

Abstract
The nucleus accumbens (NAc) is an important cerebral area involved in reward and spatial memory (Pennartz et al., 1994), but little is known about synaptic plasticity in this region. Here, electron microscopy revealed that, in the NAc, metabotropic glutamate receptors 2/3 (mGlu2/3) immunostaining was essentially associated with axonal terminals and glial processes, whereas postsynaptic dendrites and neuronal cell bodies were unstained. Electrophysiological techniques in the NAc slice preparation demonstrated that activation of mGlu2/3 with synaptically released glutamate or specific exogenous agonist, such as LY354740 (200 nm, 10 min), induced long-term depression of excitatory synaptic transmission (mGlu2/3-LTD). Tetanic-LTD and pharmacological mGlu2/3-LTD occluded each other, suggesting common mechanisms. The mGlu2/3-LTD did not require synaptic activity but depended on the cAMP–protein kinase A cascade. Selective inhibition of P/Q-type Ca2+channels with ω-agatoxin-IVA occluded the expression of mGlu2/3-LTD, and, conversely, the inhibitory effects of ω-agatoxin-IVA were abolished during mGlu2/3-LTD. Thus, mGlu2/3 play an important role in the control of use-dependent synaptic plasticity at prelimbic cortex–NAc synapses: their activation causes a form of LTD mediated by the long-lasting reduction of P/Q-type Ca2+channels contribution to transmitter release.
Keywords: nucleus accumbens, long-term depression, metabotropic glutamate receptors, mGlu2/3, presynaptic inhibition, mice, prelimbic cortex, calcium channels
The nucleus accumbens (NAc) is a structure of particular interest because of its role in connecting the limbic system to the basal ganglia and to extrapyramidal motor system, its involvement in spatial memory and adaptative processes, and its implication in the mechanisms at the origin of the rewarding properties of many drugs of abuse (Schacter et al., 1989;Pennartz et al., 1994). Glutamate is the excitatory neurotransmitter at the synapses between prelimbic cortex afferents and medium spiny neurons of the NAc (Horne et al., 1990). Pennartz and collaborators showed that high-frequency stimulation of prelimbic afferents could cause long-term depression (LTD), long-term potentiation (LTP), or no change in the gain of excitatory synaptic transmission (Pennartz et al., 1993). What determines the fate of these synapses during phasic activity of prelimbic afferents? LTP (but not LTD) induction partially depended on NMDA receptors, and both LTD and LTP were independent of dopamine receptors (Pennartz et al., 1993; Kombian and Malenka, 1994). Morphological and physiological studies have described various metabotropic glutamate receptor (mGlu) subtypes at the glutamatergic afferents to the NAc (Ohishi et al., 1993a,b; Shigemoto et al., 1993;Manzoni et al., 1997). Converging reports indicate that, during tetanic stimulation of glutamatergic synapses, activation of mGlu induces LTD (for review, see Anwyl, 1999). Among the various mGlu subtypes (for review, see Conn and Pin, 1997), mGlu2/3 play key roles in the control of LTD in hippocampal mossy fibers (Kobayashi et al., 1996; Yokoi et al., 1996; Tzounopoulos et al., 1998), the dentate gyrus (Huang et al., 1997, 1999), the amygdala (Lin et al., 2000), the prefrontal cortex (Otani et al., 1999), and the dorsal striatum (Kahn et al., 2001).
This study shows that mGlu2/3 play an important role in the control of use-dependent synaptic plasticity at prelimbic cortex–NAc synapses: their activation causes a form of LTD mediated by a reduction of P/Q-type Ca2+ channels contribution to transmitter release.
MATERIALS AND METHODS
Immunohistochemistry–electron microscopy. After being deeply anesthetized with sodium pentobarbital (50 mg/kg), mice were perfused through the ascending aorta with PBS, pH 7.4, followed by 300 ml of fixative composed of 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4. The brain was then dissected and fixed by immersion in the fixative without glutaraldehyde for 12 hr at 4°C. It was then cut sagittally with a vibratome into 40- to 50-μm-thick sections. After careful rinsing in PBS, sections were successively incubated as follows: (1) for 48 hr at 4°C with the antibody anti-mGlu2/3 (diluted 1:500; Chemicon, Temecula, CA), (2) for 12 hr at 4°C with a peroxidase-labeled Fab fragment of goat IgG anti-rabbit IgG (Biosys, Compiègne, France), diluted 1:1000, and (3) with 0.1% 3,3′ diaminobenzidine diluted in 0.05 m Tris buffer, pH 7.3, in the presence of 0.2% H2O2. The primary and secondary antibodies were diluted in PBS containing 1% BSA, 1% normal goat serum, and 0.1% saponin. Immunostained sections were either mounted in Permount and observed under a light microscope or further treated for electron microscopy. For this, they were carefully rinsed in 0.1 m cacodylate buffer, pH 7.3, and postfixed in 1% OsO4 in the same buffer. They were then dehydrated in graded concentrations of ethanol and embedded in araldite. Punches of 1.5 mm in diameter were cut through the accumbens nucleus and mounted on araldite blocks. After being cut into ultrathin sections, they were observed in an electron microscope (model H 7110; Hitachi, Tokyo, Japan) without counterstaining or with slight uranyl acetate staining.
Electrophysiology. Whole-cell patch-clamp and extracellular field recordings were made from medium spiny neurons in parasagittal slices of mouse nucleus accumbens. This method has been described previously (Manzoni et al., 1997; Robbe et al., 2001). In brief, mice (male C57BL/6, 4–6 weeks) were anesthetized with isoflurane and decapitated. The brain was sliced (300–400 μm) in the parasagittal plane using a vibratome and maintained in physiological saline at 4°C. Slices containing the NAc were stored at least 1 hr at room temperature before being placed in the recording chamber and superfused (2 ml/min) with artificial CSF (in mm: 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, 1.2 NaH2PO4, and 11 glucose) and was equilibrated with 95% O2–5% CO2. All experiments were done at room temperature. The superfusion medium contained picrotoxin (100 μm) to block GABAAreceptors. All drugs were added at the final concentration to the superfusion medium.
For field EPSPs (fEPSPs), the recording pipette was filled with a 3 m NaCl solution, and both the fEPSP slope (calculated with a least-square method) and fEPSP amplitude were measured (graphs depict amplitudes). For patch-clamp experiments, cells were visualized using an upright microscope with infrared illumination, and recordings were made with whole-cell electrodes containing the following (in mm): 128 Cs-gluconate, 20 NaCl, 1 MgCl2, 1 EGTA, 0.3 CaCl2, 2 Mg-ATP, 0.3 GTP, and 0.2 cAMP, buffered with 10 HEPES, pH 7.3. Electrode resistance was 4 MΩ, acceptable access resistance was <20 MΩ, and the holding potential was −70 mV. An Axopatch-1D (Axon Instruments, Foster City, CA) was used to record the data, which were filtered at 1–2 kHz, digitized at 5 kHz on a DigiData 1200 interface (Axon Instruments), and collected on a personal computer using ACQUIS-1 software (Bio-Logic, Claix, France). To evoke synaptic currents, stimuli (100–150 μsec duration) were delivered at 0.033 Hz through bipolar tungsten electrodes placed at the prefrontal cortex–accumbens border (Manzoni et al., 1997, 1998). Recordings were made in the rostromedial dorsal accumbens close to the anterior commissure. According to the atlas of mouse brain in stereotaxic coordinates (Paxinos and Franklin, 2001), slices were located between lateral 1.44 and 0.6 mm to the midline. Electrodes of stimulation were placed rostrally and close to the recording electrode.
Miniature and spontaneous EPSCs (mEPSCs and sEPSCs) were recorded in the presence or absence of tetrodotoxin (TTX) (300 nm), respectively, using Axoscope 1.1.1. mEPSC amplitude and inter-interval time were measured using Axograph 3.6. For this analysis, a template of mEPSCs having the width and time course of a typical synaptic event [a double exponential: f(t) = exp(−t/Rise) − exp(−t/Decay), where Rise is 0.5 msec and Decay is 3 msec] was slid along the data trace one point at a time. At each position, this template is optimally scaled and offset to fit the data, and a detection criterion is calculated. The detection criterion is the template-scaling factor divided by the goodness-of-fit at each position. An event is detected when this criterion exceeds a threshold and reaches a sharp maximum. The limit of detection was 2 pA (Manzoni and Williams, 1999; Robbe et al., 2001).
The fitting of concentration–response curves were calculated according to y = {ymax −ymin/1 + (x/EC50)n} +ymin (whereymax indicates response in the absence of agonist, ymin indicates response remaining in presence of maximal agonist concentration, xindicates concentration, EC50 indicates concentration of agonist producing 50% of the maximal response, andn indicates slope) with Kaleidagraph software (Abelbeck Software, Reading, PA). All values are given as mean ± SEM. Statistical analyses were done with the Mann–Whitney Utest, the Fisher's exact test for distribution in Figure3A, the Kolmogorov–Smirnov tests using Statview for cumulative distribution analysis of mEPCSs and sEPSCs (Abacus Concepts, Calabasas, CA), and the paired Student's t test for mean frequency and amplitude of mEPSCs and sEPSCs. p < 0.05 was taken as indicating statistical significance (*p < 0.05 and **p < 0.01).
Fig. 3.

Induction of synaptic mGlu2/3-LTD and mutual occlusion with pharmacological LTD. A, High-frequency tetanus (3 times for 1 sec at 100 Hz, 20 sec interval) can induce LTD, LTP, or no plasticity in the NAc. Summarized histogram showing the percentage of slices exhibiting LTD (defined as a inhibition of at least 10% of baseline fEPSP, 30 min after tetanus), LTP (defined as an enhancement of at least 10% of baseline fEPSP, 30 min after tetanus), or no plasticity in control conditions or after perfusion with the highly selective mGlu2/3 antagonist LY341495 (200 nm, 15 min). Blocking mGlu2/3 receptors during the tetanus clearly reduced the number of slices expressing LTD and increased the number of slices exhibiting either LTP or no change in synaptic efficacy.B, Typical experiment showing tetanus-induced LTD in control medium. The inset shows traces (average of 10 consecutive fEPSPs) taken at the points indicated. Calibration: 0.4 mV, 5 msec. C, Summary of all of the experiments comparing the effects of high-frequency stimulation of prelimbic afferents in control conditions and after blockade of mGlu2/3. D, Typical experiment showing that, after inducing the mGlu2/3-LTD with LY354740 (200 nm), the tetanus induces no more LTD (here it induces LTP). E, Summary of all of the experiments showing the occlusion of tetanus-induced LTD after induction of mGlu2/3-LTD with LY354740. F, Typical experiment showing that, after saturation of tetanus-induced LTD, LY354740 (200 nm) did not induce LTD. G, Summary of all of the experiments showing the occlusion of mGlu2/3-LTD induced with LY354740 (200 nm) after induction of tetanus-induced LTD. Tet, Tetanus (three times at 1 sec at 100 Hz). *p < 0.05; **p< 0.01.
Drugs used were as follows: (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK801),l-AP-5, picrotoxin, forskolin, and nimodipine (Sigma, St. Louis, MO); 2-amino-2-(2-carboxycyclopropan-1-yl)-3-(dibenzopyran-4-yl) propanoic acid (LY341495), (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine (LCCG1) and (2S)-α-ethylglutamate (eGlu) (Tocris Cookson, Ballwin, MO); H 89, KT 5720, and ω-agatoxin-IVA (Alexis, San Diego, CA); tetrodotoxin and ω-conotoxin-GVIA (Alomone, Jerusalem, Israel); and (+)-2-aminobicyclo-[3.1.0]hexane-2,6-dicarboxylic acid (LY354740) [gift of Drs. A. Schoepp and Monn (Eli-Lilly, Indianapolis, IN)]. Other chemicals were from the highest commercial grade available.
RESULTS
Localization of mGlu2/3 in the NAc
Examination of immunostained sections under the light microscope indicated that, throughout all of the forebrain regions examined, the organization of the mGlu2/3-immunostained structures conformed to previous descriptions in the rat (Petralia et al., 1996; Mineff and Valtschanoff, 1999) and mice (Ohishi et al., 1998; Tamaru et al., 2001). Within the nucleus accumbens, as in the surrounding regions including the striatum, mGlu2/3 labeling was essentially associated with elongated processes and with numerous dot structures dispersed between unlabeled neuronal cell bodies (Fig.1A). The examination of immunostained sections at the electron microscopic level indicated that, throughout the nucleus accumbens of C57BL/6 mice, mGlu2/3 immunostaining was essentially associated with axonal terminals and glial processes, postsynaptic dendrites and neuronal cell bodies being unstained (Fig. 1B,D). Throughout the nucleus, the mGlu2/3-immunostained glial processes closely surround both labeled and unlabeled synaptic axon terminals (Fig.1C,D). mGlu2/3-immunostained axon terminals represented ∼20% of total axon terminals (identified by their content in synaptic-like vesicles). When identified, these mGlu2/3-immunostained terminals form typical asymmetric synaptic contacts with unlabeled dendrites (Fig. 1B). These results are in agreement with a previous detailed study by Tamaru and colleagues, who have described subcellular localization and the presence of mGlu3 immunostaining in striatal presynaptic terminals of BALB/c mice (Tamaru et al., 2001).
Fig. 1.

Immunolocalization of mGlu2/3 in the nucleus accumbens. A, At the light microscope level, immunostaining is associated with elongated processes (arrows) and dot structures dispersed between unlabeled neuronal cell bodies (N). B–D, At the electron microscope level, immunostaining is essentially associated with axon terminals containing numerous synaptic-like vesicles (ax) and glial processes (gl), whereas dendritic profiles (D) always appear unstained. Note that labeled axon terminals frequently form typical synaptic contacts with unlabeled dendrites (arrowheads in B andC), whereas labeled glial processes frequently enwrap either labeled (C) or unlabeled (D) synaptic axon terminals.Asterisks in B–D indicate the unlabeled axon terminals. Scale bars: A, 25 μm;B–D, 1 μm.
LTD can be induced by specific group 2 mGlu agonist
We decided to evaluate the effects of the direct stimulation of presynaptic mGlu2/3. Conventional extracellular field recording were first chosen because of the non-invasive nature of the technique. fEPSPs were measured in mice NAc core (Manzoni et al., 1997, 1998;Robbe et al., 2001). It was found that application of the highly selective mGlu2/3 agonist LY354740 (Schoepp et al., 1997) (200 nm) for 10 min caused a rapid and strong inhibition of fEPSP, which was maximal at the end of the application of the drug (62.7 ± 3.0% of inhibition; n = 34) (Fig.2A). After washout of the drug, the amplitude of the fEPSP was only partially recovered to a stable response that was depressed at 60 min by 25.7 ± 2.5% relative to baseline value (LTD; n = 34) (Fig.2A). LTD was also triggered by another mGlu2/3 agonist, LCCG1; application of this drug at 10 μm for 5 min induced an initial inhibition of the fEPSP of 67.05 ± 5.56% (n = 8), which was followed by an LTD of 34.79 ± 7.94% (n = 8) at 60 min (data not shown).
Fig. 2.
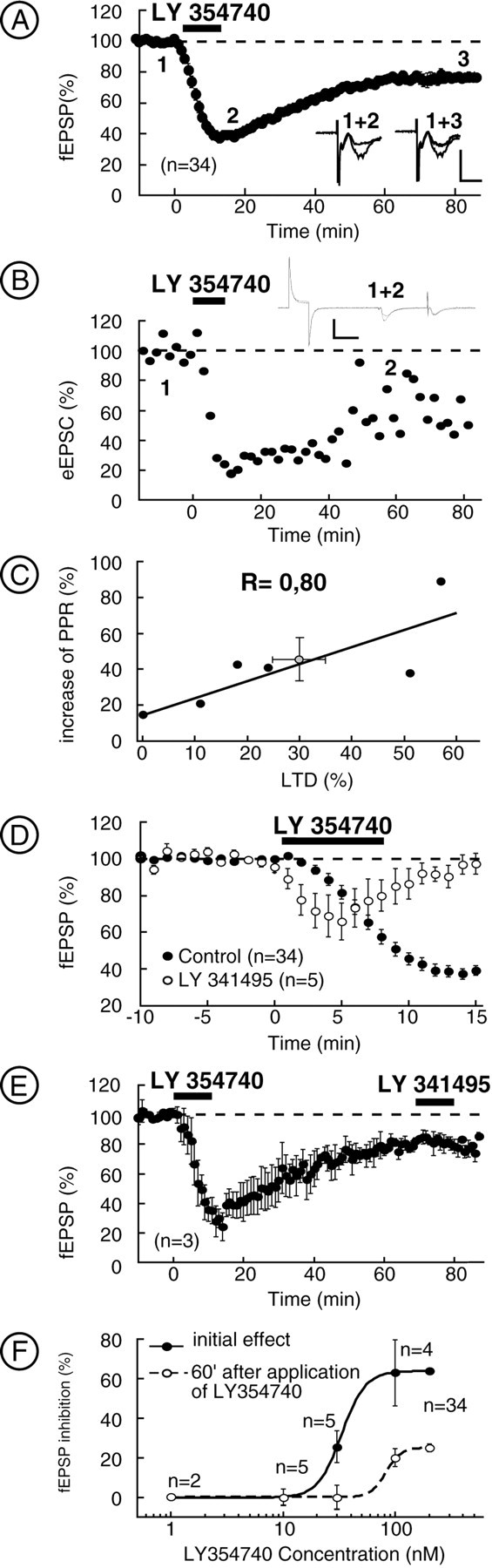
Characterization of pharmacological mGlu2/3-induced LTD. A, Summarized data showing that application of LY354740 (200 nm) resulted in an initial depression, which was followed during washout by an LTD.Inset shows traces (average of 10 consecutive fEPSPs) taken at the points indicated in a typical experiment. Calibration: 0.3 mV, 10 msec. B, Typical whole-cell experiment showing LTD induced by LY354740. Inset shows traces (average of 10 consecutive eEPSCs) taken at points indicated on the graph. Calibration: 100 pA, 25 msec. C, Inhibition of eEPSC 30 min after LY354740 is accompanied by an increase in the paired-pulse ratio. D, In presence of the group 2 mGlu antagonist LY341495 (200 nm), the initial depression induced by LY354740 was reduced and the LTD was prevented. E, Application of LY341495 (200 nm) 60 min after LTD induced by LY354740 did not reverse the LTD. F, Dose–response curves for the LY354740-induced initial inhibition of the fEPSP and for LTD.
To verify that LTD is a synaptic phenomenon and does not result from a decrease of cell excitability, we use patch-clamp recording in the whole-cell configuration. Figure 2B depicts a typical experiment, and it is clear that LTD could be obtained even when the cells were held at −70 mV, without modification of the input resistance; on average, 30 min after LY354740, excitatory EPSCs (EPSCs) measured 72 ± 11% of baseline (n= 6; data not shown). The paired-pulse ratio was increased 30 min after LY354740 by 45 ± 12% (n = 6) (Fig.2C). That increase was significantly correlated with the magnitude of LTD induced by LY354740 (r = 0.80; Spearmann's rank correlation; p < 0.0005) (Fig.2C). Together, these experiments demonstrated that, at the NAc glutamatergic synapses, direct activation of mGlu2/3 induces LTD. Moreover, the accompanying increase of paired-pulse ratio suggested a presynaptic locus of expression.
Pharmacological characterization of the mGlu2/3-LTD
To demonstrate that the effects of group 2 mGlu agonists were attributable to genuine interactions with mGlu2/3 receptors, the following experiments were performed. First, slices were treated with the highly specific mGlu2/3 antagonist LY341495 (Ornstein et al., 1998) (200 nm, 15 min before, during, and 10 min after superfusion of LY354740). In this condition, LY354740 induced a smaller initial depression of the fEPSP than in control medium (34.11 ± 10.11%, n = 5 vs 62.7 ± 3%, n = 34; p < 0.05) (Fig. 2D), consistent with the antagonist actions of LY341495 at mGlu2/3. The acute mGlu2/3 inhibition was completely reversed 10 min after washout of the agonist, and LTD was never observed. A similar result was obtained with eGlu, another selective mGlu2/3 antagonist (Pin et al., 1999): in slices treated with eGlu (200 μm), the initial LY354740 depression of fEPSP was reduced compared with control (32.31 ± 10.13%, n = 5 vs 62.7 ± 3%,n = 34; p < 0.05) and completely reversed after 5 min of washout of LY354740 (data not shown). Accordingly, in slices treated with eGlu (200 μm), the initial LCCG1 depression of fEPSP was reduced compared with control (29.2 ± 13.6%, n = 5 vs 67.05 ± 5.56%, n = 8; p < 0.05) and completely reversed after 5 min of washout of LCCG1 (data not shown). Together, these results clearly show the efficacy of group 2 antagonists (LY341495 and eGlu) at inhibiting the actions of group 2 mGlu agonists. Finally, slices were treated with 10 μm LY341495 to ensure the total blockade of mGlu2/3. In this condition, fEPSP measured 103.73 ± 5.82% of baseline at the end of LY354740 application (n = 4; data not shown) and 111.22 ± 0.88% 60 min after LY354740 application (n = 3; data not shown).
Figure 2E illustrates that superfusion of the mGlu2/3 antagonist LY341495 (200 nm) for 10 min (1 hr after agonist perfusion) did not reverse mGlu2/3-induced LTD. In a similar experiment, superfusion of eGlu (200 μm) for 10 min, 30 min after LCCG1 washout, did not reverse LCCG1-induced LTD (data not shown). These results show that mGlu2/3-induced LTD is not attributable to the incomplete washout of LY354740 or LCCG1. The EC50 values for the initial depression of the fEPSP attributable to LY354740 and LTD were 33.09 ± 0.6 nm and 79.89 ± 8.75 μm, respectively (Fig. 2F). The LCCG1-induced effects were also dose dependent, and the EC50 for the initial depression and the LTD were 1.4 ± 0.8 and 2.0 ± 0.4 μm,respectively (data not shown).
Synaptic activation of mGlu2/3 induces LTD and occludes pharmacological mGlu2/3-LTD
We next tested whether synaptically released glutamate, similar to exogenous mGlu2/3 agonist, can induce mGlu2/3 presynaptic LTD. First, we reproduced the observation of Pennartz and collaborators: in rat NAc slices, high-frequency stimulation of prelimbic cortex–NAc synapses resulted in LTP, LTD, or no change in synaptic efficacy (Pennartz et al., 1993). As summarized Figure3A, tetanic stimulation (three times for 1 sec at 100 Hz, 20 sec intervals) of prelimbic glutamatergic afferents in mice NAc slices (black bars) caused LTD in a majority of the experiments (in 60% of the slices; 15 of 25) (a typical experiment is shown Fig. 3B). In the remaining slices, tetanic stimulation caused either no change (28% of slices; 7 of 25; No Plasticity) or rarely LTP (12% of slices; 3 of 25). There was no correlation between the magnitude of basal fEPSPs and the direction of plasticity (r = 0.210). When all of the slices were averaged, tetanus induced an overall LTD of 14.05 ± 3.61% (n = 25) (Fig. 3C, black circles). Based on the aforementioned results, we reasoned that stimulation of presynaptic mGlu2/3 during intense synaptic activity should induce LTD. Indeed, perfusion of the slices with the mGlu2/3 antagonist LY341495 (200 nm, 15 min) clearly reduced the number of slices expressing LTD in response to tetanic stimulation (12.5% of the slices; one of eight; p < 0.05 vs control Fisher's exact test) (Fig. 3A) but had no effect on basal synaptic transmission (data not shown). Comparing all of the slices revealed that synaptic activation of mGlu2/3 drives the prelimbic cortex–NAc synapses to express LTD (Fig. 3C) [30 min after tetanus, fEPSP measured 85.95 ± 3.67% of baseline in control (n = 25) and 106.01 ± 8.44% of baseline in LY341495-treated slices (n = 8); p< 0.05]. Finally, we confirmed the results of Pennartz et al. and found that tetanic-LTD was independent of NMDA receptors (NMDARs) (60 and 50% of tetanized slices exhibited LTD in control and after 50 μml-APV treatment, respectively; p > 0.05; Fisher's exact test; data not shown).
Occlusion experiments were performed to test whether mGlu2/3 agonist-induced LTD and tetanus-induced LTD shared similar mechanisms. LTD was first induced by perfusion with LY354740, and, 70 min after agonist application, tetanus (three times for 1 sec at 100 Hz, 20 sec intervals) were given. Tetanus-induced LTD was never observed in slices in which mGlu2/3-LTD was already triggered (zero of seven;p < 0.05 vs control; Fisher's exact test), demonstrating the complete occlusion between these two forms of LTD (Fig. 3D, typical experiment, E, average data) [fEPSP at 30 min after measured 85.95 ± 3.67% (n = 25) in control vs 110.73 ± 7.84% (n = 7) after inducing LTD with LY354740;p < 0.05). Occlusion from mGlu2/3-LTD was also observed in slices in which tetanus-induced LTD was induced first. Figure 3F shows a typical experiment in which, after induction of tetanic-LTD (note that tetanic-LTD is already saturated after the first tetanus), LY354740 causes acute inhibition but not LTD. Figure 3G presents average data. LY354740 initial effect is reduced compared with control (46.32 ± 3.29 vs 62.71 ± 3.03%; p < 0.05) and completely reversed 20 min after washout. Together, these experiments showed the existence of common mechanisms between mGlu2/3-LTD and tetanus-induced LTD.
mGlu2/3-LTD is independent of presynaptic activity and intracellular Ca2+ levels
Compared with tetanus-induced LTD, agonist-induced mGlu2/3-LTD offered an easy and reliable assay to study the LTD of prelimbic cortex–NAc and particularly its transduction pathways. Is the simple activation of presynaptic mGlu2/3 sufficient to LTD induction? At excitatory synapses of the hippocampus (Huang et al., 1997, 1999;Tzounopoulos et al., 1998) or the amygdala (Lin et al., 2000), an additional component that depended on presynaptic terminal activity was required to produce LTD. Thus, test stimulation was stopped during perfusion and washout of LY354740 (for a total of 40 min). Figure4A clearly demonstrates that concurrent presynaptic activity was not necessary for the induction of mGlu2/3-LTD [LTD at 60 min measured 22.90 ± 5.91% (n = 6) of baseline vs 25.69 ± 2.24% (n = 34) in control; p > 0.05] (Kahn et al., 2001). To test the importance of intracellular Ca2+ levels (and especially presynaptic Ca2+), intracellular Ca2+ was lowered with BAPTA AM (50 μm). Similar to other studies using membrane-permeant Ca2+ chelators (Tzounopoulos et al., 1998; Casado et al., 2000; Lin et al., 2000), BAPTA AM markedly reduced the fEPSP (45.5 ± 7.2% of baseline in BAPTA AM; n = 4; data not shown), suggesting that this concentration effectively buffered intracellular Ca2+. In marked contrast with studies in which bath perfusion with Ca2+ chelator reduced mGlu2/3-LTD (Tzounopoulos et al., 1998; Lin et al., 2000), in the NAc, the initial depression or the LTD induced by LY354740 are unaltered [LTD at 60 min measured 36.51 ± 11.43% (n = 4) vs 25.69 ± 2.24% (n = 34) in control; p > 0.05; data not shown). Together, these results suggested that activity-dependent changes in intracellular Ca2+ are unlikely to participate in mGlu2/3-LTD.
Fig. 4.

Transduction pathways of mGlu2/3-LTD.A, mGlu2/3-LTD was not blocked in the absence of presynaptic activity. Summary of all of the experiments in which evoked synaptic stimulation was stopped during bath perfusion of LY354740 (200 nm) and 30 min after the washout. B, Summarized data showing that treatment with the adenylate cyclase activator forskolin (10 μm) inhibited the initial depression attributable to LY354740 and completely abolished mGlu-LTD.C, Summary of the effects the PKA inhibitor KT 5720 (1 μm) on basal synaptic transmission. D, Summary of all of the experiments in which the slices were preincubated at least 2 hr with the PKA inhibitor KT 5720 at 1 μm. In this condition, the initial depression of fEPSP by LY354740 was slightly decreased, and LTD at 60 min was totally prevented. E, Summary of the effects the PKA inhibitor H 89 (10 μm) on basal synaptic transmission.F, Summary of the experiments in which slices have been treated 20 min before, during, and 10 min after LY354740 application with the PKA inhibitor H 89 (10 μm). In this condition, the LTD induced by LY354740 was completely prevented at 60 min. *p < 0.05; **p < 0.01.
Role of the cAMP/protein kinase A-signaling cascade in the mGlu2/3-LTD
Because group 2 mGlu are coupled to adenylate cyclase via G-protein of the Gi/o family (Conn and Pin, 1997) and because mGlu2/3-LTD has been shown to be dependent on both group 2 mGlu activation and the cAMP–protein kinase A (PKA) cascade (Tzounopoulos et al., 1998; Huang et al., 1999; Lin et al., 2000) (but see Otani et al., 1999; Kahn et al., 2001), the involvement of the cAMP–PKA cascade in the mGlu2/3-LTD at the NAc synapses was tested. The adenylate cyclase activator forskolin (10 μm) caused a large increase of the NAc fEPSP (150.05 ± 10.24% of basal fEPSP; n = 9; data not shown), demonstrating the sensitivity of these synapses to the elevation of cAMP levels (Manzoni et al., 1998; Robbe et al., 2001). Figure 4B shows that, in slices treated with forskolin (20 min before agonist perfusion and thereafter, 10 μm), the initial depression of fEPSP induced by LY354740 was reduced compared with control [36.95 ± 7% inhibition (n = 3) vs 62.7 ± 3% (n = 34); p < 0.05], and the LTD at 60 min was completely abolished [2.15 ± 1.02% of baseline (n = 3) vs 25.69 ± 2.45% (n = 34) in control; p < 0.05]. Similar results were obtained with the LCCG1-induced LTD that was prevented in slices treated with forskolin [3.2 ± 0.8% of baseline (n = 4) vs 25.69 ± 2.45% (n = 34); p < 0.05; data not shown].
Is protein kinase A involved in the mGlu2/3-LTD? The PKA inhibitor KT 5720 (1 μm) alone depressed basal synaptic transmission (25.5 ± 5.5% inhibition; 60 min; n = 3), as shown in Figure 4C (Tzounopoulos et al., 1998). Accordingly, pretreatment of the slices for at least 2 hr with KT 5720 (1 μm) completely abolished LY354740-induced LTD at 60 min [0.8 ± 13% of baseline (n = 6) vs 25.69 ± 2.49% (n = 34); p < 0.05] (Fig. 4D). Another PKA inhibitor structurally unrelated to KT 5720, H 89 (10 μm, applied 20 min before, during, and 10 min after LY354740) also caused the inhibition of fEPSP (12.8 ± 4.82% of inhibition at 20 min;n = 15) (Fig. 4E) (Lin et al., 2000), mimicking and clearly occluding mGlu2/3-LTD (9.4 ± 6.6%,n = 4 vs 25.69 ± 2.49%, n = 34;p < 0.05) (Fig. 4F). It was also found that the LCCG1-induced LTD was blocked by treatment with H 89 (0.2 ± 8.6%; n = 3; p < 0.05; data not shown). From this set of experiments, it was concluded that activation of mGlu2/3-LTD at the NAc synapses involved the cAMP–PKA cascade. More specifically, the data suggested that mGlu2/3 activation induced LTD via the reduction of PKA activity.
At the mossy fiber synapses, both LTD and LTP are cAMP–PKA mediated (Weisskopf et al., 1993; Huang et al., 1994; Tzounopoulos et al., 1998). In contrast, we found that, in experimental conditions favoring the occurrence of LTP (i.e., in the presence of mGlu2/3 antagonist), the PKA-inhibitor KT 5720 had no effect on the tetanus-induced LTP (LTP was observed in three of eight slices in control and five of eight with KT 5720; p = 0.31; Fisher's exact test; data not shown), suggesting that modulation of the cAMP–PKA cascade is important to presynaptic LTD but not to LTP.
Role of Ca2+ channels in mGlu2/3-LTD
What is the locus of expression of mGlu2/3-LTD? Previous studies have suggested preferential interactions between mGlu2/3 and high-voltage-activated (HVA) Ca2+ channels (Chavis et al., 1994; Glaum and Miller, 1995; Choi and Lovinger, 1996;Stefani et al., 1996). Moreover, the influence of the cAMP–PKA cascade on HVA Ca2+ currents has been clearly identified (Surmeier et al., 1995; Fukuda et al., 1996; Huang et al., 1996; Kaneko et al., 1998). Of particular interest, enhanced PKA activity was shown to potentiate N-, Q-, and P-type HVA Ca2+ currents (Fukuda et al., 1996; Huang et al., 1998). We reasoned that the mGlu2/3-induced reduction of PKA activity could lead to inhibition of presynaptic HVA and reduction of synaptic efficacy. To test this hypothesis, two types of experiments were performed. We estimated the effects of selectively blocking L-, N-, or P/Q-type HVA Ca2+ channels (Manzoni et al., 1997; Robbe et al., 2001) (with 1 μm nimodipine, 1 μm ω-conotoxin-GVIA, or 200 nmω-agatoxin-IVA, respectively) on control synaptic transmission and on the expression of mGlu2/3-LTD. Figure5A shows in a representative experiment that ω-conotoxin-GVIA-sensitive Ca2+ channels (N-type) are responsible for most of the evoked transmission in mice NAc (average fEPSP inhibition measured after 15 min of ω-conotoxin-GVIA was 62.90 ± 3.62%;n = 13) (Fig. 5H) (Robbe et al., 2001). In the presence of ω-conotoxin-GVIA, neither mGlu2/3 initial depression nor LTD was modified (19.49 ± 5.46%,n = 8 vs 25.69 ± 2.49%, n = 34 in control; p > 0.05) (Fig. 5A–D). Strikingly, bath perfusion with ω-agatoxin-IVA to selectively block P/Q-type channels reduced by 29.46 ± 7.35% (n = 5) (for a typical experiment, see Fig.5B,H) synaptic transmission (Robbe et al., 2001) and completely abolished the expression of mGlu2/3-LTD (1.25 ± 4.02%, n = 8 vs 25.69 ± 2.49%, n = 34 in control; p < 0.05) (Fig. 5B–D) but not the initial depression (66.22 ± 5.11%, n = 8 vs 62.7 ± 3%,n = 34 in control; p > 0.05) (for a representative experiment, see Fig.6B). In contrast, perfusing nimodipine to selectively block L-type channels (Robbe et al., 2001) had no effect on mGlu2/3-LTD (19.62 ± 6.3%,n = 4 vs 25.69 ± 2.49%, n = 34 in control; p > 0.05) or initial depression (59.49 ± 5.71%, n = 4 vs 62.7 ± 3%,n = 34 in control; p > 0.05) (Fig.5C,D).
Fig. 5.

mGlu2/3-LTD is attributable to the reduction of P/Q-type Ca2+ channels to evoked synaptic transmission. A–D, The mGlu2/3-LTD requires P/Q-type Ca2+ channels. A, Typical experiment in which the slice was perfused with ω-conotoxin-GVIA (1 μm). The fraction of synaptic transmission insensitive to ω-conotoxin-GVIA displayed a normal form of mGlu2/3-LTD.B, Typical experiment in which the slice was perfused with ω-agatoxin-IVA (200 nm). The fraction of synaptic transmission insensitive to ω-agatoxin-IVA did not display mGlu2/3-LTD. C, Typical experiment in which the slice was perfused with nimodipine (1 μm). This treatment had no effect on the mGlu2/3-LTD. D, Summary of all of the experiments performed as above. The histogram of the mGlu2/3-LTD at 60 min reveals that blocking P/Q-type HVA Ca2+ channels suppressed mGlu2/3-LTD. E–H, P/Q-type Ca2+ channels do not contribute to synaptic transmission when mGlu2/3-LTD is induced. E–G, Typical experiments in which, after induction of mGlu2/3-LTD with LY354740 (200 nm), the slices were perfused with ω-conotoxin-GVIA (1 μm), ω-agatoxin-IVA (200 nm), and nimodipine (1 μm), respectively.H, Summary of all of the experiments performed as above. The histogram represents mean fEPSP inhibition (i.e., fEPSP contribution) induced by ω-conotoxin-GVIA, ω-agatoxin-IVA, and nimodipine and taken after 15–20 min of drug application. It reveals that P/Q-type Ca2+ channels do not contribute to synaptic transmission once mGlu2/3-LTD is induced. Conversely, N-type contribution is augmented. *p < 0.05; **p < 0.01.
Fig. 6.

mGlu2/3-LTD is reduced on Ca2+-independent spontaneous EPSCs.A, B, Spontaneous EPSCs recorded in the absence of TTX express both acute and long-term effects of mGlu2/3 activation. A, Typical experiment. Representative consecutive 1 sec current sweeps from a cell (holding potential of −70 mV) in which sEPSCs were recorded in the absence, during, or 45 min after LY354740 (200 nm). Calibration: 30 pA, 200 msec. The distribution of sEPSC inter-event intervals was altered after bath-perfusion of LY354740. B, Mean frequency of sEPSCs was equally reduced during and 45 min after LY354740 perfusion. **p < 0.01.n.s., Not significant. C,D, Action potential-independent miniature EPSCs recorded in the presence of TTX (300 nm) express acute but reduced long-term effects of mGlu2/3 activation. C, Typical experiment. Representative consecutive 1 sec current sweeps from a cell (holding potential of −70 mV) in which mEPSCs were recorded in the absence, during, or 45 min after LY354740 (200 nm). Calibration: 30 pA, 200 msec. The distribution of mEPSC inter-event intervals was altered during application of the mGlu2/3 agonist but was back to normal after 45 min. The distribution of mEPSC amplitude was unchanged after bath perfusion of LY354740.D, Mean frequency of sEPSCs was significantly more reduced during than 45 min after LY354740 perfusion. *p < 0.05; **p < 0.01.
If mGlu2/3-LTD is expressed via the inhibition of P/Q-type HVA Ca2+ channels, then one expects a modification of the relative participation of these Ca2+ channels to evoked transmission during the plateau phase of mGlu2/3-LTD.
To test this prediction and evaluate the relative contribution of N-, P/Q-, and L-type HVA Ca2+ channels during mGlu2/3-LTD, LTD was first induced by bath application of LY354740 (10 min, 200 nm), and, at the time at which LTD had reached its plateau, selective Ca2+ channels blockers were perfused. Figure 5E–H shows that the ω-conotoxin-GVIA inhibition was significantly larger during mGlu2/3-LTD than in control conditions (83.65 ± 7.65%,n = 4 after LTD vs 62.90 ± 3.62%,n = 13 in control; p < 0.05). Remarkably, ω-agatoxin-IVA, which normally reduced by 29.46 ± 7.35% (n = 5) synaptic transmission, had no effect when mGlu2/3-LTD was already induced (2.25 ± 3.8%;n = 4; p < 0.05) (Fig.5F–H). In contrast, the inhibitory effects of nimodipine were identical during mGlu2/3-LTD and in control conditions (respectively, 20.09 ± 5.84%, n = 3 and 15.28 ± 4.91%, n = 8; p > 0.05), excluding the participation of L-type HVA Ca2+ channels in the expression of LTD (Fig. 5G,H). These experiments reinforced the idea that inhibition of presynaptic P/Q-type Ca2+ channels is the principal mechanisms of expression of mGlu2/3-LTD.
mGlu2/3-LTD is reduced on mEPSCs compared with sEPSCs
If mGlu2/3-LTD is mediated by the inhibition of presynaptic P/Q-type Ca2+ channels, one predicts that action potential-dependent, spontaneously occurring EPSCs (sEPSCs) express both mGlu2/3-induced acute inhibition and LTD, whereas action potential-independent EPSCs (mEPSCs, previously shown to be Ca2+-independent; Robbe et al., 2001) only express acute mGlu2/3-LTD acute inhibition. Figure 6, A andB, shows that LY354740 (200 nm, 10 min) significantly altered the inter-event interval distributions (p < 0.05; n = 9; Kolmogorov–Smirnov test; data not shown) and reduced the mean frequency of sEPSCs during and 45 min after its application: the mean frequency in control was 2.23 ± 0.44 Hz compared with 1.01 ± 0.24 Hz during LY354740 and 1.12 ± 0.27 Hz 45 min after LY354740 (n = 9; p < 0.01). Note that there was no statistical difference between the mean frequency during application of LY354740 and after 45 min washout (p > 0.05; n = 9) (Fig.6B) or between cumulative distributions (p > 0.05; n = 9; data not shown). Amplitudes of sEPSCs were not affected during these experiments: the mean amplitude in control was 17.59 ± 8.84 pA compared with 18.26 ± 1.08 pA 10 min after LY354740 and 16.76 ± 0.96 pA 45 min after washout (n = 9;p > 0.05; paired t test) or their cumulative amplitude distributions (p > 0.05,n = 9, Kolmogorov-Smirnov test, not shown).
In marked contrast, whereas action potential-independent mEPSCs in presence of the voltage-dependent Na+channel blocker TTX (300 nm) were still highly sensitive to the acute effect of LY354740, the expression of mGlu2/3-LTD was clearly reduced. At the end of the 10 min LY354740 application, both the inter-event interval distributions (p < 0.05;n = 8) and the mean frequency of the mEPSCs were modified (mean frequency was 2.17 ± 0.57 Hz in control compared with 0.91 ± 0.28 Hz during LY354740; n = 8;p < 0.01) (Fig. 6C,D). In contrast, Figure 6, C and D, shows that the acute effects of LY354740 are partially reversed 45 min after LY354740 application: the mean frequency 45 min after LY354740 was 1.63 ± 0.52 Hz compared with 0.91 ± 0.28 Hz during LY354740 application (n = 8; p < 0.05; the cumulative distribution was also different, p < 0.05; data not shown). Amplitudes of sEPSCs were not affected during these experiments: the mean amplitude in control was 17.29 ± 0.84 pA, 18.26 ± 1.08 pA during LY354740, and 16.76 ± 0.96 pA 45 min after LY354740 (n = 8; p > 0.05 between the three conditions). Accordingly, the cumulative amplitude distributions were also identical (p > 0.05 between the three conditions; n = 8; data not shown). These results show that TTX can reduce the long-term effect of LY354740.
In addition to confirming the electron microscopy observations showing the presynaptic localization of mGlu2/3 and the whole-cell experiments on paired-pulse ratio, these experiments indicate that different effectors mediate the acute and long-term effects of mGlu2/3.
DISCUSSION
This study describes the mechanisms of mGlu2/3-LTD at the prelimbic cortex–NAc synapses: mGlu2/3 autoreceptors cause a long-lasting inhibition of presynaptic P/Q-type Ca2+ channels, which is responsible for LTD.
Synapses have developed a number of strategies to modulate their gain in response to high-frequency or simply repetitive stimulation. Short-term forms of synaptic plasticity include paired-pulse and frequency-dependent facilitation or depression (Thomson, 2000a,b;Sudhof, 2001). Noteworthy, the central role of both Ca2+ channels and mGlus in these regulations has been well identified (Scanziani et al., 1997; Wu and Saggau, 1997; Anwyl, 1999; Meir et al., 1999; Zucker, 1999; Cartmell and Schoepp, 2000; Parnas et al., 2000). It was not known, however, whether these local, short-living means of controlling synaptic gain were implicated in longer-lasting forms of synaptic plasticity.
Until now, there was no data concerning the final target of presynaptic mGlu2/3-LTD in the NAc or in other brain areas. Although reduction of Ca2+ channels is a classical intermediary of acute receptor-mediated presynaptic inhibition (Miller, 1990; Wu and Saggau, 1997), such a mechanism has never been linked to LTD. Our finding that inhibition of presynaptic P/Q-type Ca2+ channels is the principal mechanisms of expression of mGlu2/3-LTD at the prelimbic cortex–NAc synapses is in accord with the well known modulatory role of the cAMP/PKA cascade on HVA Ca2+ currents (Surmeier et al., 1995; Fukuda et al., 1996; Huang et al., 1996; Kaneko et al., 1998). It will be interesting to determine whether a reduction of presynaptic Ca2+ channel contribution is a general feature of all mGlu2/3-LTD-expressing synapses. Determining factors will certainly include the subtypes of adenylyl cyclase and Ca2+ channels linked to the transmitter release machinery at individual synapses.
This study is reminiscent of a first work of our team on mGlu regulation of glutamatergic transmission in the NAc of rat (Manzoni et al., 1997). Because we focused on acute mGlu-mediated inhibitions, the LCCG1-induced long-term effects were unfortunately overlooked. Using the highly selective mGlu2/3 agonist LY354740, we have since reproduced these results and found that, similar to the mice NAc slices, mGlu2/3-LTD can be induced in rat NAc slices: 1 hr after a 10 min perfusion with 200 nm LY354740, the fEPSP measured 64.92 ± 8.53% of baseline (n = 4) (L. Kahn and O. J. Manzoni, unpublished data).
Synaptic activity has been shown to be negatively regulated by mGlu2/3 at many central synapses of the CNS (Kobayashi et al., 1996;Yokoi et al., 1996; Tzounopoulos et al., 1998; Anwyl, 1999; Huang et al., 1999; Lin et al., 2000). The majority of these studies showed the involvement of the cAMP/PKA cascade in mGlu2/3-LTD (hippocampal mossy fibers, dentate gyrus, and amygdala). Accordingly, we found that both activation of adenylate cyclases with forskolin (Seamon and Daly, 1986) and inhibition of the PKA pathway (with H 89 or KT 5720) abolished the expression of LTD. Interestingly, mGlu2/3 can be negatively (Manzoni et al., 1992; Prézeau et al., 1992) or positively coupled to the adenylate cyclases (Conn and Pin, 1997), and analysis of genetically modified mice have shown that mice with defective cAMP-dependent protein kinases display severe deficit in hippocampal postsynaptic LTP and LTD (Brandon et al., 1995; Qi et al., 1996). Considering our observations that elevation of cAMP levels enhances NAc-fEPSP (present report, Manzoni et al., 1998; Robbe et al., 2001) and that PKA inhibitors alone reduce NAc synaptic transmission (present report;Tzounopoulos et al., 1998; Lin et al., 2000), it was concluded that inhibition of PKA activity is a necessary early step of mGlu2/3-LTD. In all cases, our data show the crucial role of the cAMP–PKA cascade in mGlu2/3-LTD at the prelimbic cortex–NAc synapses.
In conditions favoring the induction of LTP (i.e., in the presence of mGlu2/3 antagonist), it was found that LTP was completely blocked by NMDAR antagonist, confirming previous reports of a postsynaptic NMDAR-dependent LTP (three of eight slices in the presence of LY341495 alone vs zero of six in LY341495 and MK801) (Pennartz et al., 1993;Kombian and Malenka, 1994). Together with the lack of effect of KT 5720 on the tetanus-induced LTP, our study indicates that modulation of the cAMP–PKA cascade is important to presynaptic LTD but not to LTP. Thus, unlike the mossy fiber synapses that exhibit both presynaptic cAMP–PKA-mediated LTD and LTP (Weisskopf et al., 1993; Huang et al., 1994; Tzounopoulos et al., 1998), it is conceivable that prelimbic cortex–NAc synapses are not capable of expressing activity-dependent forms of LTP in response to increased PKA activity. However, considering that we consistently observed synaptic enhancement in response to forskolin application (present study; Manzoni et al., 1998;Robbe et al., 2001), another interpretation is that the experimental conditions (tetanus, external Ca2+ levels, temperature, etc… . ) were inadequate to activate the cAMP/PKA–dependent enhancing cascade.
Extending the pioneer study of Pennartz et al. (1993) who showed that tetanizing prelimbic cortex–NAc synapses induced a mixture of LTP, LTD, or no change in synaptic efficacy, we found that simple antagonism of mGlu2/3 turns “indecisive synapse” into LTP-expressing ones. What can explain the apparent discrepancy with Kombian and Malenka (1994) who reported an overall increase of fEPSPs during tetanic stimulation of prelimbic cortex afferents, whereas we observed an overall LTD of excitatory synapses (Fig. 1)? The choice of species (rat vs mice), the age of the animals, and more importantly the stimulation protocols might be of importance: we repeated the 1 sec 100 Hz tetanus three times, whereas Kombian and Malenka only applied it once. It is possible that a single tetanus, which is unable release sufficient glutamate to activate presynaptic mGlu2/3 receptors and induce presynaptic LTD, still allows the induction of postsynaptic NMDAR-dependent LTP.
Recently, evidences have been provided that glial cells contribute to the regulation of synaptic transmission by controlling glutamate concentration in the extracellular space (Rothstein et al., 1996; Oliet et al., 1997; Bergles and Jahr, 1998). Similar to what we observed in the NAc, the occurrence of mGlu2/3 on the limiting membrane of glial processes enwrapping axon terminals has been described in a number of brain regions (Petralia et al., 1996; Mineff and Valtschanoff, 1999). In the NAc, the functions of glial mGlu2/3 receptors remain to be determined, and it is possible that these receptors contribute to synaptic transmission and plasticity: mGlu2/3 may regulate glial glutamate transporters and affect extracellular glutamate levels. In all cases, the involvement of P/Q-channels in mGlu2/3-LTD indicates a predominant role of neuronal mGlu2/3 in LTD of the prelimbic cortex–NAc synapses.
How are mGlu2/3 and in particular mGlu2/3-LTD involved in the functions of the nucleus accumbens? The therapeutic potentials of mGlu2/3 drugs might provide indications in this regard. It is indeed intriguing that agonists of these receptors reduce withdrawal syndromes from nicotine (Helton et al., 1997) and opiates (Fundytus and Coderre, 1997; Fundytus et al., 1997) have anxiolytic properties (Helton et al., 1998) and can modulate drug-evoked behaviors (Cartmell et al., 1999, 2000a,b). Interestingly, chronic morphine treatment augmented mGlu-induced inhibition of neurotransmission in nucleus accumbens and the ventral tegmental area (Manzoni and Williams, 1999; Martin et al., 1999). Together with the importance of mGlu2/3 in controlling synaptic plasticity at the prelimbic cortex–NAc synapses, these observation reinforce the idea that mGlu2/3 can participate in drug-related behaviors and addiction.
Footnotes
The work in the laboratory of O.J.M. was supported by grants from Mission Interministérielle de Lutte contre la Drogue et la Toxicomanie and Ministère de la Recherche (Les Actions Concertées Incitatives Jeunes Chercheurs). We thank Dr. P. M. Lledo and Dr. P. Castillo for critical reading of this manuscript, M. Passama for the artworks, and Dr. Monn and Dr. Shoepp at Eli Lilly (Indianapolis, IN) for their generous gift of LY-354740.
Correspondence should be addressed to Dr. O. Manzoni, Centre National de la Recherche Scientifique Unité Propre de Recherche 9023, 34094 Montpellier Cedex 5, France. E-mail:manzoni@ccipe.montp.inserm.fr.
REFERENCES
- 1.Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- 2.Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral–commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, Kandel ER, McKnight GS, Idzerda RL. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1995;92:8851–8855. doi: 10.1073/pnas.92.19.8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- 5.Cartmell J, Monn JA, Schoepp DD. The metabotropic glutamate 2/3 receptor agonists LY354740 and LY379268 selectively attenuate phencyclidine versus d-amphetamine motor behaviors in rats. J Pharmacol Exp Ther. 1999;291:161–170. [PubMed] [Google Scholar]
- 6.Cartmell J, Monn JA, Schoepp DD. Attenuation of specific PCP-evoked behaviors by the potent mGlu2/3 receptor agonist, LY379268 and comparison with the atypical antipsychotic, clozapine. Psychopharmacology. 2000a;148:423–429. doi: 10.1007/s002130050072. [DOI] [PubMed] [Google Scholar]
- 7.Cartmell J, Monn JA, Schoepp DD. The mGlu(2/3) receptor agonist LY379268 selectively blocks amphetamine ambulations and rearing. Eur J Pharmacol. 2000b;400:221–224. doi: 10.1016/s0014-2999(00)00423-4. [DOI] [PubMed] [Google Scholar]
- 8.Casado M, Dieudonne S, Ascher P. Presynaptic NMDA receptors at the parallel fiber-Purkinje cell synapse. Proc Natl Acad Sci USA. 2000;97:11593–11597. doi: 10.1073/pnas.200354297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavis P, Shinozaki H, Bockaert J, Fagni L. The metabotropic glutamate receptor types 2/3 inhibit L-type calcium channels via a pertussis toxin-sensitive G-protein in cultured cerebellar granule cells. J Neurosci. 1994;14:7067–7076. doi: 10.1523/JNEUROSCI.14-11-07067.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi S, Lovinger DM. Metabotropic glutamate receptor modulation of voltage-gated Ca2+ channels involves multiple receptor subtypes in cortical neurons. J Neurosci. 1996;16:36–45. doi: 10.1523/JNEUROSCI.16-01-00036.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 12.Fukuda K, Kaneko S, Yada N, Kikuwaka M, Akaike A, Satoh M. Cyclic AMP-dependent modulation of N- and Q-type Ca2+ channels expressed in Xenopus oocytes. Neurosci Lett. 1996;217:13–16. doi: 10.1016/0304-3940(96)13055-x. [DOI] [PubMed] [Google Scholar]
- 13.Fundytus ME, Coderre TJ. Attenuation of precipitated morphine withdrawal symptoms by acute i.c.v. administration of a group II mGluR agonist. Br J Pharmacol. 1997;121:511–514. doi: 10.1038/sj.bjp.0701174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fundytus ME, Ritchie J, Coderre TJ. Attenuation of morphine withdrawal symptoms by subtype-selective metabotropic glutamate receptor antagonists. Br J Pharmacol. 1997;120:1015–1020. doi: 10.1038/sj.bjp.0701000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glaum SR, Miller RJ. Presynaptic metabotropic glutamate receptors modulate omega-conotoxin-GVIA-insensitive calcium channels in the rat medulla. Neuropharmacology. 1995;34:953–964. doi: 10.1016/0028-3908(95)00076-i. [DOI] [PubMed] [Google Scholar]
- 16.Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ. LY354740: a metabotropic glutamate receptor agonist which ameliorates symptoms of nicotine withdrawal in rats. Neuropharmacology. 1997;36:1511–1516. doi: 10.1016/s0028-3908(97)00170-6. [DOI] [PubMed] [Google Scholar]
- 17.Helton DR, Tizzano JP, Monn JA, Schoepp DD, Kallman MJ. Anxiolytic and side-effect profile of LY354740: a potent, highly selective, orally active agonist for group II metabotropic glutamate receptors. J Pharmacol Exp Ther. 1998;284:651–660. [PubMed] [Google Scholar]
- 18.Horne AL, Woodruff GN, Kemp JA. Synaptic potentials mediated by excitatory amino acid receptors in the nucleus accumbens of the rat, in vitro. Neuropharmacology. 1990;29:917–921. doi: 10.1016/0028-3908(90)90142-e. [DOI] [PubMed] [Google Scholar]
- 19.Huang CC, Hsu KS, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang CC, Wang SJ, Gean PW. Selective enhancement of P-type calcium currents by isoproterenol in the rat amygdala. J Neurosci. 1998;18:2276–2282. doi: 10.1523/JNEUROSCI.18-06-02276.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang L, Killbride J, Rowan MJ, Anwyl R. Activation of mGluRII induces LTD via activation of protein kinase A and protein kinase C in the dentate gyrus of the hippocampus in vitro. Neuropharmacology. 1999;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 22.Huang LQ, Rowan MJ, Anwyl R. mGluR II agonist inhibition of LTP induction, and mGluR II antagonist inhibition of LTD induction, in the dentate gyrus in vitro. NeuroReport. 1997;8:687–693. doi: 10.1097/00001756-199702100-00022. [DOI] [PubMed] [Google Scholar]
- 23.Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 24.Kahn L, Alonso G, Robbe D, Bockaert J, Manzoni OJ. Group 2 metabotropic glutamate receptors induced long-term depression in mouse striatal slices. Neurosci Lett. 2001;316:178–183. doi: 10.1016/s0304-3940(01)02397-7. [DOI] [PubMed] [Google Scholar]
- 25.Kaneko S, Akaike A, Satoh M. Differential regulation of N- and Q-type Ca2+ channels by cyclic nucleotides and G-proteins. Life Sci. 1998;62:1543–1547. doi: 10.1016/s0024-3205(98)00104-0. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi K, Manabe T, Takahashi T. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science. 1996;273:648–650. doi: 10.1126/science.273.5275.648. [DOI] [PubMed] [Google Scholar]
- 27.Kombian SB, Malenka RC. Simultaneous LTP of non-NMDA and LTD of NMDA-receptor-mediated responses in the nucleus accumbens. Nature. 1994;368:242–246. doi: 10.1038/368242a0. [DOI] [PubMed] [Google Scholar]
- 28.Lin HC, Wang SJ, Luo MZ, Gean PW. Activation of group II metabotropic glutamate receptors induces long-term depression of synaptic transmission in the rat amygdala. J Neurosci. 2000;20:9017–9024. doi: 10.1523/JNEUROSCI.20-24-09017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manzoni O, Prezeau L, Sladeczek F, Bockaert J. Trans-ACPD inhibits cAMP formation via a pertussis toxin-sensitive G-protein. Eur J Pharmacol. 1992;225:357–358. doi: 10.1016/0922-4106(92)90112-9. [DOI] [PubMed] [Google Scholar]
- 30.Manzoni O, Michel JM, Bockaert J. Metabotropic glutamate receptors in the rat nucleus accumbens. Eur J Neurosci. 1997;9:1514–1523. doi: 10.1111/j.1460-9568.1997.tb01506.x. [DOI] [PubMed] [Google Scholar]
- 31.Manzoni O, Pujalte D, Williams J, Bockaert J. Decreased presynaptic sensitivity to adenosine after cocaine withdrawal. J Neurosci. 1998;18:7996–8002. doi: 10.1523/JNEUROSCI.18-19-07996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin G, Przewlocki R, Siggins GR. Chronic morphine treatment selectively augments metabotropic glutamate receptor-induced inhibition of N-methyl-d-aspartate receptor-mediated neurotransmission in nucleus accumbens. J Pharmacol Exp Ther. 1999;288:30–35. [PubMed] [Google Scholar]
- 34.Meir A, Ginsburg S, Butkevich A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimoff R. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev. 1999;79:1019–1088. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- 35.Miller RJ. Receptor-mediated regulation of calcium channels and neurotransmitter release. FASEB J. 1990;4:3291–3299. [PubMed] [Google Scholar]
- 36.Mineff E, Valtschanoff J. Metabotropic glutamate receptors 2 and 3 expressed by astrocytes in rat ventrobasal thalamus. Neurosci Lett. 1999;270:95–98. doi: 10.1016/s0304-3940(99)00484-x. [DOI] [PubMed] [Google Scholar]
- 37.Ohishi H, Shigemoto R, Nakanishi S, Mizumo N. Distribution of the mRNA for a metabotropic glutamate receptors, mGluR2 in the central nervous system of the rat. Neuroscience. 1993a;53:1009–1018. doi: 10.1016/0306-4522(93)90485-x. [DOI] [PubMed] [Google Scholar]
- 38.Ohishi H, Shigemoto R, Nakanishi S, Mizumo N. Distribution of the mRNA for a metabotropic glutamate receptors, mGluR3 in the rat brain: an in situ hybridization study. J Comp Neurol. 1993b;335:252–266. doi: 10.1002/cne.903350209. [DOI] [PubMed] [Google Scholar]
- 39.Ohishi H, Neki A, Mizumo N. Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci Res. 1998;30:65–82. doi: 10.1016/s0168-0102(97)00120-x. [DOI] [PubMed] [Google Scholar]
- 40.Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 41.Ornstein PL, Arnold MB, Bleisch TJ, Wright RA, Wheeler WJ, Schoepp DD. [3H]LY341495, a highly potent, selective and novel radioligand for labeling Group II metabotropic glutamate receptors. Bioorg Med Chem Lett. 1998;8:1919–1922. doi: 10.1016/s0960-894x(98)00329-1. [DOI] [PubMed] [Google Scholar]
- 42.Otani S, Auclair N, Desce JM, Roisin MP, Crepel F. Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci. 1999;19:9788–9802. doi: 10.1523/JNEUROSCI.19-22-09788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parnas H, Segel L, Dudel J, Parnas I. Autoreceptors, membrane potential and the regulation of transmitter release. Trends Neurosci. 2000;23:60–68. doi: 10.1016/s0166-2236(99)01498-8. [DOI] [PubMed] [Google Scholar]
- 44.Paxinos G, Franklin KBJ. The mouse brain atlas in stereotaxic coordinates. Academic; San Diego: 2001. [Google Scholar]
- 45.Pennartz CMA, Ameerun RF, Groenewegen HJ, Lopes da Silva FH. Synaptic plasticity in an vitro slice preparation of the rat nucleus accumbens. Eur J Neurosci. 1993;5:107–117. doi: 10.1111/j.1460-9568.1993.tb00475.x. [DOI] [PubMed] [Google Scholar]
- 46.Pennartz CMA, Groenewegen HJ, Lopes da Silva FH. The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioral, electrophysiological and anatomical data. Prog Neurobiol. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 47.Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- 48.Pin JP, De Colle C, Bessis AS, Acher F. New perspectives for the development of selective metabotropic glutamate receptor ligands. Eur J Pharmacol. 1999;375:277–294. doi: 10.1016/s0014-2999(99)00258-7. [DOI] [PubMed] [Google Scholar]
- 49.Prézeau L, Manzoni O, Homburger V, Sladeczeck F, Curry K, Bockaert J. Characterization of a metabotropic glutamate receptor: direct negative coupling to adenylate cyclase and involvement of a pertussis toxin-sensitive G protein. Proc Natl Acad Sci USA. 1992;89:8040–8044. doi: 10.1073/pnas.89.17.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL. Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1996;93:1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 53.Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- 54.Schacter GB, Yang CR, Innis NK, Mogenson GJ. The role of hippocampal-nucleus accumbens pathway in radial-arm maze performance. Brain Res. 1989;494:339–349. doi: 10.1016/0006-8993(89)90602-1. [DOI] [PubMed] [Google Scholar]
- 55.Schoepp DD, Johnson BG, Wright RA, Salhoff CR, Mayne NG, Wu S, Cockerman SL, Burnett JP, Belegaje R, Bleakman D, Monn JA. LY354740 is a potent and highly selective group II metabotropic glutamate receptor agonist in cells expressing human glutamate receptors. Neuropharmacology. 1997;36:1–11. doi: 10.1016/s0028-3908(96)00160-8. [DOI] [PubMed] [Google Scholar]
- 56.Seamon KB, Daly JW. Forskolin: its biological and chemical properties. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1986;20:1–150. [PubMed] [Google Scholar]
- 57.Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizumo N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- 58.Stefani A, Pisani A, Mercuri NB, Calabresi P. The modulation of calcium currents by the activation of mGluRs. Functional implications. Mol Neurobiol. 1996;13:81–95. doi: 10.1007/BF02740753. [DOI] [PubMed] [Google Scholar]
- 59.Sudhof TC. The synaptic vesicle cycle revisited. Neuron. 2001;28:317–320. doi: 10.1016/s0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 60.Surmeier DJ, Bargas J, Hemmings HC, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 61.Tamaru Y, Nomura N, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGlu3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503. doi: 10.1016/s0306-4522(01)00305-0. [DOI] [PubMed] [Google Scholar]
- 62.Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000a;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- 63.Thomson AM. Molecular frequency filters at central synapses. Prog Neurobiol. 2000b;62:159–196. doi: 10.1016/s0301-0082(00)00008-3. [DOI] [PubMed] [Google Scholar]
- 64.Tzounopoulos T, Janz R, Sudhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- 65.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1993;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 66.Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 67.Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakaguchi I, Katsuura G, Shigemoto R, Ohishi H, Nomura S, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science. 1996;273:645–647. doi: 10.1126/science.273.5275.645. [DOI] [PubMed] [Google Scholar]
- 68.Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]