

Abstract
Stimulation of the brain CCK2 receptor by the C-terminal octapeptide CCK8 of cholecystokinin (CCK) negatively modulates opioid responses. This suggests the existence of physiologically relevant interactions between endogenous CCK and opioid peptides, opening new perspectives particularly in the treatment of pain or drug addiction. CCK2 receptor-deficient mice were used to analyze the incidence of this gene invalidation on opioid system. Compared with wild-type mice, mutants exhibited the following: (1) a hypersensitivity to the locomotor activity induced by inhibitors of enkephalin catabolism or by morphine; (2) a spontaneous hyperalgesia to thermal nociceptive stimulus, which was reversed by previous administration of the NMDA antagonist MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], and a large reduction in analgesic effects of endogenous or exogenous opioids; and (3) a more severe withdrawal syndrome after chronic morphine treatment. As expected, stimulation of μ, δ, and D2 receptors on brain tissue of wild-type animals induced a dose-dependent decrease in adenylate cyclase activity, whereas a striking mirror effect was observed in mutants. All of these results suggest that the absence, in knock-out mice, of the negative feedback control on the opioid system, normally performed out by CCK2 receptor stimulation, results in an upregulation of this system. These biochemical and pharmacological results demonstrate the critical role played by CCK2receptors in opioid-dependent responses.
Keywords: cholecystokinin, opioid, mutant mice, adenylyl cyclase, binding, behavior
The opioid peptides, in particular enkephalins, β-endorphin, and dynorphin, are involved in pain perception, cognitive functions, affective behaviors, and locomotion (for review, see Vaccarino et al., 1999). These endogenous effectors induce their biological effects by interacting with three major classes of targets, designated μ, δ, and κ receptors, which are widely distributed centrally and peripherally (Mansour et al., 1995). The physiological actions of opioid peptides are regulated by a number of neuromodulators among them, the sulfated C-terminal octapeptide (CCK8) of cholecystokinin (CCK) plays a crucial role (for review, see Cesselin, 1995; Wiesenfeld-Hallin et al., 1999). CCK8, the predominant form found in the CNS (Rehfeld et al., 1985), interacts with two different receptors, CCK1, which is abundant in peripheral tissues, and CCK2, which is the major type present in brain (Wank, 1995; Noble et al., 1999). Anatomical studies have shown that the distribution of CCK8 and CCK receptors is the same as that of endogenous opioid peptides and their receptors in several brain regions (for review, see Stengaard-Pedersen and Larsson, 1981; Gall et al., 1987; Skinner et al., 1997; Zhang et al., 2000). The CCK receptors modulate the opioid system in physiological processes, such as the control of pain or modulation of mood, including emotional and/or motivational responses (Crawley and Corwin, 1994; Daugé and Roques, 1995). CCK2receptor stimulation reduces the analgesic action of morphine or the endogenous opioid peptides (Noble et al., 1993) protected by the enkephalin-degrading enzyme inhibitor RB 101 ([N-[(R,S)]-2-benzyl-3[(S)(2-amino-4-methyl-thio)butyl-dithio]-1-oxo-propyl]-l-phenyl-alanine benzyl ester) (Fournié-Zaluski et al., 1992), which was shown to significantly increase the extracellular levels of enkephalins in brain structures (Daugé et al., 1996). Accordingly, CCK2 antagonists facilitate opioid-induced analgesia (Wiesenfeld-Hallin et al., 1990; Valverde et al., 1994) and antidepressant-like effects (Smadja et al., 1995) and increase the potency of RB 101 to reduce the severity of naloxone-precipitated withdrawal syndromes in rats that are chronically treated with morphine (for review, see Noble et al., 1999). Conversely, opioids stimulate CCK release via their action on δ receptors, which in-turn negatively modulate opiate neurotransmission by activating CCK2 receptors (Noble et al., 1993). Numerous actions of opioids are reversed by dopamine antagonists, in agreement with the presence of opioid and dopamine targets on the same neurons, for example in mesolimbic and striatopallidal pathways (Van der Kooy et al., 1986; Manzanedo et al., 1999; Svingos et al., 1999).
Mice lacking CCK2 receptors (Nagata et al., 1996) provide a unique tool to investigate the counteractions between CCK and opioid systems by using complementary pharmacological and biochemical approaches. Behavioral responses involving opioid receptor activation, such as locomotion, analgesia, and withdrawal syndrome, were modified in mutant mice without observed changes in the binding parameters of μ and δ opioid receptors. In addition, the amount of endogenous opioids in the whole brain of these animals was significantly increased, as suggested by the reduction in the in vivobinding of the opioid antagonist [3H]diprenorphine in knock-out mice. Finally, the adenylyl cyclase activity coupled to opioid or dopamine receptors, which is decreased in wild-type animals after activation of these targets, was strongly enhanced in mutant mice. All of these results strongly suggest that long-term deletion of the CCK2 receptor induces an upregulation of the opioidergic system.
MATERIALS AND METHODS
Animals
To isolate the mouse CCK2 receptor gene, a mouse 129sv genomic library was screened with a human CCK2 receptor cDNA probe, and the clone was inserted in a vector as described previously (Nagata et al., 1996). J1 embryonic stem cells were electroporated with the linearized targeting vector. Clones displaying evidence for homologous recombination on the disrupted CCK2receptor gene were microinjected into blastocytes of C57BL/6J females. Germ line transmission occurred from the breeding of chimeric animals with C57BL/6J mice. To homogenize the genetic background of the mice, the first generation heterozygous were bred for 10 generations, with selection for the mutant CCK2 gene at each generation. Fifth generation heterozygous were bred together to generate the CCK2 receptor-deficient mice and control mice. Mice were housed by gender and genotype. Male and female mice (3 months old) were used, and each animal was used only once. At least 1 week before the experiments, they were housed in temperature- (22 ± 1°C) and humidity- (50% ± 5%) controlled conditions, with access to food and water ad libitum. The animals were treated in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals (1985), and the experiments were controlled by the ethical committee of the faculty.
Chemicals
RB 101 was synthesized in the laboratory as described previously (Fournié-Zaluski et al., 1992) and was dissolved in the following vehicle: 10% EtOH, 10% cremophor EL, and 80% H2O. Morphine hydrochloride (Francopia, Libourne, France), naloxone, MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (Sigma, St. Quentin Fallavier, France), and naltrindole (NTI) (synthesized in the laboratory) were dissolved in 0.9% NaCl. [3H] Tyr-d.Ala-Gly-(Me)Phe-Gly-ol (DAMGO) (50 Ci/mmol) and [3H]diprenorphine (48 Ci/mmol) were from Amersham Biosciences (Les Ulis, France). [3H] naltrindole (49 Ci/mmol) was from Isotopchim (Peyruis, France). DAMGO, Tyr-d.Pen-Gly-Phe-d.Pen (DPDPE), and quinelorane were from Sigma.
Behavioral experiments
Actimeter. Mice were individually placed in plastic cages (255 × 205 cm) that were isolated from noise and under a light intensity of 5 lux. Horizontal movements of the animals were monitored by photocells. RB 101 was injected intraperitoneally (60 and 80 mg/kg in a volume of 0.1 ml/10 gm). Morphine was injected subcutaneously (2 and 6 mg/kg in a volume of 0.1 ml/10 gm). Mice were tested immediately after injection of the chemicals for 60 min.
In another experiment, mice received 0.3 mg/kg (subcutaneously) of the nonspecific opioid antagonist naloxone, 15 min before the test.
Hot-plate test. A glass cylinder was used to keep the mouse on the heated surface of the plate, which was maintained at 52 ± 0.5°C by a thermoregulated water-circulating pump. The hot-plate test was performed 20 min after injection of saline, RB 101 (80 mg/kg, i.p.), or morphine (2.5, 6, 9, and 18 mg/kg, s.c., in a volume of 0.1 ml/10 gm). NMDA antagonist (MK-801, 0.1 mg/kg, s.c.) and opioid antagonist (naloxone, 0.1 mg/kg s.c.) were administered 30 and 20 min before the hot-plate test, respectively. The jumping responses were measured. The percentage of analgesia was calculated as follows: percentage of analgesia = (test latency − control latency)/(cutoff time − control latency) × 100. The cutoff time was 240 sec. In these conditions, no tissue damage to the paws occurred. Results are expressed as means ± SEM. The number of animals in each group was between 10 and 12.
Morphine withdrawal syndrome. Wild-type and CCK2R(−/−) mice were rendered opioid dependent by repeated intraperitoneal injection of morphine twice daily at an interval of 10 hr during 6 d. On day 6, the second daily injection of morphine was omitted. The morphine dose was progressively increased as follows: day 1, 20 mg/kg; day 2, 40 mg/kg; day 3, 60 mg/kg; day 4, 80 mg/kg; and days 5 and 6, 100 mg/kg. Control mice were treated with saline under the same conditions. Withdrawal was precipitated by 1 mg/kg naloxone injected subcutaneously, 2 hr after the last morphine injection. Mice were placed in a circular clear plastic observation area (30 × 45 cm), and somatic signs were evaluated immediately after naloxone injection for a period of 30 min.
The number of jumps, paw tremors, piloerection, and chewing frequency were counted. The time of ptosis and the degree of diarrhea were also evaluated. Results are expressed as mean ± SEM. The number of animals in each group was between 10 and 12.
Biochemical experiments
In vivo binding of [3H]diprenorphine. The experiments were performed as described previously (Ruiz-Gayo et al., 1992). Mice were killed 15 min after intravenous injection of [3H]diprenorphine (15 pmol in 0.2 ml of saline), and the brains were quickly removed and placed on ice. Total brain (minus cerebellum) was homogenized for 10 sec in 10 ml of ice-cold Tris-HCl buffer, pH 7.4, with a Brinkman Polytron. Aliquots of 0.15 ml were immediately filtered through Whatman (Maidstone, UK) GF/B glass filters and rinsed twice with ice-cold buffer. Four filters were placed in a scintillation vial containing 15 ml of Wallac (PerkinElmer Life Sciences, Emeryville, CA) scintillation cocktail, and the radioactivity was counted. Each measure correspond to the mean ± SEM of one experiment made in triplicate. Total radioactivity injected in each mouse was determined by counting 0.6 ml of the brain homogenate.
Binding of μ and δ opioid ligands.Mice were killed by decapitation. Whole brain minus cerebellum was dissected and homogenized in 10 vol (milliliters per gram of wet weight tissue) of ice-cold 0.25 m sucrose using a homogenizer. After centrifugation at 1100 × g (10 min), the pellet was rehomogenized in 5 vol of 0.25m sucrose and recentrifuged at 1100 ×g (10 min). The combined supernatants were adjusted to a final dilution of 45 vol in 50 mm Tris-HCl, pH 7.4, 1 mm EDTA. The mixture was then centrifuged at 35,000 × g for 30 min at 4°C, and the supernatant was discarded. The pellet (mitochondrial and microsomal membranes) was homogenized in 5 vol of ice-cold 0.25 m sucrose. Binding was performed by incubating 100 μg of total brain membrane proteins in 50 mm Tris-HCl, pH 7.4, 1 mm EDTA at 25°C for 1 hr with [3H]DAMGO or [3H] NTI at concentrations of 0.075–0.06 and 0.01–0.02 nm, respectively. Nonspecific binding was determined by use of 2 μm naloxone. Three or more experiments were performed in triplicate using separate membrane preparations. Binding data were analyzed using the EBDA-LIGAND program (Biosoft, Stapleford, UK).
Determination of adenylyl cyclase activity. This experiment was performed as described previously (Brown et al., 1971; Noble and Cox, 1995). Mice were killed by decapitation, and their brains were rapidly removed. Tissues were homogenized into buffer (in mm: 20 Tris-HCl, pH 7.4, 2 EGTA, 1 MgCl2, and 250 sucrose) and centrifuged at 27,000 × g for 15 min at 4°C. The pellet was resuspended in fresh buffer and centrifuged again for 15 min. The supernatant was discarded, and the pellet was homogenized in 30% (w/v) ice-cold buffer (2 mm Tris-HCl, pH 7.4, and 2 mm EGTA) for the determination of adenylyl cyclase activity. Tissue homogenate (15–30 μg of protein in 10 μl) was added to assay tubes (final volume of 60 μl) containing (in mm): 80 Tris-HCl, pH 7.4, 10 theophylline, 1 MgSO4, 0.8 EGTA, 30 NaCl, 0.25 ATP, 0.01 GTP, and either the drug being tested or water. Triplicate samples for each treatment were incubated at 30°C for 5 min. The tubes were placed in boiling water for 2 min. The amount of cAMP formed was determined by addition of [3H]cAMP (final concentration of 4 nm) in citrate-phosphate buffer, pH 5.0, and a binding protein was prepared from bovine adrenal glands. Additional samples were prepared, without tissue, containing known amounts of cAMP; they served as standards for quantification. The binding reaction was allowed to reach equilibrium by incubation for 90 min at 4°C, and the assay was terminated by the addition of charcoal and centrifugation (1000 × g for 10 min, at 4°C) to separate the free tritiated cAMP from that which was bound to the binding protein. Aliquots of the supernatant containing bound cAMP were placed into scintillation vials, and Wallac scintillation cocktail was added.
Statistical analysis
Unless specified, all of the experiments were analyzed using a two-way ANOVA. If two-way ANOVA found significant differences, one-way ANOVA was performed, followed by a Dunnett's test or a Duncan's test.
RESULTS
Motor activity measured in actimeter
An increase of the spontaneous motor activity measured in actimeter for 60 min was observed in CCK2receptor-deficient mice compared with wild-type animals (p < 0.05) (Fig.1). Compared with CD1 mice (Baamonde et al., 1992), only a slight increase (not significant) was induced by intraperitoneal administration of RB 101 (60 or 80 mg/kg) in wild-type 129sv/C57BL/6. In the case of CCK2 receptor-deficient mice, intraperitoneal administration of RB 101 at 60 and 80 mg/kg resulted in an important hyperactivity (p < 0.01) (Fig.1A). The subcutaneous administration of 2 mg/kg morphine produced no effect, whereas 6 mg/kg morphine significantly increased the motor activity of both wild-type (p < 0.05) and knock-out (p < 0.01) mice. Moreover, the motor activation was found greater in mutant than in wild-type mice (p < 0.01) (Fig. 1B).
Fig. 1.
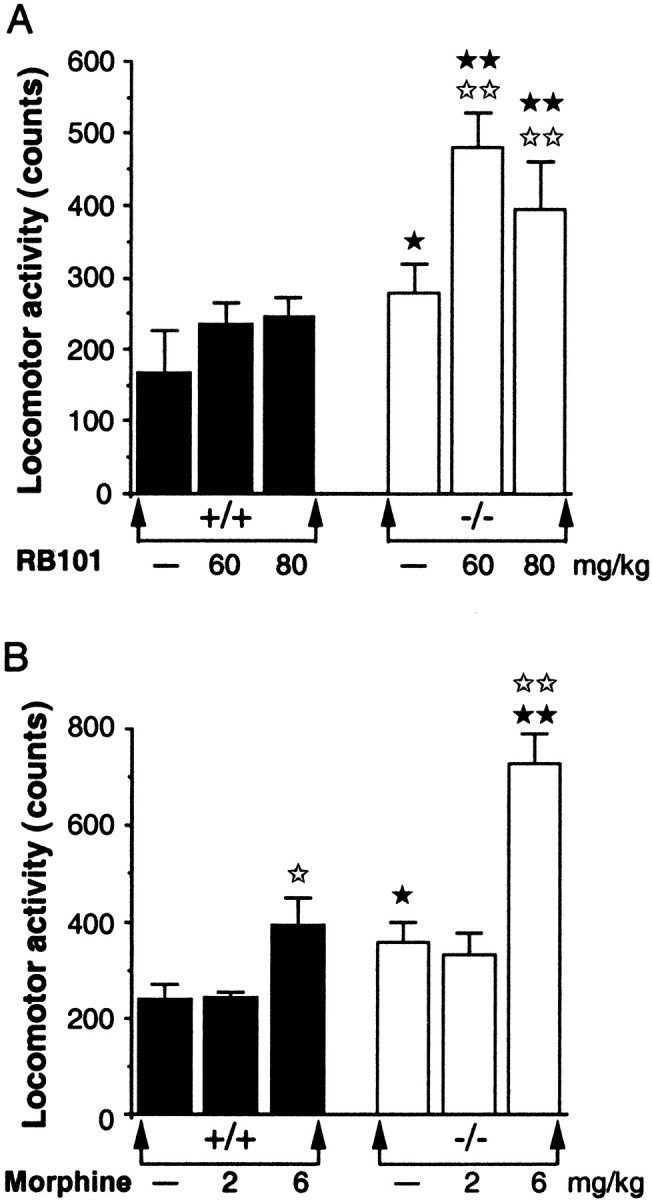
Effect of RB 101 (A) or morphine (B) on the motor activity of wild-type (+/+) (▪) and CCK2 receptor-deficient (−/−) (■) mice measured in an actimeter for 60 min. RB 101 was injected intraperitoneally (60 or 80 mg/kg). Morphine was injected subcutaneously (2 or 6 mg/kg). Mice were tested immediately after injection. Data are expressed as means ± SEM of cumulative counts. ⋆p < 0.05;⋆⋆p < 0.01 compared with the respective control groups (Dunnett's t test).★p < 0.05;★★p < 0.01 compared with the wild-type mice (Duncan's test).
The increase of spontaneous motor activity observed for 60 min in CCK2 receptor-deficient mice compared with wild-type animals was suppressed by the subcutaneous administration of the nonselective opioid antagonist naloxone (0.3 mg/kg) (p < 0.05) (Fig.2).
Fig. 2.

Effect of the nonselective opioid antagonist naloxone on the motor activity of wild-type (+/+) (▪) or CCK2 receptor-deficient (−/−) (■) mice measured in actimeter for 60 min. Naloxone was injected subcutaneously (0.3 mg/kg), 15 min before the test. Data are expressed as means ± SEM of cumulative counts. ⋆p < 0.05 compared with the respective control group (Dunnett's ttest); ★★p < 0.01 compared with the control wild-type mice (Duncan's test).
Antinociceptive responses in the hot-plate test
A decrease of the spontaneous jump latency was observed in the mutant mice compared with the wild-type mice (p< 0.01) (Fig. 3A). This hyperalgesia was reversed by the NMDA antagonist MK-801 (0.1 mg/kg, s.c.) but was not significantly modified by naloxone administration (0.1 mg/kg, s.c.) (Fig. 3C).
Fig. 3.
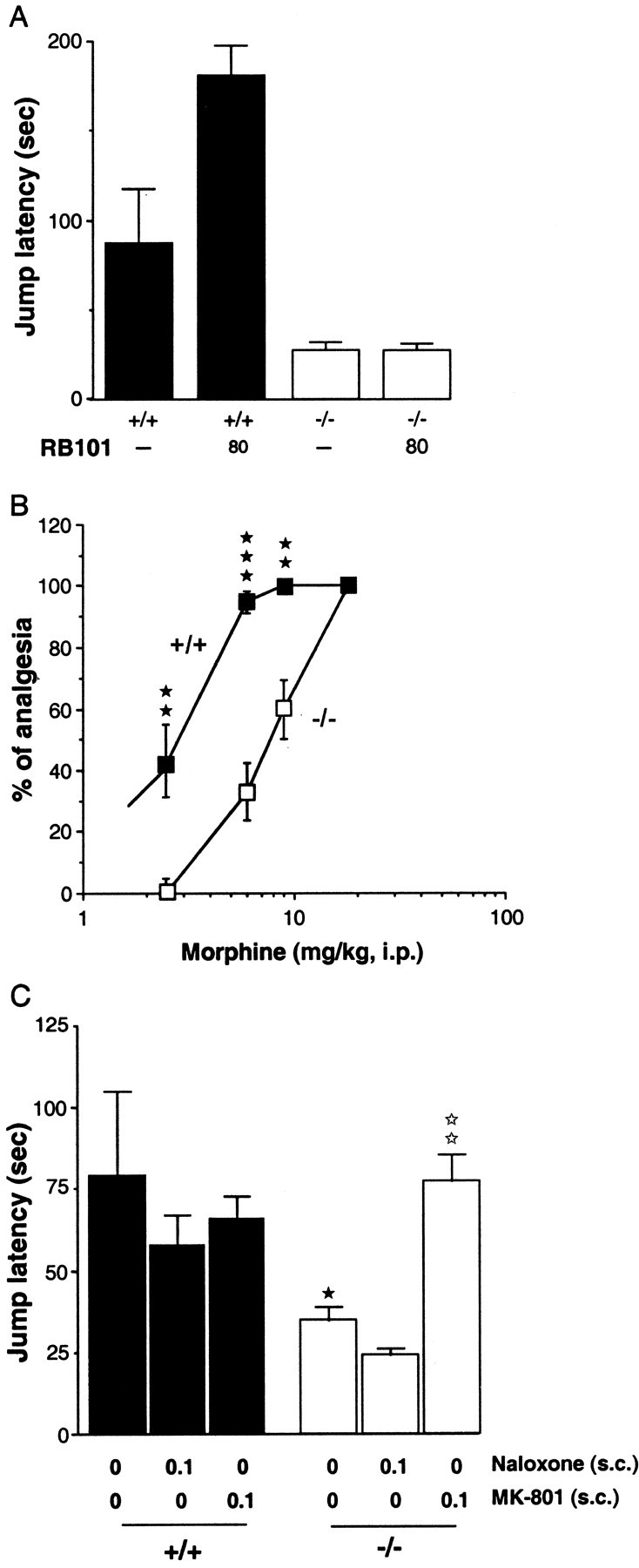
Antinociceptive responses to RB 101 (A) or morphine (B) in wild-type (+/+) (▪) and CCK2 receptor-deficient (−/−) (■) mice and effects of naloxone or MK-801 on the spontaneous hyperalgesia observed in −/− mice (C), measured in the hot-plate test. RB 101 (80 mg/kg, i.p.), morphine (2.5, 6, 9, and 18 mg/kg, s.c.), and naloxone (0.1 mg/kg, s.c.) were injected 20 min before the test. MK-801 (0.1 mg/kg, s.c.) was injected 30 min before the hot-plate test. Data are expressed as mean ± SEM of the jump latency (cutoff time, 240 sec) or as the percentage of analgesia ± SEM. A,⋆⋆p < 0.01 compared with the respective control groups (Dunnett's t test);★★p < 0.01,★★★p < 0.001 compared with the control wild-type mice (Scheffe's test). B,★★p < 0.01,★★★p < 0.001 compared with the control wild-type mice (Dunnett's t test).C, ★p < 0.05 compared with the control wild-type mice;⋆⋆p < 0.01 compared with the control CCK2 receptor-deficient mice (Scheffe's test).
The injection of 80 mg/kg RB 101 only increased the jump latency in the wild-type mice but had no effect in mutant animals (p < 0.01) (Fig. 3A). Higher doses of RB 101 could not be used because of the low solubility of this inhibitor. Morphine was able to induce analgesia in the hot-plate test in wild-type animals with an ED50 of 3 mg/kg and a maximum effect at 6 mg/kg. The dose–response curve induced by morphine in mutant animals was shifted to the right, with an ED50 of 9 mg/kg and a maximum effect at 18 mg/kg (Fig. 3B).
Withdrawal syndrome
Several behavioral manifestations of naloxone-evoked withdrawal were evaluated during a 30 min period immediately after naloxone administration (1 mg/kg, s.c.) in wild-type and mutant mice that had been chronically treated with saline or morphine (from 20 to 100 mg/kg, i.p., for 5 d) (Fig. 4).
Fig. 4.

Somatic signs of withdrawal syndrome after naloxone administration (1 mg/kg, s.c.) in wild-type (+/+; black bars) and mutant mice lacking the CCK2 receptor (−/−; white bars) chronically treated with morphine (from 20 to 100 mg/kg, i.p., for 5 d). Results are expressed as means ± SEM. The treatment is described in detail in the Materials and Methods. The number of animals per group was 10–12.⋆⋆⋆p < 0.01 compared with the respective control groups (Dunnett's t test);★★★p < 0.001 compared with the control wild-type mice (Scheffe's test).
As expected, morphine treatment induced a strong physical dependence in both strains of mice. Thus, naloxone administration precipitated the standard behavior signs of withdrawal (increase in jumps, paw tremors, piloerection, chewing, and ptosis) in morphine-treated animals but not in saline-injected control groups.
Moreover, there was a significantly greater incidence of jumps, paw tremors, and chewing in mutant than in wild-type mice, whereas the time of ptosis was longer in +/+ than in knock-out animals.
Concerning the diarrhea, wild-type animals treated with saline did not show any sign of diarrhea, in contrast to mutant mice. The incidence of this withdrawal sign was significantly increased in +/+ animals treated with morphine compared with the control group but not in mutant mice.
In vivo binding of [3H]diprenorphine
In vivo[3H]diprenorphine binding was used to evaluate the endogenous opioid levels in mice. We observed a 22% decrease in specific binding of the [3H]diprenorphine in knock-out mice compared with wild-type mice (wild-type mice, 100 ± 4.3%; mutant mice, 77.6 ± 6.7%; p < 0.05; Student'st test) (data not shown). This suggests that there is an increased extracellular amount of endogenous opioids in mutant mice that competes with [3H]diprenorphine for opioid receptors binding.
Binding of μ and δ agonists
Scatchard analysis using specific μ-(DAMGO) and δ-(NTI) radioligands showed similar plots in both +/+ and −/− mouse brains. The total amount (Bmax) of μ receptors in +/+ and −/− mouse brains was 0.080 ± 0.006 and 0.060 ± 0.005 pmol/mg protein, respectively. The total amount of δ receptors was 0.069 ± 0.007 and 0.060 ± 0.006 pmol/mg protein, respectively (data not shown). TheKD values were almost identical in wild-type and mutant mice (DAMGO, KDof 1.8 ± 0.3 and 1.1 ± 0.2 nm in wild-type and mutant mice, respectively; naltrindole,KD of 0.05 ± 0.01 and 0.04 ± 0.02 nm in wild-type and mutant mice, respectively).
Adenylyl cyclase activity
The μ-selective agonist DAMGO and the δ-selective agonist DPDPE decreased the adenylyl cyclase activity of wild-type animals in a dose-dependent manner (p < 0.01 at 10 μm for both treatments), as reported previously (for review, see Childers, 1991). In contrast, both agonists induced an increase in the adenylyl cyclase activity in mutant mice, which was statistically significant for DAMGO (p < 0.05 at 10 μm) but not for DPDPE. A ttest revealed significant differences between the responses obtained with 0.1 μm, 1 μm(p < 0.05), and 10 μm(p < 0.01) DAMGO or DPDPE in wild-type animals compared with knock-outs (Fig. 5).
Fig. 5.
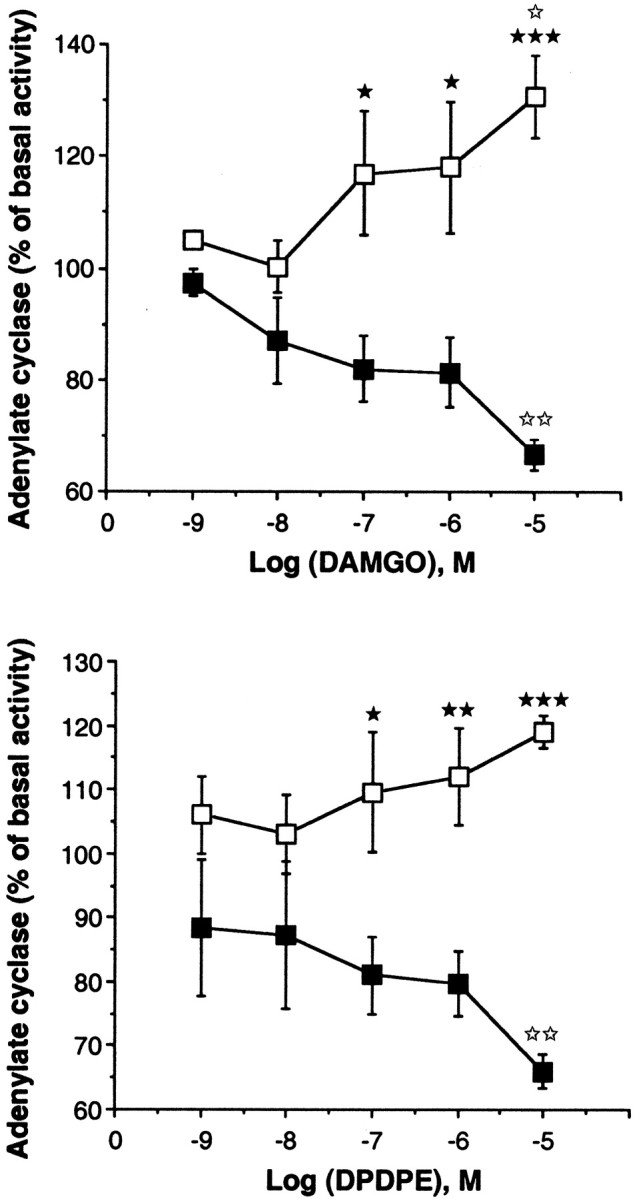
Effect of the selective μ opioid agonist DAMGO (A) and the selective δ opioid agonist DPDPE (B) on adenylyl cyclase activity in wild-type (▪) and mutant (■) mice. Results are expressed as the mean ± SEM percentage of basal adenylyl cyclase activity from five or more independent experiments, each performed in triplicate.★p < 0.05,★★p < 0.01,★★★p < 0.001 compared with the control wild-type mice (Student's t test).⋆⋆p < 0.01 compared with the mice treated with 1 nm either agonist (Dunnett's ttest).
The functionality of the dopamine D2 receptors, which are also coupled to the adenylyl cyclase via a Gi-protein (Albert et al., 1990), was also examined. The D2 agonist quinelorane induced a dose-dependent decrease in the amount of cAMP in wild-type mice (p < 0.05 at 10 nm and 1 μm; p < 0.01 at 100 nm; and p < 0.001 at 10 μm) and a dose-dependent increase in the amount of cAMP (p < 0.05 at 1 μm and p < 0.01 at 10 μm) in the knock-out mice. A t test revealed significant differences between wild-type and mutant mice at 1 and 10 μm for the D2agonist (p < 0.05 and p < 0.01, respectively) (Fig. 6).
Fig. 6.

Effect of the selective D2 agonist quinelorane on adenylyl cyclase activity in wild-type (▪) and mutant (■) mice. Results are expressed as the mean ± SEM percentage of basal adenylyl cyclase activity from five or more independent experiments, each performed in triplicate.★★p < 0.01,★★★p < 0.001 compared with the control wild-type mice (Student's t test).⋆p < 0.05,⋆⋆⋆p < 0.01,⋆⋆⋆p < 0.01 compared with the mice treated with 1 nm either agonist (Dunnett'st test).
DISCUSSION
Numerous studies have shown that CCK8 is one of the most powerful endogenous antagonists of opioid system. Accordingly, CCK2 receptor antagonists have been used to potentiate analgesia and antidepressant-like effects induced by endogenous or exogenous opioid agonists (for review, see Noble et al., 1999). It has been established that CCK2 and opioid receptors are colocalized in spinal cord and various brain structures (Gall et al., 1987), but very few studies have attempted to investigate mechanisms underlying the interactions between both systems. Deletion of the CCK2 receptor in mice provides an interesting model to explore such a modulation. Thus, in the present study, we used this model to achieve a better understanding of the role play by CCK2 receptors on opioid system at the supraspinal level.
Locomotor activity measured in an actimeter showed that mutant mice had an enhanced basal activity compared with wild-type animals. This effect was reversed by the opioid antagonist naloxone, suggesting that the endogenous opioid system plays a role in the hyperlocomotion observed. This result was not attributable to an upregulation of the μ or the δ opioid receptors in CCK2 receptor knock-out mice, because the binding parameters (KD andBmax values) of μ- and δ-selective ligands were similar in the brain of both strains of mice. Nevertheless, the lack of changes in opioid receptors density measured in the whole brain minus cerebellum cannot exclude that decrease and/or increase in Bmax values could take place with a regional selectivity. One of the possibilities accounting for the spontaneous hyperlocomotor activity observed could be an increase in the endogenous opioid agonist tone in the brain of mutant mice, because other studies have shown that selective opioid agonists increase locomotor activity in rodents (Kalivas et al., 1985;Calenco-Choukroun et al., 1991; Baamonde et al., 1992). This was investigated by measuring the in vivo binding of [3H]diprenorphine. Accordingly, the total amount of endogenous opioid ligands able to compete for [3H]diprenorphine was found enhanced in the brain of mutant compared with wild-type mice. This result could be expected, because previous studies have indirectly suggested a tonic inhibitory action of CCK through CCK2 receptor activation to diminish the release of endogenous opioid peptides (Suh et al., 1992; Noble et al., 1993; Nichols et al., 1996). This hypothesis was in good agreement with the fact that CCK2 antagonists strongly potentiate RB 101-induced antinociceptive responses and antidepressant-like effects (Valverde et al., 1994; Smadja et al., 1995). Nevertheless, one cannot exclude that the higher hyperlocomotion induced by morphine or the dual inhibitor of enkephalin-degrading enzymes RB 101 in mutant mice compared with wild-type animals also involves other regulatory processes occurring within the dopaminergic system of mutant animals, because it is well established that hyperlocomotion induced by basal or tonic stimulation of opioid receptors is related to dopamine receptor activation (Baamonde et al., 1992).
Hot-plate experiments were then performed to measure the nociceptive responses and the analgesia mediated by supraspinal mechanisms in CCK2 receptor-deficient mice. Surprisingly, mutant mice exhibited a lower nociceptive threshold than wild-type animals. This could be a consequence of the slight increase in endogenous opioid peptides, suggested by the decrease in [3H]diprenorphine binding. Indeed, it has been shown that the mixed enkephalin-degrading enzyme inhibitor RB 101 used at low doses that slightly increase the level of endogenous opioid ligands, mimicking the situation observed in knock-out mice, elicits paradoxical hyperalgesia (Willer et al., 1990; Noble et al., 1994). Another hypothesis could be an imbalance in antinociceptive–pronociceptive systems. Indeed, it has been proposed that opiates concomitantly activate both pathways and, more specifically, the NMDA-dependent pronociceptive system (Larcher et al., 1998; Célèrier et al., 2001). This is in good agreement with the results obtained in the present study, because the hyperalgesia observed was totally reversed by the NMDA-antagonist MK-801. Thus, the slight increase in endogenous opioid peptides may be involved in the enhancement of sensitivity of postsynaptic excitatory amino acid (NMDA) receptor and modulation of primary afferent neurotransmission, including nociception, leading to hyperalgesia (Cerne et al., 1992). On the other hand, mutant mice were less sensitive to the analgesic effect of endogenous (RB 101) or exogenous (morphine) opioid agonists. Although the binding characteristics of opioid receptors were not statistically different between mutant and wild-type mice, a difference in the coupling to G-proteins could not be excluded. This was investigated by studying the ability of adenylyl cyclase to inhibit cAMP production, which is one of the most important second messengers in the opioid receptor transduction pathway (for review, see Cox, 1993). Strikingly, the activation of μ and δ receptors in knock-out mice did not decrease the formation of cAMP, as observed in wild-type animals, but significantly increased cAMP production. This increase of cAMP formation may account for the lack of analgesic responses observed in mutant mice because previous studies have shown that upregulation of the cAMP pathway induces excitatory effects in dorsal root ganglion neurons leading to hyperalgesia (Cerne et al., 1992; Randic et al., 1995; Vanegas and Schaible, 2001).
These results suggest that stimulation of CCK2receptors play an important role to keep the opioid receptors coupled to the Gi-proteins with subsequent decrease in cAMP production. Several hypotheses may account for the observed increase in cAMP production induced by opioids in mutant mice. (1) Deletion of the CCK2 receptor could impair the association of Giα to the opioid receptors, thus reducing the inhibition of adenylyl cyclase activity. In good agreement with this hypothesis, blockade of the αi subunit by pertussis toxin triggers an increase in cAMP production after μ opioid receptor stimulation (Sarne et al., 1998). (2) It has been shown that phospholipase C-specific inhibitors blocked opioid receptor-mediated inhibition of adenylyl cyclase activity (Fan et al., 1998). Because CCK2 receptor stimulation induces phospholipase C activation, it could be speculated that deletion of this receptor inhibits the negative coupling to adenylyl cyclase, thus revealing another signaling pathway with an increase in cAMP production.
In addition, the D2 dopamine receptors, which interact with the CCK network and which are normally negatively coupled to adenylyl cyclase (Hökfelt et al., 1980; Albert et al., 1990;Crawley, 1991), were also found to be positively coupled to the cAMP pathway in mutant mice. Thus, as for μ and δ opioid receptors, CCK2 receptors appear to be critical in the negative coupling of D2 receptors to the adenylyl cyclase pathway. This change in the transduction processes coupled to the D2 receptors may also account for the hyperlocomotion observed in mutant animals described above.
At last, morphine was chronically administered to both strains of mice to investigate the role of CCK2 receptors in physical signs of opioid withdrawal. It appeared that the major withdrawal signs were more severe in mutant mice than in wild-type animals. Because several studies have shown that chronic morphine treatments enhance cAMP levels in wild-type animals (Nestler and Aghajanian, 1997), these results could be attributable to the cumulative increase in adenylyl cyclase activity observed in untreated mutant mice and the one triggered by the chronic morphine treatment.
In conclusion, our results show that CCK2receptors play a crucial role in the homeostasis of the opioid system at the supraspinal level. Until now, only acute or repetitive treatment over a very short period with CCK2 receptor antagonists had been used in association with opioid agonists, leading to a potentiation of the analgesic effects of opioids (Wiesenfeld-Hallin et al., 1990; Valverde et al., 1994) and to a reduction of the severity of the withdrawal syndrome (for review, seeNoble et al., 1999). However, the absence, in knock-out mice, of the negative feedback control on the opioid system, normally performed by CCK2 receptor stimulation, results in an upregulation of this system, with a positive coupling of the μ and δ opioid receptors to the adenylyl cyclase pathway. This may account for the hypersensitivity to the locomotor activity induced by morphine or RB 101, the hyposensitivity toward the antinociceptive effects of these compounds, and the more severe withdrawal syndromes observed in these genetically modified animals after chronic morphine treatment. Assays on spinal tissues in the CCK2receptor-deficient mice have to now be performed, because it is well established that CCK interacts with opioid system in the spinal cord.
Footnotes
This work has been supported by European Union Biomed II Program Grant BMH4 CT98 2267. We gratefully acknowledge C. Dupuis for expert manuscript drafting and C. Canestrelli for the animal care.
Correspondence should be addressed to Dr. Florence Noble, Département de Pharmacochimie Moléculaire et Structurale, Institut National de la Santé et de la Recherche Médicale U266–Centre National de la Recherche Scientifique Unité Mixte de Recherche 8600, Unité de Formation et de Recherche des Sciences Pharmaceutiques et Biologiques, 4 avenue de l'Observatoire, 75270 Paris Cedex 06, France. E-mail: noble@pharmacie.univ-paris5.fr.
REFERENCES
- 1.Albert PR, Neve KA, Bunzow JR, Civelli O. Coupling of cloned rat dopamine-D2 receptor to inhibition of adenylyl cyclase and prolactine secretion. J Biol Chem. 1990;265:2098–2104. [PubMed] [Google Scholar]
- 2.Baamonde A, Daugé V, Ruiz-Gayo M, Fulga IG, Turcaud S, Fournié-Zaluski MC, Roques BP. Antidepressant-type effects of endogenous enkephalins protected by systemic RB 101 are mediated by opioid delta and dopamine D1 receptor stimulation. Eur J Pharmacol. 1992;216:157–166. doi: 10.1016/0014-2999(92)90356-9. [DOI] [PubMed] [Google Scholar]
- 3.Brown BL, Albano JDM, Ekins RP, Sgherzi AM. A simple and sensitive saturation assay method for the measurement of adenosine 3′:5′-cyclic monophosphate. Biochem J. 1971;121:561–562. doi: 10.1042/bj1210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calenco-Choukroun G, Daugé V, Gacel G, Féger J, Roques BP. Opioid delta agonists and endogenous enkephalins induce different emotional reactivity than mu agonists after injection in the rat ventral tegmental area. Psychopharmacology. 1991;103:493–502. doi: 10.1007/BF02244249. [DOI] [PubMed] [Google Scholar]
- 5.Célèrier E, Laulin JP, Corcuff JB, Le Moal M, Simonnet G. Progressive enhancement of delayed hyperalgesia induced by repeated heroin administration: a sensitization process. J Neurosci. 2001;21:4074–4080. doi: 10.1523/JNEUROSCI.21-11-04074.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerne R, Jiang M, Randic M. Cyclic adenosine 3′5′-monophosphate potentiates excitatory amino acid and synaptic responses of rat spinal dorsal horn neurons. Brain Res. 1992;596:111–123. doi: 10.1016/0006-8993(92)91538-p. [DOI] [PubMed] [Google Scholar]
- 7.Cesselin F. Opioid and anti-opioid. Clin Pharmacol. 1995;9:409–433. doi: 10.1111/j.1472-8206.1995.tb00517.x. [DOI] [PubMed] [Google Scholar]
- 8.Childers S. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- 9.Cox BM. Opioid receptor-G protein interactions: acute and chronic effects of opioids. In: Herz A, editor. Opioids I, Ch 8. Springer; Berlin: 1993. pp. 145–188. [Google Scholar]
- 10.Crawley JN. Cholecystokinin-dopamine interactions. Trends Pharmacol Sci. 1991;12:232–236. doi: 10.1016/0165-6147(91)90558-a. [DOI] [PubMed] [Google Scholar]
- 11.Crawley JN, Corwin RL. Biological actions of cholecystokinin. Peptides. 1994;15:731–755. doi: 10.1016/0196-9781(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 12.Daugé V, Roques BP. Reward versus anxiety: the opposite pharmacology of opioids and CCK. In: Bradwejn J, Vasar E, editors. Cholecystokinin and anxiety from neuron to behavior, molecular biology intelligence unit, Ch 8. Springer; Berlin: 1995. pp. 151–171. [Google Scholar]
- 13.Daugé V, Mauborgne A, Cesselin F, Fournié-Zaluski MC, Roques BP. The dual peptidase inhibitor RB 101 induces a long lasting increase in the extracellular level of Met-enkephalin in the nucleus accumbens of freely moving rats. J Neurochem. 1996;67:1301–1308. doi: 10.1046/j.1471-4159.1996.67031301.x. [DOI] [PubMed] [Google Scholar]
- 14.Fan GH, Zhou TH, Zhang WB, Pei G. Suppression of phospholipase C blocks Gi-mediated inhibition of adenylyl cyclase activity. Eur J Pharmacol. 1998;341:317–322. doi: 10.1016/s0014-2999(97)01477-5. [DOI] [PubMed] [Google Scholar]
- 15.Fournié-Zaluski MC, Coric P, Turcaud S, Lucas E, Noble F, Maldonado R, Roques BP. Mixed-inhibitor-prodrug as a new approach towards systemically active inhibitors of enkephalin degrading enzymes. J Med Chem. 1992;35:2474–2481. doi: 10.1021/jm00091a016. [DOI] [PubMed] [Google Scholar]
- 16.Gall C, Lauterborn J, Burks D, Seroogy K. Co-localization of enkephalins and cholecystokinin in discrete areas of rat brain. Brain Res. 1987;403:403–408. doi: 10.1016/0006-8993(87)90085-0. [DOI] [PubMed] [Google Scholar]
- 17.Hökfelt T, Skirboll LR, Rehfeld JH, Gostein M, Markey K, Dann O. A subpopulation mesencephalic neurons projecting to limbic areas contains a cholecystokinin-like peptide: evidence from immunohistochemistry combined with retrograde tracing. Neuroscience. 1980;5:2093–2124. doi: 10.1016/0306-4522(80)90127-x. [DOI] [PubMed] [Google Scholar]
- 18.Kalivas PW, Taylor S, Miller JS. Sensitization to repeated enkephalin administration into the ventral tegmental area of the rat. I. Behavioral characterization. J Pharmacol Exp Ther. 1985;235:537–543. [PubMed] [Google Scholar]
- 19.Larcher A, Laulin JP, Célèrier E, Le Moal M, Simonnet G. Acute tolerance associated with a single opiate administration: involvement of N-methyl-d-aspartate-dependent pain facilitatory systems. Neuroscience. 1998;84:583–589. doi: 10.1016/s0306-4522(97)00556-3. [DOI] [PubMed] [Google Scholar]
- 20.Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- 21.Manzanedo C, Aguilar MA, Minarro J. The effects of dopamine D2 and D3 antagonists on spontaneous motor activity and morphine-induced hyperactivity in male mice. Psychopharmacology. 1999;143:82–88. doi: 10.1007/s002130050922. [DOI] [PubMed] [Google Scholar]
- 22.Nagata A, Ito M, Iwata N, Kuno J, Takano H, Minowa O, Chihara K, Matsui T, Noda T. G protein-coupled cholecystokinin-B/gastrin receptors are responsible for physiological cell growth of the stomach mucosa in vivo. Proc Natl Acad Sci USA. 1996;93:11825–11830. doi: 10.1073/pnas.93.21.11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nestler EJ, Aghajanian G. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 24.Nichols ML, Bian D, Ossipov MH, Malan TP, Jr, Porreca F. Antiallodynic effects of a CCK-B antagonist in rats with nerve ligation injury: role of endogenous enkephalins. Neurosci Lett. 1996;215:161–164. doi: 10.1016/0304-3940(96)12964-5. [DOI] [PubMed] [Google Scholar]
- 25.Noble F, Cox BM. Differential regulation of D1 dopamine receptor- and of A2a adenosine receptor-stimulated adenylyl cyclase by μ-, δ1-, and δ2-opioid agonists in rat caudate putamen. J Neurochem. 1995;65:125–133. doi: 10.1046/j.1471-4159.1995.65010125.x. [DOI] [PubMed] [Google Scholar]
- 26.Noble F, Derrien M, Roques BP. Modulation of opioid analgesia by CCK at the supraspinal level: evidence of regulatory mechanisms between CCK and enkephalin systems in the control of pain. Br J Pharmacol. 1993;109:1064–1070. doi: 10.1111/j.1476-5381.1993.tb13730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noble F, Fournié-Zaluski MC, Roques BP. Paradoxical analgesia induced by low doses of naloxone is not potentiated by complete inhibition of enkephalin degradation. Neuropharmacology. 1994;33:135–140. doi: 10.1016/0028-3908(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 28.Noble F, Wank A, Crawley JN, Bradwejn J, Seroogy KB, Hamon M, Roques BP. International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol Rev. 1999;51:745–781. [PubMed] [Google Scholar]
- 29.Randic M, Cheng G, Kojic L. Kappa-opioid receptor agonists modulate excitatory transmission in substantia gelatinosa neurons of the rat spinal cord. J Neurosci. 1995;15:6809–6826. doi: 10.1523/JNEUROSCI.15-10-06809.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rehfeld JF, Hansen HF, Marley PD, Stengaard-Pedersen K. Molecular forms of cholecystokinin in the brain and the relationship to neuronal gastrins. Ann NY Acad Sci. 1985;448:11–23. doi: 10.1111/j.1749-6632.1985.tb29902.x. [DOI] [PubMed] [Google Scholar]
- 31.Ruiz-Gayo M, Baamonde A, Turcaud S, Fournié-Zaluski MC, Roques BP. In vivo occupation of mouse brain opioid receptors by endogenous enkephalins: blockade of enkephalin degrading enzymes by RB 101 inhibits [3H]diprenorphine binding. Brain Res. 1992;571:306–312. doi: 10.1016/0006-8993(92)90669-z. [DOI] [PubMed] [Google Scholar]
- 32.Sarne Y, Rubovitch V, Fields A, Gafni M. Dissociation between the inhibitory and stimulatory effects of opioid peptides on cAMP formation in SK-N-SH neuroblastoma cells. Biochem Biophys Res Commun. 1998;246:128–131. doi: 10.1006/bbrc.1998.8582. [DOI] [PubMed] [Google Scholar]
- 33.Skinner K, Basbaum AI, Fields HL. Cholecystokinin and enkephalin in brain stem pain modulating circuits. NeuroReport. 1997;8:2995–2998. doi: 10.1097/00001756-199709290-00001. [DOI] [PubMed] [Google Scholar]
- 34.Smadja C, Maldonado R, Turcaud S, Fournié-Zaluski MC, Roques BP. Opposite role of CCK-A and CCK-B receptors in the modulation of endogenous enkephalins antidepressant-like effects. Psychopharmacology. 1995;128:400–408. doi: 10.1007/BF02245811. [DOI] [PubMed] [Google Scholar]
- 35.Stengaard-Pedersen K, Larsson LI. Comparative immunocytochemical localization of putative opioid ligands in the central nervous system. Histochemistry. 1981;73:89–114. doi: 10.1007/BF00493136. [DOI] [PubMed] [Google Scholar]
- 36.Suh HH, Collins KA, Tseng LF. Intrathecal cholecystokinin octapeptide attenuates the antinociception and release of immunoreactive Met-enkephalin induced by intraventricular beta-endorphin in the rat. Neuropeptides. 1992;21:131–137. doi: 10.1016/0143-4179(92)90034-t. [DOI] [PubMed] [Google Scholar]
- 37.Svingos AL, Clarke CL, Pickel VM. Localization of the delta-opioid receptor and dopamine transporter in the nucleus accumbens shell: implications for opiate and psychostimulant cross-sensitization. Synapse. 1999;34:1–10. doi: 10.1002/(SICI)1098-2396(199910)34:1<1::AID-SYN1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 38.Vaccarino AL, Olson GA, Olson RD, Kastin AJ. Endogenous opiates: 1998. Peptides. 1999;20:1527–1574. doi: 10.1016/s0196-9781(99)00166-7. [DOI] [PubMed] [Google Scholar]
- 39.Valverde O, Maldonado R, Fournié-Zaluski MC, Roques BP. Cholecystokinin B antagonists strongly potentiate antinociception mediated by endogenous enkephalins. J Pharmacol Exp Ther. 1994;270:77–88. [PubMed] [Google Scholar]
- 40.Van der Kooy D, Weinreich P, Nagy JI. Dopamine and opiate receptors: localization in the striatum and evidence for their axoplasmic transport in the nigrostriatal and striatonigral pathways. Neuroscience. 1986;19:139–146. doi: 10.1016/0306-4522(86)90011-4. [DOI] [PubMed] [Google Scholar]
- 41.Vanegas H, Schaible H. Prostaglandins and cycloxygenases in the spinal cord. Prog Neurobiol. 2001;64:327–363. doi: 10.1016/s0301-0082(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 42.Wank SA. Cholecystokinin receptors. Am J Physiol. 1995;269:G628–G646. doi: 10.1152/ajpgi.1995.269.5.G628. [DOI] [PubMed] [Google Scholar]
- 43.Wiesenfeld-Hallin Z, Xu XJ, Hugues J, Horwell DC, Hökfelt T. PD134308, a selective antagonist of cholecystokinin type B receptor, enhances the analgesic effect of morphine and synergistically interacts with intrathecal galanin to depress spinal nociceptive reflexes. Proc Natl Acad Sci USA. 1990;87:7105–7109. doi: 10.1073/pnas.87.18.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiesenfeld-Hallin Z, de Arauja Lucas G, Alster P, Xu XJ, Hökfelt T. Cholecystokinin/opioid interactions. Brain Res. 1999;848:78–89. doi: 10.1016/s0006-8993(99)01978-2. [DOI] [PubMed] [Google Scholar]
- 45.Willer JC, Le Bars D, de Broucker T. Diffuse noxious inhibitory controls in man: involvement of an opioidergic link. Eur J Pharmacol. 1990;182:347–356. doi: 10.1016/0014-2999(90)90293-f. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, de Araujo Lucas G, Elde R, Wiesenfeld-Hallin Z, Hökfelt T. Effect of morphine on cholecystokinin and mu-opioid receptor-like immunoreactivities in rat spinal dorsal horn neurons after peripheral axotomy and inflammation. Neuroscience. 2000;95:197–207. doi: 10.1016/s0306-4522(99)00419-4. [DOI] [PubMed] [Google Scholar]