

Abstract
In hippocampal and other cortical neurons, action potentials are followed by a slow afterhyperpolarization (sAHP) generated by the activation of small-conductance Ca2+-activated K+ channels and controlling spike frequency adaptation. The corresponding current, the apamin-insensitive sIAHP, is a well known target of modulation by different neurotransmitters, including acetylcholine (via M3 receptors) and glutamate (via metabotropic glutamate receptor 5, mGluR5), in CA1 pyramidal neurons. The actions of muscarinic and mGluR agonists on sIAHP involve the activation of pertussis toxin-insensitive G-proteins. However, the pharmacological tools available so far did not permit the identification of the specific G-protein subtypes transducing the effects of M3 and mGluR5 on sIAHP. In the present study, we used mice deficient in the Gαq and Gα11 genes to investigate the specific role of these G-protein α subunits in the cholinergic and glutamatergic modulation of sIAHP in CA1 neurons. In mice lacking Gαq, the effects of muscarinic and glutamatergic agonists on sIAHP were nearly abolished, whereas β-adrenergic agonists acting via Gαs were still fully effective. Modulation of sIAHP by any of these agonists was instead unchanged in mice lacking Gα11. The additional depolarizing effects of muscarinic and glutamatergic agonists on CA1 neurons were preserved in mice lacking Gαq or Gα11. Thus, Gαq, but not Gα11, mediates specifically the action of cholinergic and glutamatergic agonists on sIAHP, without affecting the modulation of other currents. These results provide to our knowledge one of the first examples of the functional specificity of Gαq and Gα11 in central neurons.
Keywords: G-protein, muscarinic, metabotropic glutamate, calcium-activated potassium current, afterhyperpolarization, CA1 pyramidal neurons
In the hippocampus, glutamatergic and cholinergic regulation of neuronal excitability is thought to play a pivotal role in learning and memory processes (Pin and Bockaert, 1995; Riedel, 1996; Segal and Auerbach, 1997; Holscher et al., 1999;Perry et al., 1999). Acetylcholine and glutamate, beside mediating fast excitatory synaptic transmission in the CNS, modulate neuronal metabolic responses by acting on metabotropic receptors (muscarinic and metabotropic glutamate receptors, mGluRs). Previous studies have focussed on the molecular and cellular mechanisms by which muscarinic and mGluR agonists regulate membrane excitability in central and peripheral neurons and have revealed their effects on several ionic currents, such as the following: (1) the M current (IM), a voltage-dependent K+ current (Brown and Adams, 1980;Halliwell and Adams, 1982); (2) a voltage-independent “leak” K+ current [IK(leak)] (Madison et al., 1987;McCormick and von Krosigk, 1992; Guerineau et al., 1994); (3) the slowly inactivating K+ currentID (Wu and Barish, 1999); (4) the delayed rectifier K+ currentIK (Zhang et al., 1992); (5) the unspecific cationic pacemaker current (Ih) (Colino and Halliwell, 1993); (6) a Ca2+-dependent, cation-nonspecific current (Crepel et al., 1994; Greene et al., 1994; Guerineau et al., 1995; Haj-Dahmane and Andrade, 1997, 1999); and (7) the Ca2+-activated K+ current responsible for the slow afterhyperpolarization (sIAHP) (Benardo and Prince, 1982; Cole and Nicoll, 1983, 1984; Charpak et al., 1990).
The signal transduction pathways mediating these muscarinic and mGluR effects are only partly understood in hippocampal as in other central neurons. In particular, the involvement and molecular identification of G-proteins has been carefully investigated in some peripheral (Hille, 1994; Brown et al., 1997), but not in central neurons. Pertussis toxin-insensitive G-proteins have been proposed to be involved in the suppression of IM, sIAHP, andIK(leak) by muscarinic agonists in CA1 neurons (Dutar and Nicoll, 1988). However, the lack of selective pharmacological tools and the difficulty of successfully applying antisense or antibody techniques, as in dissociated or cultured cells (McFadzean et al., 1994; Ikeda, 1997; Buckley et al., 2000), has so far prevented the identification of specific G-protein subtypes mediating cholinergic and glutamatergic actions in central neurons in situ. Their identification is important, because the diversity of G-protein forms is likely to be matched by a corresponding range of cellular targets and functions.
The high structural homology, overlapping distribution patterns, and similar coupling properties to downstream effectors of Gαq and Gα11 subunits (Wu et al., 1992; Offermanns et al., 1994; Exton, 1996; Offermanns, 1999) have raised the question of their functional specificity in physiological systems. In this study, we unequivocally identified the G-protein subtypes that selectively transduce the signals of muscarinic M3 and glutamatergic mGluR5receptors on sIAHP in CA1 pyramidal neurons in mouse hippocampal slices. Using knock-out mice lacking Gαq or Gα11 subunits, we showed that Gαq, but not Gα11, mediates the downregulation of sIAHP by both muscarinic and mGluR agonists. Neither Gαq nor Gα11 seem instead to be required for the slow depolarization induced by these agonists. This study provides, to our knowledge, the first evidence of precise and distinct roles of these two G-protein α subunit isoforms in physiologically relevant signaling pathways in central neurons.
MATERIALS AND METHODS
Gαq- and Gα11-deficient mice. Mice deficient in the Gαq gene (Gαq−/−) or the Gα11 gene (Gα11 −/−) were generated by targeted disruption with a neomycin gene as described previously (Offermanns et al., 1997, 1998). Gαq −/− and Gα11 −/− mice used in the experiments were obtained by mating heterozygous males and females to obtain wild-type (Gαq +/+; Gα11 +/+) and knock-out (Gαq −/−; Gα11 −/−) littermates. Mice were kept on a C57BL/6 × 129/Sv background, and genotypes were confirmed by PCR on genomic DNA from tail biopsies of each mouse. All experiments were performed in a double-blind manner, and the code was broken only after completion of the experimental work and data analysis.
Slice preparation. Acute slices were obtained from Wistar rats (18–28 d old), Gαq −/− mice (11–24 weeks old), Gα11 −/− mice (14–25 weeks old), or wild-type mice. Wild-type mice were NMRI albino mice (4–8 weeks old), C57BL/6J (8–12 weeks old) mice, or littermates of Gαq −/− or Gα11 −/− (11–25 weeks old). Throughout the text, it is specified to which group we refer according to the experiment performed. Transversal hippocampal slices (300–400 μm) were cut with either a tissue chopper (Mickle Laboratory, Gomshall, Surrey, UK) or a vibratome (Leica, Nussloch, Germany) and subsequently incubated in a humidified interface chamber at room temperature for ≥1 hr.
Electrophysiology. Tight-seal whole-cell voltage-clamp recordings were obtained using the “blind method” (Blanton et al., 1989). Patch electrodes (4–7 MΩ) were filled with an intracellular solution containing (in mm): 140 potassium methylsulfate, 10 HEPES, 2 Na2-ATP, 0.4 Na3-GTP, and 3 MgCl2(osmolarity, 280–300 mOsm), pH 7.2–7.3 with KOH. Recordings were performed in a submerged recording chamber with a constant flow of artificial CSF (ACSF) (2 ml/min) at room temperature (22–24°C). Drugs were applied in the bath solution. ACSF contained (in mm): 125 NaCl, 1.25 KCl, 2.5 CaCl2, 1.5 MgCl2, 1.25 KH2PO4, 25 NaHCO3, and 16 d-glucose [bubbled with carbogen (95% O2–5% CO2]. In all recordings, tetrodotoxin (TTX) (0.5 μm), tetraethylammonium (TEA) (5 mm), and picrotoxin (5 μm) were added to the superfusing ACSF. Whole-cell patch-clamp recordings were obtained from 90 neurons from mice and 13 from rats in acute hippocampal slices. Neurons were voltage clamped at −60 mV, and 100- to 200-msec-long depolarizing pulses to 0–30 mV were delivered every 30 sec. These pulses led to unclamped Ca2+ action currents sufficient to activate AHP currents. Slow depolarizing currents were measured as changes in the holding current under the same experimental conditions (same intracellular and extracellular solutions; holding potential of −60 mV). Series resistance was regularly monitored, and only recordings with stable series resistances ≤35 MΩ were included in this study. No series resistance compensation and no corrections for liquid junction potentials were made. Data were acquired using a patch-clamp EPC9 amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany), filtered with a −3 dB cutoff frequency at 250 Hz, sampled at 1 KHz, and stored on a Macintosh Power personal computer (Apple Computers, Cupertino, CA). Analysis was made using the programs Pulse and Pulsefit (Heka), Igor Pro 3.01 (WaveMetrics Inc., Lake Oswego, OR), and Excel (Microsoft, Seattle, WA). We analyzed the amplitude, charge transfer (area enclosed by IAHP and sIAHP), and time course of AHP currents decay. No discrepancies in the variations of these parameters on pharmacological manipulations were observed; therefore, in Results and the figures, we reported only the values concerningIAHP and sIAHP amplitudes. The amplitude ofIAHP was determined at the peak of the current, whereas that of sIAHP was determined at a point 700–800 msec after the end of the command pulse, in which a possible contamination by the partially overlappingIAHP was negligible (Stocker et al., 1999). Values are presented as mean ± SEM. For statistical analysis, the unpaired, two-tailed Student's t test was used, and differences were considered statistically significant ifp ≤ 0.05.
Drugs and solutions. TEA, carbamylcholine (carbachol, CCh), Na2-ATP, and Na3-GTP were obtained from Sigma (St. Louis, MO). TTX was from Alomone Labs (Jerusalem, Israel). Picrotoxin and isoproterenol were from Research Biochemicals (Natick, MA).trans-1-Amino-cyclopentyl-1,3-dicarboxylate (trans-ACPD) and (S)-3,5-dihydroxyphenyl-glycine (DHPG) were from Tocris Cookson (Bristol, UK). All drugs were dissolved in water, stored at +4°C or −20°C, and bath applied in the perfusing ACSF.
RESULTS
AHP currents in the mouse hippocampus
Rat CA1 pyramidal neurons have been shown to present two distinct Ca2+-dependent afterhyperpolarizing currents, contributing to the early and late spike frequency adaptation: IAHP, deactivating in hundreds of milliseconds and sensitive to the toxin apamin; and sIAHP, characterized by a slow deactivation (in the range of seconds), insensitive to apamin and sensitive to the neuromodulatory effects of a number of transmitters (Stocker et al., 1999). In mouse hippocampal pyramidal neurons, the types of AHP currents and their key features are still unexplored. In CA1 pyramidal neurons from mouse slices, short depolarizing pulses inducing Ca2+ influx elicited an apamin-sensitive IAHP in 94.9% of the cells, followed by an apamin-insensitive sIAHP in 77.8% (Fig.1A). In two different strains of wild-type mice tested, NMRI and C57BL/6, sIAHP presented an amplitude of 19.6 ± 3.5 (n = 11) and 25.6 ± 3.4 (n = 31) pA, respectively, whereas in Wistar rats, the sIAHP amplitude was substantially larger (109.8 ± 13.5 pA; n = 13) under the same experimental conditions. The time courses of decay of the two AHP currents were clearly different: the apamin-sensitiveIAHP presented an average decay time constant of 88.3 ± 4.9 msec, and the apamin-insensitive sIAHP presented an average decay time constant of 3.5 ± 0.2 sec. In most cells measured,IAHP coexisted and partially overlapped with the rising phase of sIAHP, as shown in Figure1A (left panel). The pharmacological separation by apamin (Fig. 1A) suggests that IAHP and sIAHP are distinct currents, as reported previously in rat CA1 pyramidal neurons (Stocker et al., 1999). In all CA1 pyramidal neurons from mouse, as in rat, the Ca2+ spikes and sIAHP remained stable for the duration of the recording (usually ∼2–3 hr). Additionally, sIAHP displayed a normal sensitivity to a number of neurotransmitters (see below), as observed previously in rat neurons.
Fig. 1.

A, Mouse CA1 pyramidal neurons present a Ca2+-activated, apamin-sensitive afterhyperpolarizing current (IAHP) that deactivates in the hundreds of milliseconds range, followed by a Ca2+-activated, apamin-insensitive AHP current (sIAHP), lasting seconds.IAHP is partially overlapping with the rise time of sIAHP. These currents were elicited by Ca2+ influx induced by a 100 msec depolarizing pulse to +10 mV. Traces shown were recorded from CA1 neurons of a Gα11 −/− mouse. Gα11 +/+ littermates, as well as Gαq +/+ and Gαq −/− mice, displayed similar currents (see Results). In the right panel, IAHP was blocked by 50 nm apamin, leaving sIAHPamplitude and kinetics unaffected. B, Nissl stains of the hippocampal formation from Gαq +/+ and Gαq −/− mice. No changes in the gross morphology of the hippocampus were observed in the knock-out animals when compared with their wild-type littermates.
Cholinergic and glutamatergic modulation of sIAHP is inhibited in Gαq-deficient mice
Activation of muscarinic (M3; Rouse et al., 2000; M. Krause and P. Pedarzani, unpublished observation) or metabotropic (mGluR5; Gereau and Conn, 1995) glutamate receptors leads to the suppression of the apamin-insensitive sIAHP in CA1 pyramidal neurons. The actions of both muscarinic and mGluR agonists on sIAHP have been shown to involve activation of G-proteins of the pertussis toxin-insensitive type (Dutar and Nicoll, 1988; Gerber et al., 1992; Abdul-Ghani et al., 1996a;Krause and Pedarzani, 2000). However, pertussis toxin and GDP-β-S used in those studies blocked a wide subset or all G-proteins, respectively. Therefore, this approach did not allow the identification of specific G-protein subtypes transducing the effects of M3 muscarinic and mGluR5receptor stimulation on sIAHP. We used mice deficient in the Gαq gene (Gαq −/−) to investigate the specific role of this G-protein in the cholinergic and glutamatergic modulation of sIAHP. The hippocampus of Gαq-deficient mice presented an overall structure and gross morphology indistinguishable from those of wild-type animals (Fig. 1B). Additionally, sIAHP amplitude, time course, and passive membrane properties (resting membrane potential,Vm; input resistance,Ri) were not significantly different in Gαq −/− (Vm of −71.0 ± 1.4 mV;Ri of 186 ± 9 MΩ) when compared with Gαq +/+ mice (Vm of −72.4 ± 1.2 mV;Ri of 190 ± 11 MΩ) or mice of a different strain (NMRI; Vm of −68.3 ± 1.8 mV; Ri of 173 ± 13 MΩ). The only observed difference was an intrinsically smaller amplitude of the apamin-sensitive IAHP in the absence of neuromodulatory agents in Gαq −/− mice (87.6 ± 15.9 pA in Gαq +/+,n = 25; 44.1 ± 5.0 pA in Gαq −/−, n = 19;p = 0.01).
In wild-type mice, the cholinergic agonist carbachol (5 μm) (Fig.2A,E) and the mGluR agonists DHPG (2–3 μm) (Fig.3A,E) and trans-ACPD (10–20 μm; data not shown) all produced a robust inhibition of sIAHP but did not significantly affect the apamin-sensitive IAHP. In mice lacking Gαq, the effects of carbachol and DHPG on sIAHP were strongly reduced (Fig.2C,E for CCh; Fig.3C,E for DHPG), arguing in favor of a substantial contribution of Gαq in mediating the suppression of sIAHP. The apparent small decrease in IAHP amplitude, observed in wild-type as in knock-out Gαq mice during application of these neuromodulatory agents, was most likely attributable to the partial overlap betweenIAHP and sIAHP and the concomitant suppression of sIAHP (Stocker et al., 1999).
Fig. 2.
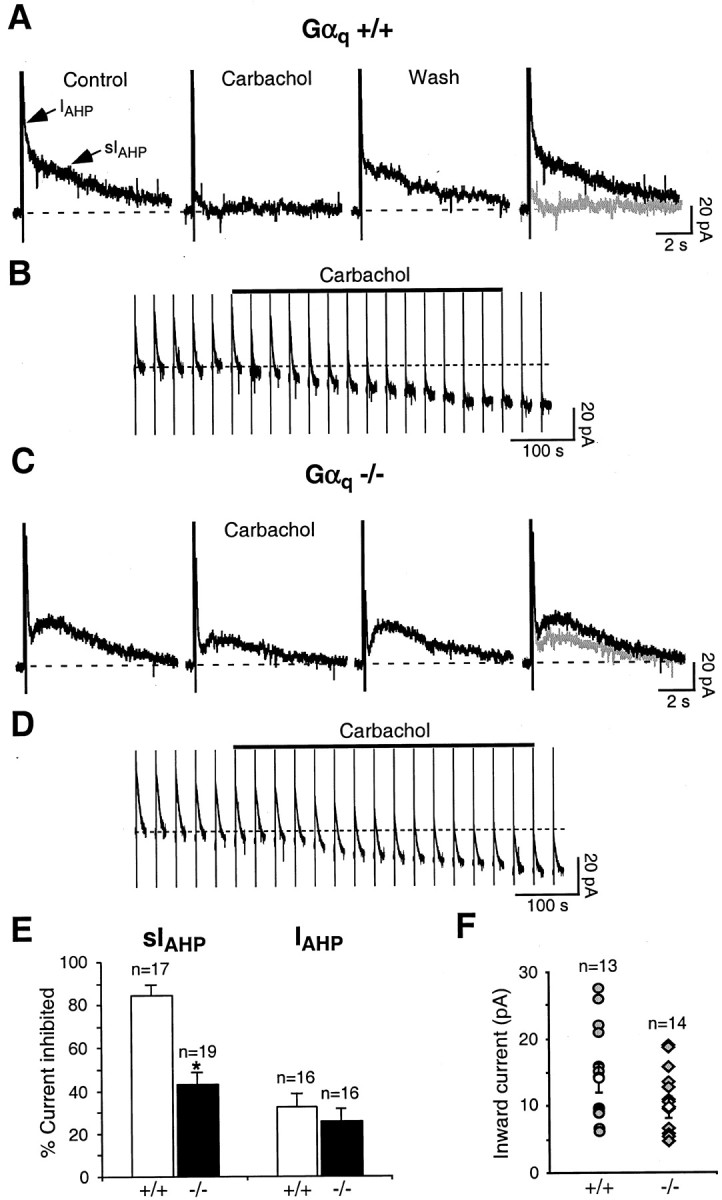
A, In Gαq +/+ mice, the cholinergic agonist carbachol (5 μm) strongly and reversibly suppressed sIAHP. B, Voltage-clamp recording of the inward current underlying the depolarizing response to carbachol (5 μM) in CA1 neurons from Gαq +/+ mice. The vertical linescorrespond to depolarizing pulses used to elicit the AHP currents once every 30 sec. The dotted line represents the baseline holding current before the application of carbachol. The carbachol application lasted 7 min (solid bar). C, In Gαq −/− mice, 5 μm carbachol induced a significantly smaller inhibition of sIAHPthan in A. In A and C, therightmost panels show superimpositions of thetraces before and during carbachol application.D, Inward current underlying the depolarizing response to carbachol (5 μm) in CA1 neurons from Gαq−/− mice. The carbachol application lasted 7.5 min (solid bar). All of the rest are as in B.E, Summary of the results obtained with carbachol in CA1 pyramidal neurons from Gαq +/+ and Gαq−/− mice. In Gαq +/+, carbachol suppressed sIAHP by 84.0 ± 4.2% (n = 17), whereas in Gαq −/−, 42.6 ± 5.6% of sIAHP was inhibited during carbachol application (n = 19). *represents a statistically significant difference (p < 0.001). In the same pools of CA1 neurons, carbachol did not significantly downregulate IAHP in Gαq +/+ mice (31.8 ± 5.9% inhibition) compared with Gαq−/− mice (25.5 ± 5.5% inhibition; n = 16;p = 0.44). F, Scatter plot summarizing the effect of 5 μm carbachol on the membrane potential of CA1 pyramidal neurons from Gαq +/+ (filled circles) and Gαq −/− (filled diamonds) animals. Carbachol elicited a mean inward shift in the holding current of 14.6 ± 2.0 pA in Gαq +/+ (open circle;n = 13) and 10.3 ± 1.4 pA in Gαq −/− mice (open diamond;n = 14; p = 0.10).
Fig. 3.
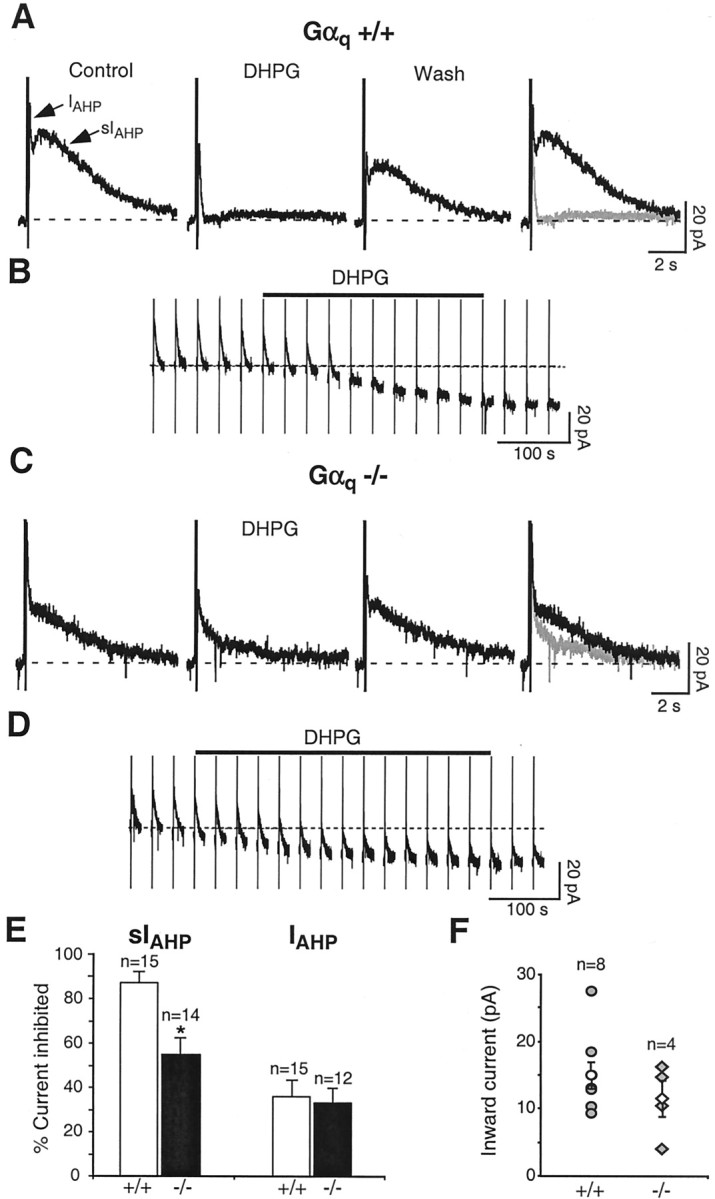
A, The group I mGluR receptor agonist DHPG (3 μm) suppressed sIAHP in a reversible manner in Gαq +/+ mice. B, Inward shift in the holding current underlying the depolarizing response to DHPG (3 μm) in CA1 neurons from Gαq +/+ mice. The vertical linescorrespond to depolarizing pulses used to elicit the AHP currents once every 30 sec. The dotted line represents the baseline holding current before the application of DHPG. The DHPG application lasted 5 min (solid bar). C, In Gαq −/− mice, 3 μm DHPG produced a significantly smaller inhibition of sIAHP. In A and C, the rightmost panels show superimpositions of the tracesbefore and during DHPG application. D, Depolarizing current elicited in response to DHPG (3 μm) in CA1 neurons from Gαq −/− mice. The DHPG application lasted 7 min (solid bar). All of the rest are as inB. E, Bar diagram summarizing the results obtained with DHPG in CA1 pyramidal neurons from Gαq +/+ and Gαq −/−. In Gαq +/+, 86.9 ± 4.4% of sIAHP was inhibited after DHPG application (n = 15), whereas in Gαq−/−, DHPG suppressed sIAHP by 54.7 ± 7.3% (n = 14). *represents a statistically significant difference (p = 0.001). In the same pools of CA1 neurons, the small inhibitory effect of DHPG onIAHP was similar in Gαq+/+ (35.9 ± 7.4% inhibition; n = 15) and Gαq −/− (32.5 ± 6.2% inhibition;n = 12; p = 0.73) mice.F, Summary of the effect of DHPG (2–3 μm) on the holding current in Gαq +/+ (filled circles) and Gαq −/− (filled diamonds) neurons. DHPG elicited a mean inward depolarizing current of 15.2 ± 2.0 pA in Gαq +/+ (open circle; n = 8) and 11.8 ± 2.7 pA in Gαq −/− (open diamond;n = 4; p = 0.33).
Beside downregulating sIAHP, activation of M3 receptors and mGluR5 leads to the generation of a depolarizing inward current in CA1 pyramidal neurons (Figs. 2B,3B) (Pitler and Alger, 1990; Gereau and Conn, 1995). The signal transduction pathway leading to this depolarizing effect is only partly known. We tested whether Gαq was mediating the generation of the depolarizing current during application of muscarinic and mGluR agonists. The inward current elicited by carbachol and DHPG was not significantly different in Gαq knock-out mice (Fig.2D,F for CCh; Fig.3D,F for DHPG) when compared with their wild-type littermates (Fig.2B,F for CCh; Fig.3B,F for DHPG). These results suggest that Gαq is not required for the muscarinic- or mGluR-induced depolarizing inward current in CA1 neurons.
Gα11 is not required for the modulation of sIAHP by muscarinic and mGluR agonists
The remaining effect of muscarinic and mGluR agonists on sIAHP in Gαq-deficient mice might be attributable to activation of Gα11, because this Gα subunit is also expressed, although to a lower level, in CA1 pyramidal neurons (Mailleux et al., 1992; Milligan, 1993; Tanaka et al., 2000) and displays a similar functional role as Gαq in other systems (Wu et al., 1992; Offermanns et al., 1994; Exton, 1996;Offermanns, 1999). To investigate the role of Gα11 in the cholinergic and metabotropic signal transduction cascades suppressing sIAHP, we recorded from CA1 neurons of mice lacking Gα11 (Gα11−/−). As in the case of the Gαq knock-outs, mice lacking Gα11 presented a normal hippocampal gross morphology and intrinsic neuronal properties indistinguishable from those of control mice (data not shown). In Gα11 −/− mice and in their wild-type littermates (Gα11 +/+), carbachol (Fig.4A,C) and DHPG (Fig. 4B,D) suppressed sIAHP to the same extent. In the same neurons, the apamin-sensitive IAHP was only marginally affected by application of muscarinic (Fig.4C) and mGluR (Fig. 4D) agonists. These results demonstrate that Gα11 is not required to transduce the cholinergic and glutamatergic signals that result in the downregulation of sIAHP. Therefore, it is unlikely that Gα11 is part of the signal cascade regulating the residual sIAHP present in Gαq −/− after cholinergic and glutamatergic receptor activation. In mice lacking Gα11, the depolarizing effects of carbachol and DHPG were indistinguishable from those observed in their wild-type littermates (data not shown), suggesting that Gα11 is not mediating the muscarinic- or mGluR-induced depolarizing inward current in CA1 neurons.
Fig. 4.

A, Left, In Gα11 +/+ mice, 5 μm carbachol strongly suppressed sIAHP. Right, In Gα11 −/−, 5 μm carbachol inhibited sIAHP to the same extent as observed in Gα11 +/+ mice. B, Left, DHPG (3 μm) induced a robust suppression of sIAHP in Gα11 +/+ mice.Right, Similarly, in Gα11 −/−, 3 μm DHPG produced a strong inhibition of sIAHP. In A andB, traces before and during carbachol and DHPG applications are shown superimposed. C, Bar diagram summarizing the results obtained with carbachol in CA1 pyramidal neurons from Gα11 +/+ and Gα11 −/− mice. In Gα11 +/+, carbachol suppressed sIAHP by 56.2 ± 9.8% (n = 5), and in Gα11, −/−, 67.3 ± 7.2% of sIAHP was inhibited during carbachol application (n = 11). The difference observed between Gα11 +/+ and Gα11 −/− mice was not statistically significant (p = 0.39). In the same pools of CA1 neurons, carbachol did not significantly affectIAHP in Gα11 +/+ compared with Gα11 −/− mice (n = 5 andn = 10, respectively). D, Bar diagram summarizing the results obtained with DHPG in CA1 pyramidal neurons from Gα11 +/+ and Gα11 −/− mice. In Gα11 +/+, 60.5 ± 14.2% of sIAHP was inhibited during DHPG application (n = 4), and in Gα11 −/−, DHPG suppressed sIAHP by 68.0 ± 9.6% (n = 9). The difference observed between Gα11 +/+ and Gα11 −/− mice was not statistically significant (p = 0.68). In the same pools of CA1 neurons, DHPG did not significantly affectIAHP in Gα11 +/+ compared with Gα11 −/− mice (n = 4 andn = 9, respectively).
The β-adrenergic modulation of sIAHPis intact in Gαq-deficient mice
Next, we wanted to test whether the reduction of the effects of cholinergic and glutamatergic agonists on sIAHP observed in Gαq-deficient mice is specific or whether supposedly unrelated modulatory pathways converging on sIAHP might be unspecifically affected in these genetically modified animals. To this purpose, we took advantage of the fact that sIAHP is strongly suppressed by monoamine transmitters (noradrenaline, serotonin, histamine, and dopamine) in rat CA1 pyramidal neurons (Nicoll, 1988; Pedarzani and Storm, 1993; Pedarzani and Storm, 1995). Among these transmitters, noradrenaline activates β-adrenergic receptors, which are coupled to Gαs-subunits and lead to the stimulation of adenylyl cyclase, increase of cAMP level, and activation of PKA, finally resulting in a complete suppression of sIAHP. We used the β-adrenergic agonist isoproterenol, which suppressed sIAHP both in wild-type (Fig.5A,C) and Gαq-deficient (Fig.5B,C) mice in an indistinguishable manner but had no significant effect on the apamin-sensitiveIAHP (Fig. 5C). We can therefore conclude that the absence of Gαqimpairs specifically cholinergic and glutamatergic signal cascades suppressing sIAHP, but it does not affect the modulation of this current by other neurotransmitters coupled to different second-messenger pathways.
Fig. 5.

A, In Gαq +/+, the β-adrenergic agonist isoproterenol (1 μm) strongly suppressed sIAHP. B, In Gαq −/−, 1 μm isoproterenol suppressed sIAHP to a similar extent as inA. In A and B, therightmost panels show superimpositions of thetraces before and during isoproterenol application.C, Summary of the results obtained with isoproterenol in CA1 pyramidal neurons from Gαq +/+ and Gαq−/− mice. In Gαq +/+, isoproterenol suppressed sIAHP by 75.0 ± 11.4% (n = 6), and in Gαq −/−, 83.6 ± 7.8% of sIAHP was inhibited during isoproterenol application (n = 10). The difference observed between Gαq +/+ and Gαq −/− mice was not statistically significant (p = 0.54). In the same pools of CA1 neurons, isoproterenol did not affectIAHP in Gαq +/+ differently from Gαq −/− mice (n = 5 andn = 9, respectively).
DISCUSSION
This study presents the first demonstration and characterization of apamin-sensitive and -insensitive AHP currents in the mouse hippocampus. Based on this, knock-out animals could be used to analyze specific transducing elements of signal cascades regulating sIAHP. The primary purpose of the present experiments was to elucidate which heterotrimeric G-proteins transduce muscarinic- and mGluR-mediated signals leading to a downregulation of sIAHP and to an enhanced excitability of hippocampal pyramidal neurons. To this purpose, we used mice lacking the Gαq or Gα11 gene. These mice did not present gross morphological abnormalities of the CNS and displayed an overall normal hippocampal morphology. In addition, intrinsic membrane properties (this study) and basic parameters of synaptic function (input–output curves and paired-pulse facilitation; Kleppisch et al., 2001) were not altered in the CA1 region. Our results demonstrate that Gαq is the main transducing element in these signal cascades leading to sIAHPsuppression, whereas Gα11 does not seem to be required. Neither Gαq nor Gα11 seem to be involved in mediating the membrane potential depolarization caused by muscarinic and mGluR agonists in CA1 pyramidal neurons.
Muscarinic and mGluR downregulation of sIAHP requires Gαq, because the effects of muscarinic and mGluR agonists were strongly reduced in neurons from Gαq knock-out mice. It is unlikely that this was a result of generalized secondary effects on transduction mechanisms arising from the absence of Gαqduring development, because muscarinic and mGluR agonists were still able to elicit the inward, depolarizing current. Furthermore, transmitters acting via other G-proteins, such as the β-adrenergic agonist isoproterenol acting via Gαs, fully suppressed sIAHP in Gαq-deficient neurons, indicating a preserved function of other signaling pathways targeting the same current.
In the absence of Gαq, muscarinic and mGluR agonists partially suppressed sIAHP(Figs. 2, 3). Our first assumption was that this could be attributable to the presence of Gα11, which is expressed at lower levels in hippocampal neurons (Mailleux et al., 1992; Milligan, 1993; Tanaka et al., 2000) and presents receptor- and effector-coupling properties very similar to Gαq (Wu et al., 1992; Offermanns et al., 1994; Exton, 1996; Offermanns, 1999). However, the results we obtained in Gα11 knock-out mice revealed that Gα11 is not required for sIAHP modulation and is therefore unlikely to substitute for Gαq in Gαq knock-out mice. This observation is further supported by the lack of compensatory overexpression of Gα11 in the hippocampus of Gαq knock-out mice, as shown recently by immunoblot experiments (Kleppisch et al., 2001). Despite this evidence, in principle the possibility remains that Gα11mediates the residual effect of muscarinic and glutamatergic agonists observed in Gαq knock-out mice. This could happen, for example, if Gα11 contributed in a small but significant way to the muscarinic or glutamatergic action, resulting in an invisible phenotype in the Gα11knock-out mice but emerging as a sufficient contribution in the Gαq knock-outs. A subtle, partial effect linked to Gα11, unmasked in Gαq knock-out mice, would be expected to be slower when compared with the Gαq-mediated effect observed in their wild-type littermates, given the lower density of the Gα11 subtype in CA1 pyramidal neurons (Mailleux et al., 1992; Milligan, 1993; Tanaka et al., 2000). Indeed, a compensatory phenomenon with altered (slower) kinetics has been reported recently for the modulation of potassium and calcium channels by GABAB and adenosine receptors in hippocampal neurons from mice lacking Gαo (Greif et al., 2000). This seems unlikely to occur in our case, because the time course of the residual muscarinic and glutamatergic effects on sIAHP was not different in Gαq knock-out mice when compared with their wild-type littermates [compare Figs.2B,D (for carbachol, 3B,D (for DHPG)]. However, the possibility of a marginal Gα11 involvement could be conclusively ruled out only by using animal models lacking both Gαq and Gα11subunits, but unfortunately a double mutant generated by conventional gene targeting is not viable (Offermanns et al., 1998). If conditional double mutants prove to be viable, they could provide a potential future approach to unequivocally answer this question.
In several mammalian cells, receptors activating Gq family members do not seem to discriminate between Gαq and Gα11(Wange et al., 1991; Wu et al., 1992; Offermanns et al., 1994), and, in reconstituted systems, both G-proteins are indistinguishable in their ability to regulate different phospholipase C (PLC) isoforms (Exton, 1996) and inward rectifier potassium channels (Lei et al., 2001). In the light of such apparent functional redundancy, the prominent involvement of Gαq and the lack of effect of Gα11 in mediating the cholinergic and glutamatergic suppression of sIAHPprovide therefore a remarkable indication of functional specificity for members of the Gq family in the CNS. In line with this view, Gαq, but not Gα11, has been shown recently to be critically involved in the induction of mGluR-dependent long-term depression in CA1 pyramidal neurons (Kleppisch et al., 2001).
The picture emerging from the results presented here is different from that which has been observed for another potassium current, the voltage-dependent IM, in the peripheral nervous system. In rat superior cervical ganglion neurons, M1 muscarinic receptors inhibitIM primarily via Gαq (Haley et al., 1998). In mouse, inhibition is also mediated by M1 receptors (Hamilton et al., 1997) but appears to involve both Gαq and Gα11 and also a pertussis toxin-sensitive G-protein (Haley et al., 2000). The suppression of sIAHP by muscarinic agonists in CA1 neurons is instead linked to M3 receptor activation (Rouse et al., 2000) and is mediated principally by Gαq, in a similar way to that observed for the M1-mediated inhibition of the N-type calcium current in sympathetic neurons (Haley et al., 2000). The preponderant involvement of Gαq in the M3- and mGluR5-mediated suppression of sIAHP presented in this study is in agreement with previous works using pharmacological approaches to show that G-proteins, and in particular pertussis toxin-insensitive ones, were essential components of the muscarinic and mGluR signal cascades converging on sIAHP (Dutar and Nicoll, 1988; Gerber et al., 1992; Abdul-Ghani et al., 1996a; Krause and Pedarzani, 2000).
Beside modulating sIAHP, muscarinic and mGluR agonists induce a depolarization of the membrane potential in CA1 neurons. The underlying current has been ascribed to the inhibition of at least two types of K+ currents: the voltage-dependent M current (Brown and Adams, 1980; Halliwell and Adams, 1982; McCormick and Williamson, 1989) and a voltage-independent leak current (Madison et al., 1987; Guerineau et al., 1994). More recently, a cation nonselective current has been proposed to be mainly responsible for the depolarization generated by muscarinic and mGluR agonists in cortical and hippocampal neurons (Crepel et al., 1994;Greene et al., 1994; Guerineau et al., 1995; Haj-Dahmane and Andrade, 1997, 1999). Interestingly, mGluR1 receptors have been shown recently to elicit an inward, depolarizing current associated with a slow excitatory postsynaptic response in a G-protein-independent manner in CA3 pyramidal neurons (Heuss et al., 1999). Our experiments show that neither the lack of Gαq nor Gα11 affect the depolarizing action of M3 muscarinic and mGluR5 receptors in CA1 pyramidal neurons. Our results are compatible with the possibility that muscarinic and mGluR agonists induce a depolarization of the membrane potential in a G-protein-independent manner in CA1 neurons, as proposed recently for CA3 neurons (Heuss et al., 1999).
The signal transduction events downstream from M3or mGluR5 and G-protein activation, leading to sIAHP suppression,IK(leak) inhibition, and activation of a depolarizing cationic conductance, are still rather unclear. Calcium/calmodulin-dependent kinase II has been shown to be involved in the muscarinic, but not in the mGluR, modulation of sIAHP (Müller et al., 1992;Pedarzani and Storm, 1996), suggesting that the two receptors might be coupled to distinct pathways. In dentate gyrus granule cells, PLC, IP3, and a tyrosine kinase have been reported to mediate the glutamatergic suppression of sIAHP (Abdul-Ghani et al., 1996a,b), whereas in CA1 pyramidal neurons neither PLC nor IP3 seem to be involved in the inhibition of this current by muscarinic or mGluR agonists (Krause and Pedarzani, 1999,2000). Instead, we obtained evidence for the involvement of a protein phosphatase (Krause and Pedarzani, 1999, 2000), but the mechanism of coupling to G-proteins needs to be elucidated. The present study provides evidence that signaling of both M3muscarinic and mGluR5 receptors converge onto the same G-protein, Gαq, to modulate sIAHP. Future experiments will show whether the residual effect on sIAHPobserved in Gαq-deficient mice is attributable to the coupling of M3 and mGluR5 to a parallel, second pathway, activated by βγ subunits, by G-proteins not belonging to the Gq family, or by a G-protein-independent mechanism.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Sonderforschungsbereich 406 (M.S., P.P.), the Human Frontier Science Program (P.P.), and the Wellcome Trust (M.S.). We are very grateful to Walter Stühmer for generous support. We thank David A. Brown for useful discussion and critical reading of this manuscript and Christina Sterner and the personnel of the transgenic animal facility of the Max-Planck-Institute for Experimental Medicine for technical assistance.
Correspondence should be addressed to Dr. Paola Pedarzani, Department of Physiology, University College London, Gower Street, London WC1E 6BT, UK. E-mail: p.pedarzani@ucl.ac.uk.
M. Krause's present address: Pharmacia Corporation, Neurobiology, 301 Henrietta Street, Kalamazoo, MI 49007.
REFERENCES
- 1.Abdul-Ghani MA, Valiante TA, Carlen PL, Pennefather PS. Metabotropic glutamate receptors coupled to IP3 production mediate inhibition of IAHP in rat dentate granule neurons. J Neurophysiol. 1996a;76:2691–2700. doi: 10.1152/jn.1996.76.4.2691. [DOI] [PubMed] [Google Scholar]
- 2.Abdul-Ghani MA, Valiante TA, Carlen PL, Pennefather PS. Tyrosine kinase inhibitors enhance a Ca2+-activated K+ current (IAHP) and reduce IAHP suppression by a metabotropic glutamate receptor agonist in rat dentate granule neurones. J Physiol (Lond) 1996b;496:139–144. doi: 10.1113/jphysiol.1996.sp021671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benardo LS, Prince DA. Cholinergic excitation of mammalian hippocampal pyramidal cells. Brain Res. 1982;249:315–331. doi: 10.1016/0006-8993(82)90066-x. [DOI] [PubMed] [Google Scholar]
- 4.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 5.Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 6.Brown DA, Abogadie FC, Allen TG, Buckley NJ, Caulfield MP, Delmas P, Haley JE, Lamas JA, Selyanko AA. Muscarinic mechanisms in nerve cells. Life Sci. 1997;60:1137–1144. doi: 10.1016/s0024-3205(97)00058-1. [DOI] [PubMed] [Google Scholar]
- 7.Buckley NJ, Abogadie FC, Brown DA, Dayrell M, Caulfield MP, Delmas P, Haley JE. Use of antisense expression plasmids to attenuate G-protein expression in primary neurons. Methods Enzymol. 2000;314:136–148. doi: 10.1016/s0076-6879(99)14100-4. [DOI] [PubMed] [Google Scholar]
- 8.Charpak S, Gähwiler BH, Do KQ, Knöpfel T. Potassium conductances in hippocampal neurons blocked by excitatory amino-acid transmitters. Nature. 1990;347:765–767. doi: 10.1038/347765a0. [DOI] [PubMed] [Google Scholar]
- 9.Cole AE, Nicoll RA. Acetylcholine mediates a slow synaptic potential in hippocampal pyramidal cells. Science. 1983;221:1299–1301. doi: 10.1126/science.6612345. [DOI] [PubMed] [Google Scholar]
- 10.Cole AE, Nicoll RA. The pharmacology of cholinergic excitatory responses in hippocampal pyramidal cells. Brain Res. 1984;305:283–290. doi: 10.1016/0006-8993(84)90434-7. [DOI] [PubMed] [Google Scholar]
- 11.Colino A, Halliwell JV. Carbachol potentiates Q current and activates a calcium-dependent non-specific conductance in rat hippocampus in vitro. Eur J Neurosci. 1993;5:1198–1209. doi: 10.1111/j.1460-9568.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- 12.Crepel V, Aniksztejn L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca2+-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- 13.Dutar P, Nicoll RA. Classification of muscarinic responses in hippocampus in terms of receptor subtypes and second-messenger systems: electrophysiological studies in vitro. J Neurosci. 1988;8:4214–4224. doi: 10.1523/JNEUROSCI.08-11-04214.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Exton JH. Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G proteins. Annu Rev Pharmacol Toxicol. 1996;36:481–509. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- 15.Gerber U, Sim JA, Gähwiler BH. Reduction of potassium conductances mediated by metabotropic glutamate receptors in rat CA3 pyramidal cells does not require protein kinase C or protein kinase A. Eur J Neurosci. 1992;4:792–797. doi: 10.1111/j.1460-9568.1992.tb00189.x. [DOI] [PubMed] [Google Scholar]
- 16.Gereau RW, Conn PJ. Roles of specific metabotropic glutamate receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J Neurophysiol. 1995;74:122–129. doi: 10.1152/jn.1995.74.1.122. [DOI] [PubMed] [Google Scholar]
- 17.Greene CC, Schwindt PC, Crill WE. Properties and ionic mechanisms of a metabotropic glutamate receptor-mediated slow afterdepolarization in neocortical neurons. J Neurophysiol. 1994;72:693–704. doi: 10.1152/jn.1994.72.2.693. [DOI] [PubMed] [Google Scholar]
- 18.Greif GJ, Sodickson DL, Bean BP, Neer EJ, Mende U. Altered regulation of potassium and calcium channels by GABAB and adenosine receptors in hippocampal neurons from mice lacking Gαo. J Neurophysiol. 2000;83:1010–1018. doi: 10.1152/jn.2000.83.2.1010. [DOI] [PubMed] [Google Scholar]
- 19.Guerineau NC, Gähwiler BH, Gerber U. Reduction of resting K+ current by metabotropic glutamate and muscarinic receptors in rat CA3 cells: mediation by G-proteins. J Physiol (Lond) 1994;474:27–33. doi: 10.1113/jphysiol.1994.sp019999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerineau NC, Bossu JL, Gähwiler BH, Gerber U. Activation of a nonselective cationic conductance by metabotropic glutamatergic and muscarinic agonists in CA3 pyramidal neurons of the rat hippocampus. J Neurosci. 1995;15:4395–4407. doi: 10.1523/JNEUROSCI.15-06-04395.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haj-Dahmane S, Andrade R. Calcium-activated cation nonselective current contributes to the fast afterdepolarization in rat prefrontal cortex neurons. J Neurophysiol. 1997;78:1983–1989. doi: 10.1152/jn.1997.78.4.1983. [DOI] [PubMed] [Google Scholar]
- 22.Haj-Dahmane S, Andrade R. Muscarinic receptors regulate two different calcium-dependent non-selective cation currents in rat prefrontal cortex. Eur J Neurosci. 1999;11:1973–1980. doi: 10.1046/j.1460-9568.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- 23.Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ. The α subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. J Neurosci. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haley JE, Delmas P, Offermanns S, Abogadie FC, Simon MI, Buckley NJ, Brown DA. Muscarinic inhibition of calcium current and M current in Gαq-deficient mice. J Neurosci. 2000;20:3973–3979. doi: 10.1523/JNEUROSCI.20-11-03973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- 26.Hamilton SE, Loose MD, Qi M, Levey AI, Hille B, McKnight GS, Odzerda RL, Nathanson NM. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc Natl Acad Sci USA. 1997;94:13311–13316. doi: 10.1073/pnas.94.24.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heuss C, Scanziani M, Gähwiler BH, Gerber U. G-protein-independent signaling mediated by metabotropic glutamate receptors. Nat Neurosci. 1999;2:1070–1077. doi: 10.1038/15996. [DOI] [PubMed] [Google Scholar]
- 28.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 29.Holscher C, Gigg J, O'Mara SM. Metabotropic glutamate receptor activation and blockade: their role in long-term potentiation, learning and neurotoxicity. Neurosci Biobehav Rev. 1999;23:399–410. doi: 10.1016/s0149-7634(98)00045-1. [DOI] [PubMed] [Google Scholar]
- 30.Ikeda SR. Heterologous expression of receptors and signaling proteins in adult mammalian sympathetic neurons by microinjection. Methods Mol Biol. 1997;83:191–202. doi: 10.1385/0-89603-495-X:191. [DOI] [PubMed] [Google Scholar]
- 31.Kleppisch T, Allmann R, Voigt V, Offermanns S. Gαq-deficient mice lack mGluR-dependent LTD but show normal LTP in the hippocampal CA1 region. J Neurosci. 2001;21:4943–4948. doi: 10.1523/JNEUROSCI.21-14-04943.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krause M, Pedarzani P. Proceedings of the 1st Göttingen Conference of the German Neuroscience Society. Verlag; Stuttgart: 1999. Suppression of the Ca2+-activated K+ current sIAHP by cholinergic and glutamatergic agonists involves a protein phosphatase and PKG in hippocampal neurons. p. 702. [Google Scholar]
- 33.Krause M, Pedarzani P. A protein phosphatase is involved in the cholinergic suppression of the Ca2+-activated K+ current sIAHP in hippocampal pyramidal neurons. Neuropharmacology. 2000;39:1274–1283. doi: 10.1016/s0028-3908(99)00227-0. [DOI] [PubMed] [Google Scholar]
- 34.Lei Q, Talley EM, Bayliss DA. Receptor-mediated inhibition of G protein-coupled inwardly rectifying potassium channels involves Gαq family subunits, phospholipase C, and a readily diffusible messenger. J Biol Chem. 2001;276:16720–16730. doi: 10.1074/jbc.M100207200. [DOI] [PubMed] [Google Scholar]
- 35.Madison DV, Lancaster B, Nicoll RA. Voltage-clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mailleux P, Mitchell F, Vanderhaeghen JJ, Milligan G, Erneux C. Immunohistochemical distribution of neurons containing the G-proteins Gq alpha/G11 alpha in the adult rat brain. Neuroscience. 1992;51:311–316. doi: 10.1016/0306-4522(92)90317-u. [DOI] [PubMed] [Google Scholar]
- 37.McCormick DA, von Krosigk M. Corticothalamic activation modulates thalamic firing through glutamate metabotropic receptors. Proc Natl Acad Sci USA. 1992;89:2774–2778. doi: 10.1073/pnas.89.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormick DA, Williamson A. Convergence and divergence of neurotransmitter action in human cerebral cortex. Proc Natl Acad Sci USA. 1989;86:8098–8102. doi: 10.1073/pnas.86.20.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McFadzean I, Caulfield MP, Vallis Y, Brown DA. Injection of antisera into cells to study G-protein regulation of channel function. Methods Enzymol. 1994;238:357–364. doi: 10.1016/0076-6879(94)38032-5. [DOI] [PubMed] [Google Scholar]
- 40.Milligan G. Regional distribution and quantitative measurement of the phosphoinositidase C-linked guanine nucleotide binding proteins G11 alpha and Gq alpha in rat brain. J Neurochem. 1993;61:845–851. doi: 10.1111/j.1471-4159.1993.tb03595.x. [DOI] [PubMed] [Google Scholar]
- 41.Müller W, Petrozzino JJ, Griffith LC, Danho W, Connor JA. Specific involvement of Ca2+-calmodulin kinase II in cholinergic modulation of neuronal responsiveness. J Neurophysiol. 1992;68:2264–2269. doi: 10.1152/jn.1992.68.6.2264. [DOI] [PubMed] [Google Scholar]
- 42.Nicoll RA. The coupling of neurotransmitter receptors to ion channels in the brain. Science. 1988;241:545–551. doi: 10.1126/science.2456612. [DOI] [PubMed] [Google Scholar]
- 43.Offermanns S. New insights into the in vivo function of heterotrimeric G-proteins through gene deletion studies. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:5–13. doi: 10.1007/s002109900030. [DOI] [PubMed] [Google Scholar]
- 44.Offermanns S, Heiler E, Spicher K, Schultz G. Gq and G11 are concurrently activated by bombesin and vasopressin in Swiss 3T3 cells. FEBS Lett. 1994;349:201–204. doi: 10.1016/0014-5793(94)00667-9. [DOI] [PubMed] [Google Scholar]
- 45.Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M, Simon MI. Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Gαq. Proc Natl Acad Sci USA. 1997;94:14089–14094. doi: 10.1073/pnas.94.25.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Offermanns S, Zhao LP, Gohla A, Sarosi I, Simon MI, Wilkie TM. Embryonic cardiomyocyte hypoplasia and craniofacial defects in Gαq/Gα11-mutant mice. EMBO J. 1998;17:4304–4312. doi: 10.1093/emboj/17.15.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- 48.Pedarzani P, Storm JF. Dopamine modulates the slow Ca2+-activated K+ current IAHP via cyclic AMP-dependent protein kinase in hippocampal neurons. J Neurophysiol. 1995;74:2749–2753. doi: 10.1152/jn.1995.74.6.2749. [DOI] [PubMed] [Google Scholar]
- 49.Pedarzani P, Storm JF. Evidence that Ca/calmodulin-dependent protein kinase mediates the modulation of the Ca2+-dependent K+ current, IAHP, by acetylcholine, but not by glutamate, in hippocampal neurons. Pflügers Arch. 1996;431:723–728. [PubMed] [Google Scholar]
- 50.Perry E, Walker M, Grace J, Perry R. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22:273–280. doi: 10.1016/s0166-2236(98)01361-7. [DOI] [PubMed] [Google Scholar]
- 51.Pin JP, Bockaert J. Get receptive to metabotropic glutamate receptors. Curr Opin Neurobiol. 1995;5:342–349. doi: 10.1016/0959-4388(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 52.Pitler TA, Alger BE. Activation of the pharmacologically defined M3 muscarinic receptor depolarizes hippocampal pyramidal cells. Brain Res. 1990;534:257–262. doi: 10.1016/0006-8993(90)90137-z. [DOI] [PubMed] [Google Scholar]
- 53.Riedel G. Function of metabotropic glutamate receptors in learning and memory. Trends Neurosci. 1996;19:219–224. doi: 10.1016/0166-2236(96)20012-8. [DOI] [PubMed] [Google Scholar]
- 54.Rouse ST, Hamilton SE, Potter LT, Nathanson NM, Conn PJ. Muscarinic-induced modulation of potassium conductances is unchanged in mouse hippocampal pyramidal cells that lack functional M1 receptors. Neurosci Lett. 2000;278:61–64. doi: 10.1016/s0304-3940(99)00914-3. [DOI] [PubMed] [Google Scholar]
- 55.Segal M, Auerbach JM. Muscarinic receptors involved in hippocampal plasticity. Life Sci. 1997;60:1085–1091. doi: 10.1016/s0024-3205(97)00051-9. [DOI] [PubMed] [Google Scholar]
- 56.Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka J, Nakagawa S, Kushiya E, Yamasaki M, Fukaya M, Iwanaga T, Simon MI, Sakimura K, Kano M, Watanabe M. Gq protein α subunits Gαq and Gα11 are localized at postsynaptic extra-junctional membrane of cerebellar Purkinje cells and hippocampal pyramidal cells. Eur J Neurosci. 2000;12:781–792. doi: 10.1046/j.1460-9568.2000.00959.x. [DOI] [PubMed] [Google Scholar]
- 58.Wange RL, Smrcka AV, Sternweis PC, Exton JH. Photoaffinity labeling of two rat liver plasma membrane proteins with [32P]gamma-azidoanilido GTP in response to vasopressin. Immunologic identification as α subunits of the Gq class of G proteins. J Biol Chem. 1991;266:11409–11412. [PubMed] [Google Scholar]
- 59.Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by α1-adrenergic receptors is mediated by the α subunits of Gq family. J Biol Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- 60.Wu RL, Barish ME. Modulation of a slowly inactivating potassium current, ID, by metabotropic glutamate receptor activation in cultured hippocampal pyramidal neurons. J Neurosci. 1999;19:6825–6837. doi: 10.1523/JNEUROSCI.19-16-06825.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang L, Weiner JL, Carlen PL. Muscarinic potentiation of IK in hippocampal neurons: electrophysiological characterization of the signal transduction pathway. J Neurosci. 1992;12:4510–4520. doi: 10.1523/JNEUROSCI.12-11-04510.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]