

Abstract
APP23 transgenic mice express mutant human amyloid precursor protein and develop amyloid plaques predominantly in neocortex and hippocampus progressively with age, similar to Alzheimer's disease. We have previously reported neuron loss in the hippocampal CA1 region of 14- to 18-month-old APP23 mice. In contrast, no neuron loss was found in neocortex. In the present study we have reinvestigated neocortical neuron numbers in adult and aged APP23 mice. Surprisingly, results revealed that 8-month-old APP23 mice have 13 and 14% more neocortical neurons compared with 8-month-old wild-type and 27-month-old APP23 mice, respectively. In 27-month-old APP23 mice we found an inverse correlation between amyloid load and neuron number. These results suggest that APP23 mice have more neurons until they develop amyloid plaques but then lose neurons in the process of cerebral amyloidogenesis. Supporting this notion, we found more neurons with a necrotic–apoptotic phenotype in the neocortex of 24-month-old APP23 mice compared with age-matched wild-type mice. Stimulated by recent reports that demonstrated neurogenesis after targeted neuron death in the mouse neocortex, we have also examined neurogenesis in APP23 mice. Strikingly, we found a fourfold to sixfold increase in newly produced cells in 24-month-old APP23 mice compared with both age-matched wild-type mice and young APP23 transgenic mice. However, subsequent cellular phenotyping revealed that none of the newly generated cells in neocortex had a neuronal phenotype. The majority were microglial and to a lesser extent astroglial cells. We conclude that cerebral amyloidosis in APP23 mice causes a modest neuron loss in neocortex and induces marked gliogenesis.
Keywords: Alzheimer's disease, amyloid, APP, Aβ, CNS, brain, transgenic mouse, stereology, neurodegeneration, stem cells, neurogenesis, gliogenesis, microglia, aging
The hallmark lesions of Alzheimer's disease (AD) brain are extracellular deposits of β-amyloid (Aβ) and intracellular neurofibrillary tangles (Probst et al., 1991; Selkoe, 1999). In addition, significant neuron and synapse loss in brain regions involved in information processing and memory acquisition have consistently been reported in AD and are thought to be morphological correlates of dementia (West et al., 1994; Gomez-Isla et al., 1996a;Morrison and Hof, 1997).
Considerable effort has been devoted to studying the relationship between amyloid plaques and AD neurodegeneration. In particular, it has remained unclear whether Aβ and/or its deposition in the parenchyma is the cause of the nerve cell loss. Whereas older studies report a poor correlation between dementia and amyloid plaques (Gomez-Isla et al., 1996b; Giannakopoulos et al., 1997), more recent studies have found stronger correlations between either total Aβ load or neuritic Aβ plaques and nerve cell loss and/or dementia (Cummings and Cotman, 1995; Knowles et al., 1998; Naslund et al., 2000).
Several transgenic mouse models of cerebral β-amyloidosis have been generated through expression of mutated amyloid precursor protein (APP) (Games et al., 1995; Hsiao et al., 1996; Sturchler-Pierrat et al., 1997; Calhoun et al., 1999; Hsia et al., 1999; Lamb et al., 1999; Van Dorpe et al., 2000). These mice develop amyloid plaques and vascular amyloid predominately in neocortex and hippocampus as they age. The amyloid plaques share many features with the amyloid deposits in AD brain. They are surrounded by activated microglia, reactive astrocytes, dystrophic synaptic boutons, and abnormally phosphorylated tau-positive neurites (Masliah et al., 1996; Sturchler-Pierrat et al., 1997;Frautschy et al., 1998; Phinney et al., 1999; Stalder et al., 1999).
To study the impact of cerebral amyloidosis on neurodegeneration, modern stereological techniques have been used to relate neuron number to amyloid burden in these APP transgenic mice (Irizarry et al., 1997a,b; Calhoun et al., 1998b; Takeuchi et al., 2000). Although in two transgenic mouse lines, Tg2576 and PDAPP mice, no significant nerve cell loss was reported in hippocampus and neocortex (Irizarry et al., 1997a,b), we have previously reported that amyloid plaque formation is accompanied by CA1 hippocampal neuron loss in 14- to 18-month-old APP23 mice. In contrast, in the same mice we could not detect any global neuron loss in neocortex, despite an amyloid burden comparable with that in the CA1 region (Calhoun et al., 1998b).
The apparent lack of neocortical neuron loss at a given age of APP transgenic mice may, however, be attributable to compensating mechanisms. In particular, it has been shown that discrete lesions to the neocortex of mice can stimulate neurogenesis (Magavi et al., 2000). It is therefore conceivable that neurogenesis partly compensates for amyloid-associated neuron loss in APP transgenic mice at the lesion sites. Thus, in the present study we have reevaluated neocortical neuron loss and have additionally assessed neurogenesis in the neocortex of adult and aged APP23 mice.
MATERIALS AND METHODS
Animals. The generation of APP23 transgenic mice is described elsewhere (Sturchler-Pierrat et al., 1997). Briefly, a murine Thy-1 promoter element was used to drive neuron-specific expression of human APP751 with the Swedish double mutation 670/671KM→NL in B6D2 mice. All mice used were from the F6 to F10 generation of backcrossing to C57BL/6 mice. The wild-type control mice were either littermate mice or nontransgenic age-matched mice from another litter of the same generation of backcrossing.
The following mice were used for neuron counting: 13 adult (six females and seven males) and 13 aged (seven females and six males) APP23 mice; 15 adult (seven females and eight males) and 11 aged (six females and five males) wild-type mice served as controls. All the adult mice were 8 months old. The aged mice ranged from 26 to 29 months with a mean age of 27 months in both groups. Additional groups of 2 to 3-month-old male APP23 mice (n = 6) and male control mice (n = 6) were also used for neuron counting. For TUNEL-labeling 4-month-old (n = 4) and 24-month-old (n = 4) male APP23 mice and equivalent numbers of male age-matched controls were used. For BrdU-labeling 4-month-old (n = 4) and 24-month-old (n = 8) male APP23 mice and equivalent numbers of male age-matched controls were used.
Histology and immunohistochemistry to assess neuron number and amyloid load. Mice were anesthetized with 2.5% isoflurane and decapitated. The brains were removed, immersion-fixed with 4% paraformaldehyde in PBS for 2 d at 4°C, and then embedded in paraffin under standard conditions (Calhoun et al., 1998a). Serial coronal sections were cut at a 25 μm microtome setting throughout the entire neocortex. Cresyl violet staining was used for neuron counting. To this end sections were deparaffinized in xylene and rehydrated through a series of graded ethanols. The slides were immersed in warm (60°C) cresyl violet solution (Merck; 0.5 gm/100 ml in dH2O with 0.3% glacial acetic acid), differentiated in ethanol, cleared in xylene, and coverslipped.
To assess amyloid load, sections were deparaffinized in xylene and then placed in 100% ethanol for 10 min followed by 30 min in methanol with 0.3% H2O2. Sections were rinsed in PBS and incubated for 1 hr in 5% goat serum. Slides were transferred to a humid chamber, and sections were incubated overnight with a polyclonal antibody to Aβ (NT11) (Sturchler-Pierrat et al., 1997) diluted 1:500 in PBS with 3% goat serum. Sections were then incubated for 60 min with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) diluted 1:200 in PBS with 3% goat serum, followed by incubation for 90 min in an avidin–biotin–peroxidase complex (Vector Laboratories) diluted 1:200 in PBS. Sections were reacted with 3,3′-diaminobenzidine (0.08%; Sigma, St. Louis, MO) and 0.03% hydrogen peroxide in PBS for 2 min, rinsed, dehydrated, cleared, and coverslipped. Congo Red staining was performed according to standard protocols and was used to assess compact amyloid.
Terminal deoxynucleotidyltransferase-mediated UTP nick end labeling. Mice were overdosed with pentobarbital and transcardially perfused with 4% paraformaldehyde in PBS. Brains were removed and post-fixed in the same fixative overnight and placed in 30% sucrose in PBS for 2 d. Brains were then frozen in 2-methylbutane at −25°C and serially sectioned on a freezing–sliding microtome at 40 μm.
The terminal deoxynucleotidyltransferase-mediated UTP nick end labeling (TUNEL) assay was performed on free-floating sections using the Apoptag In Situ cell death detection kit (Intergene, Purchase, NY) according to a recently described procedure (Biebl et al., 2000). In brief, after rinsing sections in TBS for 10 min, an ascending isopropanol series (dH2O, 70%, 90% for 2 min each) was followed by incubation in 100% isopropanol for 10 min and a descending isopropanol series (dH2O, 90%, 70%, for 2 min each). After three rinses in TBS, sections were incubated with equilibration buffer for at least 5 min at room temperature followed by TdT-reaction solution for 1 hr at 37°C and the Stop Buffer for 10 min at room temperature. To reduce background labeling the TdT-reaction solution was diluted 1:1 with TUNEL dilution buffer (Roche Diagnostics, Mannheim, Germany). With intermittent washes in TBS, sections were blocked in 3% donkey serum and 0.1% Triton X-100 in TBS for 30 min. For peroxidase detection, TUNEL-treated sections were incubated with a sheep anti-digoxigenin-FITC antibody (1:1000; Roche Diagnostics) in TBS overnight at 5°C. TUNEL labeling was visualized by incubation with biotinylated anti-sheep IgG (1:1000; Jackson ImmunoResearch, West Grove, PA) followed by the avidin–biotin peroxidase complex (Vector Laboratories) and diaminobenzidine–NiCl–H2O2. Sections were counterstained with 1% methylene green and coverslipped.
To study the phenotype of the TUNEL-labeled cells, triple-fluorescence labeling was performed. TUNEL-treated sections were incubated overnight at 5°C with combinations of the following primary antibodies: sheep anti-digoxigenin-FITC (see above), mouse monoclonal anti-NeuN (1:1000;Chemicon, Temecula, CA), rabbit polyclonal anti-S100β (1:2000; Swant, Bellinzona, Switzerland), and rat anti-CD11b (Mac-1; 1:1000, Serotec, Oxford, UK). After intermittent washes in TBS and brief fixation in 4% paraformaldehyde (15 min), primary antibodies were detected using combinations of the following secondary antibodies: FITC-labeled donkey anti-sheep IgG (to enhance the sheep anti-digoxygenin-FITC signal), RhodamineX-labeled donkey anti mouse IgG, RhodamineX-labeled donkey anti rat-IgG, and CY5-labeled donkey anti-rabbit IgG (all at 1:500 for 2 hr; Jackson ImmunoResearch. In case of high autofluorescence background, sections were incubated in 70% ethanol and 0.3% Sudan Black B (Merck, Darmstadt, Germany) and coverslipped in mounting medium with bleach protection (SlowFade; Molecular Probes, Eugene, OR) or Vectashield (Vector Laboratories). Sections were analyzed with a confocal laser scanning microscope, LSM 510, inverted Axiovert 100 M (Zeiss, Oberkochen, Germany).
BrdU labeling and cellular phenotyping. For labeling of newly generated cells, mice were given daily injections of bromodeoxyuridine (BrdU; 50 μg/gm body weight, i.p.; Sigma) for 5 consecutive days. One day or 4 weeks after the last BrdU application mice were overdosed with pentobarbital and transcardially perfused with 4% paraformaldehyde in PBS. Brains were removed and post-fixed in the same fixative overnight and placed in 30% sucrose in PBS for 2 d. Brains were then frozen in 2-methylbutane at −25°C and serially sectioned on a freezing–sliding microtome at 40 μm.
Sections were pretreated in 50% formamide in 2× SSC for 2 hr at 65°C, followed by 10 min in 2× SSC, 30 min in 2N HCl at 37°C, and 10 min in 0.1 m borate buffer. Sections were then incubated in 0.08% H2O2, followed by 0.3% Triton X-100, and blocked in 5% rabbit serum, all in TBS. Rat monoclonal antibody against BrdU (MAS250c; Accurate Ltd., Westbury, NY) was diluted 1:1000 in TBS with 2% serum and 0.3% Triton X-100. Sections were then incubated in biotinylated anti-mouse IgG followed by the avidin–biotin–peroxidase complex solution. The chromogen was Vector SG (Vector Laboratories).
To study the cellular phenotype of BrdU-labeled cells, double and triple immunofluorescence stainings were performed with a combination of antibodies to NeuN, S100β, and CD11b (see above). The secondary antibodies were Alexa488 goat anti-mouse IgG, Alexa 568 goat anti-rat IgG, and Alexa633 goat anti-rabbit IgG (1:400; Molecular Probes). CD11b/BrdU double staining was performed sequentially, i.e., sections were first reacted for CD11b, followed by BrdU pretreatment and detection. In case of high autofluorescence background, sections were treated with Sudan Black B (see above). Sections were analyzed with a confocal laser scanning microscope (see above).
Stereological analysis. Neocortical neuron number was estimated on paraffin embedded cresyl violet-stained sections, and amyloid load was estimated on Aβ-immunostained paraffin sections as previously reported (Calhoun et al., 1998b). Briefly, for both neuron number and plaque load quantification, a systematic random series of every 20th section throughout the entire neocortex was selected, yielding 10–15 sections per animal. Quantification of neuron number was done by first estimating the volume of the neocortex by the Cavalieri point-counting method (grid point area, 500 μm2; mean number of grid points, 97 ± 2.4). Numerical density of neurons was then estimated by counting the number of topmost neuronal nucleoli [using a 100×, 1.3 numerical aperture (NA) objective; on-screen magnification, 2759×] within three-dimensional optical disectors that were systematic-randomly spaced throughout the neocortex (area, 752 μm2; height, 14 μm; guard height, 4 μm; mean number of disectors sampled, 81 ± 1.8; mean number of neurons counted per disector, 2.2 ± 0.05). Only cells with typical neuronal morphology, including a clear nucleolus, were counted. Sampling was optimized to produce a coefficient of error under the observed biological variability. The product of volume times numerical density was calculated to estimate total neuron number. Anatomical regions were defined according to the Franklin and Paxinos (1997) mouse brain atlas, and reliable anatomical boundaries were established at all levels (neocortex borders: olfactory bulb/tubercle, corpus callosum/external capsule, pia mater, endopiriform nuclei, claustrum, amygdala, and subiculum). Postprocessing section thickness was measured at each disector location using a focus drive with ± 0.1 μm accuracy (Applied Scientific Instrumentation, Eugene, OR). The mean section thickness was 26.1 ± 0.6. Results reflect numbers for the right hemisphere only.
Plaque load was estimated by calculating the area fraction occupied by amyloid in two-dimensional disectors on a single focal plane (20× objective; 0.45 NA) (Calhoun et al., 1998b). The percentage of diffuse amyloid was calculated on sections double stained for Aβ and Congo Red and examined under cross-polarized light. Diffuse amyloid was defined as Aβ-positive and Congo Red-negative.
The number of TUNEL-positive cells was counted in seven or eight sections throughout the neocortex. Using a 40×, 0.75 NA objective, TUNEL-positive cells per section were counted. Because sections were not systematically sampled, no estimation of total cortical TUNEL-positive cells per hemisphere was performed.
The number of BrdU-labeled cells was determined on systematic random series of every 12th section throughout the entire neocortex (12–16 fixed-frozen sections per animal). All BrdU-labeled neocortical cells were counted using a 40×, 0.75 NA objective. Total number was calculated by multiplying the number of counted cells times the section interval 12. The results reflect numbers for the right hemisphere only. In contrast to neuron counting, no guard height was included. For reasons of counting efficiency we have not excluded this potential source of error, because qualitative analysis indicated a very robust difference in number of BrdU-labeled cells among the groups.
All stereological analysis was performed with the aid of Stereologer software and a motorized x-y-z stage coupled to a video-microscopy system (Systems Planning and Analysis, Inc., Alexandria, VA). All brains were processed and analyzed in batches of four (aged transgenic, young adult wild-type, aged wild-type, and young adult transgenic) to minimize methodological errors. Results were analyzed using ANOVA with the help of StatView 5.0.1. Indicated is the mean and the SEM. The level of significance was set at ≤ 0.05.
RESULTS
Amyloid load and neural cytoarchitecture in APP23 mice
Eight month-old APP23 mice exhibited only few amyloid plaques in the neocortex (Fig.1A). Typically, they first appeared in the frontal cortex, were very small, and of compact and congophilic nature. Diffuse amyloid was not observed in 8-month-old mice. Quantitative analysis revealed that the volume fraction occupied by amyloid ranged from 0.1 to 0.4%. In contrast, an amyloid load of 15.9–28.0% (mean, 24.1 ± 0.9%) was found in 27-month-old APP23 (Fig. 1B). In these aged mice, diffuse amyloid represented 30–40% of the total amyloid load and showed a considerable region-specific variability.
Fig. 1.

Cerebral amyloidosis in the neocortex of APP23 transgenic mice. A, In 8-month-old mice only few Aβ-immunostained amyloid deposits were found (arrows).B, In contrast, 27-month-old APP23 mice exhibited severe cerebral amyloidosis throughout the neocortex. The total amyloid load in the neocortex of the mouse shown was estimated to be 25.7%. Scale bar, 150 μm. A and B have the same magnification.
Cresyl violet staining revealed that amyloid deposits severely disrupt neuronal cytoarchitecture in the neocortex (Fig.2). In 27-month-old APP23 mice cortical layers were often barely detectable, and layer V pyramidal cells appeared in islets in between amyloid plaques with dendrites and axons leaving the cells in various directions. At higher magnification, neurons appeared displaced by the growing amyloid deposits, giving the impression of a higher density of neurons between amyloid plaques. However, at the intimate amyloid plaque periphery, neurons were largely missing. In contrast, a layer of glial cell nuclei appeared clustered around the amyloid (Fig. 2D). We have previously shown that most of these glia cells are microglia with their processes forming an intimate relationship with the amyloid fibrils (Stalder et al., 1999, 2001).
Fig. 2.
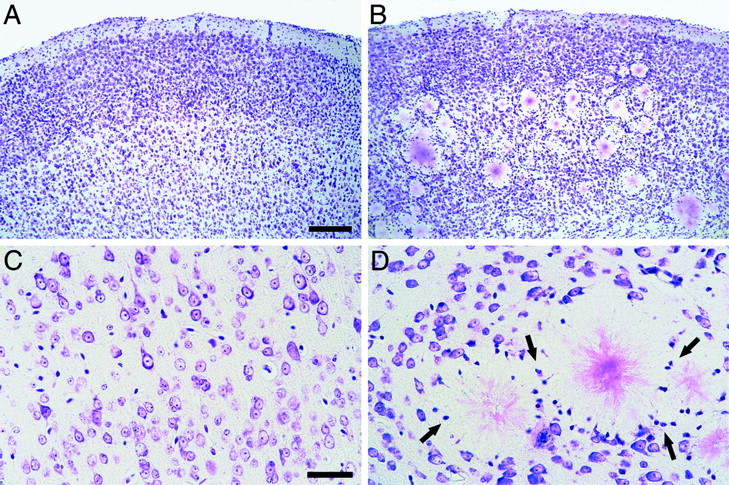
Amyloid plaques disrupt neocortical cytoarchitecture in aged APP23 mice. A, Cresyl violet staining in the neocortex of a 27-month-old wild-type mouse reveals the typical cortical cell layers. B, In contrast, in 27-month-old APP23 mice amyloid deposits disrupt the neurocytoarchitecture, and some of the layers are barely detectable.C, D, Higher magnifications of layer V neurons in a 27-month-old wild-type (C) and transgenic (D) mouse. Note the numerous glial cell nuclei (arrows in D) and the absence of neurons in the immediate vicinity of the amyloid plaques. Scale bars:A, B, 150 μm; C,D, 40 μm.
Estimation of total neocortical neuron number in 8- and 27-month-old APP23 mice
Using stereological methods we have quantified neurons in adult and aged, 8 and 27 months, APP23 mice of both sexes and in corresponding controls. ANOVA for total number of neocortical neurons per hemisphere revealed significant effects for the three main factors age (F(1,44) = 7.53; p< 0.01), genotype (F(1,44) = 6.48;p < 0.05), and sex (F(1,44) = 13.52; p < 0.001). The observation that in all groups males had on average 10% more neurons than did females was confirmed by a lack of further interactions with the factor sex. In contrast, a significant interaction was found between age and genotype (F(1,44) = 5.28; p < 0.05). Subsequent Newman–Keuls post hoc analysis revealed that the 8-month-old APP23 mice had modest but significant 13–15% increases in neocortical neuron number compared with 8-month-old wild-type mice and 27-month-old wild-type mice but also with 27-month-old APP23 mice (all p values < 0.01) (Fig.3A). No age-related loss of neocortical neuron number was found in the control mice.
Fig. 3.

Stereological estimation of total neocortical neuron number. A, Total neuron number per neocortex per hemisphere in 8- and 27-month-old APP23 and wild-type control mice. For this graph, males and females were combined (n = 11–15 per group). Results revealed that 8-month-old APP23 mice have more neurons compared with the three other groups (*p < 0.01). B, For the 27-month-old APP23 mice, linear regression analysis revealed an inverse relation between neuron number and amyloid load.
This surprising observation indicates that APP23 mice have more neurons than do wild-type mice before they develop significant cerebral amyloidosis but then lose neurons with further aging to reach the levels of wild-type mice at 27 months of age. To sustain this interpretation and because 27-month-old APP23 mice show great variability in amyloid load (see above), we wondered whether amyloid load predicts neuron number in the 27-month-old APP23 mice. Indeed, linear regression analysis (Fig. 3B) revealed a significant inverse relation between neocortical neuron number and amyloid load in the 27-month-old APP23 mice (R2(11) = 0.55; p < 0.01).
To determine whether the increase in neuron number in 8-month-old APP23 mice occurs during or after brain development, we have assessed neocortical neuron number in an additional group of young 2- to 3-month-old male APP23 mice and littermate control mice. Interestingly, we found also in these young APP23 mice 10% more neocortical neurons compared with nontransgenic littermates (6.31E6 ± 0.16E6 vs 5.79E6 ± 0.17E6; t(10) = 2.22;p = 0.05). The absolute neuron numbers in these young groups is higher compared with the adult and aged mice because only male mice were used.
Amyloid-associated cell death
Analysis of cresyl violet-stained sections of 24-month-old APP23 mice occasionally revealed neurons with a pyknotic appearance and an irregular membrane structure. Such cells were in most cases in the vicinity of amyloid plaques (Fig.4A). To identify nuclear profiles with DNA fragmentation, one of the hallmarks of apoptosis, we then used the TUNEL-labeling technique. Results revealed a approximately fourfold increase in TUNEL-positive cells per section in 24-month-old APP23 mouse neocortex compared with age-matched wild-type mice (7.2 ± 0.65 vs 1.9 ± 0.27; p< 0.001). The majority of these TUNEL-positive cells was clearly localized to amyloid plaques (Fig. 4B). We have also used immunohistochemistry using CM-1 antibody (provided by A. Srinivasan) to activated caspase-3, which is thought to be an important step in apoptotic cell death (Nicholson et al., 1995). Again we found more CM-1-positive cells in aged APP23 mice compared with wild-type mice (results not shown).
Fig. 4.

Cell death with necrotic and apoptotic appearance in the vicinity of amyloid plaques. A, Neurons with a pyknotic appearance and an irregular membrane structure (arrows) were occasionally detected in 24-month-old APP23 mice. Such neurons were almost exclusively associated with amyloid deposits. B, TUNEL-positive cells (arrowheads) in the vicinity of an amyloid plaque. Using confocal microscopy TUNEL-positive cells (red) were labeled with markers for either neurons (C), microglia (D), or astrocytes (E) (all in green). Most of the cells could not be phenotyped. Only occasionally were TUNEL-positive cells labeled for NeuN or CD11b. No double labeling was observed with S100β (Table 1). The insert in Crepresents single confocal sections of both markers. Scale bars:A, B, 40 μm; C, D, 10 μm. D and E have same magnification.
To establish the nature of these apoptotic cells, double labeling for TUNEL and NeuN, S100β, and CD11b was used (Fig. 4C–E,Table 1). However, most of the TUNEL-positive cells (77%) did not colocalize with any of these markers. Six percent were NeuN-positive and could be identified as neurons. Unexpectedly, 17% of the TUNEL-positive cells expressed CD11b and were identified as microglia. None of the TUNEL-positive cells expressed the astroglial marker S100β. The observation that the vast majority of the TUNEL-positive cells could not be identified with cell type-specific markers may be because these cells are in late stages of apoptosis (Biebl et al., 2000).
Table 1.
Phenotype of TUNEL- and BrdU-positive cells in neocortex of 24-month-old APP23 mice
| TUNEL | BrdU | |||
|---|---|---|---|---|
| # Cells analyzed | % Cells colabeled | # Cells analyzed | % Cells colabeled | |
| NeuN | 87 | 6 | 240 | 0 |
| S100β | 102 | 0 | 240 | 16 |
| CD11b | 102 | 17 | 120 | 58 |
TUNEL- and BrdU-positive cells were labeled with markers for neurons (NeuN), astrocytes (S100β), or microglia (CD11b). Number of analyzed cells and percentage of TUNEL- or BrdU-colabeled cells are indicated.
Increase in BrdU-positive cells in aged APP23 mice
Previous work has shown that apoptotic cell death can induce neurogenesis in the mouse neocortex (Magavi et al., 2000). For this reason we have studied the generation of new cells in the neocortex of 4- and 24-month-old APP23 mice and age-matched wild-type control mice.
In a first experiment mice were killed 4 weeks after the last BrdU injection to allow newly produced cells to differentiate into their final cell type (Gage, 2000). Results revealed a striking increase in BrdU-positive cells in aged APP23 mice compared with aged wild-type mice with most of the labeled cells around the amyloid deposits (Fig. 5A,B). Quantitative analysis (Fig. 6) confirmed that 24-month-old APP23 mice have a significant fourfold to sixfold increase in BrdU-labeled cells (ANOVA age × genotype:F(1,12) = 46.5; p < 0.001) in comparison with 24-month-old wild-type mice and 4-month-old APP23 (p values < 0.001).
Fig. 5.
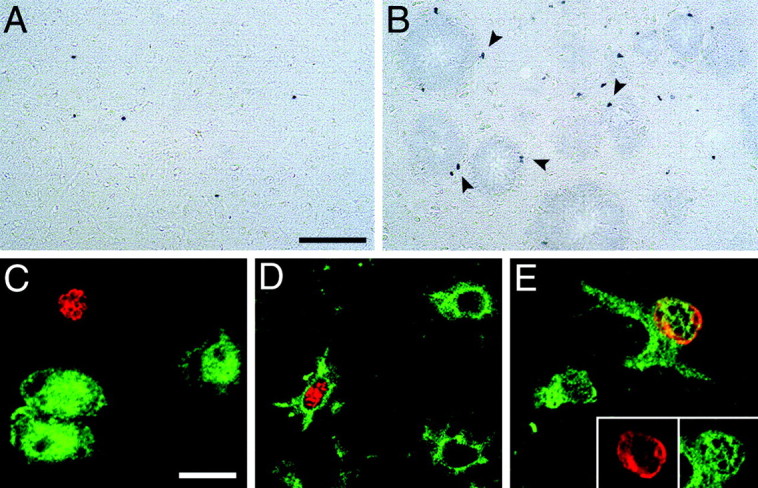
BrdU-positive cells in the neocortex of aged APP23 mice. A, Only few labeled cells were observed in the neocortex of 24-month-old wild-type mice 4 weeks after BrdU injections.B, In contrast, there was an approximately fourfold increase in labeled cells in age-matched APP23 mice (for quantitative results, see Fig. 6). Most of these cells appeared to be associated with amyloid plaques (arrowheads). To study the cellular phenotype of these cells BrdU immunofluorescence (red) was combined with NeuN (C) CD11b (D) and S100β (E) (all ingreen), and colocalization was assessed with confocal microscopy. None of the BrdU-labeled cells were positive for NeuN (C). In contrast, the majority of the BrdU-labeled cells revealed colocalization with the microglia marker CD11b (D). Colocalization was also found with the astrocytic marker S100 β (E). Theinsert in E represents single confocal sections of both markers. Quantitative phenotyping is summarized in Table 1. Scale bars: A, B, 75 μm;C–E, 10 μm.
Fig. 6.

Increase in BrdU-positive cells in the neocortex of aged APP23 mice. Total BrdU-positive cells in the neocortex of young (4-month-old) and aged (24-month-old) wild-type and APP23 mice. Numbers are for one hemisphere only. Note the approximately fourfold increase in BrdU-labeled cells in the aged APP23 mice compared with all three other groups (*p values < 0.001).
To determine whether the newly generated cells in aged APP23 mice are in fact neurons, BrdU-labeling was combined with the neuronal marker NeuN (Fig. 5C, Table 1). However, although 240 BrdU-positive cells (60/mouse) have been studied for colocalization with NeuN, we could not detect any neocortical colabeled cell. Subsequent colabeling with S100β and CD11b revealed 58% of the labeled cells as microglia and 16% as astrocytes, whereas the remaining cells could not be allocated to one of these cell types (Fig. 5D,E, Table1).
We have also studied an additional group of 24-month-old APP23 mice and age-matched wild-type control mice that were killed 1 d after the last BrdU injection. A significant approximately fourfold increase in BrdU-positive cells in APP23 neocortex was detected, when compared with control mice (5.21E4 ± 1.0E4 vs 1.47E4 ± 2.3E3;t test: p = 0.01). This increase is very similar to that observed in 24-month-old APP23 mice killed 4 weeks after the last BrdU injection. These results favor the hypothesis that glial proliferation is upregulated in APP23 mice. However, an effect on cell survival should not be ruled out because BrdU was injected over 5 d, and considerable death of newly generated cells may have occurred within this period.
DISCUSSION
In the present study we have reinvestigated neocortical neuron loss in APP transgenic mice by comparing two large homogeneous groups of adult and aged APP23 mice of both genders. For the adult group we chose 8-month-old mice, an age at which cerebral amyloidosis starts to develop. It has previously been shown that aging renders the brain vulnerable to Aβ toxicity (Geula et al., 1998) and thus we used aged APP23 mice that were 26–29 months old, which is beyond the mean life span of C57BL/6 mice (Jucker and Ingram, 1997).
The present estimate of total number of neurons in the mouse neocortex was ∼5.5E6. This result is consistent with our previous estimate (Calhoun et al., 1998b). Furthermore, the present findings show that males have a robust 10% increase in neuron number compared with females. This observation is interesting in view of a 16% gender difference that has been reported in humans (Pakkenberg and Gundersen, 1997). In wild-type murine neocortex we did not find an age-related neuron loss, whereas in humans, albeit with a larger cohort, a 10% neuron loss with aging was reported (Pakkenberg and Gundersen, 1997).
Most significant for the present investigation, we have found that young and adult APP23 mice have 10–15% more neurons compared with wild-type control mice. This observation was surprising, but in light of previous results not entirely unexpected. APP and/or its secreted form sAPP have been implicated in cell growth and cell survival, and in the protection of neurons against excitotoxicity (Saitoh et al., 1989;Milward et al., 1992; Mattson et al., 1993; Roch et al., 1994;Smith-Swintosky et al., 1994; Perez et al., 1997). An increased number of synapses and augmented neuroprotection to excitotoxic injuries has also been reported in APP transgenic mice (Mucke et al., 1994; Masliah et al., 1997). The present observation of an increased neocortical neuron number in adult APP23 mice adds to the evidence that APP overexpression in transgenic mice has growth-promoting and neuroprotective features. If this increase in neuron number is a more general phenomenon of APP overexpression, it may also occur in hippocampus of APP23 mice. This, in turn, raises the possibility that the neuron loss in CA1 of APP23 mice is in fact greater than previously reported (Calhoun et al., 1998a). Although APP overexpression is a likely cause, it cannot be ruled out that the increase in neuron number in APP23 mice is an effect of the transgene insertion site. Thus, it will be important to replicate our observation in other APP transgenic mice with similar genetic backgrounds and APP expression levels. In addition, it may be interesting to examine whether the overexpression of APP in Down's syndrome also leads to an increased neuron number.
Given that APP23 mice have 10–15% more neocortical neurons when they start to develop cerebral amyloidosis, our results suggest that they lose approximately this percentage of neurons in the course of cerebral amyloidogenesis. The neuron loss in aged APP23 mice is in line with the increased appearance of neurons with a necrotic and/or apoptotic phenotype and with the inverse correlation observed between neuron number and amyloid load.
In two other transgenic mouse lines, Tg2576 mice and PDAPP mice, no significant nerve cell loss was reported in hippocampus and neocortex (Irizarry et al., 1997a,b). This difference compared with the APP23 mouse line may be attributable to the lower amyloid burden reported in Tg2576 mice (Irizarry et al., 1997b) and the more diffuse nature of amyloid deposits in PDAPP mice (Masliah et al., 1996; Irizarry et al., 1997a). Neuron number in the frontal cortex has recently also been assessed in Tg2576 mice that were crossed with mutant PS1-overexpressing mice. Such mice also reveal a high (compact) plaque load, similar to that in APP23 mice. However only one to four mice were analyzed per group, and thus a 15% loss may not have been detectable (Takeuchi et al., 2000). Furthermore, we have reported age-dependent hemorrhagic stroke in APP23 mice, which may contribute to neurodegeneration (Winkler et al., 2001).
The neuron loss in neocortex of APP23 mice appears modest, but in fact exceeds the 2–6% global neuron loss reported in AD neocortex using similar stereological methodology (Regeur et al., 1994; Bundgaard et al., 2001). In contrast, 32% neuron loss has been reported in AD if the entorhinal cortex is analyzed separately, and up to 90% neuron loss was observed when individual laminae of the entorhinal cortex were analyzed, emphasizing that the neuropathic manifestations of AD are region-specific (Gomez-Isla et al., 1996a). Unfortunately, entorhinal cortex neuron number could not be assessed in APP23 mice because the massive amyloid deposition in aged APP23 prevented the reliable identification of the anatomical borders of the entorhinal cortex, an essential prerequisite for unbiased quantification. Nevertheless, qualitative neuron loss is also clearly observed in the entorhinal cortex of aged APP23 mice, although it probably does not reach the extent reported in humans (Calhoun et al., 1998b). Consistently, it has been reported that Aβ neurotoxicity in vivo is species-dependent with a much higher toxicity in primates compared with rodents (Geula et al., 1998). Moreover, APP overexpression per se is neuroprotective against amyloid-induced neurotoxicity (as discussed above). Thus, it will be important to assess neurodegeneration in mouse models of cerebral amyloidosis that do not overexpress APP (Iwata et al., 2000; Popp et al., 2000).
The view that estimation of total neuron numbers is suited to determine the extent of neuronal degeneration has recently been challenged by the observation of neurogenesis in mouse and rat neocortex after targeted apoptotic lesions and focal cerebral ischemia (Gu et al., 2000; Magavi et al., 2000). In one of these studies, it was demonstrated that some of the newly produced neurons in the vicinity of the lesion site extend axons into the denervated region and thus, appear to replenish damaged neuronal circuits (Magavi et al., 2000). These observations, together with recent findings that overexpression of APP may enhance proliferation of neural stem cells (Ohsawa et al., 1999) and that neocortical stem cells have the potential to differentiate into neuronsin vitro (Palmer et al., 1999), suggest that neocortical neuron number in APP23 transgenic mice might be viewed as the result of the dynamic equilibrium achieved between the continuous loss and birth of neurons.
However, in the present study we found no evidence for neurogenesis either in wild-type mice (confirming previous studies, e.g., Kuhn et al., 1997) or in APP23 mice with a massive amyloid load. Because we have counted a total of 240 cells in four mice we cannot exclude that neurogenesis occurs with a prevalence of <0.5%. In contrast, 1–2% and 3–6% of BrdU-positive cells colocalized with NeuN after targeted apoptosis and ischemic stroke, respectively (Gu et al., 2000; Magavi et al., 2000). Thus, we conclude that neurogenesis does not occur, or is an extremely rare event, in response to cerebral amyloidosis in APP23 mice. It will be important to determine whether the same is true in AD.
The finding that 58% of the BrdU-labeled cells were in fact microglia substantiates and extends our previous preliminary observation (Bornemann et al., 2001) and indicates a significant number of newly produced microglial cells predominantly around amyloid plaques. Because BrdU has a half life of ∼2 hr (Phuphanich and Levin, 1985) and was injected once daily for 5 d, we estimate that ∼2 million new microglia are produced per month in the neocortex of aged APP23 mice. At present it is not clear whether these newly produced microglia are the product of mitotic microglia or recruited macrophage. It could even be considered that they originate from neural stem cells. Furthermore, the fate of amyloid-associated microglia is not well understood. Using DNA fragmentation labeling in situ, degenerating microglial cells have been previously described around amyloid in the neocortex of AD patients (Lassmann et al., 1995). Similarly, results of the present study suggest apoptotic microglial cell death around amyloid plaques in APP23 mice. It is tempting to speculate that the continuous production and death of microglia play an important role in cerebral amyloidosis and in AD.
In conclusion, the present results demonstrate that cerebral amyloidosis in the neocortex of aged APP23 transgenic mice causes a modest but significant neuron loss and marked gliogenesis. The contribution of these changes to the reported cognitive impairment of APP23 mice (Kelly et al., 1999) and for AD pathogenesis and therapy remains to be evaluated.
Footnotes
This work was supported by grants to M.J. from the VerUm Foundation (Foundation for Behavior and Environment, Munich, Germany) and the Swiss National Science Foundation (31-44526.95 and 31-56753.99). We thank Drs. M. Tolnay, A. Probst (Institute of Pathology, Basel, Switzerland), M. Dubois-Dauphin (University Hospital, Geneva, Switzerland), A. Srinivasan (Idun Pharmaceuticals, La Jolla, CA), and D. Abramowski and C. Sturchler-Pierrat (Novartis, Basel, Switzerland) for helpful discussions and experimental support. We also acknowledge the professional help of T. Schürch, H. Zysset, and C. Mistl (Institute of Pathology, Basel, Switzerland) with photography and histology.
Correspondence should be addressed to Dr. Mathias Jucker, Institute of Pathology, University of Basel, Schönbeinstrasse 40, CH-4003 Basel, Switzerland. E-mail: mjucker@uhbs.ch.
M. Calhoun's present address: Kastor Neurobiology of Aging Laboratories, Mount Sinai School of Medicine, New York, NY 10029.
REFERENCES
- 1.Biebl M, Cooper CM, Winkler J, Kuhn HG. Analysis of neurogenesis and programmed cell death reveals a self- renewing capacity in the adult rat brain. Neurosci Lett. 2000;291:17–20. doi: 10.1016/s0304-3940(00)01368-9. [DOI] [PubMed] [Google Scholar]
- 2.Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bundgaard MJ, Regeur L, Gundersen HJ, Pakkenberg B. Size of neocortical neurons in control subjects and in Alzheimer's disease. J Anat. 2001;198:481–489. doi: 10.1046/j.1469-7580.2001.19840481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calhoun ME, Kurth D, Phinney AL, Long JM, Hengemihle J, Mouton PR, Ingram DK, Jucker M. Hippocampal neuron and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol Aging. 1998a;19:599–606. doi: 10.1016/s0197-4580(98)00098-0. [DOI] [PubMed] [Google Scholar]
- 5.Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998b;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 6.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings BJ, Cotman CW. Image analysis of beta-amyloid load in Alzheimer's disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 8.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1997. [Google Scholar]
- 9.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 10.Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- 11.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 12.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 13.Giannakopoulos P, Hof PR, Michel JP, Guimon J, Bouras C. Cerebral cortex pathology in aging and Alzheimer's disease: a quantitative survey of large hospital-based geriatric and psychiatric cohorts. Brain Res Brain Res Rev. 1997;25:217–245. doi: 10.1016/s0165-0173(97)00023-4. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Isla T, Price JL, McKeel DW, Morris JC, Jr, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996a;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, Perls TT, Lipsitz LA, Hyman BT. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer's disease. Ann Neurol. 1996b;39:62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- 16.Gu W, Brannstrom T, Wester P. Cortical neurogenesis in adult rats after reversible photothrombotic stroke. J Cereb Blood Flow Metab. 2000;20:1166–1173. doi: 10.1097/00004647-200008000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 19.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997a;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997b;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major Abeta1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 22.Jucker M, Ingram DK. Murine models of brain aging and age-related neurodegenerative diseases. Behav Brain Res. 1997;85:1–26. doi: 10.1016/s0166-4328(96)02243-7. [DOI] [PubMed] [Google Scholar]
- 23.Kelly PH, Hunziker D, Schlecht HP, Carver K, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B. Progressive impairment in amyloid precursor protein transgenic mouse line APP23. Soc Neurosci Abstr. 1999;25:1291. [Google Scholar]
- 24.Knowles RB, Gomez-Isla T, Hyman BT. Abeta associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol. 1998;57:1122–1130. doi: 10.1097/00005072-199812000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH. Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J Neurosci. 1997;17:5820–5829. doi: 10.1523/JNEUROSCI.17-15-05820.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamb BA, Bardel KA, Kulnane LS, Anderson JJ, Holtz G, Wagner SL, Sisodia SS, Hoeger EJ. Amyloid production and deposition in mutant amyloid precursor protein and presenilin-1 yeast artificial chromosome transgenic mice. Nat Neurosci. 1999;2:695–697. doi: 10.1038/11154. [DOI] [PubMed] [Google Scholar]
- 27.Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM. Cell death in Alzheimer's disease evaluated by DNA fragmentation in situ. Acta Neuropathol. 1995;89:35–41. doi: 10.1007/BF00294257. [DOI] [PubMed] [Google Scholar]
- 28.Magavi SS, Leavitt BR, Macklis JD. Induction of neurogenesis in the neocortex of adult mice. Nature. 2000;405:951–955. doi: 10.1038/35016083. [DOI] [PubMed] [Google Scholar]
- 29.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996;16:5795–5811. doi: 10.1523/JNEUROSCI.16-18-05795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L. Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience. 1997;78:135–146. doi: 10.1016/s0306-4522(96)00553-2. [DOI] [PubMed] [Google Scholar]
- 31.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 32.Milward EA, Papadopoulos R, Fuller SJ, Moir RD, Small D, Beyreuther K, Masters CL. The amyloid protein precursor of Alzheimer's disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- 33.Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- 34.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 35.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 36.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 37.Ohsawa I, Takamura C, Morimoto T, Ishiguro M, Kohsaka S. Amino-terminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. Eur J Neurosci. 1999;11:1907–1913. doi: 10.1046/j.1460-9568.1999.00601.x. [DOI] [PubMed] [Google Scholar]
- 38.Pakkenberg B, Gundersen HJ. Neocortical neuron number in humans: effect of sex and age. J Comp Neurol. 1997;384:312–320. [PubMed] [Google Scholar]
- 39.Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–8497. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez RG, Zheng H, Van der Ploeg LHT, Koo EH. The β-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17:9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phuphanich S, Levin VA. Bioavailability of bromodeoxyuridine in dogs and toxicity in rats. Cancer Res. 1985;45:2387–2389. [PubMed] [Google Scholar]
- 43.Popp A, Hartmann E, Hagelschuer I, Duff K, Wirak D, Unsworth C, Baumann KH, Koenig G. β-Amyloid plaques In amyloid precursor protein knock-in mice. Neurobiol Aging. 2000;21:S223. [Google Scholar]
- 44.Probst A, Langui D, Ulrich J. Alzheimer's disease: a description of the structural lesions. Brain Pathol. 1991;1:229–239. doi: 10.1111/j.1750-3639.1991.tb00666.x. [DOI] [PubMed] [Google Scholar]
- 45.Regeur L, Jensen GB, Pakkenberg H, Evans SM, Pakkenberg B. No global neocortical nerve cell loss in brains from patients with senile dementia of Alzheimer's type. Neurobiol Aging. 1994;15:347–352. doi: 10.1016/0197-4580(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 46.Roch JM, Masliah E, Roch-Levecq AC, Sundsmo MP, Otero DA, Veinbergs I, Saitoh T. Increase of synaptic density and memory retention by a peptide representing the trophic domain of the amyloid beta/A4 protein precursor. Proc Natl Acad Sci USA. 1994;91:7450–7454. doi: 10.1073/pnas.91.16.7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saitoh T, Sundsmo M, Roch JM, Kimura N, Cole G, Schubert D, Oltersdorf T, Schenk DB. Secreted form of amyloid-β protein precursor is involved in the growth regulation of fibroblasts. Cell. 1989;58:615–622. doi: 10.1016/0092-8674(89)90096-2. [DOI] [PubMed] [Google Scholar]
- 48.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 49.Smith-Swintosky VL, Pettigrew CL, Craddock SD, Culwell AR, Rydel RE, Mattson MP. Secreted forms of β-amyloid precursor protein protects against ischemic brain injury. J Neurochem. 1994;63:781–784. doi: 10.1046/j.1471-4159.1994.63020781.x. [DOI] [PubMed] [Google Scholar]
- 50.Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stalder M, Deller T, Staufenbiel M, Jucker M. 3D- Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging. 2001;22:427–434. doi: 10.1016/s0197-4580(01)00209-3. [DOI] [PubMed] [Google Scholar]
- 52.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–339. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van Den Haute C, Mercken M, Moechars D, Laenen I, Kuiperi C, Bruynseels K, Tesseur I, Loos R, Vanderstichele H, Checler F, Sciot R, Van Leuven F. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am J Pathol. 2000;157:1283–1298. doi: 10.1016/S0002-9440(10)64644-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 56.Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, Jucker M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J Neurosci. 2001;21:1619–1627. doi: 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]