

Abstract
In this report we describe pertussis toxin-induced reversible encephalopathy dependent on monocyte chemoattractant protein-1 (MCP-1) overexpression (PREMO), a novel animal model that exhibits features of human encephalopathic complications of inflammatory disorders such as viral meningoencephalitis and Lyme neuroborreliosis as well as the mild toxic encephalopathy that commonly precedes relapses of multiple sclerosis (MS). Overexpression of the mouse MCP-1 gene product (classically termed JE) in astrocytes, the major physiological CNS cellular source of MCP-1, failed to induce neurological impairment. Unexpectedly, transgenic (tg) mice overexpressing MCP-1 at a high level (MCP-1hi) manifested transient, severe encephalopathy with high mortality after injections of pertussis toxin (PTx) plus complete Freund's adjuvant (CFA). Surviving mice showed markedly improved function and did not relapse during a prolonged period of observation. Tg mice that expressed lower levels of MCP-1 were affected minimally after CFA/PTx injections, and tg expression of other chemokines failed to elicit this disorder. The disorder was significantly milder in mice lacking T-cells, which therefore play a deleterious role in this encephalopathic process. Disruption of CC chemokine receptor 2 (CCR2) abolished both CNS inflammation and encephalopathy, identifying CCR2 as a relevant receptor for this disorder. Proinflammatory and type 1 cytokines including TNF-α, IL-1β, IFN-γ, IL-2, RANTES, and IP-10 were elevated in CNS tissues from mice with PREMO. These studies characterize a novel model of reversible inflammatory encephalopathy that is dependent on both genetic and environmental factors.
Keywords: chemokines, chemokine receptors, mice, gene targeting, macrophages, pertussis toxin
The product of the murine monocyte chemoattractant protein-1 (MCP-1) gene (also termed JE), was identified initially as a monocyte-specific chemoattractant (Rollins, 1991, 1997), but it also attracts and activates T-cells, dendritic cells, mast cells, and basophils (Taub et al., 1996; Gunn et al., 1997; Siveke and Hamann, 1998). Recent results indicated a role for MCP-1 in the formation and evolution of both innate and adaptive immune reactions. In particular, MCP-1 promotes T helper type 2 (Th2) T-cell development by enhancing secretion of type 2 cytokines, including interleuken-4 (IL-4), IL-5, and IL-10, and by antagonizing IL-12 secretion (Chensue et al., 1996; Karpus et al., 1997, 1998; Gu et al., 2000; Matsukawa et al., 2000). Administration of MCP-1 in a murine lipopolysaccharide (LPS) endotoxemia model was protective and reduced levels of cytokines such as tumor necrosis factor (TNF) and IL-12 while increasing IL-10 (Zisman et al., 1997). Neutralizing antibodies to MCP-1 caused enhanced mortality in mice with LPS-induced endotoxemia, along with precisely complementary cytokine changes. These results indicated that MCP-1, under defined circumstances, drives a type 2 cytokine response or produces potent anti-inflammatory effects. However, contrasting results also have been obtained. For example, mice lacking MCP-1 exhibited decreased severity of experimental autoimmune encephalomyelitis (EAE), with diminished Th1 cytokine production in the CNS (Huang et al., 2001). Therefore, the precise role of MCP-1 in the pathophysiology of CNS inflammation remains uncertain. This question is worthy of consideration, because high CNS levels of a potential human orthologue to JE, MCP-1 [termed CC chemokine ligand 2 (CCL2) in the systematic nomenclature], have been documented in a large number of clinical neurological disorders, including viral and bacterial meningoencephalitis, brain injury, and HIV-associated dementia (Berman et al., 1996; Schmidtmayerova et al., 1996; Spanaus et al., 1997;Conant et al., 1998; McManus et al., 1998).
One potential rationalization for differing roles of MCP-1 or CCL2 in different contexts might involve tissue-specific chemokine receptor usage. Chemokines signal via high-affinity G-protein-coupled receptors, of which ∼18 have been well characterized in both humans and rodents (Murphy et al., 2000) (http://cytokine.medic.kumamoto-u.ac.jp/CFC/CK/Chemokine.html). Several chemokines bind productively to more than one receptor, and individual receptors often recognize more than one chemokine, indicating the functional versatility and potential for redundancy in the chemokine system. Mice deficient for MCP-1 or its leukocyte receptor CC chemokine receptor 2 (CCR2) exhibited significant phenotypic discrepancies in immune and host defense responses (Boring et al., 1997; Gu et al., 1997, 2000; Kurihara et al., 1997; Warmington et al., 1999; Sato et al., 2000; Kim et al., 2001). In part, these observations have been explained by the presence of multiple ligands for CCR2, but the possibility of alternative receptor(s) for MCP-1 (Luther and Cyster, 2001) also has been raised. With the use ofin vitro cultures it was shown that rat astrocytes and arterial smooth muscle cells that do not express CCR2 responded to MCP-1 (Heesen et al., 1996; Schecter et al., 1997). Similar results have been reported in studies of human and rat microglia (Cross and Woodroofe, 1999a,b). Therefore, it is plausible that CNS-specific effects of MCP-1 could be transduced via a putative receptor on critical and unique resident cells such as astrocytes, arterial smooth muscle cells of the blood–brain barrier (BBB), or microglia.
Organ-specific effects of MCP-1 in the mammalian CNS are also conceivable because of its distinctive anatomical features. The CNS is sheltered from the cellular and molecular elements of the bloodstream by a BBB composed of nonfenestrated endothelial cells with tight junctions and a continuous basement membrane densely invested with astrocytic processes. A myelin basic protein (MBP) promoter element was used in previous experiments to direct a transient burst of MCP-1 expression by oligodendrocytes during postnatal weeks 2 and 3, corresponding to the developmental schedule of CNS myelin protein production (Gow et al., 1992; Fuentes et al., 1995). MBP-MCP-1 transgenic (tg) mice developed prominent perivascular monocyte infiltrates of the CNS without additional manipulation. This result indicated that parenchymal CNS chemokine could signal to circulating cells via an intact BBB. Intraperitoneal LPS hugely augmented the leukocyte infiltrates. No adverse consequences of transgene expression or monocyte infiltration of the CNS were observed in these studies (Fuentes et al., 1995).
These studies established that overexpression of MCP-1 in the CNS mediates selective recruitment of monocytes (Fuentes et al., 1995). However, astrocytes are the major cellular source of MCP-1 and CCL2 during a wide spectrum of neurological disorders and models, including CNS injury (Berman et al., 1996; Glabinski et al., 1996); multiple sclerosis (MS) (McManus et al., 1998) and EAE (Hulkower et al., 1993;Ransohoff et al., 1993); infections (Spanaus et al., 1997), (Sprenger et al., 1996), virus-associated neuropathologies (Conant et al., 1998;Sanders et al., 1998), and cerebrovascular disorders (Gaetani et al., 1998). Given the complex character of CNS inflammation and of MCP-1 itself, uncertainty persists whether astrocyte-derived MCP-1 is protective or destructive. We addressed this question by placing a MCP-1 transgene under control of a promoter fragment derived from the gene encoding human glial fibrillary acidic protein (GFAP), thereby targeting expression to CNS astrocytes (Brenner et al., 1994; Chiang et al., 1996; Owens et al., 2001). Overexpression of MCP-1 in astrocytes mediated little leukocyte migration into the CNS in unmanipulated mice. Surprisingly, injection with pertussis toxin (PTx) and complete Freund's adjuvant (CFA) led to intense leukocyte infiltration in one tg mouse line (MCP-1hi), resulting in rapid-onset, transient encephalopathy with elevated CNS levels of interferon-γ (IFN-γ) and IL-2. This model disorder was designated pertussis toxin-induced reversible encephalopathy dependent on monocyte chemoattractant protein-1 overexpression (PREMO). Manifestations of PREMO were absent in MCP-1hi tg mice lacking CCR2 (Boring et al., 1997, 1998; Kurihara et al., 1997), identifying the relevant MCP-1 receptor for this model and suggesting that the disorder was dependent on the action of MCP-1. The PREMO disease phenotype was attenuated significantly in MCP-1hi tg mice that were deficient for recombination activation gene-1 (RAG-1) (Mombaerts et al., 1992), indicating a detrimental proinflammatory role for T-cells. PREMO indicates that MCP-1 overexpression can prime the CNS for macrophage-mediated inflammation and BBB disruption, with dramatic deleterious effects on physiological function. The PREMO model provides an opportunity to address mechanisms of these effects. Furthermore, this model disorder displays a polarized type 1 cytokine profile, supporting the hypothesis that the specific character of a MCP-1-dependent inflammatory response is defined by the context in which it occurs.
MATERIALS AND METHODS
Generation of mice with astrocyte-targeted overexpression of MCP-1. The transgene was constructed by cloning mouse MCP-1 gene under the control of human GFAP promoter (see Fig. 1a). Microinjection of isolated transgene fragment into SWXJ (H-2q,s) eggs was performed by following a standard procedure. SWXJ (H-2q,s) mice were generated by mating SWR/J (H-2q) females with SJL/J (H-2s) males at the Jackson Laboratory (Bar Harbor, ME). Southern blot of tail DNA for fragments of human GFAP gene was used to identify the tg founders (see Fig. 1b). The detailed sequences of the primers used in PCR-based analyses of tail DNA included hgfapf, 5′-TTC CTG GGC ACA GGC TGA ATA GAG-3′, and hgfapr, 5′-ATT GAG CAG GGG GCT TGC ATT G-3′.
Fig. 1.
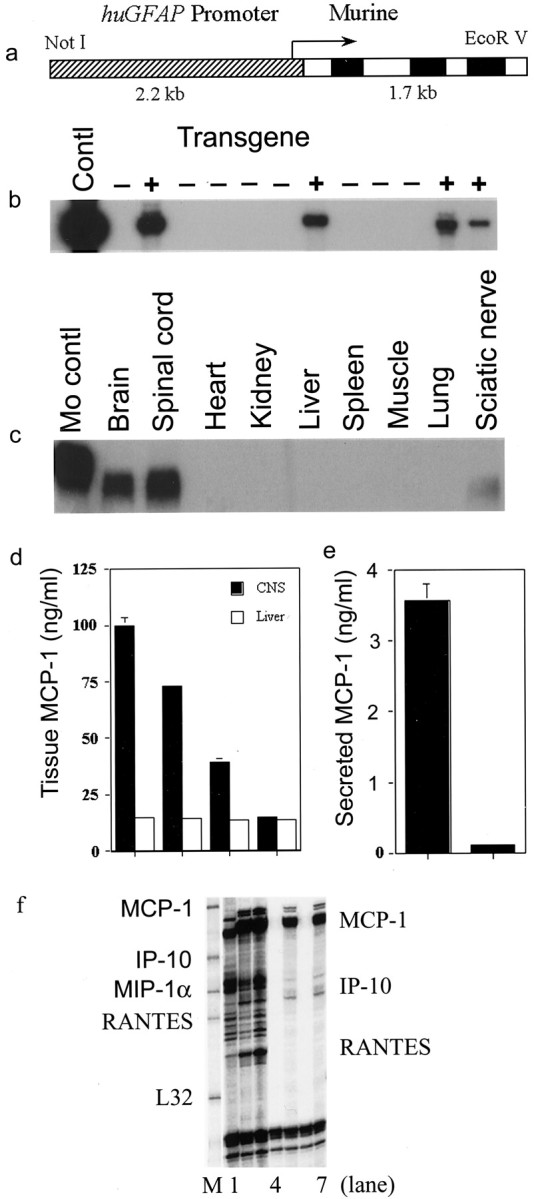
Establishment of transgenic mice overexpressing MCP-1 directed by GFAP promoter. a, Construct of a human GFAP promoter driving the expression of murine MCP-1. b, Southern blot demonstrating samples of DNA that contain elements of human GFAP promoter. c, Northern blot showing targeted MCP-1 gene expressions specifically in tissue homogenates of brain, spinal cord, and sciatic nerve. Experiments were performed as previously described in Huang et al. (2001). d, Different levels of MCP-1 expressions in tissue homogenates of CNS from three lines of MCP-1 transgenic mice, i.e., MCP-1hi,me, and low. MCP-1 levels were measured according to Karpus et al. (1998). e, Compared with astrocytes from nontransgenic littermate control mice, astrocytes from MCP-1 tg+ mice secreted significantly higher levels of MCP-1. Astrocytes were obtained and cultured by following the protocol described previously in Han et al. (2001). Levels of MCP-1 were determined via ELISA kits from R & D Systems. f, Comparable levels of MCP-1 expression in spinal cords from MCP-1hitg+ non-EAE and tg− EAE mice.Lane 1, MCP-1 tg− wild-type mouse with EAE; lanes 2, 3, MCP-1hitg+ with EAE; lanes 4, 6, tg− naive mice; lanes 5, 7, MCP-1hi tg+ with no challenge.
Mice. SWXJ (H-2q,s) huGFAP-MCP-1 tg+ mice were used to develop huGFAP-MCP-1 tg+ mice on SJL (H-2s), SWR (H-2q), SWXJ, and SJL×C57BL/6 genetic backgrounds. Briefly, a SWXJ tg+ male mouse was mated with a SWXJ tg− female mouse. The offspring were examined for the expression of the transgene (see above). The strain differentiation of SJL, SWR, and SWXJ was performed with flow cytometry as described below. Approximately 50% of offspring were tg+; 25, 50, and 25% were on SJL, SWXJ, and SWR backgrounds, respectively. An MCP-1hi tg+ male mouse on SJL background was mated to a CCR2 gene-deficient (CCR2–/–) mouse on C57BL/6 (B6) background. The resultant offspring with genotypes of MCP-1 tg+ mice heterozygous for CCR2 (MCP-1hitg+·CCR2–/+) and MCP-1hitg−·CCR2–/+were intercrossed further to generate MCP-1hitg+·CCR2–/–, MCP-1hitg+·CCR2+/+, MCP-1hitg+·CCR2–/+, and CCR2–/–,–/+, and +/+mice. MCP-1hi tg+mice that were deficient for RAG1 (RAG1–/–) on SJL×B6 background were generated in a similar manner, and the B6-RAG-1–/– mice were obtained commercially from The Jackson Laboratory (Bar Harbor, ME). Primers ccr2, 5′-GTG TGT GCA GGT TCC AAT GGA G-3′, and ccr2ko, 5′-GGA AGA CAA TAG CAG GCA TGC-3′, amplified the CCR2 null allele, whereas primers ccr2 and ccr2wt, 5′-CCT TCA TCA AGC TCT TGG-3′, amplified theCCR2 wild-type allele (see Fig. 2c). Primers ragkof, 5′-CGC TAC CGG TGG ATG TGG AAT GTG-3′, and ragkor, 5′-ATG ACT GTG AAA GAG AAA CGA ACG-3′, amplified the RAG1 mutant allele, whereas primers ragwtf, 5′-GAT CGA CGT GAA GGC AGA TG-3′, and ragwtr, 5′-GTC TCT TCC TCT TGA GTC CC-3′, amplified the wild-typeRAG1 allele (see Fig. 2b). All of the experimental groups included in the current studies were compared with their corresponding littermate control mice. Animal experimental procedures were performed in accordance with National Institutes of Health guidelines on animal care. All mice were maintained in pathogen-free conditions in the animal facilities of The Cleveland Clinic Foundation.
Fig. 2.

PREMO in huGFAP-MCP-1hitg+ mice. Intravenous PTx injections alone or in combination with subcutaneous CFA induced PREMO in huGFAP-MCP-1hi tg+, but not in tg−, mice. Shown is time-lapse photography 4 d after PTx/CFA injection revealing hunched posture and lack of exploratory behavior in a tg+ mouse compared with the normal tg− mouse.
Determination of MCP-1 levels in the circulation, CNS homogenates, and astrocyte culture supernatants. Serum samples were collected by tail vein puncture and kept at –80°C until assay. Serum MCP-1 levels were examined with a commercially obtained ELISA kit (R & D Systems, Minneapolis, MN). Concentrations of MCP-1 in CNS homogenates were measured as previously described (Karpus et al., 1995,1998). Astrocytes were isolated from CNS tissues from mice at postnatal day 2 (P2) and cultured in RPMI 1640 with 10% fetal calf serum as described previously (Han et al., 2001). Supernatants were collected and kept at –80°C until assay.
Induction and monitoring of PREMO in huGFAP-MCP-1hitg mice. Mice 8–10 wk of age were injected intravenously with 500 ng of PTx (Sigma-Aldrich, St. Louis, MO) and subcutaneously with 0.2 ml of CFA (Invitrogen, San Diego, CA) containing 400 μg ofMycobacterium tuberculosis (Difco, Detroit, MI). At day 2 after induction the mice were administered an intravenous injection of PTx to stimulate inflammatory responses. All mice were weighed, examined, and graded daily in a double-blinded manner by J. Wang and T. T. He. The score criteria were as follows: 0, no disease; 1, hyporeactive or hyper-reactive to tactile stimuli, occasionally with seizures, usually accompanied by weight loss (10–15%); 2, abnormal gait with diminished righting reflex, further dehydrated with weight loss >15%; 3, single or bilateral hindlimb paresis, with or without sphincter dysfunction; 4, stuporous and primarily motionless but responsive to stimuli; 5, death or moribund state so that humane death is required. Subcutaneous hydration was given if necessary.
RNA preparation and analysis of mRNA levels of chemokines, cytokines, chemokine receptors by RNase protection assay. Mice were anesthetized with isoflurane and perfused through the left ventricle with ice-cold PBS. Tissues were harvested and immediately snap frozen in liquid nitrogen. Samples were kept at –80°C until RNA extraction. Total RNA was extracted with the use of Trizol reagent (Invitrogen) according to the manufacturer's instructions. Concentrations of RNA were determined by ultraviolet spectroscopy at 260 nm. Levels of cytokines, chemokines, and chemokine receptors were measured by RNase protection assay (RPA) with template sets andin vitro transcription kits obtained from BD PharMingen (San Diego, CA).
Cytokine mRNA quantitation by the use of real-time reverse transcriptase-coupled PCR. CNS RNAs from huGFAP-MCP-1 tg+ mice and their non-tg littermate controls were prepared as above, and levels of specific cytokine mRNAs were determined by using a LightCycler system (Roche Molecular Biochemicals, Indianapolis, IN) with primers, amplification parameters, and data analysis, as described previously (Huang et al., 2001).
Histology. Brains and spinal cords were dissected rapidly after intracardiac perfusion with ice-cold PBS, followed by 4.0% paraformaldehyde solution. The 8-μm-thick paraffin sections or frozen sections were stained with hematoxylin and eosin (H&E). Immunohistochemical staining for mouse IgG in CNS tissue was performed by using biotinylated anti-mouse IgG (H+L; Vector Laboratories, Burlingame, CA) and avidin–biotin complex (ABC) system (Vector Laboratories), followed by incubation with peroxidase substrate DAB (Vector Laboratories).
Flow cytometry. Single cells from the CNS tissues (brain plus spinal cord) and peripheral blood mononuclear cells were isolated by following the procedures described previously (Fife et al., 2000;Huang et al., 2001). After blocking with CD16/CD32 Fc Block (BD PharMingen) in fluorescence-activated cell-sorting (FACS) buffer, we stained the cells for surface markers by using antibodies directly conjugated with fluorochromes. Antibodies used in the current study were obtained from BD PharMingen and were as follows: anti-CD4-FITC, anti-CD8-PE, anti-MHC class II antigen IAs-PE (Clone 10-3.6), anti-IAq-FITC, anti-CD45-Cy-chrome, and anti-TCR β chain-Cy. All antibodies were pretitrated with mouse peripheral blood.
Statistical analyses. The Mann–Whitney U test was used to compare levels of cytokine and chemokine expressions. Disease incidence in different groups of mice was compared by the χ2 test. All p values were two-tailed; a p value < 0.05 was considered significant.
RESULTS
Generation and characterization of mice overexpressing MCP-1 in astrocytes
The murine MCP-1 gene was placed under control of the huGFAP promoter by molecular cloning in the huGFAP-MCP-1 plasmid. Microinjection of purified huGFAP-MCP-1 fusion gene fragment into fertilized eggs of SWXJ (H-2q,s) mice resulted in four tg founders, which were identified by Southern blot of tail DNA by using 32P-labeled fragments of the huGFAP gene (Fig. 1b). Subsequently, a PCR-based genotyping protocol that used tail DNA was established and shown to correspond perfectly with the results obtained by Southern blotting. Three of four founders transmitted the transgene to progeny, all of which expressed transgene-directed mRNA and protein as confirmed by Northern blot analysis (Fig. 1c) and ELISA of CNS lysates (Fig. 1d) and supernatants of astrocyte cultures (Fig. 1e) from transgenic mouse lines. These three lines of tg mice displayed different levels (MCP-1hi, me, and low) of MCP-1 expression in the CNS as well as in peripheral nerve, but not in other tissues (Fig. 1c). Expression of the transgene in peripheral nerve (such as sciatic) was expected, because GFAP is expressed by nonmyelinating Schwann cells (Brenner et al., 1994). Levels of CNS MCP-1 expression in huGFAP-MCP-1hi tg mice were comparable with those observed in CNS tissues from mice with EAE (Fig. 1f). Aside from sparse inflammatory aggregates, MCP-1 tg mice exhibited neither overt pathological changes in CNS (see below) nor neurological impairment during 6 months of observation. Although young adult GFAP-MCP-1 tg mice exhibited much less spontaneous inflammation than MBP-MCP-1 animals, the lack of spontaneous neurological symptoms and signs were compatible with results reported previously for MBP-MCP-1 mice (Fuentes et al., 1995).
PREMO in huGFAP-MCP-1hi tg mice
CCL2 plays an enigmatic role in the human disorder MS, being reduced in the CSF during attacks but highly expressed in brain lesions (McManus et al., 1998; Sorensen et al., 1999; Van Der Voorn et al., 1999). To analyze the function of intracerebral MCP-1 in EAE, we immunized huGFAP-MCP-1hitg+ mice and littermate controls with antigen (PLP peptide 139–151) emulsified in CFA. Mice also received intravenous PTx at the time of immunization and 48 hr after, as previously described (Huang et al., 2001). Unexpectedly, all huGFAP-MCP-1hi tg+mice, but no littermate controls, developed impressive neurological impairment with high mortality 3–5 d after immunization. Surviving mice recovered and subsequently exhibited severe EAE at a time point typical of nontransgenic animals (our unpublished observations).
In clinical disorders such as HIV-associated dementia, patients harboring increased levels of CNS CCL2 are exposed to systemic inflammatory stimuli, often with transient functional decompensation (Conant et al., 1998). We considered the possibility that neurological signs in huGFAP-MCP-1hitg+ mice with early onset in <1 week after challenge represented encephalopathy, rather than autoimmune demyelination. To address this possibility, we challenged tg and wild-type littermate control mice with CFA/PTx in the absence of peptide antigen (Figs. 2, 3). We observed that huGFAP-MCP-1hitg+ mice challenged with PTx and CFA exhibited signs of encephalopathy including stupor, seizure, weight loss, sphincter dysfunction, jumping or rolling, and limb weakness (Fig. 2) with onset at day 3–5 after induction and high mortality rate (Fig. 3). Mice that survived PREMO recovered gradually and significantly, exhibiting no relapses after the initial attack during 60 d of observation (as shown in Fig. 3 for SWXJ mice). We also investigated myelin antigen responses by using spleen and lymph node cells from huGFAP-MCP-1hi tg+mice with PREMO and found no significant antigen-specific recall responses (our unpublished observations). The major residual signs of PREMO were reduced body weight and slower righting reflex compared with age-matched control mice.
Fig. 3.

High death rate and clinical course of PREMO in SWXJ huGFAP-MCP-1hi tg+ mice.a, Thirty-two of 56 SWXJ huGFAP-MCP-1hitg+ mice died of PREMO within 2 wk after the onset of PREMO compared with nil in their tg− littermate control mice; p < 0.01. b, PREMO was induced in MCP-1hi mice, but not in MCP-1meand low tg+ and tg−mice. HuGFAP-MCP-1hi, me, andlow tg+ and tg− mice were immunized with PTx and CFA, weighed, and scored daily for PREMO. Shown are PREMO scores (mean ± SD) of mice in each group;p < 0.01 compared between MCP-1hitg+ and others (MCP-1me andlow tg+ and tg−mice). n = 56, 19, 31, and 45 in groups of MCP-1hi, me, and lowtg+ and tg− mice, respectively.
These signs did not occur in littermate controls. Minimal signs of PREMO were observed in two other lines of MCP-1 tg+ mice, huGFAP-MCP-1low and me(Fig. 3). Mice of identical SWXJ background strain that overexpressed MIP-1α (macrophage inflammatory protein-1α) or GRO-α (growth-regulated oncogene-α) under control of the huGFAP expression cassette did not exhibit PREMO-like symptoms after receiving CFA/PTx (data not shown). These findings suggested that the occurrence of PREMO might be relatively specific for MCP-1 and required a threshold level of CNS overexpression.
PREMO caused by presence of the huGFAP-MCP-1 transgene was equally severe in several mouse strains.
In subsequent experiments the huGFAP-MCP-1hitransgene was placed on the background of varying strains, including SJL, SWR, SWXJ, and SJL×B6. All mice that expressed the transgene showed similar signs and disease course after exposure to CFA/PTx. Regardless of background strain, disease incidence was 100% in huGFAP-MCP-1hi tg+mice (Table 1). These results were consistent with the hypothesis that the principal genetic requirement for PREMO was high-level overexpression of MCP-1 in the CNS.
Table 1.
PREMO in huGFAP-MCP-1hi tg mice on different genetic backgrounds
| Strains | MCP-1 tg | Number of mice | |
|---|---|---|---|
| Tested | With PREMO (%) | ||
| SJL | + | 28 | 28 (100)* |
| − | 24 | 0 (0) | |
| SWR | + | 23 | 23 (100)* |
| − | 21 | 0 (0) | |
| SWXJ | + | 56 | 56 (100)* |
| − | 45 | 0 (0) | |
| SJL×B6 | + | 46 | 46 (100)* |
| − | 40 | 0 (0) | |
GFAP, Glial fibrillary acidic protein; MCP-1, monocyte chemoattractant protein 1; tg, transgenic.
p< 0.01 compared with littermate tg− controls, respectively.
Environmental requirements for PREMO
CFA and PTx, the adjuvants used for induction of murine EAE, exert selective proinflammatory effects and mediate BBB disruption. We evaluated the requirements for PREMO by examining the results of challenge with either CFA or PTx individually, staphylococcal enterotoxin B (SEB), and LPS as well. Mice receiving injections of PTx without CFA displayed a milder disease course with incidence of 96% (24 of 25), lower death rate, and shorter duration (data not shown). HuGFAP-MCP-1hi mice injected with CFA alone (n = 12), staphylococcal enterotoxin B (SEB;n = 9), or LPS (n = 8) showed no neurological signs. These data suggested that PTx was required for the induction of PREMO and that CFA could serve as a cofactor.
Histology and leukocyte infiltrates in the CNS of SWXJ mice with PREMO
The CNS of huGFAP-MCP-1hitg+ mice exhibited rare, small aggregates of mononuclear cells (Fig.4a,b). The brains and spinal cords of mice with PREMO contained numerous large mononuclear infiltrates (Fig. 4c,d). These infiltrates were primarily perivascular, with modest invasion of the parenchyma. PREMO tissues demonstrated extensive immunoglobulin deposition throughout the CNS white matter and meninges (Fig. 4i,j). By flow cytometric analysis the CNS infiltrates associated with PREMO were composed of hematogenous monocytes and CD4+ and CD8+ T-cells (Fig.5a,c,e). Leukocytes in CNS tissues from mice with PREMO expressed high levels of MHC class II molecules (Fig. 5a). Few CD19+B cells were detected in CNS tissues from huGFAP-MCP-1hi tg+mice with PREMO (data not shown). The acute course of PREMO and the lack of CD19+ cell accumulation in PREMO CNS suggest that the massive deposition of IgG resulted from BBB disruption. In surviving mice with functional restoration of the BBB the major CNS pathology was perivascular inflammation (Fig.5g,h). These observations indicated that MCP-1 prepared the CNS for intense leukocyte infiltration accompanied by BBB disruption in response to challenge with CFA/PTx.
Fig. 4.

CNS inflammation and blood–brain barrier disruption in huGFAP-MCP-1hi tg+ mice with PREMO. Although minute infiltrate was found in CNS from MCP-1hitg+ naive mice (a, b, arrowhead), H&E staining showed large amounts of infiltrates surrounding blood vessels and in the parenchyma in tissues from huGFAP-MCP-1hitg+ mice injected with PTx and CFA in the presence of CCR2 (c, d). Neither significant infiltrate nor perivascular inflammation (arrows indicating vessels) was observed in their huGFAP-MCP-1hitg+ littermate control mice on CCR2 mutant background (e, f). Mice recovered from PREMO contained perivascular infiltrates in the CNS (g, h). Immunoperoxidase histochemistry revealed abundant extravasated IgG in the cerebellar white matter, meninges (g, i), and spinal cord (h, j) of huGFAP-MCP-1hi tg+ mice (g, h), but not tg−(i, j) mice, 4 d after receiving injections of PTx plus CFA. Note positive IgG staining in peripheral nerve elements of the tg− control mouse at bottom left. a, c, e, g, Sections of brain stems, original × 100; b, d, f, h, sections of spinal cords, original × 50; i, k, brain sections, original × 25; j, l, spinal cords, original × 100.
Fig. 5.
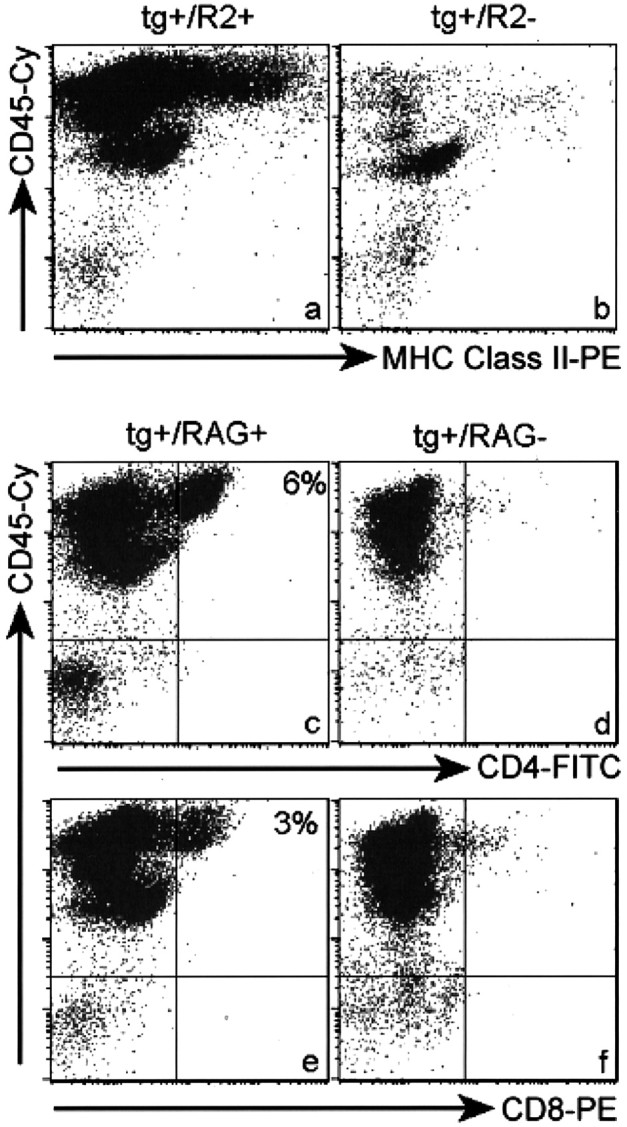
Flow cytometric analyses of CNS infiltrates in huGFAP-MCP-1hi tg+ mice with PREMO. Shown are high levels of MHC class II antigen expression on numerous leukocytes (CD45high) isolated from CNS tissues of intracardially perfused MCP-1hitg+·CCR2+mice with PREMO (a). Disruption of CCR2 abolishes the influx of leukocytes in MCP-1hi tg+mice with PREMO (b). CNS tissues from MCP-1hitg+·CCR2−mice injected with PTx plus CFA contained mainly CD45low microglia, virtually the same as those from MCP-1 tg− mice injected with PTx plus CFA (data not shown). Albeit with the lower prevalence and milder manifestation (Fig.7, Table 2), PREMO could be induced by injections of PTx plus CFA in MCP-1hitg+·RAG1−mice. Note the presence of CD45high leukocytes and the absence of both CD4+ (d) and CD8+ (f) T-cells in CNS tissues from MCP-1hitg+·RAG1−mice with PREMO compared with those in MCP-1hitg+·RAG1+ CNS tissues (c, e). Flow cytometric analyses of CNS cells from CNS tissues of huGFAP-MCP-1hi tg+-untreated mice and their wild-type controls were akin to b.
Cytokine and chemokine expression in the CNS of huGFAP-MCP-1hi mice with PREMO
The instructive role of MCP-1 toward T-cell cytokine expression has appeared to be context-dependent. To address effects of CNS-specific overexpression on inflammatory pathology, we examined the cytokine profile in tissues from mice with PREMO. In addition to abundant expression of the MCP-1 transgene, PREMO tissues contained significantly elevated levels of mRNA encoding mediators of innate immunity like TNF-α and IL-1β as well as T-cell cytokines such as the prototype type 1 mediator IFN-γ and IL-2. Levels of TNF-β showed a trend toward a decrease in huGFAP-MCP-1hi tg+mice with PREMO (p = 0.1) (Fig.6). We confirmed increased levels of IFN-γ message and protein in CNS tissues by real-time RT-PCR and ELISA (data not shown). Consistent with a type 1 cytokine environment, we also detected elevated levels of mRNA encoding RANTES/CCL5 (Fig. 6) and interferon-γ-inducible protein, 10 kDa (IP-10; data not shown). Neither IL-4 nor IL-10 was detected in CNS tissue from mice with PREMO (data not shown).
Fig. 6.

Increased levels of proinflammatory and Th1 cytokines in CNS tissues from huGFAP-MCP-1hitg+ mice with PREMO. a, RPA showing cytokine expression in samples of CNS tissues from MCP-1hitg+ and MCP-1hi tg−mice injected with PTx plus CFA. b, Quantitative analyses of inflammatory cytokine expression. M, Marker;tg, transgenic; +, tg-positive; −, tg-negative.
CC chemokine receptor 2 is essential for the induction of PREMO in huGFAP-MCP-1hi mice
Initial characterization of PREMO suggested that high-level CNS expression of MCP-1 along with specific environmental stimuli from injections of PTx and CFA was required for disease expression. PREMO was unambiguous and severe in one tg line that expressed the transgene at the highest level, leading to the hypothesis that a threshold level of MCP-1 was required for disease expression. However, there remained the unlikely possibility that insertional mutagenesis could account for this phenotype. We addressed directly whether MCP-1 was a major mediator of PREMO by generating and analyzing huGFAP-MCP-1hi tg mice lacking CCR2, the monocyte receptor for MCP-1. HuGFAP-MCP-1hitg+·CCR2−mice (transgenic but lacking CCR2) exhibited neither neurological impairment nor weight loss after injections with PTx plus CFA (Fig.7, Table 2). Concurrent analysis of littermate huGFAP-MCP-1hitg+·CCR2+excluded background strain effects, because these mice developed PREMO equally as severe as the index SWXJ strain. Flow cytometry (Fig. 5b) and histologic examination of CNS tissue sections (data not shown) demonstrated the absence of CNS inflammatory infiltrates in huGFAP-MCP-1hitg+·CCR2−mice that received CFA/PTx. These data indicated that the PREMO phenotype was associated closely with biological functions exerted by MCP-1, although participation of other MCPs (of which mice express three) and alternative MCP-1 receptors may have played contributing roles. Although additional proinflammatory cytokines (Fig. 6) well may be involved in the current model, the complete abrogation of PREMO in CCR2−/− mice demonstrated that CCR2 is the principal MCP-1 receptor in PREMO and that action toward other putative receptors on arterial smooth muscle cells or astrocytes is insufficient to cause this disorder.
Fig. 7.

Failure of PREMO induction in huGFAP-MCP-1hi tg+ mice lacking CCR2 and attenuated PREMO in mice deficient for recombination activation gene 1 (RAG1). a, Compared with the high death rate (25 of 46, 54%) in SJL×B6 MCP-1hitg+ mice with intact CCR2, none of the MCP-1hitg+·CCR2−and MCP-1hitg+·RAG1−mice injected with PTx plus CFA died of PREMO; p < 0.01. b, SJL×B6 MCP-1hitg+ mice developed PREMO similar to that in other strains, e.g., SWXJ (Fig. 4), SWR, and SJL (data not shown). Although MCP-1hitg+·RAG1−mice exhibit an attenuated PREMO, MCP-1hitg+·CCR2−mice were free from PREMO. Shown are PREMO scores (mean ± SD) in each group. MCP-1hitg+·RAG1−mice that remained healthy (18 of 31; see Table 2) were excluded.
Table 2.
PREMO in SJL×B6 huGFAP-MCP-1hi tg+mice in the presence or absence of CCR2 or RAG1
| CCR2 | RAG1 | Number of mice | |
|---|---|---|---|
| Tested | With PREMO (%) | ||
| + | + | 21 | 21 (100) |
| − | + | 32 | 0 (0)* |
| + | + | 25 | 25 (100) |
| + | − | 31 | 13 (42)* |
GFAP, Glial fibrillary acidic protein; CCR, CC chemokine receptor; tg, transgenic.
p < 0.01 compared with littermate controls with intact CCR2 and RAG1, respectively.
PREMO in huGFAP-MCP-1hi tg+ mice is inducible but less severe in the absence of T-cells
The presence of T-cell-derived and type 1 cytokines in CNS tissues of mice with PREMO raised the question of whether T-cells might be implicated in disease pathogenesis. To address this issue, we generated huGFAP-MCP-1hitg+ mice on RAG1–/– background (huGFAP-MCP-1hitg+·RAG1−). Littermate control huGFAP-MCP-1hitg+·RAG1+mice developed full-blown PREMO with high mortality, whereas huGFAP-MCP-1hitg+·RAG1−mice exhibited a diminished form of PREMO without mortality (0 of 31) and significantly lower incidence (Table 2). HuGFAP-MCP-1hitg+·RAG1−mice with PREMO recovered earlier and more completely than did their littermate controls (Fig. 7). As expected, CNS infiltrates of huGFAP-MCP-1hitg+·RAG1−mice with PREMO were devoid of both CD4+and CD8+ T-cells (Fig.5d,f). These results indicated that T-cells promoted the severity of PREMO. However, T-cells were not essential for disease occurrence, indicating that PREMO was an inflammatory reaction dependent either on MCP-1-recruited monocytes or on the action of MCP-1 toward resident CNS cells.
DISCUSSION
In this report we describe PREMO, a novel model disorder that exhibits features of human encephalopathic inflammatory disorders such as viral meningoencephalitis, Lyme neuroborreliosis, neurological manifestations of Sjögren's syndrome, and the mild toxic encephalopathy that commonly precedes relapses of MS. The essential components of PREMO may provide some insight into the mechanisms that underlie the disorder. PREMO requires the specific stimulus provided by PTx, markedly augmented by CFA. CCR2-mediated monocyte responses are essential for PREMO. T-cells play an auxiliary role in this disorder, which is characterized by high intrathecal production of type 1 cytokines and disruption of the BBB.
Three novel insights emerged from initial characterization of PREMO. First, it was shown that MCP-1 expression primes the CNS tissue for a deleterious inflammatory reaction that proceeds in the absence of T-cell autoimmunity. This finding may be of interest given that CCL2, a closely homologous human chemokine, is highly expressed in clinical neuroinflammatory disorders. Second, the critical role of CCR2 in PREMO demonstrates for the first time that this receptor transduces the essential signals for MCP-1-induced inflammation in the CNS. Although it remains plausible that non-CCR2 receptors can mediate aspects of neuroinflammation, the dominant role clearly is played by the monocyte receptor. Third, in the appropriate context MCP-1 directs a type 1-biased inflammatory reaction. This observation indicates that it might not be a useful generalization to consider MCP-1 as a type 2 mediator. Rather, it appears likely that MCP-1 plays a critical role at the interface of innate and adaptive immunity, facilitating the development of both type 1 and type 2 responses. In the case of the CNS it has been proposed that there is a tissue-specific bias toward type 1 reactions, possibly because of specific attributes of microglia as antigen-presenting cells (Krakowski and Owens, 1997; Aloisi et al., 1999). Therefore, it is not surprising that MCP-1-mediated inflammation in the CNS could be expressed as a type 1 reaction.
Overexpression of cytokines in tg mice has proven to be an incisive tool for investigating the pathogenesis of neurological disorders. PREMO differs from other models of disease generated by targeting cytokine transgenes to the CNS. In previous reports either TNF (Probert et al., 1995) or IL-3 (Chiang et al., 1996) was overexpressed, producing demyelination and neurological signs without further challenge. By contrast, PREMO required induction by systemic inflammation, and demyelination was not characteristic of this disorder. Tg mice that produced high levels of KC in the CNS (Tani et al., 1996) exhibited spontaneous inflammatory infiltrates and delayed neurodegenerative features.
A different murine model that encompassed both genetic and environmental features used MBP-specific T-cell receptor (TCR) transgenes. Mice housed in nonsterile conditions developed demyelination and typical EAE (Goverman et al., 1993). Interestingly, injections of PTx also could provoke EAE in MBP-TCR tg mice.
Aged mice that expressed IL-12 under control of the murine GFAP promoter (GF-IL-12 mice) developed spontaneous inflammatory demyelination (Pagenstecher et al., 2000). Young adult GF-IL-12 mice that were challenged with PTx/CFA developed mild neurological signs including ruffled fur, hunched posture, and general inanition (Lassmann et al., 2001). PTx/CFA-challenged GF-IL-12 mice expressed high levels of type I cytokines in the CNS, accompanied by abundant inflammatory infiltrates. Neurological disease in young adult GF-IL-12 mice most nearly resembles PREMO, differing primarily in its severity. The high expression of type 1 cytokines in both disorders supports the hypothesis that these products are integral to the clinical expression of encephalopathy.
The current studies, taken in the context of a previous report (Fuentes et al., 1995), show clearly that overexpression of MCP-1 in the CNS generates inflammatory pathology. In particular, CNS inflammation was observed in DBA/2×C57BL/6 (D2B6) mice that expressed MCP-1 under regulation of the MBP promoter (Fuentes et al., 1995) as well as SJL/J×SWR (SWXJ) mice that expressed MCP-1 under control of the GFAP promoter (the present studies). Furthermore, systemic inflammatory challenge augmented the pathology in both cases. These findings were consistent in the two models. The details of spontaneous pathology and stimulus-evoked pathology differed significantly between the two models. These differences could have arisen from variations between the models, which were substantial. First, transgene construction was different (using a cDNA in one case and the complete gene in another). Second, MBP and GFAP direct very different patterns of expression in regard to tempo, localization, and magnitude. Further, GFAP expression is upregulated by inflammation, whereas MBP is not. Finally, background genetic strain may have played a role in the variable stimulus-evoked inflammatory responses in the two models. As one example, LPS induced a brisk inflammatory response in MBP-MCP-1 D2B6 mice but only a minimal and inconsistent reaction in huGFAP-MCP-1 SWXJ transgenics. This difference may be explained by the documented difference between the two mouse strains in LPS sensitivity (Bohatschek et al., 2001).
Elevated levels of MCP-1 and CCL2 have been reported in both clinical and model neurological disorders. In patients with HIV-associated encephalitis and dementia (Schmidtmayerova et al., 1996; Sanders et al., 1998) and viral and pyogenic meningitis, impressively increased levels of CCL2 have been demonstrated consistently (Lopez-Cortes et al., 1995; Sprenger et al., 1996; Spanaus et al., 1997). Elevated expression of CCL2 has been found in acute and chronic MS plaques (McManus et al., 1998; Simpson et al., 1998), but reduced CCL2 CSF levels accompany attacks of disease (Sorensen et al., 1999). In patients with herpes simplex virus encephalitis, elevated CSF CCL2 correlates directly with the subsequent loss of functional independence (Rosler et al., 1998). MCP-1 is elevated rapidly and robustly in the CNS of animals with acute EAE (Hulkower et al., 1993; Ransohoff et al., 1993; Juedes et al., 2000) and in spontaneous attacks of chronic-relapsing EAE (Glabinski et al., 1997). Expression of MCP-1 precedes the influx of inflammatory cells to sites of penetrating mechanical (Berman et al., 1996; Glabinski et al., 1996, 1998) and cryogenic (Grzybicki et al., 1998) injury to the brain. Expression of MCP-1 occurs 6 hr after the onset of cerebral ischemia (Kim et al., 1995) and persists for days (Wang et al., 1995; Gourmala et al., 1997). Increased levels of MCP-1 expression are evident as early as 1 hr after reperfusion in rats with transient forebrain ischemia (Yoshimoto et al., 1997). Increased MCP-1 expression also has been documented in lymphocytic choriomeningitis (Asensio and Campbell, 1997) and in acute and chronic encephalomyelitis caused by mouse hepatitis virus (MHV) (Lane et al., 1998), mouse adenovirus-type 1 (Charles et al., 1999), and Theiler's murine encephalomyelitis virus (TMEV) (Ransohoff et al., 2002). The near-universal induction of MCP-1 and CCL2 after neurological insult suggests that this chemokine plays a central role in the physiology of neuroinflammation.
PREMO in huGFAP-MCP-1hitg+mice was typified by high mortality and severe clinical manifestations. Moreover, PREMO was induced successfully in several different mouse strains without significant variation in severity or incidence. PREMO required induction with a systemic inflammatory challenge. Given the anatomical isolation of the CNS behind the BBB and the fact that leukocyte extravasation is a multistep process (Luster, 1998), it is possible that naive, young huGFAP-MCP-1hitg+mice housed in a pathogen-free environment exhibited minimal CNS leukocyte infiltrates and no neurological impairment. Elevated levels of MCP-1 in the circulation (our unpublished data) could desensitize or downregulate the corresponding receptor(s) on leukocytes in the absence of systemic stimuli. PTx, which was essential for the induction of PREMO, sensitizes the BBB to disruption in the presence of inflammation (Linthicum et al., 1982). However, BBB disruption alone was not sufficient to provoke PREMO, because injections of CFA, which induces the leak of serum proteins across the BBB, failed to mediate disease (Rabchevsky et al., 1999). PTx enhances both Th1 and Th2 immune responses (Vistica et al., 1986; Ryan et al., 1998) and delayed-type hypersensitivity responses (Sewell et al., 1983, 1984). The production of IFN-γ (Sewell et al., 1986) is enhanced in the presence of PTx, which also augments the expression of costimulators such as B7-1 and B7-2 on antigen-presenting cells and CD28 on T-cells (Ryan et al., 1998). PTx prevents the induction of peripheral anergy in encephalitogenic T-cells (Kamradt et al., 1991). Proinflammatory cytokines such as TNF-α and IL-1β produced in CNS during PREMO are likely to increase BBB permeability and stimulate the secretion of chemokines by CNS parenchymal cells, thus amplifying the inflammatory cascade (Huang et al., 2000).
HuGFAP-MCP-1hi tg+mice with PREMO displayed inflammatory responses typified by a type 1 proinflammatory cytokine profile, suggesting that in this model MCP-1 may chemoattract Th1 T-cells or direct T-cell polarization toward type 1 cytokine production. Alternatively, murine CCL5, which was found at significantly elevated levels in the CNS of huGFAP-MCP-1hi tg+mice with PREMO (Fig. 7), might enhance the secretion of Th1 cytokines (Kim et al., 1998; Sin et al., 1999). HuGFAP-MCP-1hi tg+mice lacking CCR2 were completely resistant to PREMO, providing strong evidence that CCR2 plays a major role in this model.
PREMO was attenuated significantly in MCP-1hitg+·RAG1−mice, indicating that the functions of monocytes and T-cells in this model of CNS inflammation were cooperative. Reduced levels of IFN-γ were reported previously in kidneys (Tesch et al., 1999) and CNS (Huang et al., 2001) of MCP-1-null mice with inflammatory or autoimmune pathologies. Taking those results into consideration, our current data suggest that the instructive role of MCP-1 in adaptive immunity is dependent on the specific context of the inflammatory reaction. In summary, the PREMO model provides a platform for dissection of mechanisms that underlie MCP-1-associated encephalopathy and may uncover novel strategies for ameliorating the impact of clinical neuroinflammatory disorders.
Footnotes
This work was supported by National Institutes of Health Grant 2RO1 NS32151-09 to R.M.R. We gratefully acknowledge the support of the Williams Family Foundation for Multiple Sclerosis Research, the Nancy Davis Center Without Walls, and the Multiple Sclerosis Women's Committee. D.H. is a scholar of the Morgenthaler Family Foundation.
Correspondence should be addressed to Dr. Richard M. Ransohoff, Department of Neurosciences NC30, Lerner Research Institute, The Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195. E-mail: ransohr@ccf.org.
REFERENCES
- 1.Aloisi F, Ria F, Columba-Cabezas S, Hess H, Penna G, Adorini L. Relative efficiency of microglia, astrocytes, dendritic cells, and B cells in naive CD4+ T-cell priming and Th1/Th2 cell restimulation. Eur J Immunol. 1999;29:2705–2714. doi: 10.1002/(SICI)1521-4141(199909)29:09<2705::AID-IMMU2705>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 2.Asensio VC, Campbell IL. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol. 1997;71:7832–7840. doi: 10.1128/jvi.71.10.7832-7840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berman JW, Guida MP, Warren J, Amat J, Brosnan CF. Localization of monocyte chemoattractant peptide-1 expression in the central nervous system in experimental autoimmune encephalomyelitis and trauma in the rat. J Immunol. 1996;156:3017–3023. [PubMed] [Google Scholar]
- 4.Bohatschek M, Werner A, Raivich G. Systemic LPS injection leads to granulocyte influx into normal and injured brain: effects of ICAM-1 deficiency. Exp Neurol. 2001;172:137–152. doi: 10.1006/exnr.2001.7764. [DOI] [PubMed] [Google Scholar]
- 5.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, Charo IF. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knock-out mice. J Clin Invest. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2–/– mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 7.Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charles PC, Chen X, Horwitz MS, Brosnan CF. Differential chemokine induction by the mouse adenovirus type-1 in the central nervous system of susceptible and resistant strains of mice. J Neurovirol. 1999;5:55–64. doi: 10.3109/13550289909029746. [DOI] [PubMed] [Google Scholar]
- 9.Chensue SW, Warmington KS, Ruth JH, Sanghi PS, Lincoln P, Kunkel SL. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen-induced granuloma formation: relationship to local inflammation, Th cell expression, and IL-12 production. J Immunol. 1996;157:4602–4608. [PubMed] [Google Scholar]
- 10.Chiang CS, Powell HC, Gold LH, Samimi A, Campbell IL. Macrophage/microglial-mediated primary demyelination and motor disease induced by the central nervous system production of interleukin-3 in transgenic mice. J Clin Invest. 1996;97:1512–1524. doi: 10.1172/JCI118574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cross AK, Woodroofe MN. Chemokine modulation of matrix metalloproteinase and TIMP production in adult rat brain microglia and a human microglial cell line in vitro. Glia. 1999a;28:183–189. [PubMed] [Google Scholar]
- 13.Cross AK, Woodroofe MN. Chemokines induce migration and changes in actin polymerization in adult rat brain microglia and a human fetal microglial cell line in vitro. J Neurosci Res. 1999b;55:17–23. doi: 10.1002/(SICI)1097-4547(19990101)55:1<17::AID-JNR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 14.Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:899–905. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA. Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol. 1995;155:5769–5776. [PubMed] [Google Scholar]
- 16.Gaetani P, Tartara F, Pignatti P, Tancioni F, Baena R, De Benedetti F. Cisternal CSF levels of cytokines after subarachnoid hemorrhage. Neurol Res. 1998;20:337–342. doi: 10.1080/01616412.1998.11740528. [DOI] [PubMed] [Google Scholar]
- 17.Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, Ransohoff RM. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol. 1996;156:4363–4368. [PubMed] [Google Scholar]
- 18.Glabinski AR, Tani M, Strieter RM, Tuohy VK, Ransohoff RM. Synchronous synthesis of α- and β-chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am J Pathol. 1997;150:617–630. [PMC free article] [PubMed] [Google Scholar]
- 19.Glabinski AR, Tuohy VK, Ransohoff RM. Expression of chemokines RANTES, MIP-1α, and GRO-α correlates with inflammation in acute experimental autoimmune encephalomyelitis. Neuroimmunomodulation. 1998;5:166–171. doi: 10.1159/000026333. [DOI] [PubMed] [Google Scholar]
- 20.Gourmala NG, Buttini M, Limonta S, Sauter A, Boddeke HW. Differential and time-dependent expression of monocyte chemoattractant protein-1 mRNA by astrocytes and macrophages in rat brain: effects of ischemia and peripheral lipopolysaccharide administration. J Neuroimmunol. 1997;74:35–44. doi: 10.1016/s0165-5728(96)00203-2. [DOI] [PubMed] [Google Scholar]
- 21.Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T-cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 22.Gow A, Friedrich VL, Jr, Lazzarini RA. Myelin basic protein gene contains separate enhancers for oligodendrocyte and Schwann cell expression. J Cell Biol. 1992;119:605–616. doi: 10.1083/jcb.119.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grzybicki D, Moore SA, Schelper R, Glabinski AR, Ransohoff RM, Murphy S. Expression of monocyte chemoattractant protein (MCP-1) and nitric oxide synthase-2 following cerebral trauma. Acta Neuropathol (Berl) 1998;95:98–103. doi: 10.1007/s004010050770. [DOI] [PubMed] [Google Scholar]
- 24.Gu L, Rutledge B, Fiorillo J, Ernst C, Grewal I, Flavell R, Gladue R, Rollins B. In vivo properties of monocyte chemoattractant protein-1. J Leukoc Biol. 1997;62:577–580. doi: 10.1002/jlb.62.5.577. [DOI] [PubMed] [Google Scholar]
- 25.Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature. 2000;404:407–411. doi: 10.1038/35006097. [DOI] [PubMed] [Google Scholar]
- 26.Gunn MD, Nelken NA, Liao X, Williams LT. Monocyte chemoattractant protein-1 is sufficient for the chemotaxis of monocytes and lymphocytes in transgenic mice but requires an additional stimulus for inflammatory activation. J Immunol. 1997;158:376–383. [PubMed] [Google Scholar]
- 27.Han Y, He T, Huang DR, Pardo CA, Ransohoff RM. TNF-α mediates SDF-1α-induced NF-κB activation and cytotoxic effects in primary astrocytes. J Clin Invest. 2001;108:425–435. doi: 10.1172/JCI12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heesen M, Tanabe S, Berman MA, Yoshizawa I, Luo Y, Kim RJ, Post TW, Gerard C, Dorf ME. Mouse astrocytes respond to the chemokines MCP-1 and KC, but reverse transcriptase-polymerase chain reaction does not detect mRNA for the KC or new MCP-1 receptor. J Neurosci Res. 1996;45:382–391. doi: 10.1002/(SICI)1097-4547(19960815)45:4<382::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 29.Huang D, Han Y, Rani MR, Glabinski A, Trebst C, Sorensen T, Tani M, Wang J, Chien P, O'Bryan S, Bielecki B, Zhou ZL, Majumder S, Ransohoff RM. Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol Rev. 2000;177:52–67. doi: 10.1034/j.1600-065x.2000.17709.x. [DOI] [PubMed] [Google Scholar]
- 30.Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hulkower K, Brosnan CF, Aquino DA, Cammer W, Kulshrestha S, Guida MP, Rapoport DA, Berman JW. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J Immunol. 1993;150:2525–2533. [PubMed] [Google Scholar]
- 32.Juedes AE, Hjelmstrom P, Bergman CM, Neild AL, Ruddle NH. Kinetics and cellular origin of cytokines in the central nervous system: insight into mechanisms of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Immunol. 2000;164:419–426. doi: 10.4049/jimmunol.164.1.419. [DOI] [PubMed] [Google Scholar]
- 33.Kamradt T, Soloway PD, Perkins DL, Gefter ML. Pertussis toxin prevents the induction of peripheral T-cell anergy and enhances the T-cell response to an encephalitogenic peptide of myelin basic protein. J Immunol. 1991;147:3296–3302. [PubMed] [Google Scholar]
- 34.Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1α in the pathogenesis of the T-cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. 1995;155:5003–5010. [PubMed] [Google Scholar]
- 35.Karpus WJ, Lukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol. 1997;158:4129–4136. [PubMed] [Google Scholar]
- 36.Karpus WJ, Kennedy KJ, Kunkel SL, Lukacs NW. Monocyte chemotactic protein 1 regulates oral tolerance induction by inhibition of T helper cell 1-related cytokines. J Exp Med. 1998;187:733–741. doi: 10.1084/jem.187.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JJ, Nottingham LK, Sin JI, Tsai A, Morrison L, Oh J, Dang K, Hu Y, Kazahaya K, Bennett M, Dentchev T, Wilson DM, Chalian AA, Boyer JD, Agadjanyan MG, Weiner DB. CD8-positive T-cells influence antigen-specific immune responses through the expression of chemokines. J Clin Invest. 1998;102:1112–1124. doi: 10.1172/JCI3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, Welch KM. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol. 1995;56:127–134. doi: 10.1016/0165-5728(94)00138-e. [DOI] [PubMed] [Google Scholar]
- 39.Kim Y, Sung S, Kuziel WA, Feldman S, Fu SM, Rose CE., Jr Enhanced airway Th2 response after allergen challenge in mice deficient in CC chemokine receptor-2 (CCR2). J Immunol. 2001;166:5183–5192. doi: 10.4049/jimmunol.166.8.5183. [DOI] [PubMed] [Google Scholar]
- 40.Krakowski ML, Owens T. The central nervous system environment controls effector CD4+ T-cell cytokine profile in experimental allergic encephalomyelitis. Eur J Immunol. 1997;27:2840–2847. doi: 10.1002/eji.1830271115. [DOI] [PubMed] [Google Scholar]
- 41.Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lane TE, Asensio VC, Yu N, Paoletti AD, Campbell IL, Buchmeier MJ. Dynamic regulation of α- and β-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J Immunol. 1998;160:970–978. [PubMed] [Google Scholar]
- 43.Lassmann S, Kincaid C, Asensio VC, Campbell IL. Induction of type 1 immune pathology in the brain following immunization without central nervous system autoantigen in transgenic mice with astrocyte-targeted expression of IL-12. J Immunol. 2001;167:5485–5493. doi: 10.4049/jimmunol.167.9.5485. [DOI] [PubMed] [Google Scholar]
- 44.Linthicum DS, Munoz JJ, Blaskett A. Acute experimental autoimmune encephalomyelitis in mice. I. Adjuvant action of Bordetella pertussis is due to vasoactive amine sensitization and increased vascular permeability of the central nervous system. Cell Immunol. 1982;73:299–310. doi: 10.1016/0008-8749(82)90457-9. [DOI] [PubMed] [Google Scholar]
- 45.Lopez-Cortes LF, Cruz-Ruiz M, Gomez-Mateos J, Viciana-Fernandez P, Martinez-Marcos FJ, Pachon J. Interleukin-8 in cerebrospinal fluid from patients with meningitis of different etiologies: its possible role as neutrophil chemotactic factor. J Infect Dis. 1995;172:581–584. doi: 10.1093/infdis/172.2.581. [DOI] [PubMed] [Google Scholar]
- 46.Luster AD. Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 47.Luther SA, Cyster JG. Chemokines as regulators of T-cell differentiation. Nat Immunol. 2001;2:102–107. doi: 10.1038/84205. [DOI] [PubMed] [Google Scholar]
- 48.Matsukawa A, Lukacs NW, Standiford TJ, Chensue SW, Kunkel SL. Adenoviral-mediated overexpression of monocyte chemoattractant protein-1 differentially alters the development of Th1 and Th2 type responses in vivo. J Immunol. 2000;164:1699–1704. doi: 10.4049/jimmunol.164.4.1699. [DOI] [PubMed] [Google Scholar]
- 49.McManus C, Berman JW, Brett FM, Staunton H, Farrell M, Brosnan CF. MCP-1, MCP-2, and MCP-3 expression in multiple sclerosis lesions: an immunohistochemical and in situ hybridization study. J Neuroimmunol. 1998;86:20–29. doi: 10.1016/s0165-5728(98)00002-2. [DOI] [PubMed] [Google Scholar]
- 50.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 51.Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- 52.Owens T, Wekerle H, Antel J. Genetic models for CNS inflammation. Nat Med. 2001;7:161–166. doi: 10.1038/84603. [DOI] [PubMed] [Google Scholar]
- 53.Pagenstecher A, Lassmann S, Carson MJ, Kincaid CL, Stalder AK, Campbell IL. Astrocyte-targeted expression of IL-12 induces active cellular immune responses in the central nervous system and modulates experimental allergic encephalomyelitis. J Immunol. 2000;164:4481–4492. doi: 10.4049/jimmunol.164.9.4481. [DOI] [PubMed] [Google Scholar]
- 54.Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor α. Proc Natl Acad Sci USA. 1995;92:11294–11298. doi: 10.1073/pnas.92.24.11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rabchevsky AG, Degos JD, Dreyfus PA. Peripheral injections of Freund's adjuvant in mice provoke leakage of serum proteins through the blood–brain barrier without inducing reactive gliosis. Brain Res. 1999;832:84–96. doi: 10.1016/s0006-8993(99)01479-1. [DOI] [PubMed] [Google Scholar]
- 56.Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- 57.Ransohoff RM, Wei T, Pavelko KD, Lee JC, Murray PD, Rodriguez M. Chemokine expression in the central nervous system of mice with a viral disease resembling multiple sclerosis: roles of CD4+ and CD8+ T cells and viral persistence. J Virol. 2002;76:2217–2224. doi: 10.1128/jvi.76.5.2217-2224.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rollins BJ. JE/MCP-1: an early-response gene encodes a monocyte-specific cytokine. Cancer Cells. 1991;3:517–524. [PubMed] [Google Scholar]
- 59.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 60.Rosler A, Pohl M, Braune HJ, Oertel WH, Gemsa D, Sprenger H. Time course of chemokines in the cerebrospinal fluid and serum during herpes simplex type 1 encephalitis. J Neurol Sci. 1998;157:82–89. doi: 10.1016/s0022-510x(98)00061-6. [DOI] [PubMed] [Google Scholar]
- 61.Ryan M, McCarthy L, Rappuoli R, Mahon BP, Mills KH. Pertussis toxin potentiates Th1 and Th2 responses to co-injected antigen: adjuvant action is associated with enhanced regulatory cytokine production and expression of the co-stimulatory molecules B7-1, B7-2, and CD28. Int Immunol. 1998;10:651–662. doi: 10.1093/intimm/10.5.651. [DOI] [PubMed] [Google Scholar]
- 62.Sanders VJ, Pittman CA, White MG, Wang G, Wiley CA, Achim CL. Chemokines and receptors in HIV encephalitis. AIDS. 1998;12:1021–1026. [PubMed] [Google Scholar]
- 63.Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, Kuziel WA, Ahuja SS. CC chemokine receptor (CCR) 2 is required for Langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, B cell outgrowth, and sustained neutrophilic inflammation. J Exp Med. 2000;192:205–218. doi: 10.1084/jem.192.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, Giesen PL, Nemerson Y, Taubman MB. Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. J Biol Chem. 1997;272:28568–28573. doi: 10.1074/jbc.272.45.28568. [DOI] [PubMed] [Google Scholar]
- 65.Schmidtmayerova H, Nottet HS, Nuovo G, Raabe T, Flanagan CR, Dubrovsky L, Gendelman HE, Cerami A, Bukrinsky M, Sherry B. Human immunodeficiency virus type 1 infection alters chemokine β peptide expression in human monocytes: implications for recruitment of leukocytes into brain and lymph nodes. Proc Natl Acad Sci USA. 1996;93:700–704. doi: 10.1073/pnas.93.2.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sewell WA, Munoz JJ, Vadas MA. Enhancement of the intensity, persistence, and passive transfer of delayed-type hypersensitivity lesions by pertussigen in mice. J Exp Med. 1983;157:2087–2096. doi: 10.1084/jem.157.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sewell WA, Munoz JJ, Scollay R, Vadas MA. Studies on the mechanism of the enhancement of delayed-type hypersensitivity by pertussigen. J Immunol. 1984;133:1716–1722. [PubMed] [Google Scholar]
- 68.Sewell WA, de Moerloose PA, McKimm-Breschkin JL, Vadas MA. Pertussigen enhances antigen-driven interferon-γ production by sensitized lymphoid cells. Cell Immunol. 1986;97:238–247. doi: 10.1016/0008-8749(86)90394-1. [DOI] [PubMed] [Google Scholar]
- 69.Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of monocyte chemoattractant protein-1 and other β-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol. 1998;84:238–249. doi: 10.1016/s0165-5728(97)00208-7. [DOI] [PubMed] [Google Scholar]
- 70.Sin JI, Kim JJ, Arnold RL, Shroff KE, McCallus D, Pachuk C, McElhiney SP, Wolf MW, Pompa-de Bruin SJ, Higgins TJ, Ciccarelli RB, Weiner DB. IL-12 gene as a DNA vaccine adjuvant in a herpes mouse model: IL-12 enhances Th1-type CD4+ T-cell-mediated protective immunity against herpes simplex virus-2 challenge. J Immunol. 1999;162:2912–2921. [PubMed] [Google Scholar]
- 71.Siveke JT, Hamann A. T helper 1 and T helper 2 cells respond differentially to chemokines. J Immunol. 1998;160:550–554. [PubMed] [Google Scholar]
- 72.Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, Qin S, Rottman J, Sellebjerg F, Strieter RM, Frederiksen JL, Ransohoff RM. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103:807–815. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spanaus KS, Nadal D, Pfister HW, Seebach J, Widmer U, Frei K, Gloor S, Fontana A. C-X-C and C-C chemokines are expressed in the cerebrospinal fluid in bacterial meningitis and mediate chemotactic activity on peripheral blood-derived polymorphonuclear and mononuclear cells in vitro. J Immunol. 1997;158:1956–1964. [PubMed] [Google Scholar]
- 74.Sprenger H, Rosler A, Tonn P, Braune HJ, Huffmann G, Gemsa D. Chemokines in the cerebrospinal fluid of patients with meningitis. Clin Immunol Immunopathol. 1996;80:155–161. doi: 10.1006/clin.1996.0109. [DOI] [PubMed] [Google Scholar]
- 75.Tani M, Fuentes ME, Peterson JW, Trapp BD, Durham SK, Loy JK, Bravo R, Ransohoff RM, Lira SA. Neutrophil infiltration, glial reaction, and neurological disease in transgenic mice expressing the chemokine N51/KC in oligodendrocytes. J Clin Invest. 1996;98:529–539. doi: 10.1172/JCI118821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taub DD, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Chemokines and T lymphocyte activation. I. β-Chemokines costimulate human T lymphocyte activation in vitro. J Immunol. 1996;156:2095–2103. [PubMed] [Google Scholar]
- 77.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Faslpr mice. J Exp Med. 1999;190:1813–1824. doi: 10.1084/jem.190.12.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Der Voorn P, Tekstra J, Beelen RH, Tensen CP, Van Der Valk P, De Groot CJ. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol. 1999;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vistica BP, McAllister CG, Sekura RD, Ihle JN, Gery I. Dual effects of pertussis toxin on lymphoid cells in culture. Cell Immunol. 1986;101:232–241. doi: 10.1016/0008-8749(86)90200-5. [DOI] [PubMed] [Google Scholar]
- 80.Wang X, Yue TL, Barone FC, Feuerstein GZ. Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke. 1995;26:661–665. doi: 10.1161/01.str.26.4.661. [DOI] [PubMed] [Google Scholar]
- 81.Warmington KS, Boring L, Ruth JH, Sonstein J, Hogaboam CM, Curtis JL, Kunkel SL, Charo IR, Chensue SW. Effect of C-C chemokine receptor 2 (CCR2) knock-out on type-2 (schistosomal antigen-elicited) pulmonary granuloma formation: analysis of cellular recruitment and cytokine responses. Am J Pathol. 1999;154:1407–1416. doi: 10.1016/S0002-9440(10)65394-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshimoto T, Houkin K, Tada M, Abe H. Induction of cytokines, chemokines, and adhesion molecule mRNA in a rat forebrain reperfusion model. Acta Neuropathol (Berl) 1997;93:154–158. doi: 10.1007/s004010050596. [DOI] [PubMed] [Google Scholar]
- 83.Zisman DA, Kunkel SL, Strieter RM, Tsai WC, Bucknell K, Wilkowski J, Standiford TJ. MCP-1 protects mice in lethal endotoxemia. J Clin Invest. 1997;99:2832–2836. doi: 10.1172/JCI119475. [DOI] [PMC free article] [PubMed] [Google Scholar]