microglia activated by LPS or Aβ1–42; and (5) the superoxide mimetic MnTMPyP, which decreases peroxynitrite generation by scavenging superoxide, partially blocks the toxicity of activated microglia. However, we cannot rule out that the neurotoxicity is mediated by a presently unknown short-lived molecule other than peroxynitrite, which requires NO and superoxide to be formed and which is neutralized by peroxynitrite-specific decomposition catalysts.
The high second-order rate constant for the reaction between peroxynitrite and FeTMPyP (5 × 107m/sec) enables this efficient decomposition catalyst to protect mammalian cells against high doses of peroxynitrite (Salvemini et al., 1998). The effectiveness of FeTMPyP in blocking peroxynitrite toxicity was confirmed by its protection of neurons against SIN-1, which generates peroxynitrite. Yeast lacking the cytosolic SOD1 show severe growth defects in the nonfermentable carbon source ethanol. This is a particularly appropriate system to test the specificity of the peroxynitrite and superoxide decomposition catalysts and scavengers because high cytosolic levels of superoxide are the primary cause of the growth defects. The possibility that FeTMPyP protects neurons by scavenging superoxide was ruled out by showing that FeTMPyP did not reverse the defects of yeast SOD mutants (Fig. 5). In contrast, the SOD mimetic MnTMPyP was effective in reversing superoxide toxicity in yeast.
The lack of a protective effect of SOD against the neurotoxicity of activated microglia suggest that peroxynitrite is generated within microglia and therefore out of the reach of the extracellular SOD protein. However, in view of the effect of SOD in completely abolishing the EPR DMPO-OH signal caused by superoxide and/or peroxynitrite released by activated microglia (Fig. 1C), there are several possible scenarios. (1) The DMPO-OH adduct is only generated by superoxide through the decomposition of DMPO-OOH, but most ONOO− is generated within the microglia. In this case, the extracellular SOD would prevent the superoxide-dependent generation of DMPO-OH but would not prevent the generation of the neurotoxic peroxynitrite within the microglia. (2) The DMPO-OH adduct is generated by both superoxide and peroxynitrite, but the contribution of DMPO-OH by peroxynitrite is below the detectable level. Furthermore, peroxynitrite can both generate and decompose DMPO-OH and, in the absence of superoxide-generated DMPO-OH, may abolish the quartet signal. (3) The DMPO-OH adduct is only generated by peroxynitrite formed from the reaction of superoxide and NO outside microglia. In this scenario, SOD may prevent the generation of the levels of peroxynitrite required to generate a stable DMPO-OH signal but may be unable to prevent the generation of levels of peroxynitrite sufficient to kill neurons after a 48 hr exposure. (4) FeTMPyP and FeTPPS may protect neurons by decomposing peroxynitrite in the proximity of the neuronal targets in which the concentration of the decomposition catalyst is high but that of the short-lived peroxynitrite is much lower than inside microglia. Thus, additional studies are necessary to demonstrate whether peroxynitrite is generated inside microglia, extracellularly, or both.
Although NO has been implicated in Aβ1–42toxicity, its role is controversial because the NO donorS-nitroso-N-acetyl-penicillamine (SNAP) was reported by others to be neuroprotective against Aβ toxicity (Troy et al., 2000; Wirtz-Brugger and Giovanni, 2000). These differences may be explained by the presence in these studies of Aβ and of an NO donor, in the absence of microglia. Whereas microglial activation results in peroxynitrite generation, the concentration of superoxide in neurons exposed to Aβ and an NO donor in the absence of activated microglia may be too low to generate toxic levels of peroxynitrite. The subtoxic levels (100 μm) of the NO donor SNAP used in these studies may actually protect the cells against the superoxide generated in neurons treated with Aβ in the absence of microglia (Keller et al., 1998; Longo et al., 2000). In fact, NO activates guanylate cyclase and increases the generation of cGMP, which protects against cell death (Kim et al., 1999; Wirtz-Brugger and Giovanni, 2000). NO has also been shown to protect mammalian cells independently of cGMP by inhibiting complex IV activity (Beltran et al., 2000) or by inhibiting other respiratory enzymes (Paxinou et al., 2001). Thus, low levels of NO may play a protective role in the absence of superoxide.
TNF-α, a cytokine released by activated microglia, can also be both neurotoxic (Chao et al., 1993; Wood, 1995) and neuroprotective (Barger et al., 1995). Our studies suggest that TNF-α does not play a major role in the toxicity of activated microglia because the inhibition of TNF-α secretion by pentoxifylline did not prevent LPS neurotoxicity. Peroxynitrite is a short-lived molecule with a half-life of <2 sec, whereas TNF-α is a relatively long-lived protein. Therefore, the pattern of cell death in neurons confined to an area within ∼1 mm of the microglia (Fig. 6D) is not consistent with a major role for TNF-α in acute neurotoxicity. However, TNF-α may be more toxic at higher concentrations or during longer or chronic treatments. In fact, TNF-α from monocytes and microglia stimulated with 60 μm Aβ1–40 or Aβ25–35 causes neuronal apoptosis (Combs et al., 2001). Whereas in our coculture system the neurons are coincubated with activated microglia during the treatment with LPS or 5 μm Aβ, Combs et al. (2001) have removed the media from cultures of activated microglia and transferred it to neuronal cultures. This method would result in the decomposition of most of the short-lived peroxynitrite. Thus, the low dose of Aβ1–42 (5 μm) used in our experiments induces both peroxynitrite and TNF-α secretion, but neurotoxicity is mediated by peroxynitrite. However, higher doses of Aβ or longer treatments may induce the expression of neurotoxic levels of TNF-α. This may explain the role of both NO and TNF-α in the neurotoxicity of microglia treated with Aβ1–42 or Aβ 25–35 in which neurons and microglia were cocultured and treated with a higher concentration of Aβ1–42 (12 μm) for a longer time (72 hr) than in our experiments (Meda et al., 1995).
In summary, our results with cell-culture models suggest that peroxynitrite is the major mediator of the neurotoxicity of microglia activated by LPS or Aβ and indicate that it may also play an important role in the toxicity of the chronically activated microglia in AD brains. These findings support the development of drugs that protect neurons against the toxicity of specific molecules, such as peroxynitrite, without interfering with the normal functions of glial cells and neurons.
C.E.F. and V.D.L. contributed equally to this work.
This work was supported by National Institutes of Health Grants AG13499 (to C.E.F.) and AG01028 (to V.D.L.) and the John Douglas French Alzheimer's Foundation (to V.D.L.). We thank John Martin and George Fenimore for their generous donations.
Correspondence should be addressed to V. D. Longo, Division of Biogerontology, Andrus Gerontology Center, University of Southern California, 3715 McClintock Avenue, Los Angeles, CA 90089-0191. E-mail:vlongo@usc.edu.
REFERENCES
- 1.Adamec E, Yang F, Cole GM, Nixon RA. Multiple-label immunocytochemistry for the evaluation of nature of cell death in experimental models of neurodegeneration. Brain Res Brain Res Protoc. 2001;7:193–202. doi: 10.1016/s1385-299x(01)00072-1. [DOI] [PubMed] [Google Scholar]
- 2.Augusto O, Gatti RM, Radi R. Spin-trapping studies of peroxynitrite decomposition and of 3-morpholinosydnonimine N-ethylcarbamide autooxidation: direct evidence for metal-independent formation of free radical intermediates. Arch Biochem Biophys. 1994;310:118–125. doi: 10.1006/abbi.1994.1147. [DOI] [PubMed] [Google Scholar]
- 3.Banker G, Goslin K. Developments in neuronal cell culture. Nature. 1988;336:185–186. doi: 10.1038/336185a0. [DOI] [PubMed] [Google Scholar]
- 4.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behl C. Amyloid beta-protein toxicity and oxidative stress in Alzheimer's disease. Cell Tissue Res. 1997;290:471–480. doi: 10.1007/s004410050955. [DOI] [PubMed] [Google Scholar]
- 6.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 7.Beltran B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S. The effect of nitric oxide on cell respiration: A key to understanding its role in cell survival or death. Proc Natl Acad Sci USA. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. beta-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem. 1999;274:15493–15499. doi: 10.1074/jbc.274.22.15493. [DOI] [PubMed] [Google Scholar]
- 9.Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 10.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 12.Chao CC, Molitor TW, Hu S. Neuroprotective role of IL-4 against activated microglia. J Immunol. 1993;151:1473–1481. [PubMed] [Google Scholar]
- 13.Chiang KT, Switzer CH, Akali KO, Fukuto JM. The role of oxygen and reduced oxygen species in nitric oxide-mediated cytotoxicity: studies in the yeast Saccharomyces cerevisiae model system. Toxicol Appl Pharmacol. 2000;167:30–36. doi: 10.1006/taap.2000.8970. [DOI] [PubMed] [Google Scholar]
- 14.Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- 15.Colton CA, Chernyshev ON, Gilbert DL, Vitek MP. Microglial contribution to oxidative stress in Alzheimer's disease. Ann NY Acad Sci. 2000;899:292–307. doi: 10.1111/j.1749-6632.2000.tb06195.x. [DOI] [PubMed] [Google Scholar]
- 16.Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci. 1999;19:928–939. doi: 10.1523/JNEUROSCI.19-03-00928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Combs CK, Karlo JC, Kao SC, Landreth GE. β-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–2412. [PubMed] [Google Scholar]
- 19.Estevez AG, Spear N, Manuel SM, Barbeito L, Radi R, Beckman JS. Role of endogenous nitric oxide and peroxynitrite formation in the survival and death of motor neurons in culture. Prog Brain Res. 1998a;118:269–280. doi: 10.1016/s0079-6123(08)63214-8. [DOI] [PubMed] [Google Scholar]
- 20.Estevez AG, Spear N, Manuel SM, Radi R, Henderson CE, Barbeito L, Beckman JS. Nitric oxide and superoxide contribute to motor neuron apoptosis induced by trophic factor deprivation. J Neurosci. 1998b;18:923–931. doi: 10.1523/JNEUROSCI.18-03-00923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flavin MP, Zhao G, Ho LT. Microglial tissue plasminogen activator (tPA) triggers neuronal apoptosis in vitro. Glia. 2000;29:347–354. [PubMed] [Google Scholar]
- 22.Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, Li X, Kirkpatrick JB. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int. 1995;27:119–137. doi: 10.1016/0197-0186(95)00067-i. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin JL, Uemura E, Cunnick JE. Microglial release of nitric oxide by the synergistic action of beta-amyloid and IFN-gamma. Brain Res. 1995;692:207–214. doi: 10.1016/0006-8993(95)00646-8. [DOI] [PubMed] [Google Scholar]
- 25.Gralla EB, Valentine JS. Null mutants of Saccharomyces cerevisiae Cu,Zn superoxide dismutase: characterization and spontaneous mutation rates. J Bacteriol. 1991;173:5918–5920. doi: 10.1128/jb.173.18.5918-5920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 27.Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hogg N, Darley-Usmar VM, Wilson MT, Moncada S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem J. 1992;281:419–424. doi: 10.1042/bj2810419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huygen IC. Reaction of nitrogen dioxide with Griess type reagents. Anal Chem. 1970;42:407–409. doi: 10.1021/ac60285a018. [DOI] [PubMed] [Google Scholar]
- 30.Ii M, Sunamoto M, Ohnishi K, Ichimori Y. beta-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res. 1996;720:93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- 31.Ischiropoulos H, Zhu L, Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys. 1992;298:446–451. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 32.Keller JN, Kindy MS, Holtsberg FW, St. Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim YM, Chung HT, Kim SS, Han JA, Yoo YM, Kim KM, Lee GH, Yun HY, Green A, Li J, Simmons RL, Billiar TR. Nitric oxide protects PC12 cells from serum deprivation-induced apoptosis by cGMP-dependent inhibition of caspase signaling. J Neurosci. 1999;19:6740–6747. doi: 10.1523/JNEUROSCI.19-16-06740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klegeris A, McGeer PL. Rat brain microglia and peritoneal macrophages show similar responses to respiratory burst stimulants. J Neuroimmunol. 1994;53:83–90. doi: 10.1016/0165-5728(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 35.Klegeris A, McGeer PL. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res. 1997;49:229–235. doi: 10.1002/(sici)1097-4547(19970715)49:2<229::aid-jnr11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 36.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 37.Koppal T, Drake J, Yatin S, Jordan B, Varadarajan S, Bettenhausen L, Butterfield DA. Peroxynitrite-induced alterations in synaptosomal membrane proteins: insight into oxidative stress in Alzheimer's disease. J Neurochem. 1999;72:310–317. doi: 10.1046/j.1471-4159.1999.0720310.x. [DOI] [PubMed] [Google Scholar]
- 38.Longo VD, Gralla EB, Valentine JS. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J Biol Chem. 1996;271:12275–12280. doi: 10.1074/jbc.271.21.12275. [DOI] [PubMed] [Google Scholar]
- 39.Longo VD, Viola KL, Klein WL, Finch CE. Reversible inactivation of superoxide-sensitive aconitase in Abeta1–42-treated neuronal cell lines. J Neurochem. 2000;75:1977–1985. doi: 10.1046/j.1471-4159.2000.0751977.x. [DOI] [PubMed] [Google Scholar]
- 40.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. Complexes of NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 41.Marla SS, Lee J, Groves JT. Peroxynitrite rapidly permeates phospholipid membranes. Proc Natl Acad Sci USA. 1997;94:14243–14248. doi: 10.1073/pnas.94.26.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 43.McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997;17:2284–2294. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 45.Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem. 1998;273:15646–15653. doi: 10.1074/jbc.273.25.15646. [DOI] [PubMed] [Google Scholar]
- 46.Morgan TE, Laping NJ, Rozovsky I, Oda T, Hogan TH, Finch CE, Pasinetti GM. Clusterin expression by astrocytes is influenced by transforming growth factor beta 1 and heterotypic cell interactions. J Neuroimmunol. 1995;58:101–110. doi: 10.1016/0165-5728(94)00194-s. [DOI] [PubMed] [Google Scholar]
- 47.Paxinou E, Weisse M, Chen Q, Souza JM, Hertkorn C, Selak M, Daikhin E, Yudkoff M, Sowa G, Sessa WC, Ischiropoulos H. Dynamic regulation of metabolism and respiration by endogenously produced nitric oxide protects against oxidative stress. Proc Natl Acad Sci USA. 2001;98:11575–11580. doi: 10.1073/pnas.201293198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur J Pharmacol. 1991;207:367–368. doi: 10.1016/0922-4106(91)90014-9. [DOI] [PubMed] [Google Scholar]
- 49.Rozovsky I, Morgan TE, Willoughby DA, Dugichi-Djordjevich MM, Pasinetti GM, Johnson SA, Finch CE. Selective expression of clusterin (SGP-2) and complement C1qB and C4 during responses to neurotoxins in vivo and in vitro. Neuroscience. 1994;62:741–758. doi: 10.1016/0306-4522(94)90473-1. [DOI] [PubMed] [Google Scholar]
- 50.Salvemini D, Wang ZQ, Stern MK, Currie MG, Misko TP. Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc Natl Acad Sci USA. 1998;95:2659–2663. doi: 10.1073/pnas.95.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S. Activation of NADPH oxidase in Alzheimer's disease brains. Biochem Biophys Res Commun. 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 52.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer's disease. Biochim Biophys Acta. 2000;1502:139–144. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka M, Sotomatsu A, Yoshida T, Hirai S, Nishida A. Detection of superoxide production by activated microglia using a sensitive and specific chemiluminescence assay and microglia-mediated PC12h cell death. J Neurochem. 1994;63:266–270. doi: 10.1046/j.1471-4159.1994.63010266.x. [DOI] [PubMed] [Google Scholar]
- 55.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Dyke K. The possible role of peroxynitrite in Alzheimer's disease: a simple hypothesis that could be tested more thoroughly. Med Hypotheses. 1997;48:375–380. doi: 10.1016/s0306-9877(97)90031-1. [DOI] [PubMed] [Google Scholar]
- 57.Wirtz-Brugger F, Giovanni A. Guanosine 3 ′,5′-cyclic monophosphate mediated inhibition of cell death induced by nerve growth factor withdrawal and beta-amyloid: protective effects of propentofylline. Neuroscience. 2000;99:737–750. doi: 10.1016/s0306-4522(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 58.Wood PL. Microglia as a unique cellular target in the treatment of stroke: potential neurotoxic mediators produced by activated microglia. Neurol Res. 1995;17:242–248. doi: 10.1080/01616412.1995.11740321. [DOI] [PubMed] [Google Scholar]
- 59.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 60.Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, Lipton SA. Cytokine-stimulated, but not HIV-infected, human monocyte-derived macrophages produce neurotoxic levels of l-cysteine. J Immunol. 2000;164:4265–4270. doi: 10.4049/jimmunol.164.8.4265. [DOI] [PubMed] [Google Scholar]