

Abstract
c-Jun is considered a major regulator of both neuronal death and regeneration. Stress in primary cultured CNS neurons induces phosphorylation of c-Jun serines 63 and 73 and increased c-Jun protein. However, total c-Jun N-terminal protein kinase (JNK) activity does not increase, and no satisfactory explanation for this paradox has been available. Here we demonstrate that neuronal stress induces strong activation of JNK2/3 in the presence of constitutively and highly active JNK1. Correspondingly, neurons from JNK1−/−mice show lower constitutive activity and considerably higher responsiveness to stress. p38 activity can be completely inhibited without effect on c-Jun phosphorylation, whereas 10 μmSB203580 strongly inhibits neuronal JNK2/3, stress-induced c-Jun phosphorylation, induced c-Jun activity, and neuronal death in response to trophic withdrawal stress. Neither constitutive JNK1 activity nor total neuronal JNK activity were significantly affected by this concentration of drug. Thus, neuronal stress selectively activates JNK2/3 in the presence of mechanisms maintaining constitutive JNK1 activity, and this JNK2/3 activity selectively targets c-Jun, which is isolated from constitutive JNK1 activity.
Keywords: JNK, isoforms, c-Jun, stress, cerebellar granule neurons, p38
The transcription factor c-Jun is believed to be a critical mediator of the responses of neurons to stress, being implicated in both survival of axotomized dorsal root ganglion neurons and death of other neurons under a variety of circumstances (Herdegen et al., 1997). Activity of c-Jun is potently regulated by phosphorylation of its N-terminal region, and in cell lines this phosphorylation is mediated by the c-Jun N-terminal kinase (JNK) family implicated in a wide range of diseases (Kyriakis et al., 1994; Kyriakis and Avruch, 2001). At least 10 isoforms of JNK are expressed from three genes. These exhibit differences in specificity toward substrates and binding proteins (Gupta et al., 1996). Knock-outs revealed distinct functions of the different gene products (Yang et al., 1997; Kuan et al., 1999), yet evidence for selective activation of endogenous JNKs is absent. A recent investigation concluded that tissue differences in JNK isoform activation merely correspond to the relative abundance of the isoforms (Finch et al., 2001). The mechanisms regulating c-Jun are unknown in differentiating CNS neurons, because total JNK activity does not correlate with c-Jun regulation (Watson et al., 1998; Coffey et al., 2000). This has led to some confusion over the identity of signal pathways regulating c-Jun, the phosphorylation of which is reportedly required for apoptosis in cerebellar granule neurons, a widely used model for studies of neuronal death and development. The only explanation reported is the recent proposal that p38 is the c-Jun kinase in these cells (Yamagishi et al., 2001), yet p38 isoforms are very poor kinases of c-Jun (Goedert et al., 1997), and a previous report detected no p38 activation under these conditions (Watson et al., 1998).
Here we investigate mechanisms regulating c-Jun in response to different neuronal stresses. We demonstrate an induction in specific phosphorylation of c-Jun, indicating a genuine change in c-Jun kinase-phosphatase balance. We had previously reported that functionally distinct pools of JNK coexist in cerebellar granule neurons, including a developmentally regulated pool of specific activity considerably greater than JNK from cell lines. Thus, when total JNK activity is measured, this active pool masks a minor stress-responsive pool that has preferential access to c-Jun substrate in the nucleus (Coffey et al., 2000). Here we explore the possibility that a specific JNK isoform could explain stress-induced c-Jun regulation that precedes neuronal death. We find that JNK1 shows elevated constitutive activity yet is localized to cytoplasmic structures. JNK2/3 is diffuse and inactive in resting neurons, but is strongly activated by stress. Furthermore, the drug SB203580 at 10 μm blocks most of the neuronal JNK2/3 activity, in vivo c-Jun phosphorylation and transcriptional activity, and neuronal death, but not JNK1 activity. Importantly, 1 μm is not sufficient for any of these effects, although 1 μm SB203580 blocks p38 activityin vivo. Finally we demonstrate using a c-Jun promoter reporter assay that JNK also contributes to withdrawal stress-induced c-Jun promoter activity. In conclusion, neuronal stress selectively activates JNK2/3 isoforms, which have preferential access to c-Jun despite the presence of constitutive JNK1 activity.
MATERIALS AND METHODS
Molecular cloning and plasmid construction. Coding sequences for the MAPK kinase kinase 1 (MEKK1) kinase domain (amino acids 1174–1493, “MEKK1Δ”) and full-length p38δ were obtained by PCR from rat cerebellar granule neuron cDNA. The 10 green fluorescent protein (GFP)-JNK isoforms and pEGFP-MEKK1Δ were prepared as described for pEBG-JNK1α1 (Coffey et al., 2000), except that pEGFP-C1 was used for GFP fusions. The rat p38δ sequence was inserted into pcDNA3 after subcloning into pGEM-Teasy. pRL-EF1 was prepared by removing the cytomegalovirus (CMV) promoter from pRL-CMV and replacing it with EF1 promoter, obtained by PCR from pEBG plasmid. pEGFP-C1, pEFGP-F, and pDsRed-C1 were obtained from Clontech (Palo Alto, CA), pcDNA3 was obtained from Invitrogen (San Diego, CA), and pGEM-Teasy, pRL-CMV, and pRL-SV40 were obtained from Promega (Madison, WI). pSG424-c-Jun (6–89), pSG424-ATF2(1–109), pcDNA3-MKK6E, and pcDNA3- JNK interacting protein (JIP)-JBD were generous gifts of Martin Dickens (University of Leicester, Leicester, UK), and pMT108, 111, 131 and 161 were generous gifts of Dirk Bohmann (European Molecular Biology Laboratory, Heidelberg, Germany). These were used to prepare pcDNA3-GAL4-c-Jun(6–89), -ATF2(1–109), and -c-Jun(5–105) wt, S63/73A, T91/93A and 58, 62, 63, 73, 89, 91, 93A by PCR-based methods. pEBG-p38α, Tam-67, and pCMV were kind gifts of Bruce Mayer (University of Connecticut Health Center School of Medicine, Farmington, CT), Michael Birrer (National Institutes of Health, National Cancer Institute, Bethesda, MD), and Sander van den Heuvel (MGH, Boston, MA), respectively.
Cell culture and staining. Primary cultures of cerebellar granule neurons and cell lines were prepared and maintained as described previously (Courtney et al., 1997). Neurons were cultured to maturity (8–13 d in vitro) before use. Immunofluorescent staining was performed as previously described (Coffey et al., 2000). For cell death assay, cells treated as described were stained with 4 μg/ml Hoechst 33342, fixed, and scored on the basis of nuclear morphology, pyknotic nuclei being taken to indicate cell death. Neurons were obtained from rat in all experiments except in Figure 3, in which neurons from wild-type and JNK1−/− mouse were compared.
Fig. 3.

Regulation of total JNK revealed in neurons from JNK1−/− mouse. Cerebellar granule neurons were prepared from JNK1−/− or wild-type mice and treated as in Figure 1, trophic support being withdrawn for 0–240 min, as indicated above the corresponding lanes. Phospho-JNK, total JNK, and c-Jun, detected by immunoblotting, are indicated by the arrows. An increase in phospho-JNK level was observed in JNK1−/−lysates. Mean values of specific JNK activation (phosphoJNK/JNK quantitated from blots), normalized to the initial levels, are indicated below the corresponding phospho-JNK lanes. Data shown is representative of two or three replicates (wild-types) and three replicates (JNK1−/−).
SDS-PAGE, immunoblotting and quantification. Cells were stimulated as indicated, washed in ice-cold PBS, and lysed with 1× Laemmli sample buffer [62.5 mm Tris-HCl, pH 6.8, 1% SDS (w/v), 5% 2-mercaptoethanol, 10% glycerol (v/v), and 0.001% bromophenol blue (w/v)]. Samples were resolved by 10% SDS-PAGE and transferred by semidry transfer onto nitrocellulose. Nitrocellulose was blocked with 5% milk in either TBS–Tween 20 (0.1%) or PBS–Tween 20 (0.05%). Antibodies used were as follows: mouse anti-c-Jun (Transduction Laboratories, Lexington, KY; 0.5 μg/ml), mouse anti-phosphoserine63 c-Jun (Santa Cruz Biotechnology, Santa Cruz, CA; 1 μg/ml), rabbit anti-phosphoserine73 c-Jun, rabbit anti-phospho-p38, rabbit anti-phospho-JNK (New England Biolabs, Beverly, MA: 1:1000 in each case), rabbit anti-phospho- extracellular signal-regulated kinase (ERK) (Promega; 1:20,000), mouse anti-JNK1 (PharMingen, San Diego, CA; G151–333, 0.5 μg/ml), rabbit anti-JNK2/3 (“SAPK1a”; Upstate Biotechnology, Lake Placid, NY; 14–258, 0.2 μg/ml), rabbit pan-JNK antibody (“JNK3”; Upstate Biotechnology; 1 μg/ml), mouse anti-pan-ERK and pan-p38 (“pan-ERK” and “p38”; Transduction Laboratories, 1:5000 in each case), mouse anti-GFP (Clontech; 0.2 μg/ml), and mouse anti-flag (M2; Sigma, St. Louis, MO; 0.3 μg/ml). Blots were developed using the enhanced chemiluminescence detection method. Films were preflashed with a “sensitize” preflash unit according to manufacturer's instructions (Amersham Biosciences, Arlington Heights, IL) so that signals were recorded on the film above the nonlinear response range. Multiple exposures were taken at different times, so that nonsaturated ECL film (Amersham) was used for quantitation. In cases in which dynamic range of the film was insufficient, fainter bands were quantitated from longer exposures and more intense bands from shorter exposures. In all cases there was at least one band that could be quantitated on at least two different films, and greater overlap permitted verification of the quantification. This allowed calculation of the responses as a percentage of control. Films were digitized by densitometry with a flatbed transparency scanner and quantified by image analysis software developed by the authors. Although great care was taken to ensure accurate quantitation of immunoreactivity, it is acknowledged that there are many potential sources of error, such as differences in antibody affinity. However, the ratios of phosphoERK1:phosphoERK2, phosphoJun63:cJun, and phosphoJun73:cJun were found to be constant for much of the time courses (0–120′, 60′-240′ and 120′-240′, respectively) in Figure 1, despite large changes in the levels of each. This strongly suggests that errors in quantitation are minimal.
Fig. 1.

Withdrawal of trophic support rapidly induces c-Jun phosphorylation without corresponding activation of total JNK.A, Cerebellar granule neurons were deprived of trophic support by incubating the cells with medium free of serum and without additional KCl for the times indicated, and lysates were immunoblotted for total c-Jun, phosphoserine 63 c-Jun (“63P”), phosphoserine 73 c-Jun (“73P”), activated, dually phosphorylated JNK (“P-JNK”) and ERK (“P-ERK”), pan-ERK, and pan-JNK, as indicated by thearrows. Representative data are shown. Thetopmost band detected by anti-phosphoserine 63 c-Jun is nonspecific; it is constitutively present and does not co-migrate with c-Jun immunoreactivity. Arrowheads on theright of the blots indicate molecular mass marker (42 kDa). B, Top panel, Blots from replicate experiments as shown in A were scanned, and intensities of bands corresponding to those indicated by the arrows to theleft of the blots were quantitated from unsaturated films from replicate experiments, normalized to the maximum values, and scaled to percentage of initial (control) values. Multiple exposures were taken with preflashed films to avoid nonlinear response, saturated, or undetectably faint bands that may occur in any individual exposure. Means ± SEM are shown (n = 3–6). Values significantly different from 0′ control levels (pairedt test; p < 0.05 or better) are indicated by * (for c-Jun), # (c-Jun73P) or † (c-Jun63P).Bottom panel, The data were normalized to the levels of c-Jun at each time point, demonstrating that regulation of specific c-Jun phosphorylation rises to a plateau (the 60′ JunP73 peak being not significantly above the plateau) before total c-Jun levels change.C, P-JNK and P-ERK levels were quantitated as inB. P-JNK isoforms are resolved as top (54 kDa) and bottom (46 kDa) bands, and ERK1 and 2 are resolved as top (44 kDa) and bottom bands (42 kDa), respectively, as indicated by the arrows. The P-JNK exposure shown is a long exposure saturated in the 46 kDa bands to demonstrate the slight regulation of the fainter 54 kDa bands. Values significantly different from 0′ control levels (pairedt test; p < 0.05 or better) are indicated by *. D, Cerebellar granule neurons were deprived of trophic support for the times indicated above the figure as in A, but in the presence of 50 μm of the selective MEK1/2 inhibitor PD98059 to prevent ERK activation, added one hour before withdrawal of trophic support in each case. No inhibitor was added to the control (0′). Lysates were immunoblotted with antibodies recognizing c-Jun and the activated dually phosphorylated form of ERK.
Preparation of recombinant proteins. Recombinant c-Jun and ATF2 substrates were prepared as previously described (Courtney and Coffey, 1999; Coffey et al., 2000) from plasmids pGEX-Jun(5–89) and ATF2(1–109), generous gifts of Jim Woodgett (Ontario Cancer Institute, Toronto, Canada) and Roger Davis (University of Massachusetts, Worcester, MA), respectively. Recombinant activated and flag-tagged murine p38α, αAF, β, γ, and δ were prepared by transfection of COS-7 cells with pcDNA-flag-p38 plasmids, kind gifts of Jiahuai Han (The Scripps Research Institute, La Jolla, CA) (Zhao et al., 1999), followed by stimulation with 10 μg/ml anisomycin for 30 min.
Protein kinase assays. Cerebellar granule neurons were stimulated as described, washed twice with PBS, and lysed in 500 μl of lysis buffer (20 mm HEPES, pH 7.4, 2 mm EGTA, 50 mmβ-glycerophosphate, 1 mm DTT, 1 mm Na3VO4, 1% Triton X-100, 10% glycerol, 50 mm NaF, 1 mm benzamidine, 1 μg/ml aprotonin, leupeptin, and pepstatin, and 100 μg/ml PMSF). Homogenized and precleared supernatants for immune-complex kinase assay were incubated with antibodies specific for JNK1 (G151–333; PharMingen) or JNK2/3 (14–258; Upstate Biotechnology) followed by protein G and protein A Sepharose, respectively. Immobilized kinases were washed as previously described (Coffey et al., 2000). Active recombinant JNKs (see above) were sequestered from COS-7 cell lysates using S-hexylglutathione agarose. Kinase activity toward GST-cJun(5–89) was measured as previously described (Coffey et al., 2000), with the addition of SB203580 as indicated. For in-gel kinase assay, cleared lysates were incubated at 4°C for 3 hr withS-hexylglutathione agarose-conjugated GST-c-Jun(5–89). Beads were washed as above, lysed in Laemmli sample buffer, and run on 10% SDS-PAGE in which 250 μg/ml GST-c-Jun(5–89) or GST alone had been polymerized. After electrophoresis, gels were washed with 20% 2-propanol in 50 mm Tris-HCl, pH 8.0, for 1 hr at room temperature followed by 1 hr in 100 ml of buffer A (50 mm Tris-HCl, pH 8.0, 0.5 mmDTT) with continuous agitation. Proteins were denatured for 1 hr in 6m guanidine-HCl in buffer A and renatured by incubation for 16 hr at 4°C in buffer A containing 0.04% Tween 40 with four changes of buffer. For the kinase assay, the gel was preincubated in kinase buffer (in mm: 40HEPES-HCl, pH 8.0, 2 DTT, 0.1 EGTA, and 5 Mg acetate) at room temperature with agitation and then changed to kinase buffer supplemented with 50 μCi 32P-γ-ATP and 50 μm ATP and incubated for 1 hr. The gel was then washed six times with 5% (w/v) TCA and 1% sodium pyrophosphate, dried, and c-Jun-associated kinase activity was analyzed by autoradiography.
RT-PCR. RNA was isolated from stimulated cerebellar granule neurons, and c-jun and β-actin messages were detected by RT-PCR, by procedures and with controls as previously described (Coffey et al., 2000).
Reporter gene expression. GAL4-fusion protein activity was measured as previously described (Coffey et al., 2000). Thec-jun promoter reporter construct used was JC6-luc, a generous gift of Ron Prywes (Columbia University, New York, NY). Firefly luciferase responses were normalized using the dual luciferase assay (Promega) using renilla luciferase plasmids pRL-CMV, pRL-SV40, or pRL-EF1 with similar results. The GAL4 reporters used had promoters SV40 (pSG424) or CMV (pcDNA3), and the use of different promoters did not affect the responses to withdrawal stress.
RESULTS
Withdrawal of trophic support induces rapid c-Jun phosphorylation followed by gradual increase in total c-Jun protein
The transcription factor c-Jun is a major stress-activated protein in neuronal cells and is likely to coordinate transcriptional programs in response to stress. Induction of c-Jun is believed to mediate neuronal death in a variety of circumstances (Yang et al., 1997;Behrens et al., 1999; Crocker et al., 2001). Cultured cerebellar granule neurons can be maintained in culture under depolarizing conditions in the presence of serum (Thangnipon et al., 1983); withdrawal of this trophic support is reported to increase both phospho-Jun and total c-Jun levels. Although JNK is generally considered the kinase that phosphorylates c-Jun, JNK activity is reported not to change under these conditions (Watson et al., 1998). Therefore, we first examined if the phospho-c-Jun:c-Jun ratio changes or if phospho-Jun increases merely follow the increase in total protein.
Immunoblotting reveals rapid changes in c-Jun protein mobility within 15′ of withdrawal of trophic support (Fig. 1A, top panel), and up to three or four immunoreactive bands are detected. Such retarded mobility is consistent with increased phosphorylation (Papavassiliou et al., 1995; Ui et al., 1998), and parallel detection of phospho-c-Jun species confirms this (Fig.1A, second and third panels). Once again, up to three or four bands are detected for each phospho-antibody, because the precise mobility depends on phosphorylation of at least Ser63, Ser73, Thr91, and Thr93, each of which occurs in response to stress. Thus, the large mobility shifts absolutely require phosphorylation of Ser63 and Ser73, but the actual amount of shift depends on multiple sites (Ui et al., 1998). The changes in phosphorylation and mobility progress with time and clearly precede the increase in total c-Jun protein that begins after 60′-120′ (Fig.1A,B). The ratio of phospho-c-Jun forms to total c-Jun levels rise to a plateau at 60′, and then c-Jun protein level begins to rise (Fig. 1C). This indicates either a specific increase in phosphorylation or decrease in dephosphorylation of c-Jun during this early phase of withdrawal-evoked signaling.
JNK and ERK are the best characterized kinases for c-Jun Ser-63/73, although JNK is considerably more active in phosphorylating the c-Jun transactivating domain than ERK (Pulverer et al., 1991; Kyriakis et al. 1994). ERK and JNK activation was investigated in the same samples using phospho-specific “anti-active” antibodies. Changes in JNK activation were barely detectable, but a rapid peak of ERK activation followed by inhibition of this kinase is detected (Fig.1A, bottom two panels, and quantitated data shown inC). The possibility that the rapid ERK activation contributed to c-Jun phosphorylation was investigated by repeating the experiment in the presence of the ERK pathway inhibitor PD98059 (Alessi et al., 1995). The ERK activation was eliminated, but c-Jun was still phosphorylated and accumulated (Fig. 1). Thus, neither ERK activation nor total JNK activity can explain the dramatic c-Jun response.
JNK2/3 isoforms are selectively activated by withdrawal of trophic support from cerebellar neurons in the continued presence of high JNK1 activity
We reported that stress in cerebellar neurons regulates a minor pool of JNK with preferential access to c-Jun. Regulation of this pool is not easily detected in whole-cell lysates because of a constitutively active differentiation-associated pool of JNK (Coffey et al., 2000). We previously speculated that these different pools correspond to different JNK isoforms, which may explain why total JNK activity correlates poorly with c-Jun regulation after withdrawal of trophic support (Fig. 1). We identified isoform-specific JNK antibodies by constructing expression plasmids for the 10 known JNK isoforms, expressing them in COS-7 cells, and screening commercial, putative JNK isoform-specific antibodies with these lysates (Fig.2A) (JNK1α2 and 2β1 isoforms not shown). JNK1- and JNK2/3-specific antibodies (PharMingen monoclonal G151–333 and Upstate Biotechnology polyclonal 14–258) were identified as specific for the four JNK1 and the six JNK2/3 splice variants, respectively. These JNK1 and JNK2/3 antibodies also showed specificity by immunoprecipitation (data not shown). “JNK3” antibodies tested recognized all isoforms to a similar extent and were subsequently used as panJNK antibodies.
Fig. 2.
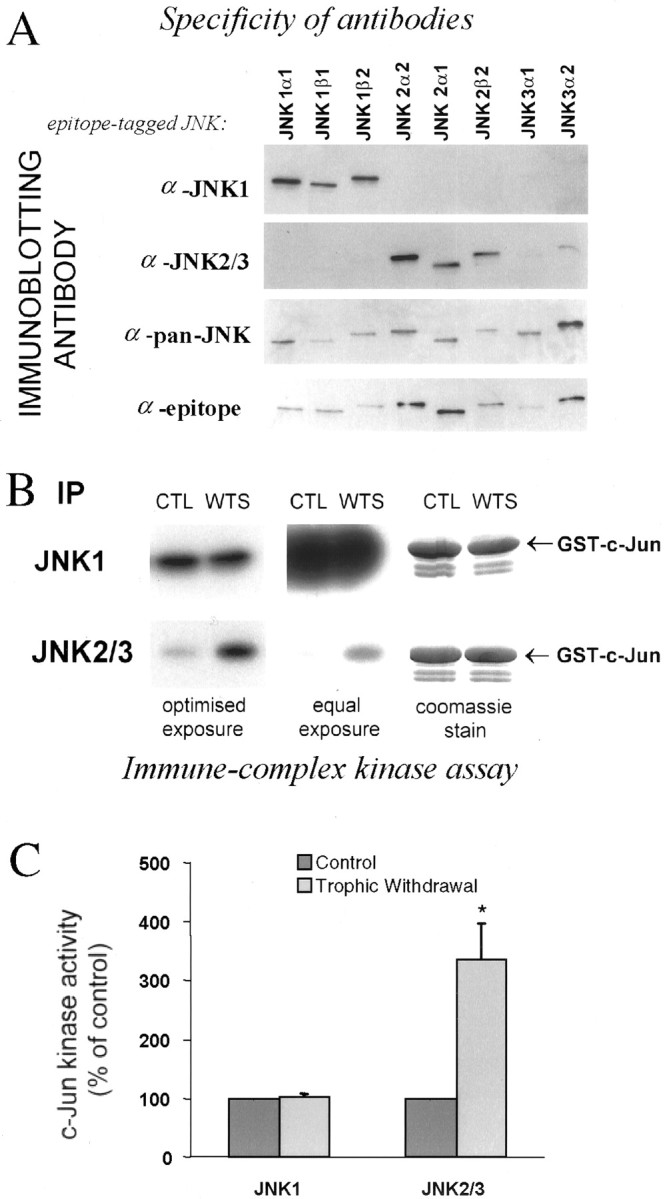
Selective activation of JNK2/3 by withdrawal of trophic support in the persistent presence of high constitutive JNK1 activity. A, JNK isoforms were expressed in COS-7 cells, and selective JNK1 and JNK2/3 antibodies were identified by immunoblotting as indicated. The bottom paneldemonstrates JNK isoform loading by blotting for the epitope tag of the JNK constructs. B, Trophic support was withdrawn from cerebellar neurons for 2 hr, JNK1 and JNK2/3 were immunoprecipitated from the cells with antibodies used in A, and activity was measured by in vitro kinase assay with c-Jun substrate. The center panel shows equal exposure time to the gels, the left panel shows nonsaturated film, and the right panel shows the Coomassie-stained gel, indicating equal substrate loading. C, Quantitated activity is displayed as a percentage of control activity (n = 4). An * in the histogram indicates significant difference from control by paired t test (p < 0.05).
We treated neurons as in Figure 1 for 2 hr, which gave a maximal and stable increase in phospho-Jun:c-Jun ratio, and immunoprecipitated and assayed JNK1 and JNK2/3 from parallel cell lysates using the antibodies we have demonstrated to be isoform-specific (Fig. 2B). High constitutive JNK1 activity was detected, whereas activity of JNK2/3 was very low (right panel). We detected strong JNK2/3 activation in response to the stress without detectable effect on JNK1 (Fig.2B, left panel, C). We repeated the JNK1 assay on samples 15 min after stimulation to investigate whether JNK1 activation might potentially contribute to c-Jun phosphorylation at an earlier time point; however no changes were detected at this time point either (data not shown). This confirms our previous hypothesis that the stress-activated pool of JNK in cerebellar granule neurons corresponds to specific isoforms. The huge difference in basal activities of JNK1 and 2/3 immunoprecipitates reflects in part differences between the antibodies. However, the regulation of JNK2/3 and lack of regulation of either JNK1 or total phospho-JNK suggests there is a genuine difference between JNK1 and JNK2/3 activity in the neurons (see Fig.6C). JNK1 activity can be considered high as both total JNK, and JNK1 activity in neurons are considerably higher than JNK activity from non-neuronal origin (Coffey et al., 2000).
Fig. 6.

SB203580 inhibits c-Jun phosphorylation and JNK2/3 immunoprecipitated from cerebellar granule neurons. A,SB203580 was added to neurons 1 hr before either no treatment (left lanes) or 30′ withdrawal stress (top panel) or anisomycin (10 μg/ml) treatment (bottom panel). c-Jun phosphorylation was detected by mobility shift, demonstrating that ≥10 μmSB203580 inhibits the mobility shift in both cases. B,HeLa cells were transfected with empty vector or plasmids encoding constitutively active MKK6 and p38α, together with plasmids encoding GAL4-ATF2 and the GAL4-driven luciferase reporter pGL3-G5E4Δ38 and pRL-CMV as internal control. Cells were grown for 24 hr after transfection with DMSO carrier with or without SB203580 as indicated, cells were lysed and normalized luciferase expression activity data assayed by dual luciferase assay is shown (mean ± SEM;n = 3). C, JNK1 and JNK2/3 were immunoprecipitated with specific antibodies (compare Fig.2A) from cells withdrawal-stressed for 2 hr, and immune-complex kinase assay performed in the presence of concentrations of SB203580 as shown (top panel). Center panel shows Coomassie-stained gels, indicating equal substrate loading. Bottom panel shows meaned data (±SEM) from replicates, demonstrating high sensitivity of JNK2/3 to SB203580 (IC50 ≈ 3 μm). Results with a pan-JNK antibody are also shown, demonstrating that total JNK has properties consistent with JNK1 (compare Fig. 2B, right panel) An asterisk denotes significant difference from control (p < 0.05 or better; paired t test).
To further substantiate our observations that non-JNK1 isoforms are specifically regulated but swamped by constitutive JNK1 activity in assays of total lysates, we prepared cerebellar granule neurons from wild-type or JNK1 knock-out mice and repeated the experiments shown in Figure 1A. Whereas neurons from wild-type mouse also show negligible activation of total JNK in response to withdrawal of trophic support, neurons from JNK1−/− mice had lower basal activation, and a trophic withdrawal-evoked activation of total JNK was revealed for the first time (Fig. 3). The neurons from JNK1−/− did not show lower c-Jun levels (and cells were not resistant to death, not shown) when compared with wild-type mice, although the c-Jun antibodies used recognized the mouse protein somewhat less well than rat protein. This data supports our hypothesis that cerebellar neurons possess a high basal JNK1 activity that does not phosphorylate c-Jun in vivo in response to withdrawal of trophic support, but masks inin vitro assay the lower JNK2/3 activity that is activated by this stimulus.
The finding that JNK2/3 is preferentially activated by withdrawal of trophic support may have specific relevance for c-Jun regulation, because recombinant JNK2 has a higher affinity for c-Jun than JNK1, which may be physiologically relevant (Kallunki et al., 1994). Thus, although JNK1 is constitutively active, the regulation of JNK2/3 might be expected to have a disproportionate effect on c-Jun. However, isoform differences in affinities for c-Jun may only partly explain the inability of highly active JNK1 to phosphorylate c-Jun and provide no explanation for the difference in regulation of JNK1 and 2/3 in the neurons. Immunofluorescence staining with the isoform antibodies (Fig.4A) indicates that JNK1 is strongly retained in the cytoplasm in punctate structures, especially visible along processes, whereas JNK2/3 is more diffuse and considerably more localized to the nucleus, which is where c-Jun is expected to be located. The preferential localization of JNK2/3 with c-Jun could contribute to the selective targeting of c-Jun by JNK2/3, whereas the cytoplasmic retention of JNK1 in spite of its activity suggests that mechanisms exist to exclude JNK1 from the nucleus and thus from c-Jun. The isoform-specific granularity suggests a differential association with intracellular structures; it is possible that this could contribute to the observed isoform-specific regulation. The diffuse JNK2/3 staining could also be the result of nonspecific recognition of some other antigen by the antibody; however, we did not observe any nonspecific bands on immunoblots developed with this antibody. Levels of JNK1 and JNK2/3 in cytoplasm and nucleus were unchanged in response to withdrawal of trophic support, indicating no overall nuclear translocation in response to this stress (Fig.4B).
Fig. 4.
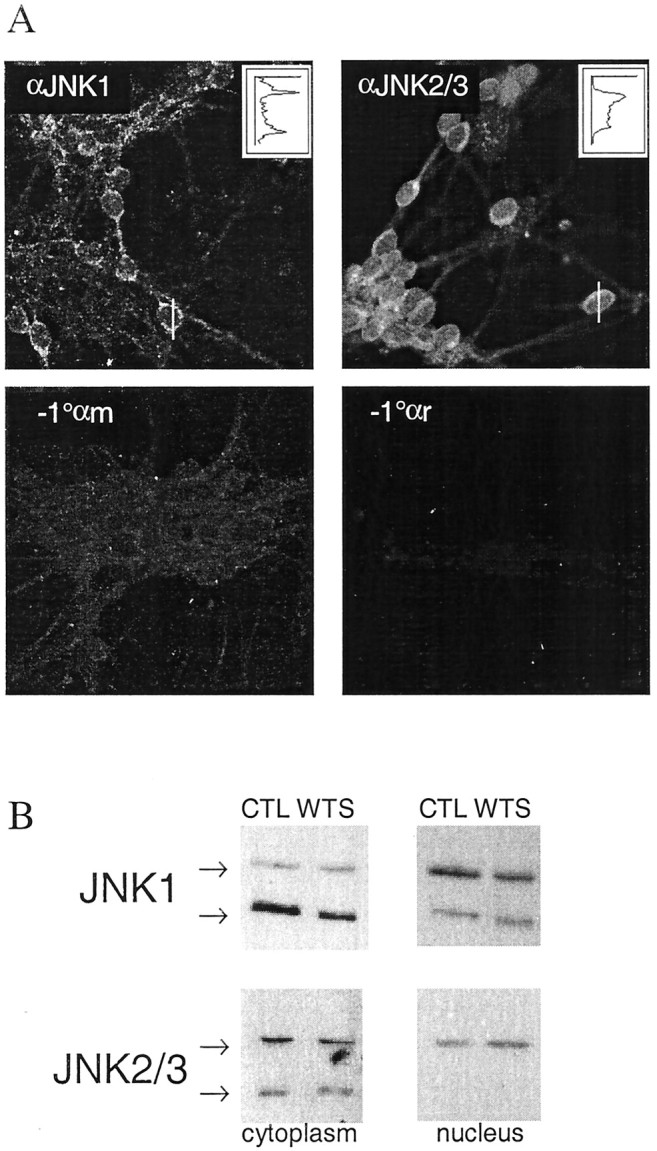
Compartmentalization and granularity of JNK isoform localization. A, Cerebellar granule neurons were fixed and stained with JNK isoform-specific antibodies or without primary antibody as shown. Confocal scans are shown. The fluorescence intensity profiles along the white linesare shown in the inset line graphs (arbitrary scale), demonstrating the extranuclear localization of JNK1 and the relative abundance of JNK2/3 in the nucleus. Note also the granularity of JNK1 staining along processes and the relatively diffuse JNK2/3 staining.B, Immunoblot detection of JNK1 and JNK2/3 from cytoplasmic and nuclear fractions indicate no change in levels in either compartment in response to 2 hr withdrawal of trophic support. Representative blots from three replicates are shown. The nuclear JNK1 blots required longer exposure time than the corresponding cytoplasmic blot, in accordance with the relative distribution shown by immunofluorescent staining in Figure 4A.
p38 is activated by withdrawal of trophic support from cerebellar neurons
The regulation of the stress-activated MAP kinase p38 in response to this particular stress is controversial. One report found no response, by using antibody raised against a Xenopussequence (Rouse et al., 1994) similar to mammalian α and β, but quite different from γ and δ isoforms. A second report using a commercial antibody of undefined specificity showed an increase in activity (Yamagishi et al., 2001). As the isoform specificity of antibodies may cause different results to be obtained, we tested a phospho-specific “anti-active” p38 antibody for isoform specificity. Isoforms of p38 were expressed in COS-7 cells and activated, and the ability of the phospho-specific antibody to detect all isoforms was demonstrated (Fig.5A, β isoform not shown). Immunoblotting lysates of neurons treated as in Figure 1 with this phospho-p38 antibody demonstrated that a p38 isoform was indeed activated by withdrawal of trophic support (Fig. 5B). The identity of the isoform is unknown, but the retarded mobility of γ and β isoforms (Fig. 5A) (data not shown) (Li et al., 1996) together with our detection by RT-PCR of α and δ but not β or γ in these neurons (data not shown) suggests that both α and δ isoforms are present and either one or both are activated by trophic withdrawal stress.
Fig. 5.

Activation of p38 is detected by a phospho-specific p38 antibody that recognizes active forms of all p38 species. COS-7 cells were transfected with pcDNA3 vectors containing flag-tagged p38α, γ, δ, or as controls, p38αAF, which cannot be phosphorylated or no insert (“pcDNA3”), and anisomycin-treated to activate p38. The ability of anti-phospho-p38 to recognize the activated isoforms was determined by immunoblotting (top panel), and expression levels verified by flag immunoblotting (bottom panel). The flag-tag of the constructs caused retarded mobility, allowing them to be distinguished from endogenous anisomycin-activated p38 as shown.B, Lysates of cerebellar granule neurons stimulated as in Figure 1 were immunoblotted with phospho-p38 antibody and pan-p38 antibody as shown. The arrows on the leftof the blots indicate the proteins, and the arrowhead on the right indicates the 42 kDa Mr marker. Quantitated data (means ± SEM; n = 3–4) were normalized and scaled to percentage of initial (control) values. The * represents a significant difference from control (p < 0.05 or better; pairedt test).
Sensitivity of stress-induced c-Jun phosphorylation, p38, total JNK, JNK1, and JNK2/3 to SB203580
Both JNK2/3 and an unidentified p38 isoform are activated during neuronal c-Jun phosphorylation. Some isoforms of the JNK2/3 group have high affinity for c-Jun (Kallunki et al., 1994) and appear most likely to be responsible for the phosphorylation, yet p38 has recently been proposed to directly phosphorylate c-Jun in these cells (Yamagishi et al., 2001). This is somewhat surprising because (1) all known p38 isoforms are poor kinases for c-Jun (Goedert et al., 1997;Kumar et al., 1997), (2) we could not induce detectable c-Jun phosphorylation with p38-immunoprecipitated from neurons, and (3) in-gel assay showed activation of c-Jun-associating kinases at molecular weights of JNKs not p38 (data not shown)
We addressed this issue more directly with SB203580, a compound rigorously tested for selectivity. Lower concentrations selectively block p38α/β, and higher concentrations block some JNK isoforms, in particular JNK2 and JNK3 isoforms, but ERK and p38γ/δ are unaffected (Cuenda et al., 1995; Kumar et al., 1997; Whitmarsh et al., 1997; Davies et al., 2000) (E. T. Coffey, V. Hongisto, J. Cao, and M. J. Courtney, unpublished data). This compound does not affect MAP kinase phosphorylation state but inhibits the ability of activated kinases to phosphorylate their substrates (Kumar et al., 1999), thus, its actions can be observed by investigating substrate phosphorylation and activity, not with phospo-specific MAPK antibodies. A brief (30′) withdrawal of trophic support was used because this induces a clear reproducible c-Jun phosphorylation mobility shift, without any increase in c-Jun protein that might hinder the detection of increased specific phosphorylation (Fig. 1). SB203580 at 1 μm had no effect on this phosphorylation, but 10 μm dramatically inhibited the response (Fig. 6A, top panel). To investigate if this behavior was specific to trophic withdrawal or a more general property of the neuronal stress response, we used anisomycin, an independent stress frequently used to activate JNKs. The induced c-Jun phosphorylation was again prevented by 10 μm but not 1 μmSB203580 (Fig. 6A, bottom panel). SB203580 at 1 μm is sufficient to completely inhibit p38α-evoked ATF2 activation in vivo, as measured by GAL4 reporter assay (Fig. 6B), consistent with our previous report using p38α and the specific substrate Mef2A in vivo in cerebellar granule neurons (Coffey et al., 2000). The β isoform is equally sensitive, and γ/δ are insensitive (Stein et al., 1997; Davies et al., 2000). As block of p38 isoforms could not explain the effects on c-Jun, and the compound has been reported to inhibit select JNK isoforms (Whitmarsh et al., 1997) we tested the drug on total JNK, JNK1, and JNK2/3 immunoprecipitated from neurons treated as in Figure 2B. Whereas total JNK and JNK1 were only partly inhibited by concentrations of at least 100 μm SB203580, 10 μminhibited ∼70% of neuronal JNK2/3 activity (Fig. 6C). These data are additional evidence against a role for JNK1 in c-Jun phosphorylation changes at the 30 min time point and once again support our hypothesis that JNK2/3 specifically targets c-Jun during neuronal stress in the presence of constitutively elevated JNK1 activity.
Withdrawal stress induces GAL4-c-Jun transcriptional activity, which is sensitive to SB203580 but does not involve p38 isoforms
Phosphorylation of c-Jun is associated with increased transcriptional activation capacity, although it has not been demonstrated whether withdrawal of trophic support from cerebellar neurons has any effect on c-Jun transcriptional activity. To investigate this issue, we cotransfected GAL4-c-Jun(6–89) with a GAL4-driven luciferase reporter, followed by withdrawal of trophic support. This stimulus induced an increase in expression from the reporter plasmid, indicating increased c-Jun transcriptional activity (Fig. 7A). The addition of 10 μm SB203580 during withdrawal of trophic support strongly reduced the induction of reporter. Ser-63/73 are required for activation of c-Jun by JNK, although Thr-91/93 have also been proposed to contribute (Papavassiliou et al., 1995). Therefore, we investigated the requirements of phosphoryation sites for the activation of c-Jun shown in Figure 7A. Point mutants of the longer GAL4-c-Jun(5–105) were prepared and expressed, and activation was assayed as above. Ser-63/73 were required for maximal activation by withdrawal of trophic support, but mutation of Thr-91/93 has little effect (Fig. 7B). As expected, the “seven ala” mutation that has been previously shown to protect neurons from death induced by withdrawal of trophic support also prevents activation by withdrawal of trophic support (Fig. 7B). Notably, the basal activity from wild-type GAL4-c-Jun and the seven ala mutant are not significantly different, suggesting that basal phosphorylation of the GAL4-c-Jun is very low, consistent with the low endogenous c-Jun phosphorylation shown in Figure 1.
Fig. 7.
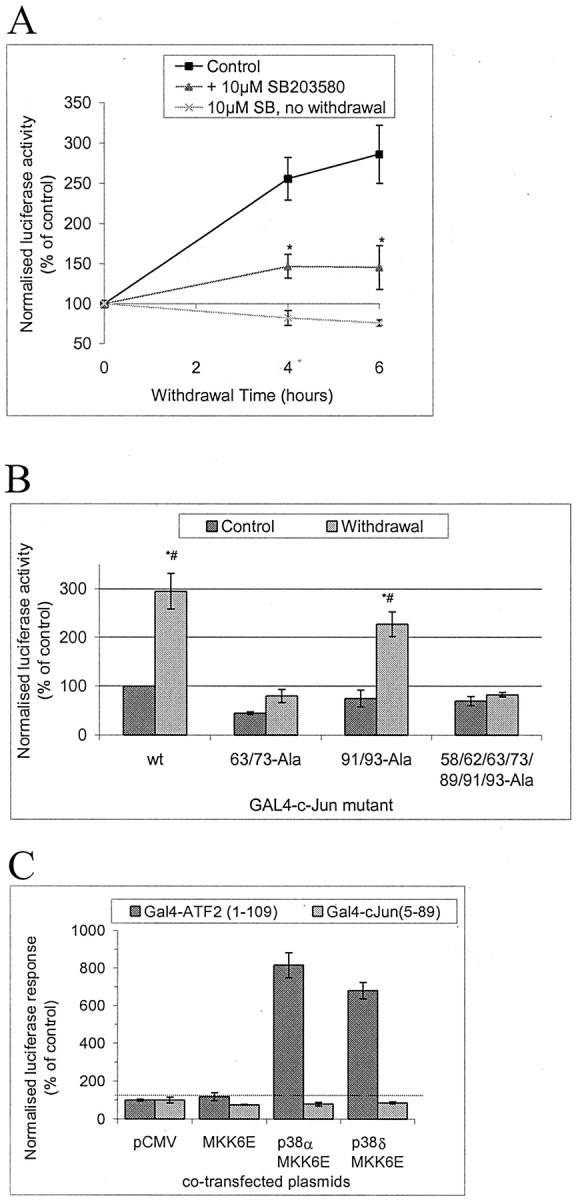
Withdrawal of trophic support induces transcriptional activation of GAL4-c-Jun in a manner sensitive to 10 μm SB203580 and dependent on phosphorylation sites serines 63/73; p38 isoforms cannot activate it. A,Cerebellar granule neurons were transfected with GAL4-luciferase reporter, GAL4-c-Jun(6–89) fusion construct, Renilla luciferase internal control, and empty vector (pCMV) to bring total DNA to 4 μg/well of a 12 well plate. Cells were deprived of trophic support in the presence or absence of 10 μm SB203580 (added 1 hr before withdrawal of trophic support) for the times shown or treated with SB203580 without withdrawal of trophic support alone for an equivalent time. This was achieved by transfecting all samples at the same time, withdrawing trophic support at different times, and lysing all samples at the same time, thus that all samples had the same amount of time to express the transfected plasmids. Firefly luciferase activity was normalized to the Renilla luciferase internal standard. Normalized activity levels were expressed as a percentage of values from samples with continued trophic support. Means ± SEMs (n = 3–5) are shown. An * indicates that the values in the presence of SB203580 are significantly different from in the absence of the drug (p < 0.05; pairedt test). B, Cerebellar granule neurons were transfected as in A but with GAL4 fused to c-Jun (5–105) wild-type “wt” or Ser/Thr→Ala point mutants as shown. Neither GAL4-c-Jun(5–105) construct in which Ser63/73 is mutated to Ala is significantly activated by withdrawal of trophic support; both wt and Thr91/93Ala are activated to similar extents (2.9- and 3.0-fold, respectively). Means ± SEM (n = 4) are shown. An * indicates a significant increase in comparison with control value with wt, a # indicates significant difference from control using the same GAL4 fusion protein (p ≤ 0.05; pairedt test). C, The possibility of c-Jun activation in A by p38 was investigated by transfecting neurons inA but with GAL4 fusions of either c-Jun(6–89) as inA or ATF2(1–109) as a control p38 substrate, and cotransfected with plasmids as shown to express and activate specific isoforms of p38. Data shown represents mean ± range (n = 2).
It is important to note that withdrawal-induced activation shown in Figure 7A was obtained with a GAL4-c-Jun(6–89) construct, and therefore this minimal N-terminal region typically used to assay JNK (e.g., in Fig. 2) is sufficient to detect the withdrawal response. We did not obtain larger induction with GAL4 fusions of longer fragments of c-Jun (Fig. 7B) (data not shown for c-Jun(5–223) constructs), suggesting this minimal region may be sufficient for the full c-Jun transactivation response to withdrawal stress.
p38 has been proposed to post-translationally activate c-Jun in response to withdrawal of trophic support (Yamagishi et al., 2001), and we concluded that p38 α or δ are activated (see above). We therefore tested the ability of these kinases to activate either GAL4-c-Jun(6–89), the minimal sequence sufficient to demonstrate withdrawal stress-induced activation, or GAL4-ATF2(1–109), a known substrate (Fig. 7C). As expected both isoforms of p38 were able to activate Gal4-ATF2, but neither α nor δ had any effect on GAL4-c-Jun(6–89) (Fig. 6C). This indicates that the p38 isoforms activated in cerebellar granule neurons (Fig. 5B) cannot explain the c-Jun regulation that could be reproduced in the GAL4 assay.
Withdrawal stress induction of c-jun promoter activation requires JNK
The c-Jun activation in response to withdrawal of trophic support consists of an early phase of increased c-Jun phosphorylation followed by a later increase in c-Jun protein (Fig. 1). Our results suggest JNK2/3 mediates the early phase of the response, i.e., increased c-Jun phosphorylation, but it is not known what signaling pathways contribute to the later phase. Treatment of cells with SB203580 inhibits both the phosphorylation and the increase in total levels of c-Jun protein induced by a 4 hr withdrawal of trophic support (Fig.8A, compare Fig. 1). In contrast, the RNA polymerase inhibitor actinomycin D efficiently prevents the increase in c-Jun protein without preventing the c-Jun phosphorylation (Fig. 8A). We investigated the later phase of the response further by measuring changes in c-junmRNA levels by RT-PCR. Withdrawal of trophic support increases the levels of c-jun PCR product, and the presence of 10 μm SB203580 has a partial effect on this response (Fig. 8B). This suggests that isoforms of either p38 or JNK could potentially contribute to regulation ofc-jun mRNA levels under these conditions. We then used ac-jun promoter reporter construct (Clarke et al., 1998) together with JIP-JBD, a construct that inhibits JNK in these neurons (Dickens et al., 1997; Coffey et al., 2000). The ability of inhibitory constructs to fully block a response requires high levels of cotransfection. To assess the cotransfection efficiency we achieve with cerebellar granule neurons, we coexpressed two fluorescent protein constructs of similar intensity but colocalized to different parts of the cell; the cytoplasmic and nuclear DsRed-C1 and the membrane-localized EGFP-F. Transfected neurons expressed both plasmids with very high efficiency (data not shown), suggesting that virtually every transfected neuron is likely to express all plasmids added to the cells as a mixture. Therefore we cotransfected c-junpromoter construct with or without JIP-JBD. As shown in Figure8C, the c-jun promoter was activated by withdrawal of trophic support, and the activation was inhibited by JIP-JBD. This suggests that JNK is required for transcriptional upregulation of c-jun mRNA at the promoter level by withdrawal of trophic support from cerebellar granule neurons.
Fig. 8.

Withdrawal-stress induced c-Jun mRNA and protein are inhibited by SB203580, and activation of c-Jun promoter is prevented by the JNK inhibitor JIP-JBD in cerebellar granule neurons. A, Cerebellar granule neurons were withdrawal-stressed in the presence or absence of SB203580 (10 μm) and/or actinomycin D (5 μm) added 1 hr before withdrawal of trophic support as indicated. Actinomycin D, which targets RNA synthesis, inhibits the withdrawal-induced accumulation of c-Jun, whereas SB203580 inhibits both the accumulation and the phosphorylation-associated mobility shift of c-Jun. B,SB203580 (10 μm) or carrier was added 1 hr before withdrawal of trophic support as indicated. After 3 hr, RNA was isolated, and actin and c-jun mRNA were amplified by RT-PCR and detected on agarose gels with ethidium bromide. A representative experiment is shown. C, Neurons were cotransfected with c-Jun promoter-driven luciferase reporter plasmid (pGL3-JC6), Renilla luciferase internal standard, and 1 μg of either empty vector (pCMV) or JIP-JBD to inhibit JNK signaling as shown. After 20 hr, neurons were withdrawal-stressed for 4 hr where indicated. Reporter induction was calculated as described in the legend to Figure 7(n = 3). The JNK inhibitor prevented induction of c-jun promoter reporter expression by withdrawal of trophic support. * and # indicate significant differences from empty vector-transfected, control, and withdrawal-stressed values, respectively (p < 0.05 or better; pairedt test).
Neuronal cell death is not inhibited by concentrations of SB203580 that inhibit p38
The data presented indicate that withdrawal of trophic support selectively activates the JNK2/3 isoforms as well as p38 α and/or δ (Figs. 2, 3, 5). JNK2/3 activated by different neuronal stresses selectively targets c-Jun for phosphorylation and activation via ser-63/73 (Figs. 1A, 6A–C,7B). The effect of SB203580 on c-Jun phosphorylation can be explained by selective inhibition of JNK2/3 (Fig. 2C). Because the c-Jun regulation has been shown to be required for neuronal death, we investigated if the amount of SB203580 required to inhibit the death corresponded to inhibition of JNK2/3 (≥10 μm), p38α (≤1 μm) (Fig. 6B) (Coffey et al., 2000), or p38δ (insensitive; Kumar et al., 1997; Davies et al., 2000). Trophic support was withdrawn from neurons, and cell death was measured as the percentage of nuclei displaying pyknosis, typical of apoptotic cell death (Fig.9A). This trophic withdrawal-induced neuronal death is a caspase-dependent death (Gerhardt et al., 2001) that can be reduced by the dominant-negative c-Jun construct “Tam-67” (V. Hongisto et al., unpublished observations). We find that SB203580 also partially prevents the cell death at concentrations closely corresponding to the inhibition of JNK2/3 (Fig, 8B, compare Fig. 5C). This suggests that inhibition of JNK2/3 but not p38α may be necessary to prevent this form of neuronal cell death.
Fig. 9.

Neuronal death induced by withdrawal of trophic support is reduced by SB203580. Cerebellar granule neurons were cultured in medium containing serum and 20 mm additional KCl. Death induced by withdrawal of trophic support was evoked by incubating the cells with medium free of serum and without additional KCl, in the presence or absence of SB203580 added 1 hr before withdrawal of trophic support. DNA was stained with Hoechst 33342 24 hr later. A, Fluorescence images of Hoechst 33342: DNA complexes show healthy nuclei in controls. Withdrawal of trophic support induced pyknosis of over half the nuclei in a manner largely prevented by 10 μm SB203580. B, Percentage of neurons present scored as alive (means ± SEMs;n = 4) is shown. An * indicates that survival with SB203580 is significantly higher than without (p < 0.001 by paired ttest).
DISCUSSION
c-Jun has for many years served as a marker of survival of axotomized neurons, and more recently c-Jun activation has been implicated in neuronal cell death in the CNS (Herdegen et al., 1997;Behrens et al., 1999; Kuan et al., 1999; Crocker et al., 2001). Although the contrasting functions attributed to c-Jun activity in these cases remain to be explained, a central role for c-Jun regulation in the response to neuronal stress is generally accepted. Despite the abundance of evidence for JNK as the major regulator of c-Jun, several reports have called into question the role of JNK in stress-induced c-Jun regulation in the most abundant neuronal type of the mammalian brain, the cerebellar granule neuron (Watson et al., 1998; Yamagishi et al., 2001).
We show here that withdrawal of trophic support from cerebellar granule neurons potently increases phosphorylation of c-Jun before the subsequent increase in c-Jun protein, demonstrating that increased c-Jun phosphorylation cannot be attributed to increased total c-Jun levels. c-Jun kinase:phosphatase balance genuinely changes, resulting in a maintained threefold increase in specific c-Jun phosphorylation at Ser-63 and Ser-73. In contrast, we detect only a very minor, somewhat variable change in total JNK activity, consistent with previous negative results (Gunn-Moore et al., 1998; Watson et al., 1998;Yamagishi et al., 2001). JNKs are expressed from three genes, each product producing low and high molecular weight splice variants (46–49 and 54–57 kDa, respectively). We have previously suggested that the different JNK pools in neurons may represent different JNK isoforms. Here we show that although a very large amount of activity can be immunoprecipitated with JNK1-specific antibodies, this activity is not responsive to trophic-withdrawal stress. In sharp contrast, JNK2/3-specific antibodies immunoprecipitate very little activity in resting neurons, but this activity increases sharply after withdrawal of trophic support. This is the first report of selective and differential activation of the product of different JNK genes i.e., induced activation of JNK2/3 in the presence of high constitutive activity of JNK1. This suggests that JNKs must be strictly associated with different upstream activating components, which in turn is likely to restrict the menu of substrates available to each of these kinases. Activation by trophic withdrawal of total JNK is revealed in JNK1−/− neurons, which have a lower total JNK activation level under basal conditions. We cannot exclude the possibility that a minor subpool of JNK1 is also activated by stress in wild-type neurons. However, any action such a pool may have on c-Jun is redundant because c-Jun activation still occurs in JNK1−/−neurons. Furthermore, these observations demonstrate that measurement of total JNK activity in wild-type neurons is misleading, appearing to demonstrate a lack of JNK activation during a specific functional response, such as c-Jun phosphorylation or cell death. Statements about the involvement of JNK based on an assay of composite JNK activity should therefore be treated with great caution.
Previous studies with recombinant JNKs suggested JNK2 may have physiologically significant higher affinity for c-Jun than JNK1 (Kallunki et al., 1994). We found that GST-c-Jun can be used to selectively enrich endogenous evoked c-Jun phosphorylating activity from neurons under withdrawal stress (data not shown). This activity has molecular weights in in-gel kinase assay corresponding to JNK not p38, but the activation of p38 by withdrawal stress is controversial (Watson et al., 1998; Yamagishi et al., 2001). We detect increased phosphoryation of the TGY motif of p38s corresponding to α and/or δ, but were unable to phosphorylate c-Jun in vitro with p38 immunoprecipitated from the neurons, and p38 α and δ were unable to activate c-Jun, although they activated ATF2. The effects of JNK2/3 and p38α can be distinguished by their sensitivity to pyridinyl imidazoles such as the rigorously tested and selective inhibitor SB203580 (Whitmarsh et al., 1997; Davies et al., 2000). Although 1 μm SB203580 strongly inhibits p38α activity against multiple substrates in cerebellar granule neurons, in HeLa cells and in vitro (Coffey et al., 2000) (Fig.6B) (data not shown), 10 μmis required to block recombinant JNK2 isoforms (Whitmarsh et al., 1997) and stress-activated endogenous JNK2/3 from neurons (Fig.6C). The ability of 10 but not 1 μmof SB203580 to block both c-Jun phosphorylation in response to different stresses and to block increased c-Jun transactivation capacity strongly suggests that p38α is not responsible for the c-Jun activation and that the JNK2/3 groups, which we now know are stress-responsive in neurons, are good candidates. A novel activation of c-Jun by Abl tyrosine kinase was recently reported (Barila et al., 2000). This pathway is unlikely to contribute in the present case, because the Abl inhibitor CGP57148 (Buchdunger et al., 1996) did not prevent the c-Jun response described here. Altogether, these data suggest that JNK2/3 isoforms in cerebellar neurons have preferential access to c-Jun as well as being selectively activated by stress (Fig.9). In addition, it demonstrates the importance of distinguishing JNK isoforms to understand stress responses in neuronal cells. Why the constitutive JNK1 activity is selectively prevented from phosphorylating c-Jun in the nucleus under resting conditions remains to be resolved. The retention of JNK1 in punctate structures in the cytoplasm may well be one reason (Figs. 4, 10). Other possible reasons exist. JIP1 has 100-fold higher affinity for JNK1 than c-Jun and can inhibit c-Jun phosphorylation by JNK (Dickens et al., 1997). In these neurons JIP1 colocalizes with c-Jun in the nucleus (Coffey et al., 2000), suggesting that any JNK1 that does enter the nucleus may still be unable to access c-Jun. Interestingly, the JNK2 isoforms with highest sensitivity to SB203580 are also those isoforms that have lowest affinity for JIP-1 (Whitmarsh et al., 1997; Yasuda et al., 1999). However it is unclear whether JIPs, which potentiate JNK activation in vitro (Whitmarsh et al., 1998), promote signaling to c-Jun in vivo or compete with c-Jun for binding of JNK sufficiently to actually direct JNK signaling to other pathways.
Fig. 10.

Scheme depicting proposed JNK and p38 regulation in stressed cerebellar neurons. Stresses, e.g., withdrawal of trophic support, selectively activate JNK2/3. This leads to c-Jun phosphorylation and activation of promoters of stress responsive genes such as c-jun itself, ultimately leading to the response of the neuron to stress. A p38 isoform is also activated, but its role is unknown. In contrast, JNK1 is constitutively active; in spite of this, it is predominantly associated with cytoplasmic structures and unable to phosphorylate c-Jun in the nucleus.
The c-Jun response to stress in neurons consists not only of increasedc-jun-specific phosphorylation but also a delayed increase in c-jun RNA and c-Jun protein. JNK and p38 can activate a number of transcription factors reported to contribute toc-jun promoter activity, including c-Jun itself as well as ATF2 and the Mef2 family. The increase in c-jun mRNA is partly sensitive to SB203580, suggesting that p38α and/or JNK2/3 may potentially contribute to this later phase response. The ability of overexpression of the JIP-JNK binding domain to prevent stress-inducedc-jun promoter activity indicates that JNK activation is an essential requirement for this response. This suggests that the novel, isoform-selective activation of JNK2/3 described here also contributes to c-jun promoter activity, as would be expected subsequent to activation of c-Jun. However, the incomplete block by SB203580 of both the c-jun mRNA increase and subsequent death suggests that other pathways in addition to the SB203580-sensitive JNK2/3s are on their own sufficient to partly increase c-jun mRNA levels and induce cell death in stressed neurons. In support of this, we find that the dominant-negative c-Jun “Tam-67” is also not completely protective (V. Hongisto, M. J. Courtney, and E. T. Coffey, unpublished observations). In addition, the substantial impact on JNK2/3 activity, c-Jun phosphorylation, transactivation, and survival by 10 μm SB203580, the lack of effect of lower concentrations of SB203580, which are sufficient to inhibit p38α/βin vivo but not JNK, suggest that conclusions drawn about a role of p38 isoforms based on this drug and the apparent lack of JNK regulation should be treated with some skepticism, particularly because p38 has confusingly been attributed roles in both neuronal death and neuronal survival (Xia et al., 1995; Kawasaki et al., 1997; Mao et al., 1999; Kikuchi et al., 2000; Okamoto et al., 2000;Yamagishi et al., 2001).
In conclusion, we have demonstrated that cerebellar neurons coexpress distinct, differentially localized pools of JNK1 and JNK2/3 with high constitutive activity and very low activity, respectively. The constitutively active JNK1 is unable to cause phosphorylation of c-Jun in the cells, whereas the minor JNK2/3 pool is selectively activated by stress and has preferential access to and thus a profound effect on c-Jun. The two isoforms are therefore members of distinct signaling pathways. We propose that the JNK1 and JNK2/3 pools in neuronal cells are coupled with different signaling cascades and that this can result in responsiveness to separate stimuli, access to some substrates, and exclusion from others.
Footnotes
This work was supported by the Academy of Finland (project grants 41340 and 44190, and the life 2000 program grants 50037 and 49949) and the University of Kuopio. We thank Martin Dickens (University of Leicester, Leicester, UK), John Kyriakis [Massachusetts General Hospital (MGH), Boston, MA], Dirk Bohmann (European Molecular Biology Laboratory, Heidelberg, Germany), Jim Woodgett (Ontario Cancer Institute, Toronto, Canada), Roger Davis (University of Massachusetts, Worcester, MA), Bruce Mayer (Children's Hospital, Boston, MA), Sander van den Heuvel (MGH, Boston, MA), Michael Birrer (National Institutes of Health, National Cancer Institute, Bethesda, MD), Jiahuai Han (The Scripps Research Institute, La Jolla, CA), and Ron Prywes (Columbia University, New York, NY) for providing reagents, Roger Davis and Richard Flavell (Yale University School of Medicine, New Haven, CT) for JNK1−/− mice, and Martin Dickens for helpful discussion.
Correspondence should be addressed to Michael J. Courtney, Molecular Signaling Laboratory, Department of Neurobiology, A. I. Virtanen Institute, Neulaniementie 2, University of Kuopio, P. O. Box 1627, Kuopio, FIN-70211, Finland. E-mail:courtney@messi.uku.fi.
REFERENCES
- 1.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD098059 is a specific inhibitor of the activation of MAPKK in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 2.Barila D, Mangano R, Gonfloni S, Kretzschmar J, Moro M, Bohmann D, Superti-Furga G. A nuclear tyrosine phosphorylation circuit: c-Jun as an activator and substrate of c-Abl and JNK. EMBO J. 2000;19:273–281. doi: 10.1093/emboj/19.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 4.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, Lydon NB. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- 5.Clarke N, Arenzana N, Hai T, Minden A, Prywes R. EGF induction of the c-jun promoter by a Rac pathway. Mol Cell Biol. 1998;18:1065–1073. doi: 10.1128/mcb.18.2.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ. Dual roles for JNK in developmental and stress responses in cerebellar granule neurons. J Neurosci. 2000;20:7602–7613. doi: 10.1523/JNEUROSCI.20-20-07602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courtney MJ, Coffey ET. The mechanism of Ara-C-induced apoptosis of differentiating cerebellar granule neurons. Eur J Neurosci. 1999;11:1073–1084. doi: 10.1046/j.1460-9568.1999.00520.x. [DOI] [PubMed] [Google Scholar]
- 8.Courtney MJ, Akerman KE, Coffey ET. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci. 1997;17:4201–4211. doi: 10.1523/JNEUROSCI.17-11-04201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crocker SJ, Lamba WR, Smith PD, Callaghan SM, Slack RS, Anisman H, Park DS. c-Jun mediates axotomy-induced dopamine neuron death in vivo. Proc Natl Acad Sci USA. 2001;98:13385–13339. doi: 10.1073/pnas.231177098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 11.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 13.Finch A, Davis W, Carter WG, Saklatvala J. Analysis of MAPK pathways used by interleukin 1 in tissues in vivo: activation of hepatic JNK 1 and 2:and MKK 4 and 7. Biochem J. 2001;353:275–281. doi: 10.1042/0264-6021:3530275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerhardt E, Kugler S, Leist M, Beier C, Berliocchi L, Volbracht C, Weller M, Bahr M, Nicotera P, Schulz JB. Cascade of caspase activation in potassium-deprived cerebellar granule neurons: targets for treatment with peptide and protein inhibitors of apoptosis. Mol Cell Neurosci. 2001;17:717–731. doi: 10.1006/mcne.2001.0962. [DOI] [PubMed] [Google Scholar]
- 15.Goedert M, Cuenda A, Craxton M, Jakes R, Cohen P. Activation of the novel stress-activated protein kinase SAPK4 by cytokines and cellular stresses is mediated by SKK3 (MKK6): comparison of its substrate specificity with that of other SAP kinases. EMBO J. 1997;16:3563–3571. doi: 10.1093/emboj/16.12.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunn-Moore FJ, Tavare JM. Apoptosis of cerebellar granule cells induced by serum withdrawal, glutamate or beta-amyloid, is independent of Jun kinase or p38 MAPK activation. Neurosci Lett. 1998;250:53–56. doi: 10.1016/s0304-3940(98)00438-8. [DOI] [PubMed] [Google Scholar]
- 17.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 18.Herdegen T, Skene P, Bahr M. The c-Jun transcription factor - bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997;20:227–231. doi: 10.1016/s0166-2236(96)01000-4. [DOI] [PubMed] [Google Scholar]
- 19.Kallunki T, Su B, Tsigelny I, Sluss HK, Derijard B, Moore G, Davis R, Karin M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activaiton and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- 21.Kikuchi M, Tenneti L, Lipton SA. Role of p38 MAPK in axotomy-induced apoptosis of rat retinal ganglion cells. J Neurosci. 2000;20:5037–5044. doi: 10.1523/JNEUROSCI.20-13-05037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Jiang MS, Adams JL, Lee JC. Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 MAPK. Biochem Biophys Res Commun. 1999;263:825–831. doi: 10.1006/bbrc.1999.1454. [DOI] [PubMed] [Google Scholar]
- 25.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 26.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase sub-family of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 27.Li Z, Jiang Y, Ulevitch RJ, Han J. The primary structure of p38 gamma: a new member of p38 group of MAPKs. Biochem Biophys Res Commun. 1996;228:334–340. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- 28.Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- 29.Okamoto S, Krainc D, Sherman K, Lipton SA. Antiapoptotic role of the p38 MAPK-Mef2 transcription factor pathway during neuronal differentiation. Proc Natl Acad Sci USA. 2000;97:7561–7566. doi: 10.1073/pnas.130502697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papavassiliou AG, Treier M, Bohmann D. Intramolecular signal transduction in c-Jun. EMBO J. 1995;14:2014–2019. doi: 10.1002/j.1460-2075.1995.tb07193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAPKs. Nature. 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 32.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 33.Thangnipon W, Kingsbury A, Webb M, Balazs R. Observations on rat cerebellar cells in vitro: influence of substratum, potassium concentration and relationship between neurones and astrocytes. Brain Res. 1983;313:177–189. doi: 10.1016/0165-3806(83)90215-8. [DOI] [PubMed] [Google Scholar]
- 34.Stein B, Yang MX, Young DB, Janknecht R, Hunter T, Murray BW, Barbosa MS. p38–2:a novel MAPK with distinct properties. J Biol Chem. 1997;272:19509–19517. doi: 10.1074/jbc.272.31.19509. [DOI] [PubMed] [Google Scholar]
- 35.Ui M, Sonobe MH, Ito T, Murakami M, Okazaki S, Takada M, Sato T, Iba H. Biochemical and functional analysis of highly phosphorylated forms of c-Jun protein. FEBS Lett. 1998;429:289–294. doi: 10.1016/s0014-5793(98)00618-8. [DOI] [PubMed] [Google Scholar]
- 36.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitmarsh AJ, Yang SH, Su MS, Sharrocks AD, Davis RJ. Role of p38 and JNK MAPK in the activation of ternary complex factors. Mol Cell Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 39.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAPK on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 40.Yamagishi S, Yamada M, Ishikawa Y, Matsumoto T, Ikeuchi T, Hatanaka H. p38 MAPK regulates low potassium-induced c-Jun phosphorylation and apoptosis in cultured cerebellar granule neurons. J Biol Chem. 2001;276:5129–5133. doi: 10.1074/jbc.M007258200. [DOI] [PubMed] [Google Scholar]
- 41.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 42.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of MAPK scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, Olson EN, Ulevitch RJ, Han J. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999;19:21–30. doi: 10.1128/mcb.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]