

Abstract
The dystrophin glycoprotein complex (DGC) is a multimolecular complex that links the extracellular matrix to the cytoskeleton. The DGC is present at the skeletal neuromuscular junction and required for its maturation and maintenance. Members of the DGC are also expressed in brain. We used cultured hippocampal neurons to analyze the distribution, regulation, and role in synaptogenesis of the major transmembrane component of the DGC, dystroglycan; one of its extracellular ligands, agrin; and one of its cytoskeletal binding partners, dystrophin. α-Dystroglycan, β-dystroglycan, and dystrophin clustered at a subset of inhibitory synapses containing GABAAR subunits α1, α2, and γ2, and the inhibitory receptor anchoring protein gephyrin. DGC components were not detected at excitatory glutamatergic synapses. Dystroglycan is the first identified adhesive macromolecule at mature GABA synapses. Developmentally, dystroglycan clustered at synaptic loci after synaptic vesicles, GABAAR, and gephyrin, the latter being closely associated with GABAAR at all stages of synaptogenesis analyzed. Analysis of gephyrin −/−, agrin −/−, andmdx mouse hippocampal neurons in culture indicated that synaptic clustering of dystroglycan occurs independently of gephyrin, agrin, and dystrophin. In dystroglycan-deficient neurons, cultured from a conditional mutant strain, GABAergic synapses differentiated with clusters of gephyrin and GABAAR apposed to synaptic terminals, but these synapses did not contain detectable dystrophin. Thus the DGC is not essential for GABAergic synaptogenesis but is likely to function in modulating inhibitory synapses or conferring specialized properties on a subset of them.
Keywords: synaptogenesis, GABA receptor, gephyrin, dystrophin, agrin, mdx
The development of central inhibitory GABAergic synapses involves the formation of high-density clusters of GABAA receptors (GABAARs) opposite GABAergic terminals. The mechanisms of inhibitory synaptic differentiation are not well understood, especially in comparison with molecular events at the mammalian skeletal neuromuscular junction (NMJ) (for review, see Sanes and Lichtman, 1999) and at central excitatory glutamatergic synapses (for review, see Sheng and Pak, 1999; Garner et al., 2000). At the NMJ, nerve-derived agrin signaling through MuSK is necessary for the formation of acetylcholine receptor (AChR) clusters opposite nerve terminals. The dystrophin-associated glycoprotein complex (DGC) is necessary for stabilization of AChR clusters and morphological maturation of the postsynaptic apparatus (Deconinck et al., 1997; Grady et al., 1997, 2000; Adams et al., 2000; Jacobson et al., 2001). At glutamatergic synapses, a number of scaffolding proteins including PSD-95, GKAP, Shank, GRIP, and PICK1 function in receptor localization within the postsynaptic cell. The trans-synaptic signaling molecules are not yet well defined but may include neuroligins, cadherins, ephrins and Eph receptors, and Narp (Craig and Lichtman, 2001; Klein, 2001). At GABAergic synapses, gephyrin is concentrated within the postsynaptic domain and is reported to be essential for synaptic clustering of GABAAR α2 and γ2 subunits in hippocampal neurons (Kneussel et al., 1999). However, gephyrin is only partially required for synaptic clustering of GABAAR α2 and γ2 subunits in retina and spinal cord and is not required for synaptic clustering of other α subunits (Fischer et al., 2000; Kneussel et al., 2001). The trans-synaptic signaling molecules are not yet defined for GABAergic synapses.
A role for dystrophin in GABAergic synaptogenesis was recently suggested by Kneussel et al. (1999). Dystrophin is a large cytoskeletal protein of the α−actinin/β−spectrin family. Mutations in dystrophin result in Duchenne and Becker muscular dystrophies (Hoffman and Kunkel, 1989). The DGC provides a transmembrane linkage between the extracellular matrix and the cytoskeleton of muscle fibers (for review, see Henry and Campbell, 1999; Blake and Kroger, 2000). A core component of the DGC is dystroglycan, which is composed of an extracellular α subunit and a transmembrane β subunit, derived by proteolytic cleavage and glycosylation of a single precursor (Ibraghimov-Beskrovnaya et al., 1992). Dystroglycan binds dystrophin and utrophin intracellularly and binds several matrix molecules including agrin, laminin, and perlecan extracellularly. Targeted deletion of dystroglycan in mice leads to a deficiency in formation of basement membranes and early embryonic lethality (Williamson et al., 1997), showing that the DGC has widespread functions.
Evidence is also accumulating for a function of the DGC at central neuronal synapses. Dystrophin and dystroglycan are concentrated in photoreceptor terminals, and DMD patients and mdx3cv mice with reduced expression of dystrophin isoforms show an altered electroretinogram indicating impaired synaptic transmission under conditions of dark adaptation (Blake and Kroger, 2000). DGC components dystroglycan, dystrophin, the short dystrophin isoforms Dp140 and Dp71, dystrobrevins, and syntrophins are abundant in brain, and some are concentrated in postsynaptic density fractions (Kim et al., 1992; Blake et al., 1999; Moukhles and Carbonetto, 2001). Dystroglycan binds two cell surface proteins thought to be involved in synaptogenesis: agrin and neurexin (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Sugita et al., 2001). Previous immunolocalization studies suggested a concentration of dystrophin at a subset of postsynaptic sites (Lidov et al., 1990) and dystroglycan at excitatory spine synapses (Tian et al., 1997; Zaccaria et al., 2001). A recent study found a selective association of dystrophin with GABAergic synapses (Knuesel et al., 1999). Kneussel et al. (1999) further reported a 38% reduction in immunofluorescent clusters of GABAAR α2 subunit in hippocampal tissue ofmdx mice, which lack the long form of dystrophin. These data all suggest that the DGC may function as a trans-synaptic signal for some aspect of central neuron synaptic differentiation.
We explore here the role of the DGC complex in synaptic differentiation by analysis of wild-type and mutant hippocampal neurons in culture. We found that the DGC is selectively associated with a subset of inhibitory GABAergic synapses but is not detectable at excitatory glutamatergic synapses. Dystroglycan clustered at synaptic loci after synaptic vesicles, GAD, GABAAR, and gephyrin. Gephyrin, agrin, and dystrophin were not required for synaptic clustering of dystroglycan. Despite the potential of dystroglycan as a synaptogenic cell adhesion protein, we show using mutant mouse cultures that dystroglycan is not required for synaptic clustering of GAD, gephyrin, or major GABAAR subtypes. Thus the DGC is a gephyrin-independent complex associated with the maturation of a subset of inhibitory synapses.
MATERIALS AND METHODS
Genotyping. The agrin-null mice were generated by a ∼9 kb DNA deletion in the agrin gene corresponding to most of the agrin coding region (from the BamHI site in exon 6 downstream of exon 33), which was replaced by the neomycin resistance gene (Lin et al., 2001). In the gephyrin −/− mice, the sequences responsible for initiating transcription and translation and exon 1 of the gephyrin gene were deleted and replaced with the neomycin resistance gene (Feng et al., 1998). Animals were maintained as heterozygotes on a C57/129 hybrid background. Embryos were genotyped by PCR amplification as described previously (Feng et al., 1998;Lin et al., 2001). Mdx mice (C57BL/10ScSn) were purchased from Jackson Laboratories (Bar Harbor, ME). Conditional mutant DG lox/lox mice were generated by introduction of a loxP-neo cassette in the first intron and a loxP site in the second intron of the mouse DG gene (Saito et al., 2001; Moore et al., 2002). The DG lox/lox mice are viable and fertile and exhibited no phenotypic abnormalities.
Cell culture. Primary cultures were prepared from embryonic hippocampi using previously described methods (Goslin et al., 1998). Rat hippocampi were dissected at embryonic day (E) 18, mouse hippocampal cultures were prepared from individual embryos at E17. Tissues were dissociated by trypsinization and trituration and plated at 4000 cells per square centimeter onto poly-l-lysine-coated glass coverslips. After cells were allowed to attach in minimal essential medium (MEM) with 10% horse serum, they were transferred and maintained up to 5 weeks by growing them over rat glial feeders in serum-free MEM with N2 supplements. Cytosine arabinoside was added after 2 d to inhibit glial proliferation. The neurons were used at 7–35 d after plating for immunocytochemical staining.
Neuron infection with adenovirus-Cre. The defective adenovirus vector expressing Cre was obtained from F. L. Graham (McMaster University, Hamilton, Canada) (Anton and Graham, 1995). The virus was propagated and amplified in 293 cells, a permissive human cell line that supplies the E1 function intrans, and purified by CsCl gradient centrifugation (Graham and Prevec, 1995). The titer (2 × 1010 pfu/ml) was determined by plaque assay. At 7 d in culture, wild-type and DG lox/lox neurons on coverslips were transferred from their home dishes into a sterile incubation chamber. Adenovirus-Cre was diluted in warm conditioned culture medium, and neurons were infected with a multiplicity of infection (MOI) 10 for 1 hr at 37°C. Most of the virus-containing medium was then removed, and coverslips were transferred back to their home dishes over glial feeders and maintained for another 12 d before immunostaining.
Antibodies. The following mouse monoclonal antibodies were used: GAD6 against glutamic acid decarboxylase (1:2; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), mAb7a against gephyrin (5 μg/ml; Cedarlane, Hornby, Canada), VIA4.1 against α−dystroglycan (20 μg/ml, IgG1; Upstate Biotechnology, Lake Placid, NY), 43DAG1/8D5 against β−dystroglycan (1:25, IgG2a; Novocastra Laboratories, Newcastle, UK), Dys-2, which recognizes the last 17 C-terminal amino acids common to all the forms of dystrophin (Dy8/6C5, 1:20, IgG1; Novocastra Laboratories), and microtubule-associated protein 2 (MAP2; Chemicon International, Temecula, CA). Rabbit antibodies were used against synapsin I (0.4 μg/ml; Chemicon), SynGAP (1:1000; Affinity Bioreagents), and glutamic acid decarboxylase 65 and 67 kDa (GAD, 1:200; Chemicon). A human antibody against gephyrin was used (CSF861, 1:100; gift from P. de Camilli, Yale University) (Butler et al., 2000) that gave an identical pattern of immunoreactivity as mAb7a. Antibodies raised in the guinea pig against the GABAAR γ2 subunit (1:2000), α2 subunit (1:3000), and α1 subunit (1:2000) were gifts from J.-M. Fritschy (University of Zurich, Switzerland) (Fritschy and Mohler, 1995). A rat antibody against NCAM was also used (H28, 1:200) (Gennarini et al., 1984). Primary antibodies were visualized with fluorochrome-conjugated secondary antibodies (2.5 μg/ml; Vector Laboratories, Burlingame, CA) or with biotin-conjugated secondary antibody (2.5 μg/ml) followed by fluorochrome-conjugated streptavidin (500 ng/ml). The fluorochromes used were fluorescein, Texas Red, and 7-amino-4-methylcoumarin-3-acetic acid. For simultaneous detection of monoclonal primary antibodies, Alexa Fluor 488 goat anti-mouse IgG1 (1:1000; Molecular Probes, Eugene, OR) and Alexa Fluor 594 goat anti-mouse IgG2a (1:200; Molecular Probes) subtype-specific secondary antibodies were used.
Immunocytochemistry. Coverslips used for β−dystroglycan immunostaining were fixed with methanol for 10 min at −20°C. For other antigens, neurons were fixed with warmed 4% paraformaldehyde/4% sucrose in PBS for 15 min at room temperature and permeabilized for 5 min with 0.25% Triton X-100 in PBS. For surface GABAAR detection, living neurons were incubated with antibodies against GABAARγ2 or GABAARα2 extracellular epitopes (1:1000) for 1 hr at 37°C in conditioned culture medium, washed, and fixed. Nonspecific staining was blocked for 30 min in 10% BSA at 37°C, and neurons were incubated in 3% BSA overnight at room temperature for primary antibodies and for 45 min at 37°C for secondary antibodies. For multiple-labeling experiments, antibodies were incubated simultaneously. Coverslips were mounted in Tris-HCl, glycerol, polyvinyl alcohol with 2% 1.4-diazobicyclo[2.2.2] octane. Fluorescence and phase-contrast images were captured with a Photometrics series 250 cooled CCD camera mounted on a Zeiss Axioscope microscope with a 63× 1.4 numerical aperture lens using Metamorph imaging software. Images were prepared for printing using Adobe Photoshop 5 software.
Quantitative analysis. The number of synaptic gephyrin, GABAARγ2, and β−dystroglycan puncta per 100 μm dendrite length and the proportion of clusters that were synaptic were determined on neurons immunolabeled for synapsin, GABAARγ2, and either gephyrin or β−dystroglycan after 1, 2, 3, and 5 weeks in culture using Metamorphimaging software (Rao et al., 2000). One dendrite per neuron was chosen, and images were subjected to a user-defined intensity threshold to select clusters. GABAARγ2, gephyrin, or β−dystroglycan clusters were classified as synaptic if they were apposed to synapsin-immunoreactive puncta. Apposition was determined by first generating a binary mask from the thresholded synapsin image and widening the regions representing the presynaptic puncta by one pixel all around. Any cluster in the thresholded GABAARγ2, gephyrin, or β−dystroglycan images that had any pixel overlap with the binarized dilated synapsin mask was considered synaptic. A second mask containing the synaptic GABAARγ2 clusters was then compared with the thresholded gephyrin or β−dystroglycan images to determine the percentage of synaptic GABAARγ2 clusters exhibiting pixel overlap (colocalization) with either gephyrin or β−dystroglycan. Similarly, a mask containing the synaptic gephyrin or β−dystroglycan clusters was compared with the thresholded GABAARγ2 image to determine percentage of clusters colocalized. The percentage of β−dystroglycan clusters colocalized with gephyrin, α−dystroglycan, dystrophin, or SynGap was measured from double-labeling experiments at 3 weeks. Clusters were defined by visually thresholding all images. The thresholded β−dystroglycan image was then used to generate a binary mask, and all clusters that exhibited any pixels above threshold in the paired images were counted as colocalized. Surface areas of clusters and of the entire dendrite regions were also recorded. All measures were obtained from 20 neurons, 10 each from two independent cultures. Mean values ± SEM were calculated using StatView F.4.11 software (Abacus Concepts, Inc.).
RESULTS
β−Dystroglycan is concentrated at a subset of inhibitory GABAergic synapses in cultured hippocampal neurons
To test for association between the DGC and synapses, the distribution of the essential DGC component β-dystroglycan was determined in relation to several synaptic antigens by double- and triple-label immunofluorescence in cultured hippocampal neurons (Fig.1). These cultures develop functional inhibitory and excitatory synapses with molecularly specialized clusters of GABAAR opposite GABAergic terminals and AMPA and NMDA receptor opposite glutamatergic terminals (Craig et al., 1994; Rao et al., 1998). β-Dystroglycan formed clusters along the soma and dendrites of many but not all of the neurons. The pattern of β−dystroglycan clusters was reminiscent of the pattern of GABAergic synapses, including a selective association with proximal versus distal dendrites (Benson and Cohen, 1996). Indeed, most β−dystroglycan clusters colocalized with synapsin (Fig.1A). By quantitative measures, which may yield a low estimate because some clusters may have fallen below threshold, a similar percentage (65 ± 5%) of β−dystroglycan clusters exhibited overlap with synapsin puncta as did clusters of GABAARγ2 (72 ± 4%) or gephyrin (63 ± 3%). Furthermore, synaptic β−dystroglycan clusters also colocalized with GABAARγ2 (70 ± 7% colocalization) (Fig. 1A), indicating a specific concentration at GABAergic synapses. A concentration of β−dystroglycan at GABAergic synapses was also visually apparent in neurons double-labeled for β−dystroglycan and GAD or gephyrin (Fig.1B,C).
Fig. 1.

β−Dystroglycan is concentrated at a subset of inhibitory GABAergic synapses. Rat hippocampal neurons were fixed at 3 weeks in culture and immunostained for combinations of β-dystroglycan (β−DG, all panels,green) and either GABAARγ2 (A, red) and synapsin (A4,blue) or the synthetic enzyme for GABA, GAD, a marker of GABAergic terminals (B, red), or the inhibitory postsynaptic scaffolding protein gephyrin (C,red), or the excitatory postsynaptic marker SynGAP (D, red). β−Dystroglycan formed clusters colocalized with GABAARγ2, GAD, and gephyrin. Almost all β−dystroglycan clusters localized to inhibitory synapses (arrowheads, yellow puncta inA3, B2, C2;white puncta in A4), but not all inhibitory synapses contained concentrations of β−dystroglycan (arrows, red puncta in A3,B2, C2; pink puncta inA4). β−dystroglycan (D,arrowheads) was not detected at excitatory glutamatergic synapses labeled with SynGAP (D, arrows; note the absence of yellow puncta in D3). Scale bar, 10 μm.
The close association between β−dystroglycan and GABAAR, gephyrin, and GAD strongly suggests a selective accumulation of β−dystroglycan at inhibitory synapses and not excitatory synapses. This idea was confirmed by comparing the distribution of β−dystroglycan with that of SynGAP or GKAP, PSD-95-binding proteins that are selectively localized to excitatory glutamatergic synapses (Naisbitt et al., 1997; Chen et al., 1998; Kim et al., 1998). We observed SynGAP or GKAP clusters and β−dystroglycan clusters in distinct nonoverlapping distributions along dendrites (Fig. 1D) (and data not shown). Only 13 ± 4% of β−dystroglycan clusters colocalized with SynGAP. In contrast, 78 ± 3% of β−dystroglycan clusters colocalized with gephyrin. In these same dendrites, β−dystroglycan occupied only 0.9 ± 0.1% of the total surface area, SynGAP occupied 4.4 ± 3.3%, and gephyrin occupied 2.8 ± 0.2%. The apparent colocalization between β−dystroglycan and SynGAP could represent random overlap or a low level of true colocalization, as reported previously for PSD-95 with GAD (9.6% colocalization) in these cultures (Rao et al., 2000). However, the colocalization of β−dystroglycan with gephyrin is clearly nonrandom and is similar to that found previously for GABAAR with GAD (68% colocalization) (Rao et al., 2000). Thus β−dystroglycan is preferentially associated with inhibitory synapses in hippocampal neurons. Dystroglycan is the first identified adhesive macromolecule at mature GABAergic synapses.
Many clusters of GAD, gephyrin, and GABAARγ2 were not colocalized with detectable β−dystroglycan (Fig.1A–C, arrows) (for quantification, see Table 1), indicating that high concentrations of β−dystroglycan were present only at a subset of inhibitory synapses. β−Dystroglycan was found at GABAergic synapses on both pyramidal neurons and interneurons. Because GABAA receptors are pentameric heteroligomers derived from combinations of at least 17 different subunits (Hevers and Luddens, 1998), we tested whether β−dystroglycan might associate with a particular receptor composition. We found β−dystroglycan clusters colocalized with a subset of clusters of GABAAR subunits α1, α2, γ2, and β2/3 (Figs. 1, 3, 4) (and data not shown), indicating no particular association with any of these subunits.
Table 1.
Quantification of the developmental synaptic accumulation of GABAARγ2, gephyrin, and β-dystroglycan
| 1 week | 2 weeks | 3 weeks | 5 weeks | |
|---|---|---|---|---|
| Number of synaptic clusters/100 μm dendrite length | ||||
| GABAARγ2 subunit | 6 ± 1 | 18 ± 1 | 23 ± 2 | 32 ± 2 |
| Gephyrin | 6 ± 1 | 21 ± 2 | 29 ± 4 | 29 ± 2 |
| β-dystroglycan | 2 ± 0.5 | 4 ± 1 | 9 ± 1 | 12 ± 2 |
| % of synaptic GABAARγ2 colocalized with: | ||||
| Gephyrin | 64 ± 5 | 84 ± 3 | 86 ± 3 | 75 ± 3 |
| β-dystroglycan | 5 ± 2 | 14 ± 4 | 28 ± 4 | 35 ± 4 |
| % of postsynaptic marker colocalized with GABAARγ2 | ||||
| Gephyrin | n.d. | n.d. | 76 ± 3 | 82 ± 4 |
| β-dystroglycan | n.d. | n.d. | 70 ± 7 | 74 ± 5 |
Neurons were fixed after 1, 2, 3, or 5 weeks in culture and triple stained for synapsin, GABAARγ2, and either gephyrin or β-dystroglycan. The quantitative analysis was performed on digital images using Metamorph imaging software. Ten neurons from two independent experiments were analyzed for each combination of markers. Results are expressed as mean ± SEM; n.d., not determined.
Fig. 3.
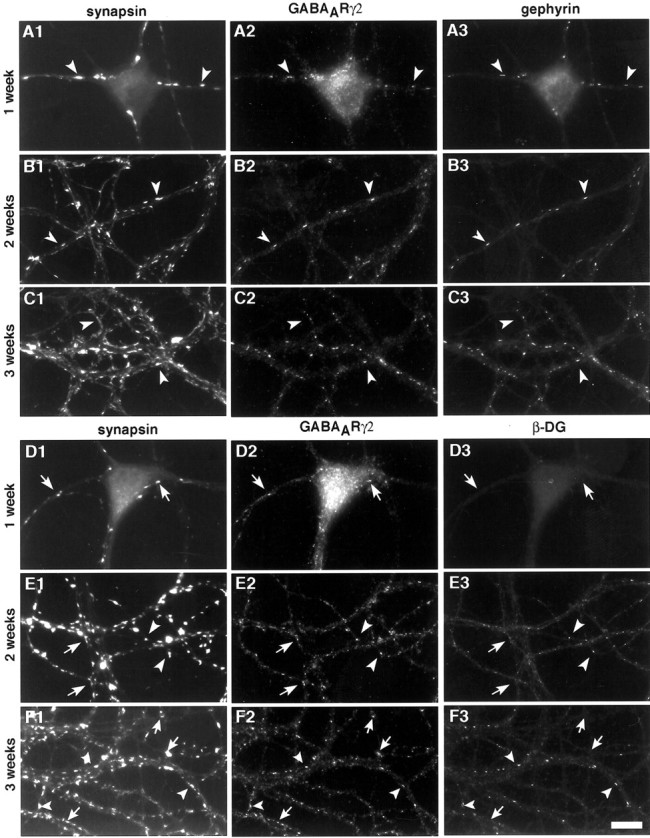
Gephyrin coclusters with GABAAR at synaptic sites very early during synaptogenesis, whereas β−dystroglycan accumulates at a subset of synaptic GABAAR clusters only late in development. Cultures were fixed at 1 (A, D), 2 (B, E), and 3 weeks (C, F) and triple-stained for synapsin (A1–F1), GABAARγ2 (A2–F2), and gephyrin (A3– C3) or β−dystroglycan (D3–F3). The number of gephyrin and GABAARγ2 clusters increased in parallel during in vitro development. At all stages in culture, GABAARγ2 clusters apposed to presynaptic sites were colocalized with gephyrin (arrowheads). At 1 week in culture, almost all postsynaptic GABAARγ2 clusters were devoid of detectable β−dystroglycan. At 2 weeks, β−dystroglycan was clustered at a few synaptic sites. The number of synaptic β−dystroglycan clusters increased between 2 and 3 weeks in culture. The proportion of GABAARγ2 coclustered with β−dystroglycan increased in parallel. Arrowheadsindicate GABAARγ2 colocalized with gephyrin or β−dystroglycan; arrows show GABAARγ2 clustering independently of β−dystroglycan. Scale bar, 10 μm.
Fig. 4.
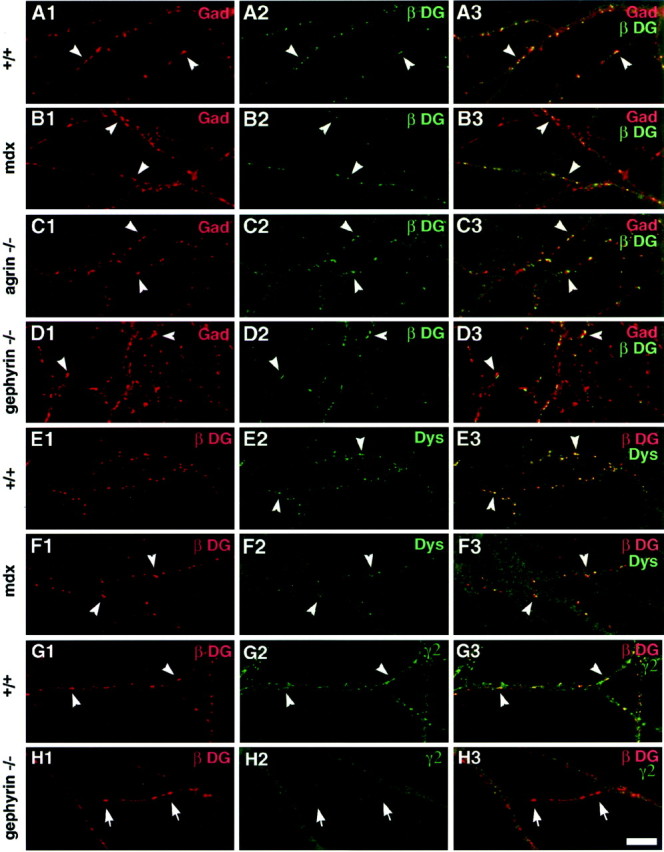
Agrin, dystrophin, and gephyrin are not required for localization of dystroglycan to GABA synapses. Individual cultures were grown from hippocampi of wild-type (A,E, G), mdx (B, F), agrin −/− (C), and gephyrin −/− (D, H) mice and analyzed after 17–19 d. Neurons were immunolabeled for combinations of GAD (red,A–D) and β−dystroglycan (green, A–D); β−dystroglycan (red, E,F) and dystrophin (green,E, F); or GABAARα2 (red, G, H) and β−dystroglycan (green, G,H). In mdx, agrin −/−, and gephyrin −/− neurons, clusters of β−dystroglycan apposed to GAD were observed as in wild-type neurons (yellow puncta in A3,B3, C3, D3). Even inmdx neurons, clusters of some dystrophin isoforms were still observed with the dys2 antibody (F2), and these colocalized with β−dystroglycan (yellowpuncta in F3). In wild-type neurons, β−dystroglycan always colocalized with GABAARγ2 (yellow puncta in G3), but in the absence of gephyrin, β−dystroglycan clusters were often found without punctate GABAARγ2 immunoreactivity (red puncta in H3). In all images,arrowheads indicate colocalization;arrows in H show β−dystroglycan clustering independently of GABAARγ2. Scale bar, 10 μm.
α−Dystroglycan, β−dystroglycan, and dystrophin colocalize at GABAergic synapses
We tested for the presence of other components of the DGC in the hippocampal neurons and detected α−dystroglycan and dystrophin (Fig.2) but not utrophin (data not shown). α−Dystroglycan and dystrophin appeared to colocalize with β−dystroglycan (Fig. 2A,B). Quantitation yielded colocalization values of 66 ± 3 and 63 ± 4% for β−dystroglycan with α−dystroglycan and dystrophin, respectively. The concentration of α−dystroglycan at GABAergic synapses was confirmed visually by colocalization with GABAARα1 and synapsin (Fig. 2C). These results suggest that α−dystroglycan, β−dystroglycan, and dystrophin form a complex at GABAergic synaptic sites.
Fig. 2.

α-Dystroglycan and dystrophin form part of the DGC at GABAergic synases. Hippocampal neurons were fixed at 3–5 weeks in culture and immunostained for combinations of β-dystroglycan (A, red) and α-dystroglycan (A, green), β-dystroglycan (B, red), and dystrophin (B, green), or α-dystroglycan (C, green), GABAARα1 (C, red), and synapsin (C, blue). α-Dystroglycan and dystrophin completely colocalized with β-dystroglycan (arrowheads, yellow puncta inA3, B3). α-Dystroglycan also colocalized with GABAARα1 (arrowheads, yellow puncta inC3) at synapses (white puncta inC4) on the interneuron shown in C. Note the absence of α-dystroglycan at some synaptic GABAARα1 (arrows, pinkpuncta in C4). Scale bar, 10 μm.
Dystroglycan accumulates at GABAergic synapses late in development, after accumulation of synaptic vesicles, gephyrin, and GABAARs
We next determined the developmental time course of association of β−dystroglycan with GABAergic synapses and compared this with the time course of synaptic accumulation of gephyrin. We used triple-label immunocytochemistry with antibodies to synapsin, GABAARγ2, and either gephyrin or β−dystroglycan and analyzed the distribution patterns both qualitatively (Fig. 3) and quantitatively (Table 1).
At 1 week in culture, a few neurons displayed GABAARγ2 and gephyrin clusters apposed to presynaptic terminals around somata and dendrites (Fig. 3A). The density of postsynaptic gephyrin and GABAARγ2 clusters increased in parallel, rapidly during the second week and more slowly during the third week in culture (Fig. 3B,C). Most synaptic gephyrin and GABAARγ2 clusters colocalized at all stages analyzed; the colocalization values in Table 1 represent an underestimate because some clusters for each antigen fell below the threshold for quantitation. The early association of gephyrin with developing synaptic GABAAR is consistent with a function of gephyrin in synapse assembly and initial GABAAR clustering.
The developmental localization profile was very different for β−dystroglycan. At 1 week in culture, very little β−dystroglycan immunoreactivity was detected, and synaptic GABAARγ2 clusters were rarely colocalized with β−dystroglycan (Fig. 3D). Over the next 2 weeks, progressively more β−dystroglycan clusters were detected, and more GABAARγ2 clusters were colocalized with β−dystroglycan (Fig.3E,F). Quantitative analysis indicated a steady continuous developmental increase in synaptic β−dystroglycan for at least 2 additional weeks (to 5 weeks in vitro; data not shown), in contrast to the earlier rapid increase and later plateau levels of synaptic gephyrin. However, even at this late stage of development many synaptic GABAARγ2 clusters were devoid of detectable β−dystroglycan. Thus dystroglycan appeared to be associated with only a subset of mature GABAergic synapses. It is not clear whether β−dystroglycan would eventually associate at high level with all mature GABAergic synapses or whether it associates with a distinct subpopulation. It is also possible that dystroglycan concentrates at all GABAergic synapses, but the levels at some synapses may fall below the detection threshold.
Genetic analysis of the requirements for synaptic clustering of the dystrophin glycoprotein complex
We tested major dystroglycan binding partners and components of GABAergic synapses for their role in synaptic localization of dystroglycan. We first analyzed hippocampal cultures frommdx mice, which lack the full-length form of dystrophin, a major intracellular binding partner for dystroglycan. Visually,mdx neurons exhibited normal accumulations of β-dystroglycan at GAD-labeled synapses (Fig.4B) compared with wild type (Fig. 4A). Immunofluorescence with a monoclonal antibody that recognizes both full-length and short forms of dystrophin was greatly reduced, although some immunoreactivity was colocalized with β−dystroglycan (Fig. 4F). Thus some short forms of dystrophin as well as full-length dystrophin are complexed with dystroglycan at GABAergic synapses. Furthermore, full-length dystrophin is not essential for assembly of the DGC at GABAergic synapses.
Agrin is a major dystroglycan-binding protein expressed by central neurons as well as at the NMJ. We showed previously that agrin isoforms containing the z splice insert, which are required for neuromuscular synaptogenesis, are not required for synaptic accumulation of GABAARβ2/3 opposite GAD-positive terminals (Serpinskaya et al., 1999). However, some z-minus agrin was expressed by the hypomorphic mutants used in our previous study, and it is known that z-minus agrin binds more strongly to dystroglycan than does z-plus agrin (Sugiyama et al., 1994). We therefore analyzed hippocampal neurons from a newly generated agrin mutant that is a complete null for all forms of agrin (Lin et al., 2001). As reported previously, synapse formation proceeded on schedule in agrin −/− mutants. Specifically, agrin −/− neurons showed normal accumulations of β−dystroglycan at GAD-labeled synapses (Fig. 4C). Thus, although agrin binds dystroglycan, it is not required for accumulation of the DGC at GABAergic synapses.
The postsynaptic anchoring protein gephyrin appears at synapses earlier than dystroglycan and is reportedly essential for synaptic accumulation of GABAAR α2 and γ2 subunits in hippocampal culture (Kneussel et al., 1999). We cultured embryonic hippocampal neurons from gephyrin −/− mice for 17–19 d and then assayed for dystroglycan and dystrophin localization to GABA synapses (Fig.4D,H) (and data not shown). Accumulation of GAD-positive GABAergic presynaptic specializations, at sites distinct from excitatory postsynaptic specializations, occurred normally in the gephyrin −/− neurons. We found that gephyrin was not required for accumulation of dystroglycan or dystrophin at GABAergic synapses (Fig. 4D) (and data not shown). β−Dystroglycan immunoreactivity in the gephyrin −/− neurons was indistinguishable from wild type (Fig.4A,D). In the absence of gephyrin, many β−dystroglycan clusters were found lacking detectable immunoreactivity for GABAARγ2, a finding not common in wild-type neurons (Fig. 4G,H). In addition, low levels of GABAARγ2 clustering persist, opposing some GAD-positive terminals in the absence of gephyrin (data not shown), and in these cases β−dystroglycan colocalized with some GABAARγ2 clusters. Thus the DGC appears to be completely independent of gephyrin: it neither requires gephyrin for accumulation at GABA synapses nor compensates for the loss of gephyrin, at least not by any change in association of dystrolgycan with GABAAR clusters.
Dystroglycan is required for association of dystrophin but not for differentiation of GABAergic synapses
Although a DGC containing short dystrophin isoforms and/or utrophin can form in the absence of full-length dystrophin (Blake and Kroger, 2000), it is thought that no DGC can form in the absence of dystroglycan. Thus to test more directly the potential function of the DGC in GABAergic synaptogenesis, we analyzed dystroglycan −/− neurons in culture (Figs. 5,6). Because dystroglycan −/− mice die early in embryogenesis before development of the brain (Williamson et al., 1997), we used a conditional dystroglycan mutant (Saito et al., 2001; Moore et al., 2002). In these mice, lox sites have been inserted into introns that flank the first coding exon of the dystroglycan gene. These insertions have no detectable effect on dystroglycan expression but act as recognition sites for Cre recombinase. Introduction of this recombinase leads to excision of the first exon, generating a protein-null mutant allele. We therefore prepared cultures from hippocampi of conditional mutant heterozygotes and then treated the cultures with adenovirus-Cre to inactivate dystroglycan. For the experiments described here, we wanted to assess roles of dystroglycan in synaptogenesis per se, separate from possible roles in earlier steps, such as differentiation or process outgrowth. We therefore introduced the adenovirus-Cre at 7 d in culture, subsequent to attachment of neurons to the substrate and initial outgrowth of axons and dendrites but before the time at which dystroglycan is detected at GABA synapses in control neurons. As controls, we used neurons that bore the conditional allele but were not treated with adenovirus and virus-treated neurons from wild-type mice.
Fig. 5.
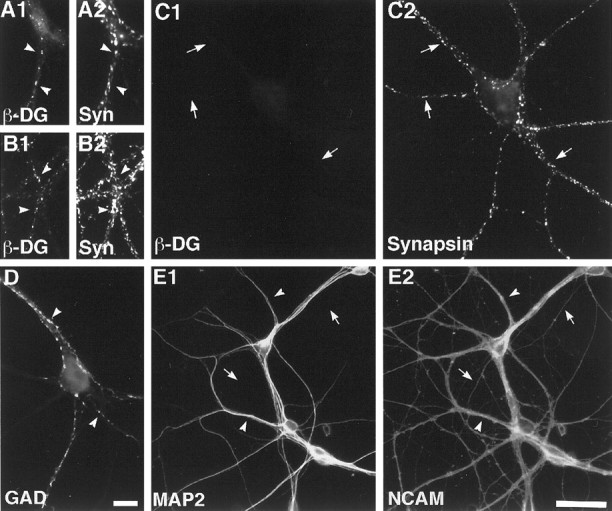
Dystroglycan is not required for the differentiation of neuronal dendritic and axonal compartments and synaptic terminals. Hippocampal neurons were cultured from wild-type (A) or DG lox/lox (B–E) mice, incubated at 7 d in culture with adenovirus-Cre (A, C–E) or not (B), and analyzed at 17–19 d. Neurons were immunolabeled for combinations of β−dystroglycan (A1,B1, C1) and synapsin (A2,B2, C2), GAD (D), MAP2 (E1), and NCAM (E2). Synaptic β−dystroglycan clusters (arrowheads) were detected in wild-type neurons treated with adenovirus-Cre (A) and in DG lox/lox neurons not treated with virus (B), but not in most DG lox/lox neurons treated with adenovirus-Cre (C, arrows; representative of 94% of the neurons). Presynaptic terminals (C, arrows) including GABA synapses (D, arrowheads) formed normally in the absence of dystroglycan. Dystroglycan was also not required to elaborate dendrites (arrowheads) and axons (E, arrows). Scale bar, 10 μm.
Fig. 6.
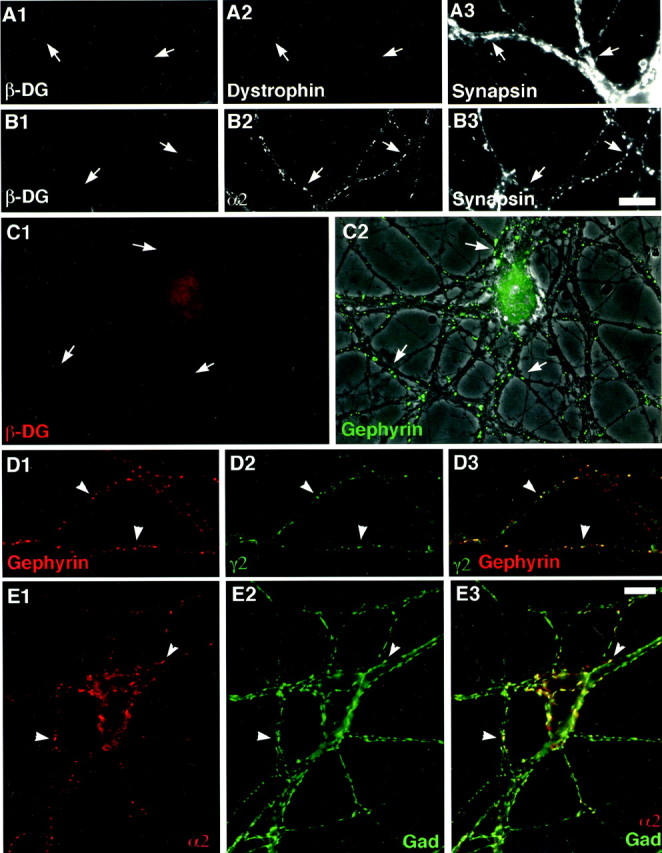
Dystroglycan is essential for association of dystrophin but not for differentiation of GABA synapses. Hippocampal neurons were cultured from DG lox/lox mice, incubated at 7 d in culture with adenovirus-Cre, and analyzed at 17–19 d. Neurons were immunolabeled for combinations of β−dystroglycan (A1), dystrophin (A2), and synapsin (A3); β−dystroglycan (B1), GABAARα2 (B2), and synapsin (B3); β−dystroglycan (red,C1) and gephyrin (green,C2) overlaid on a phase-contrast image; gephyrin (red, D) and GABAARγ2 (green, D); or GABAARα2 (red, E) and GAD (green, E). In the absence of dystroglycan, dystrophin did not form synaptic clusters (A), but GABAAR subunits, gephyrin, GAD, and synapsin formed colocalized clusters (B–E) around somata and dendrites (C) indicating GABAergic synaptic differentiation. Arrows show absence of β-dystroglycan (A–C) and dystrophin (A) at synapses and GABAAR (B) and gephyrin (C) clustering in the absence of β-dystroglycan; arrowheads indicate GABAAR colocalized with gephyrin (D) and GAD (E). Scale bar, 10 μm.
Adenovirus-Cre treatment (at MOI 10) of wild-type neurons did not affect dystroglycan expression (Fig. 5A), whereas the same treatment effectively excised dystroglycan from the vast majority of cells in culture [6% of cells exhibited dystroglycan clusters after adenovirus-Cre treatment vs 84% in sister cultures without virus-mediated excision (Fig. 5, C vs B)]. The absence of dystroglycan from neurons after excision at 1 week in culture is consistent with the result (Fig. 3) that these neurons have accumulated little if any dystroglycan at earlier times. The dystroglycan −/− neurons continued to elaborate axons and dendrites for an additional 10–12 d (analysis at 17–19 d in vitro) (Fig. 5E). Moreover, the neurons formed numerous synapsin-rich presynaptic nerve terminals, including some that were GABAergic (GAD-positive) (Fig. 5C,D). Thus, although we cannot rule out the possibility that low levels of dystroglycan are essential for initiation of neuronal differentiation, formation of elaborate processes and differentiation of nerve terminals can occur in its absence.
In view of these results, we were able to use the Cre-expressing DG lox/lox cultures to ask whether dystroglycan was necessary for formation or maintenance of the postsynaptic membrane at GABAergic synapses. We observed no dystrophin clusters in Cre-treated neurons (Fig. 6A) except for very few colocalized with the remaining dystroglycan. This defect was more striking than that documented above for mdx mice, indicating that dystroglycan is necessary for concentration of both full-length and short forms of dystrophin at GABAergic synapses. However, no other differences in GABAergic synaptic differentiation were observed. Cre-excised DG lox/lox cultures formed colocalized accumulations of synaptic vesicles, GAD, gephyrin, and GABAAR α1, α2 and γ2 (Fig. 6B–E) (α1 data not shown). The percentage of DG lox/lox neurons that exhibited synaptic clusters of GABAAR α2 was 94% without adenovirus treatment and 89% after adenovirus-Cre excision (data from the same neurons described above that showed the 78% loss of dystroglycan immunoreactivity). The amount of adenovirus required to effectively excise dystroglycan resulted in some degree of toxicity for wild-type neurons as well as DG lox/lox. Thus rigorous quantitative analyses were precluded, and we cannot rule out the possibility of a small reduction in the density of synaptic GABAAR clusters attributable to loss of dystroglycan. Nonetheless, these results indicate that dystroglycan is not a required signaling component for the fundamental aspects of GABAergic synaptogenesis, formation of GABAAR clusters opposite GABAergic terminals.
DISCUSSION
We report here four major results concerning the regulation and function of the DGC in central neurons. First, α-dystroglycan, β-dystroglycan, and dystrophin are concentrated at a subset of central neuron inhibitory GABAergic synapses. Dystroglycan is the first adhesive macromolecule identified at mature GABA synapses. Second, dystroglycan accumulates at GABAergic synapses late in development, after clustering of synaptic vesicles, GAD, gephyrin, and GABAAR. Third, gephyrin, agrin, and full-length dystrophin are all dispensable for localization of dystroglycan to GABA synapses. Fourth, dystroglycan −/− neurons develop GABAergic synapses containing clusters of GABAAR and gephyrin opposite GAD-labeled terminals. The only detectable defect in the absence of dystroglycan was the absence of dystrophin clusters at GABA synapses. Thus the DGC is not essential for assembly of GABAergic synapses but is likely to function in synaptic modulation.
Molecular assembly of GABAergic synapses
Our data indicate the following sequence of GABAergic synaptic assembly: presynaptic vesicle clusters, then gephyrin and GABAAR, and then the DGC. In cultured spinal neurons, clustering of the GABAAR opposite inhibitory terminals was observed before the accumulation of gephyrin (Dumoulin et al., 2000). We have also observed some synaptic GABAAR clusters in gephyrin −/− hippocampal neurons (our unpublished data). Thus, in contrast to the early report of Kneussel et al. (1999) of a complete absence of GABAAR clusters in gephyrin −/− hippocampal neurons, our data agree with the recent analyses of retinal cultures and spinal cord (Fischer et al., 2000; Kneussel et al., 2001) indicating the presence of some albeit reduced GABAAR clusters in gephyrin −/− neurons. Interestingly, analysis of cortical cultures from GABAAR γ2 −/− mice revealed a large reduction in synaptic clustering of gephyrin as well as GABAAR α1 and α2 (Essrich et al., 1998). However, direct binding between gephyrin and GABAAR has not been observed (Meyer et al., 1995), and the presence of gephyrin at a synapse is not sufficient to induce accumulation of GABAAR (Levi et al., 1999). One attractive idea was that gephyrin organized the GABAergic postsynaptic membrane via interaction with the DGC, but our results have excluded this possibility. Thus the precise role of gephyrin in GABAergic synaptogenesis remains to be defined, particularly how gephyrin functions to induce or maintain the normal synaptic density of some GABAAR subunits and how this contributes to synaptic function.
The DGC detectably associated with only a subset of GABAergic synapses and only late in development. The DGC complex at GABA synapses includes α-dystroglycan, β-dystroglycan, full-length dystrophin, and dystrophin short forms (Figs. 2, 4). Full-length dystrophin has also been found at GABAergic synapses in hippocampal tissue (Knuesel et al., 1999). In mdx neurons lacking full-length dystrophin, dystroglycan and some dystrophin short forms still localized to GABAergic synapses (Fig. 4). In contrast, in neurons lacking dystroglycan, dystrophin isoforms were not detected at GABA synapses (Fig. 6). Thus dystroglycan is an essential component for formation of the DGC complex at GABA synapses, but dystrophin is not. Furthermore, neither agrin nor gephyrin was required for assembly of the DGC at GABA synapses (Fig. 4). We could find no evidence of a genetic interaction between gephyrin and dystroglycan in the sense that either assembled at GABA synapses in the absence of the other, and neither could compensate for loss of the other. Dystroglycan did not rescue GABAAR clustering in the absence of gephyrin, and gephyrin did not rescue dystrophin clustering in the absence of dystroglycan.
Function of the dystrophin-associated glycoprotein complex at GABAergic synapses
The presence of the adhesive macromolecule dystroglycan at GABA synapses raised the intriguing possibility that it might function as a key trans-neuronal signal for synaptogenesis. Although we could detect dystroglycan accumulated at only a subset of mature synapses, it might be present at lower levels at developing GABAergic synapses. The findings that α-dystroglycan binds agrin and α- and β-neurexins via their extracellular domains (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Sugita et al., 2001) further supported this possibility. Agrin is essential for synaptogenesis at the NMJ (Sanes and Lichtman, 1999; Lin et al., 2001), and acute inhibition of agrin expression alters the morphological development of hippocampal neurons in culture (Bose et al., 2000). The apparent absence of phenotype in hippocampal cultures from agrin mutant mice (Fig. 4) (Serpinskaya et al., 1999) could result from compensatory mechanisms. Neurexins are a large family of alternatively spliced neuron-specific surface proteins implicated in synaptic specificity (Missler et al., 1998). However, we find here that dystroglycan is not essential for GABAergic synaptogenesis; GABAAR clustered opposite GABA terminals in the absence of dystroglycan (Fig.6). These results do not rule out roles for agrin or neurexin, both of which have alternative receptors. For example, β-neurexins also bind neuroligin, an excitatory postsynaptic protein that can induce presynaptic specializations in contacting axons (Song et al., 1999;Scheiffele et al., 2000). Moreover, it is still possible that dystroglycan is required for genesis of a small subset of GABA synapses; better methods for excising dystroglycan without long-term side effects will be required to test this possibility. Compensatory mechanisms that may mask a function for dystroglycan seem unlikely, because dystroglycan was excised after 1 week in culture rather than from the onset of development. Functional redundancy may occur for other components of the DGC, but this also seems unlikely for dystroglycan, as demonstrated with respect to its roles in non-neuronal cells (Williamson et al., 1997).
The other possibility, which we favor, is that dystroglycan plays a modulatory rather than an organizing function at many GABAergic synapses. The cytoplasmic C terminus of β-dystroglycan interacts with the Src homology 3 domains of Grb2, an adaptor protein involved in signal transduction and cytoskeletal reorganization (Yang et al., 1995). Evidence from brain synaptosomes indicates a Grb2-mediated interaction between β-dystroglycan and focal adhesion kinase, a tyrosine kinase involved in intracellular transduction pathways (Cavaldesi et al., 1999). Furthermore, Grb2 and dystrophin compete for binding to β-dystroglycan (Russo et al., 2000). These interactions suggest that dystroglycan may be part of a dynamically regulated signal transduction pathway.
Taken together, our data suggest that the DGC forms a transmembrane linkage containing dystrophin, dystrophin short forms, β-dystroglycan, and α-dystroglycan, which binds agrin and α- and β-neurexins. The simplest model is that neurexins and agrin are on the presynaptic side, and dystroglycan and dystrophin are postsynaptic at GABA synapses. However, there is little direct evidence (Lidov et al., 1990) to support this model, and the possibility that the DGC may be presynaptic needs to be tested by ultrastructural localization. Similar to its function at the NMJ (Adams et al., 2000; Grady et al., 2000; Jacobson et al., 2001), the DGC may be involved in regulating GABAAR stability or other aspects of postsynaptic response. Alternatively, similar to its function in photoreceptors (Blake and Kroger, 2000), the DGC may be involved in regulating GABA release. Finally, given the transmembrane nature of the complex, the DGC may be involved in regulating coordinated aspects of presynaptic and postsynaptic plasticity at GABA synapses.
Footnotes
This work was supported by National Institutes of Health Grants NS34448 and NS33184. M.D.H. was supported by a National Research Service Award grant, and K.P.C is an investigator of the Howard Hughes Medical Institute. We thank Dr. Jean-Marc Fritschy for gifts of antibodies and Huaiyang Wu for excellent technical assistance.
Correspondence should be addressed to Dr. Ann Marie Craig, Department of Anatomy and Neurobiology, Washington University School of Medicine, 660 South Euclid, Campus Box 8108, St. Louis, MO 63110. E-mail:acraig@pcg.wustl.edu.
M. D. Henry's present address: Millenium Pharmaceuticals Inc., Cambridge, MA 02139.
REFERENCES
- 1.Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anton M, Graham FL. Site-specific recombination mediated by an adenovirus vector expressing the Cre recombinase protein: a molecular switch for control of gene expression. J Virol. 1995;69:4600–4606. doi: 10.1128/jvi.69.8.4600-4606.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benson DL, Cohen PA. Activity-independent segregation of excitatory and inhibitory synaptic terminals in cultured hippocampal neurons. J Neurosci. 1996;16:6424–6432. doi: 10.1523/JNEUROSCI.16-20-06424.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blake DJ, Kroger S. The neurobiology of Duchenne muscular dystrophy: learning lessons from muscle? Trends Neurosci. 2000;23:92–99. doi: 10.1016/s0166-2236(99)01510-6. [DOI] [PubMed] [Google Scholar]
- 5.Blake DJ, Hawkes R, Benson MA, Beesley PW. Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol. 1999;147:645–658. doi: 10.1083/jcb.147.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bose CM, Qiu D, Bergamaschi A, Gravante B, Bossi M, Villa A, Rupp F, Malgaroli A. Agrin controls synaptic differentiation in hippocampal neurons. J Neurosci. 2000;20:9086–9095. doi: 10.1523/JNEUROSCI.20-24-09086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: a heteromeric complex related to the dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- 8.Butler MH, Hayashi A, Ohkoshi N, Villmann C, Becker CM, Feng G, De Camilli P, Solimena M. Autoimmunity to gephyrin in stiff-man syndrome. Neuron. 2000;26:307–312. doi: 10.1016/s0896-6273(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 9.Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 10.Cavaldesi M, Macchia G, Barca S, Defilippi P, Tarone G, Petrucci TC. Association of the dystroglycan complex isolated from bovine brain synaptosomes with proteins involved in signal transduction. J Neurochem. 1999;72:1648–1655. doi: 10.1046/j.1471-4159.1999.721648.x. [DOI] [PubMed] [Google Scholar]
- 11.Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- 12.Craig AM, Lichtman JW. Synapse formation and maturation. In: Cowan WM, Sudhof TC, Stevens CF, editors. Synapses. The Johns Hopkins UP; Baltimore: 2001. pp. 571–612. [Google Scholar]
- 13.Craig AM, Blackstone CD, Huganir RL, Banker G. Selective clustering of glutamate and gamma-aminobutyric acid receptors opposite terminals releasing the corresponding neurotransmitters. Proc Natl Acad Sci USA. 1994;91:12373–12377. doi: 10.1073/pnas.91.26.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deconinck AE, Potter AC, Tinsley JM, Wood SJ, Vater R, Young C, Metzinger L, Vincent A, Slater CR, Davies KE. Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J Cell Biol. 1997;136:883–894. doi: 10.1083/jcb.136.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dumoulin A, Levi S, Riveau B, Gasnier B, Triller A. Formation of mixed glycine and GABAergic synapses in cultured spinal cord neurons. Eur J Neurosci. 2000;12:3883–3892. doi: 10.1046/j.1460-9568.2000.00271.x. [DOI] [PubMed] [Google Scholar]
- 16.Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- 17.Feng G, Tintrup H, Kirsch J, Nichol MC, Kuhse J, Betz H, Sanes JR. Dual requirement for gephyrin in glycine receptor clustering and molybdoenzyme activity. Science. 1998;13:1321–1324. doi: 10.1126/science.282.5392.1321. [DOI] [PubMed] [Google Scholar]
- 18.Fischer F, Kneussel M, Tintrup H, Haverkamp S, Rauen T, Betz H, Wassle H. Reduced synaptic clustering of GABA and glycine receptors in the retina of the gephyrin null mutant mouse. J Comp Neurol. 2000;427:634–648. doi: 10.1002/1096-9861(20001127)427:4<634::aid-cne10>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 19.Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;14:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- 20.Garner CC, Nash J, Huganir RL. PDZ domains in synapse assembly and signalling. Trends Cell Biol. 2000;10:274–280. doi: 10.1016/s0962-8924(00)01783-9. [DOI] [PubMed] [Google Scholar]
- 21.Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 22.Gennarini G, Hirn M, Deagostini-Bazin H, Goridis C. Studies on the transmembrane disposition of the neural cell adhesion molecule N-CAM. The use of liposome-inserted radioiodinated N-CAM to study its transbilayer orientation. Eur J Biochem. 1984;142:65–73. doi: 10.1111/j.1432-1033.1984.tb08251.x. [DOI] [PubMed] [Google Scholar]
- 23.Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1998. pp. 339–370. [Google Scholar]
- 24.Grady RM, Merlie JP, Sanes JR. Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol. 1997;136:871–882. doi: 10.1083/jcb.136.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- 26.Graham FL, Prevec L. Methods for construction of adenovirus vectors. Mol Biotechnol. 1995;3:207–220. doi: 10.1007/BF02789331. [DOI] [PubMed] [Google Scholar]
- 27.Henry MD, Campbell KP. Dystroglycan inside and out. Curr Opin Cell Biol. 1999;11:602–607. doi: 10.1016/s0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 28.Hevers W, Luddens H. The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol Neurobiol. 1998;18:35–86. doi: 10.1007/BF02741459. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman EP, Kunkel LM. Dystrophin abnormalities in Duchenne/Becker muscular dystrophy. Neuron. 1989;2:1019–1029. doi: 10.1016/0896-6273(89)90226-2. [DOI] [PubMed] [Google Scholar]
- 30.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 31.Jacobson C, Cote PD, Rossi SG, Rotundo RL, Carbonetto S. The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J Cell Biol. 2001;152:435–450. doi: 10.1083/jcb.152.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- 33.Kim TW, Wu K, Xu JL, Black IB. Detection of dystrophin in the postsynaptic density of rat brain and deficiency in a mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 1992;89:11642–11644. doi: 10.1073/pnas.89.23.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein R. Excitatory Eph receptors and adhesive ephrin ligands. Curr Opin Cell Biol. 2001;13:196–203. doi: 10.1016/s0955-0674(00)00197-6. [DOI] [PubMed] [Google Scholar]
- 35.Kneussel M, Brandstatter JH, Laube B, Stahl S, Muller U, Betz H. Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J Neurosci. 1999;19:9289–9297. doi: 10.1523/JNEUROSCI.19-21-09289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kneussel M, Brandstatter JH, Gasnier B, Feng G, Sanes JR, Betz H. Gephyrin-independent clustering of postsynaptic GABA(A) receptor subtypes. Mol Cell Neurosci. 2001;17:973–982. doi: 10.1006/mcne.2001.0983. [DOI] [PubMed] [Google Scholar]
- 37.Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM. Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur J Neurosci. 1999;11:4457–4462. doi: 10.1046/j.1460-9568.1999.00887.x. [DOI] [PubMed] [Google Scholar]
- 38.Levi S, Chesnoy-Marchais D, Sieghart W, Triller A. Synaptic control of glycine and GABA(A) receptors and gephyrin expression in cultured motoneurons. J Neurosci. 1999;19:7434–7449. doi: 10.1523/JNEUROSCI.19-17-07434.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature. 1990;348:725–728. doi: 10.1038/348725a0. [DOI] [PubMed] [Google Scholar]
- 40.Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- 41.Meyer G, Kirsch J, Betz H, Langosch D. Identification of a gephyrin binding motif on the glycine receptor beta subunit. Neuron. 1995;15:563–572. doi: 10.1016/0896-6273(95)90145-0. [DOI] [PubMed] [Google Scholar]
- 42.Missler M, Fernandez-Chacon R, Sudhof TC. The making of neurexins. J Neurochem. 1998;71:1339–1347. doi: 10.1046/j.1471-4159.1998.71041339.x. [DOI] [PubMed] [Google Scholar]
- 43.Moore SA, Saito F, Chen J, Michele DE, Henry M, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature, in press. [DOI] [PubMed]
- 44.Moukhles H, Carbonetto S. Dystroglycan contributes to the formation of multiple dystrophin-like complexes in brain. J Neurochem. 2001;78:824–834. doi: 10.1046/j.1471-4159.2001.00466.x. [DOI] [PubMed] [Google Scholar]
- 45.Naisbitt S, Kim E, Weinberg RJ, Rao A, Yang FC, Craig AM, Sheng M. Characterization of guanylate kinase-associated protein, a postsynaptic density protein at excitatory synapses that interacts directly with postsynaptic density-95/synapse-associated protein 90. J Neurosci. 1997;17:5687–5696. doi: 10.1523/JNEUROSCI.17-15-05687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rao A, Cha EM, Craig AM. Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J Neurosci. 2000;20:8344–8353. doi: 10.1523/JNEUROSCI.20-22-08344.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russo K, Di Stasio E, Macchia G, Rosa G, Brancaccio A, Petrucci TC. Characterization of the beta-dystroglycan-growth factor receptor 2 (Grb2) interaction. Biochem Biophys Res Commun. 2000;274:93–98. doi: 10.1006/bbrc.2000.3103. [DOI] [PubMed] [Google Scholar]
- 49.Saito F, Moore SA, Henry MD, Messing A, Cohn RD, Williamson RA, Wrabetz L, Feltri ML, Campbell KP. Schwann cell specific ablation of dystroglycan gene: a role for dystroglycan/laminin interaction in peripheral nervous system. Soc Neurosci Abstr. 2001;27:105.6. [Google Scholar]
- 50.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 51.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 52.Serpinskaya AS, Feng G, Sanes JR, Craig AM. Synapse formation by hippocampal neurons from agrin-deficient mice. Dev Biol. 1999;205:65–78. doi: 10.1006/dbio.1998.9112. [DOI] [PubMed] [Google Scholar]
- 53.Sheng M, Pak DT. Glutamate receptor anchoring proteins and the molecular organization of excitatory synapses. Ann NY Acad Sci. 1999;868:483–493. doi: 10.1111/j.1749-6632.1999.tb11317.x. [DOI] [PubMed] [Google Scholar]
- 54.Song JY, Ichtchenko K, Sudhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sugita S, Saito F, Tang J, Satz J, Campbell K, Sudhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sugiyama J, Bowen DC, Hall ZW. Dystroglycan binds nerve and muscle agrin. Neuron. 1994;13:103–115. doi: 10.1016/0896-6273(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 57.Tian M, Jacobson C, Gee SH, Campbell KP, Carbonetto C, Jucker M. Dystroglycan in the cerebellum is a laminin α2-chain binding protein at the glial-vascular interface and is expressed in Purkinje cells. Eur J Neurosci. 1997;8:2739–2747. doi: 10.1111/j.1460-9568.1996.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 58.Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- 59.Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- 60.Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 2001;104:311–324. doi: 10.1016/s0306-4522(01)00092-6. [DOI] [PubMed] [Google Scholar]