

Abstract
Perturbations in the intestinal microbiome are implicated in inflammatory bowel disease (IBD). Studies of treatment-naive patients have identified microbial taxa associated with disease course and treatment efficacy. In order to gain a mechanistic understanding of how the microbiome impacts gastrointestinal health, we need to move from census to function. Bacteria including those that adhere to epithelial cells as well as several Clostridium species can alter differentiation of T helper 17 cells and regulatory T cells. Similarly, microbial products such as short-chain fatty acids and sphingolipids also influence immune responses. Metagenomics and culturomics have identified strains of Ruminococcus gnavus and adherent invasive Escherichia coli that are linked to IBD and gut inflammation. Integrated analysis of multi-omics data, including metagenomics, metatranscriptomics, and metabolomics, with measurements of host response and culturomics have great potential in understanding the role of the microbiome in IBD. In this Review, we highlight current knowledge of gut microbial factors linked to IBD pathogenesis and discuss how multi-omics data from large-scale population studies in health and disease have been used to identify specific microbial strains, transcriptional changes, and metabolic alterations associated with IBD.
Table of contents blurb
Perturbations in the intestinal microbiome are implicated in inflammatory bowel disease (IBD). In this Review, Xavier and colleagues highlight current knowledge of gut microbial factors linked to IBD pathogenesis and discuss how multi-omics data from large-scale population studies in health and disease have been used to identify specific microbial strains, transcriptional changes and metabolic alterations associated with IBD.
Introduction
The intestinal microbiota has a major role in human health, including the maturation and education of host immune responses, protection against enteric pathogen proliferation, and response to or modification of specific drugs. Host physiology can be altered at the cellular level by microbiome-induced cell signaling, proliferation and neurotransmitter biosynthesis1,2, leading to mucosal and systemic alterations and thereby affecting homeostasis, barrier function, innate and adaptive immune responses and metabolism2–4. Through microbial metabolites and their impact on dietary breakdown, the microbiota provides energy and vitamins for the host5. Furthermore, it alters therapeutic drug availability6 and modifies bile salts impacting a variety of host functions6. With such a broad range of effects on host physiology and its role in the induction, education and function of the immune system, it is unsurprising that the microbiota is implicated in gut-related diseases. Examples include inflammatory bowel disease (IBD) and metabolic disorders7–10 as well as diseases that affect other systems including atherosclerosis11–13, autism14, asthma15,16, and type I diabetes17,18. Although the microbiome is dysregulated in these conditions, causal roles have yet to be determined in most diseases, and whether the microbiome drives disease or is altered in response to disease pathology remains unclear.
IBD, which includes Crohn’s disease (CD) and ulcerative colitis (UC), is a model for the study of microbiome-related diseases (Box 1). IBD risk is linked to 200 host genetic loci, most of which are associated with key immunological pathways including innate immunity (for example, the sensor NOD2 detects bacterial peptidoglycans), immune responses (for example, IL23R allele rs11209026 provides protective effect in CD), and autophagy (for example, the product of ATG16L1 affects autophagy in Paneth cells and goblet cells)19–21. Since 1950, a drastic increase of IBD has been observed in Western countries. Incidence rates of IBD have been stable or falling since 1990; however, disease burden remains high with prevalence surpassing 0.3% in the Western world. As many newly industrialized countries are becoming more westernized and urbanized, IBD rates are rising there as well22. These patterns suggest that other factors besides host genetics must be driving changes in disease prevalence. Environmental and gut microbial influences affect the host immune response and have been linked to IBD onset and disease progression. Recent studies found that host factors associated with industrialization, such as body mass index, glycemic response, high-density lipoprotein cholesterol, and lactose consumption, have dominant roles in shaping the microbiota23,24. Effects of specific environmental factors on IBD development have also been examined, including smoking, diet, medications, circadian rhythm, and stress. In particular, increased IBD risk was associated with early childhood exposure to antibiotics25. Interestingly, in some cases disparate effects on CD and UC have been observed, suggesting unresolved heterogeneity in IBD.
Box 1: Features of inflammatory bowel disease.
Inflammatory bowel disease (IBD) is a term that is used to describe chronic inflammatory disorders of the gastrointestinal (GI) tract that manifest in two major forms — Crohn’s disease (CD) and ulcerative colitis (UC). Traditionally, CD and UC are defined by clinical, histological, endoscopic, and radiological features138. The location and extent of inflammation along the GI tract distinguishes UC and CD; UC inflammation extends proximally from the rectum and is restricted to the colon, whereas CD may affect any site in the Gl tract but commonly occurs in the terminal ileum and may be discontinuous. CD is histologically characterized by macrophage aggregates that often form non-caseating granulomas, whereas UC presents with micro-abscesses composed of neutrophils within the lamina propria and crypts. Macroscopically, extensive mucosal ulcerations develop in UC. Mucosal lesions in CD are often found over Peyer’s patches (small masses of lymphatic tissue in the ileum). In addition to these features, main immune cell involvement differs between CD and UC. CD is dominated by a T helper 1 cell phenotype and production of interferon-γ and IL-2, whereas UC is primarily a T helper 2 cell phenotype with production of TGF-β and IL-5 but not IL-4. IBD treatment regimens vary based on disease type, extent and severity.
IBD onset and disease course are associated with a combination of genetic, host (Box 2), and microbial (Box 3) factors. Precise pathophysiologies for IBD, however, remain elusive. Although CD and UC are clinically distinct, it is unknown whether they are also pathologically distinct or are part of a singular spectrum. Recent multi-omics studies such as the ones highlighted in this Review have greatly contributed to a more comprehensive understanding of the pathogenesis of IBD.
In addition to these factors, changes in the gut microbiome influence IBD susceptibility. A lack of early childhood exposure to microorganisms due to cleaner living, urbanization, and the widespread usage of antibiotics affects immune education and maturation (known as the hygiene hypothesis). This lack of early childhood exposure is hypothesized to lead to a loss of negative regulatory pathways resulting in overactive immune responses to the commensal intestinal microbiota. Serological markers are associated with disease course and IBD phenotypes, providing further evidence for the microbiome’s role. A combination of a mannan epitope of Saccharomyces cerevisiae (gASCA), the atypical perinuclear anti-neutrophil cytoplasmic antibody (pANCA) and laminaribioside (ALCA) can differentiate between healthy individuals, IBD patients, and patients with non-IBD gastrointestinal inflammation26. Furthermore, differences in gASCA and pANCA levels can distinguish between CD and UC, while increasing antibody responses against gASCA, ALCA, chitobioside, mannobioside, and outer membrane porins are associated with more complicated disease behaviour and surgery requirement in CD26,27. In UC, anti-S. cerevisiae antibody (ASCA) immunoglobulin A (IgA), ANCA, anti-flagellin antibodies, and anti-outer membrane porin C (OmpC) are associated with disease severity. Additionally, ASCA IgA and OmpC are associated with the later requirement of colectomy, and higher ASCA IgA levels are linked to refractory disease in UC28. Further evidence supporting a causal role of the microbiome in IBD are the findings that transfer of fecal microbiota from mice with colitis to healthy mice is sufficient to induce colitis29–34 and that many genetically susceptible mice do not spontaneously develop colitis in germ-free facilities35–37.
Progress to date indicates that IBD is a polymicrobial disease with a combination of various gut microbial factors, abnormal immune responses and a weakened intestinal mucosal barrier leading to aberrant host–microbial interactions38. In this Review we highlight current knowledge of gut microbial factors linked to IBD pathogenesis. We discuss how multi-omics data from large-scale population studies in health and disease have been used to identify specific microbial strains from metagenomic data, transcriptional changes from metatranscriptomic data, and metabolic alterations from metabolomic data. When integrated with host-derived data, analyses can link these microbial changes to the host in order to predict aberrant host–microbial interactions. Furthermore, much effort is currently being focused on creating large collections of strains isolated from individuals with altered mucosal ecosystems. These collections together with culturomics techniques will be invaluable to validate the colitogenic potential of disease-associated strains as well as to identify pathogenic factors. Thus, the gut microbiome’s contribution to disease onset, activity and the development of complications needs to be taken into consideration in order to treat IBD effectively, achieve long-lasting remission and reset the host–microbial balance.
Microbial immunomodulation in IBD
IBD is believed to result from aberrant immune responses to commensal bacteria in genetically susceptible hosts that disrupt the host–microbial balance (Figure 1). The symbiotic relationship between the gut microbiome and the host is foremost protected by the intestinal epithelial barrier: a mucus bilayer and cellular junctions within the intestinal epithelium that form a physical barrier to contain the microbiota. Moreover, secretion of antimicrobial peptides, such as defensins produced by Paneth cells, goblet cells and other types of epithelial cells, creates a chemical barrier against invading microorganisms (Box 2). IgA also has a crucial role in maintaining homeostasis at mucosal surfaces. Two distinct types of humoral immunity were proposed to coexist in the gastrointestinal mucosa, where IgA can elicit high-affinity responses, for example in the context of pathogens and vaccines, or be polyreactive and bind to a broad but taxonomically distinct subset of the microbiota39. IgA can mediate potent anti-inflammatory functions through interaction with the C-type lectin receptor SIGNR1 on dendritic cells, which induces immune tolerance via regulatory T cell (Treg) expansion. IgA antibodies also have a crucial role in the prevention of tissue damage in autoimmune and inflammatory diseases40. The microbiome in turn influences IgA levels in the gut mucosa through degradation, potentially disrupting homeostasis41. In IBD, there is an increased coating of intestinal microbiota by IgA antibodies.
Figure 1: Inflammatory bowel disease and the microbiota.
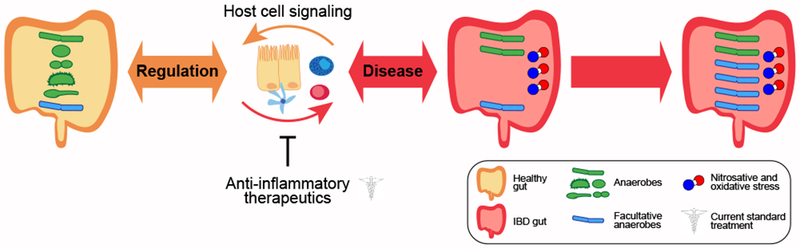
In health, gut bacterial composition (anaerobes and facultative anaerobes) is maintained in balance with host cell physiology. Alterations in gut microbiome composition during disease include reduced microbial diversity and expansion of facultative anaerobes due to increased nitrosative and oxidative stress in the gut. Current standard treatments, such as 5ASA mesalamine, corticosteroids, immunomodulators, and anti-TNFα biologic therapy, focus on treating and controlling disease symptoms, in particular chronic inflammation.
Box 2. Host physiological factors associated with inflammatory bowel disease.
Local factors
Changes in host physiology are observed within the gut during IBD.
Barrier function: The intestinal barrier in inflammatory bowel disease (IBD) is leaky due to changes in epithelial tight junctions, increased rates of apoptosis, erosion events, and ulcerations in the intestinal lining139.
Paneth cells: These specialized intestinal epithelial cells produce antimicrobial peptides, which regulate the gut microbiota. Changes in the numbers and function of Paneth cells have been noted in Crohn’s disease (CD) patients, and functional consequences of these abnormalities have been demonstrated in mice109,140–142. Paneth cell dysfunction is associated with mutations in the IBD-associated host genes NOD2 and Atg16L141.
Goblet cells: Goblet cells, specialized intestinal epithelial cells that secrete mucin, are commonly depleted in IBD143. The mucin layer is thinner in IBD, and mouse mutants lacking Muc2 develop chronic colitis144. Goblet cells also secrete cytokines, deliver antigens to dendritic cells, and affect barrier function145.
Fecal calprotectin: Quantification of this abundant neutrophil protein in stool serves as a readout of gut inflammation. Upon activation or cell death, neutrophils release calprotectin, which is then stable in stool for several days. Fecal calprotectin levels correlate with histological examination and disease severity, particularly in ulcerative colitis (UC).
Systematic factors
Evidence of a host response to IBD is also found systemically, such as in serum.
Microbial antibodies: One of the most commonly measured antibodies, anti-Saccharomyces cerevisiae antibody (ASCA), is used to distinguish CD from UC patients during diagnosis, with CD patients being more likely to have higher levels of ASCA than UC patients104.
Inflammatory markers: The most commonly used inflammatory marker is C-reactive protein (CRP). CRP is produced by the liver and released into the blood in response to inflammatory cytokines accumulating at the site of infection. Due to a short half-life, serum CRP levels rapidly decrease upon the cessation of inflammation. CRP correlates well with disease activity for most patients with UC or CD, although CD patients tend to have higher CRP levels relative to UC patients146.
Cytokines: Inflammation associated with IBD is largely driven by an imbalance between pro-inflammatory and anti-inflammatory cytokines. The role of cytokines in IBD pathogenesis has been recently reviewed147.
Defects of the epithelial barrier have been observed in IBD. Although host IBD-associated genes, such as FUT2 and C1orf10642, have roles in mucosal barrier function, several mechanisms involve gut microorganisms themselves. For example, short-chain fatty acids (SCFAs), particularly butyrate, produced by gut bacteria promote Treg development and enhance mucus production from goblet cells to strengthen the mucosal barrier43,44. SCFAs activate cells via G protein-coupled receptors (GPCRs), such as GPR41 and GPR43, leading to chemokine and cytokine production which regulate protective immunity and tissue inflammation45. Bacterial indole metabolites such as indoleacrylic acid (IA) and indole 3-propionic acid — the latter of which is produced by the intestinal commensal Clostridium sporogenes — regulate intestinal barrier function through the xenobiotic sensor pregnane X receptor46. Produced by Peptostreptococcus species, IA promotes intestinal epithelial barrier function and mitigates inflammatory responses. The biosynthetic gene cluster for IA is decreased in the gut metagenomes of IBD patients, perhaps contributing to barrier dysfunction47. Decreased tryptophan metabolism, reduced SCFA levels, and compromised epithelial barrier compound the detrimental effects of IBD.
Microbial communities have an important role in the maturation and education of the host immune system. Infant gut colonization and the early-life microbiome have long-lasting effects48,49 and are strongly influenced by delivery mode and feeding. Lower diversity and delayed colonization of Bacteroidetes, for example, are linked to delivery by caesarean section and are associated with reduced T helper 1 (Th1) cell responses in the first two years of life50. T cell subtypes have crucial roles in sensing inflammation and ensuring appropriately timed and localized immune responses. The importance of the microbiota for T cell development and immune tolerance has been studied in mouse models. Germ-free mice have fewer CD4+ Tregs in their colons than conventional mice51. CD4+ Tregs express the Foxp3 transcription factor and have a crucial role in the maintenance of immune homeostasis. Further, early life exposure to microorganisms is required in mice to prevent an accumulation of invariant natural killer T (iNKT) cells, which predispose mice to increased morbidity in models of IBD and allergic asthma52. Specifically, either colonization of neonatal mice with conventional microbiota from specific pathogen-free mice52 or monocolonization with a sphingolipid-expressing strain of Bacteroides fragilis53 is sufficient to prevent the accumulation of iNKT cells. Exposure of adult germ-free mice to these microorganisms is insufficient to decrease levels of iNKT cells52,53, supporting a body of literature that suggests early life exposure to microorganisms regulates adult immune functions and disease susceptibility54,55. Colonizing germ-free mice with fecal microbiota from individuals with IBD increases the number of intestinal Th17 cells and decreases the number of RORγt+ Tregs, providing evidence for disease mechanisms involving the gut microbiome in humans. Further, the induced proportions of Th17 and RORγt+ Tregs accounted for colitis severity in Rag1−/− mice lacking adaptive immunity56 and were predictive of human disease status.
In mouse models, segmented filamentous bacteria (SFB) are primary drivers of Th17 responses34,57. Rag1−/− mice have higher levels of SFB than immunocompetent mice58. SFB levels in these immunocompromised mice further increase when the STAT3-dependent innate immune response is compromised, indicating that the murine immune system continues to restrict these closely associated bacteria even in the absence of a functional adaptive immune response58. These findings provided a unique framework to translate mouse studies to human disease. One study isolated 20 bacterial strains from a UC patient based on their ability to induce Th17 cells in mice and showed that adhesion to epithelial cells is a common mechanism used by intestinal microorganisms to activate host Th17 responses59. One host mechanism that restricts SFB independently of the adaptive immune system is the production of α-defensins by Paneth cells, a cell type whose function is diminished in CD. SFB abundances are increased in mouse models of autoimmune polyendocrinopathy candidiasis ectodermal dystrophy, and the host was additionally seropositive for autoantibodies against an enteric α-defensin60. SFBs are typically only found in mice, and the specific human counterpart is unknown.
Microbial factors affect Treg differentiation as well. Tregs are negative regulators of inflammation, and commensal species such as B. fragilis61 and species from Clostridium cluster XIVa, IV, and XVIII51,62 can stimulate the differentiation of Tregs. Alterations of T cell subtypes can have long-lasting effects, where Treg depletion leads to non-remitting destructive disease even after restoration of normal Treg numbers in mouse models of arthritis. Microbial imbalances in IBD likely disrupt regulatory processes that suppress inflammation. For example, a pronounced depletion of Clostridiales organisms was observed in treatment-naive patients with new-onset CD and UC28,63.
These studies present evidence that the gut microbiome is a crucial component for a healthy immune system; however, a comprehensive understanding of mechanisms mediating host–microbial interactions is currently incomplete. To this end, several extensive longitudinal IBD studies were initiated with the aim of linking taxonomic and functional changes in the gut microbiome to IBD pathogenesis.
The gut microbiome in IBD pathology
Early studies determined differences in the microbiota of IBD patients based on 16S ribosomal RNA (rRNA) amplicon sequencing of stool and biopsy samples28,63,64. Collectively, these studies found a decrease in gut microbial diversity in IBD patients65,66, including a decrease in Firmicutes with a depletion of Clostridium cluster IV and XIV species65,67–69 and an increase in Enterobacteriaceae species70–72 (Figure 1). Some, but not all, studies identified changes in Bacteroides, Bifidobacteria, or Lactobacilli species65,67–70,72,73. In pediatric IBD patients, microbial shifts were detected at earlier time points in biopsies compared to stool63. Studies examining bacteria associated with tissue biopsies from IBD patients found more mucosa-associated bacteria in IBD than in controls69,70,73,74 and significant differences in the abundances of tissue-associated bacteria between inflamed and non-inflamed sites67,75. Defining distinct association networks of taxa from intestinal biopsies of CD and UC patients, one study identified Blautia, Faecalibacterium and Ruminococcus species as probable keystone taxa in CD and UC76. They further linked disturbances of Lachnospiraceae and Ruminococcaceae species with relapsing disease, poor response to anti-TNFα therapy and disease recurrence after surgical interventions in CD patients.
16S rRNA amplicon studies have limited taxonomic resolution and predominantly identified family-level or genus-level but rarely species-level associations. Although taxonomic imbalances in IBD have been described, functional disruptions may have a greater impact. However, amplicon studies do not reveal information about the metabolic pathways encoded by the microbial community. To address this possibility and circumvent the limitations described above, metagenomic sequencing of stool samples showed that metabolic pathway abundances were more consistently perturbed. The microbiomes of IBD patients encode more oxidative stress and nutrient transport pathways and fewer pathways related to carbohydrate metabolism and amino acid synthesis63,77. Metagenomic studies have been largely limited to stool samples due to a high host to microbial DNA ratio present in biopsies that makes sequencing total DNA an inefficient way to profile microbial communities. Nevertheless, these early studies indicate that important insights can be gained through metagenomic data.
Several large cohort studies (Table 1) have now used metagenomic sequencing on human stool samples to characterize species and strain-level differences as well as functional alterations in IBD. Comparing IBD, irritable bowel syndrome (IBS), and healthy controls, a study78 analyzed metagenomic profiles of stool samples from 1,792 individuals (355 IBD patients from the NLIBD cohort, of which 208 patients were diagnosed with CD, 126 patients with UC, and 21 patients with IBD-unclassified or indeterminate; 412 IBS patients; and 1,025 controls from the LifeLinesDeep cohort). Overall, 219 taxa from various taxonomic levels (including 152 species) were associated with CD, and 102 taxa (including 93 species) were associated with UC. Profiles were similar between both IBD subtypes, with 87 of the UC-associated taxa also associated with CD. Families with the highest number of decreased taxa in CD were Lachnospiraceae (n=21 taxa) and Ruminococcaceae (n=17), whereas the highest number of increased taxa belonged to the Enterobacteriaceae family (n=8). Taxa decreased in UC were largely from the Bacteroidaceae family (n=5), and taxa increased in UC were from the Lachnospiraceae family (n=11). Furthermore, significantly reduced strain diversity in beneficial species such as Faecalibacterium prausnitzii and Roseburia intestinalis were observed, and bacterial growth rates of B. fragilis and Escherichia coli were increased in CD patients compared to controls. The abundance of antibiotic resistance genes was increased in IBD and correlated with Escherichia species and Bacteroides species abundances. Functional differences were observed in the microbiome of IBD patients, in particular in the synthesis of amino acids, neurotransmitters, and vitamins, as well as the regulation of mineral absorption and the degradation of complex carbohydrates. Pathways related to SCFAs and L-arginine synthesis, which have important roles in maintaining intestinal barrier function and inflammation-associated immunosuppression, were perturbed in IBD.
Table 1.
Summary of human stool metagenomic studies to date.
| Title of paper | Year published | Number of metagenomic or metatranscriptomic datasets | SRA or BioProject identifier | Ref |
|---|---|---|---|---|
| Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment | 2012 | 11 | PRJNA175224 | 77 |
| The treatment-naive microbiome in new-onset Crohn’s disease | 2014 | 43 | PRJNA237362; PRJNA205152 | 63 |
| Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s Disease | 2015 | 369 | SRP057027 | 80 |
| Increased intestinal microbial diversity following fecal microbiota transplant for active Crohn’s disease | 2016 | 53 | PRJNA321058 | 161 |
| A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients | 2017 | 267 | PRJNA385949 | 81 |
| Gut microbiome function predicts response to anti-integrin biologic therapy in inflammatory bowel diseases | 2017 | 175 | PRJNA384246 | 114 |
| Dynamics of metatranscription in the inflammatory bowel disease gut microbiome | 2018 | 300/78 | PRJNA389280 | 94 |
| Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome | 2018 | 1792 | LifeLines DEEP (upon request): European Genome-Phenome Archive EGAS00001001704 UMCG IBD (upon request): EGAD00001004194 Maastricht IBS (MIBS): EGAS00001001924 |
78 |
| Gut microbiome structure and metabolic activity in inflammatory bowel disease | 2019 | 222 | PRJNA400072 | 79 |
SRA, sequence read archive.
In a study investigating changes in metabolic activity of the IBD microbiota, one study performed metagenomic and untargeted metabolomics profiling of 220 samples from an American discovery cohort (PRISM) and a Dutch validation cohort (LifeLines DEEP and NLIBD)79. Metagenomic analysis revealed a pronounced separation of CD and non-IBD patients, whereas UC patients were more heterogeneous. In total, 50 species were differentially abundant in IBD, of which 35 were elevated in controls indicating a general loss of diversity in IBD patients. In particular, Roseburia hominis, Dorea formicigenerans and Ruminococcus obeum were depleted in IBD. In CD patients, an enrichment of R. gnavus, E. coli and Clostridium clostridioforme was observed, whereas Bifidobacterium breve and Clostridium symbiosum were uniquely enriched in UC. Functional metagenomic analysis identified 568 differentially abundant enzymes, the majority of which could be ascribed to a single species. For example, E. coli dominated 200 of the differentially abundant enzymes, owing in part to the species’ strong enrichment in IBD and exceptionally thorough functional annotations. Whereas some predicted functions may indicate mechanistic links between the microbiome and IBD, others may simply indicate changes in the abundances of genomes encoding those functions. Overall, 246 differentially abundant enzymes were not dominated by a single species, suggesting that their enrichment is the result of a community-level shift in functional potential and therefore is of greater mechanistic significance. Examples include a magnesium-importing ATPase and an ethanolamine ammonia-lyase. As metabolites constitute a direct measurement of functional activity, their quantification may be more effective in identifying putative mechanistic associations. Metabolic alterations identified in this and other studies will be discussed further in a later section.
In addition to species-level and functional characterizations of the gut microbiome in IBD patients, temporal changes are important to monitor. Interindividual differences account for ~50% of the variation observed in taxonomic community composition28. Further, less than half of IBD patients respond to conventional treatment strategies, and disease is characterized by periodic bouts of inflammation separated by periods of remission. For these reasons, it is particularly important to conduct longitudinal studies that capture changes of microbial features and loss of response to treatment over time. To date, there have been only a handful of longitudinal, metagenomic studies that associated microbial species with IBD as described in more detail below.
One study focused on a treatment-naive pediatric CD cohort and monitored patients over the first 8 weeks of treatment with either anti-TNFα therapy or enteral nutrition80. Enteral nutrition consists of nutritionally complete liquid diets presenting simpler, often predigested amino acids, fatty acids, and oligosaccharides that can be absorbed quickly by the host and reduce the metabolic activity of the microbiota. Using metagenomic sequencing of gut microbiota from healthy individuals and CD patients, the authors found that changes in microbial genes distinguished between CD patients: decreases in selenocompound metabolism pathways and increases in microbial genes encoding glycerophospholipid metabolism, aminobenzoate degradation, sulfur relay systems, and glutathione metabolism were predictive of more profound microbial shifts within CD patients. By sequencing the entire genetic content of the samples instead of amplifying a specific bacterial gene (such as the 16S rRNA gene), they further showed that IBD was correlated with higher levels of fungal and human DNA. After one week of therapy, the authors were able to predict which patients would achieve remission based on their gut microbiome; however, gut microbial composition of CD patients remained altered over the course of the study80.
Another study examined the microbiome of patients with established CD or UC in a meta-analysis of two studies with longitudinal data80,81. In addition to examining taxonomic differences, they binned bacteria by their oxygen utilization capabilities as facultative or obligate anaerobes, which can and cannot utilize oxygen as a terminal electron acceptor. The authors found that facultative anaerobes are overrepresented in IBD patients, and oxygen utilization explained eight out of nine species that were consistently differentially abundant between the two studies. This supports previously published hypotheses that the microbial shifts associated with IBD are in part due to increased aerobicity in the inflamed gut63,77. The only species that was not explained by oxygen utilization was R. gnavus, an aerotolerant obligate anaerobe that transiently dominates the IBD microbiota during periods of increased disease severity. Such dynamics of bacterial abundances can only be captured in longitudinal studies. Furthermore, metagenomic data allowed the researchers to distinguish between two strain-specific clades of R. gnavus, one of which is found in both controls and IBD patients, whereas the other is only present in the IBD gut81. As the field transitions from amplicon studies towards metagenomic studies, our knowledge of strain diversity within a given species will increase.
Although these studies identified differentially abundant species in IBD, many microorganisms identified across studies differ in presence or absence, or abundance, making it challenging to identify common patterns. Figure 2 is a phylogenetic representation summarizing all species associations identified across these metagenomic studies. Colored leaves of the phylogenetic tree represent differentially abundant species identified in at least two of the studies. The outer rings summarize study-specific results. While this visualization highlights the broad spectrum of IBD-associated bacterial species from all major taxonomic groups typically found in the gut, it also reveals phylogenetic patterns. Species from the Proteobacteria phylum, such as E. coli, are generally increased in IBD. Actinobacteria are generally decreased, with the majority of these associations identified in the NLIBD cohort. Actinobacteria have been observed to be highly prevalent in the gut microbiomes of healthy Dutch individuals compared to American individuals23,82. Most Bacteroidetes species identified in at least two studies were depleted in IBD, in particular several Alistipes species. Although Firmicutes include increased and decreased species in IBD, the phylogenetic analysis highlights that patterns are largely clade-specific, with Eubacteria species and Ruminococcaceae consistently decreased and Streptococci species and Lactobacilli species increased. Furthermore, Lachnospiraceae species including Clostridia showed opposing trends, suggesting species-specific interactions with the host immune system.
Figure 2. Phylogenetic tree of bacterial species associated with inflammatory bowel disease.
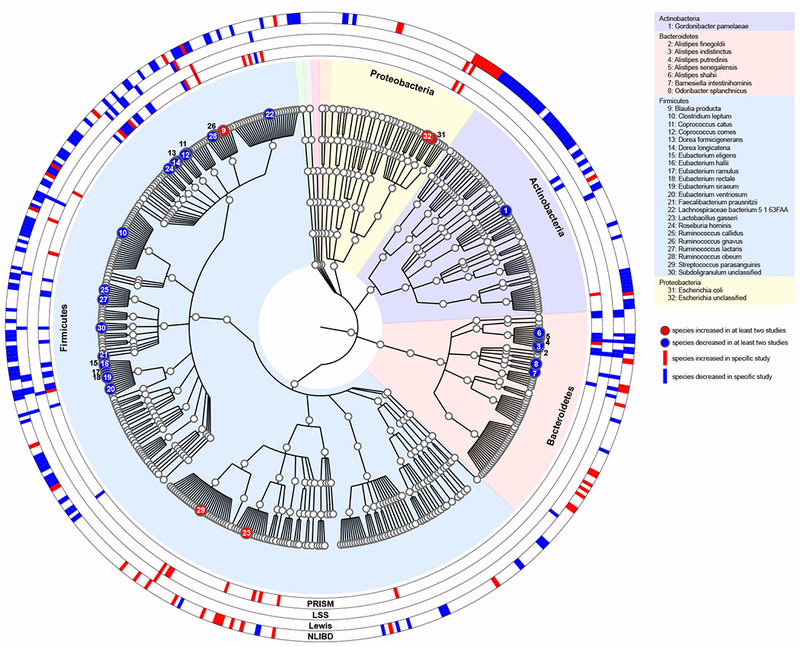
Multiple studies have implicated bacterial species in inflammatory bowel disease (IBD); however, results differ between studies. The association results from metagenomic studies have been summarized in a phylogenetic tree to highlight common pattern across studies. The phylogenetic tree (generated using the software GraPhlAn)160 was constructed based on all bacterial species identified in samples from PRISM79 (n=159), LSS81 (n=271), Lewis80,81 (n=368), and NLIBD in combination with LLDeep controls78 (n=1,380) and includes all species that were detected in at least 20 samples (nspecies=726). Colored tree leaves indicate species that were differentially abundant (false discovery rate <0.1) in at least two studies, with 6 increased (red circles) and 26 decreased (blue circles) species. The outer rings indicate study-specific results and highlight all microorganisms identified in the respective study (Supplementary Table 1 lists the differentially abundant species for each study, respectively). Background colours indicate all species that belong to the same phylum.
Communication between microorganisms and the host is not unidirectional. Just as microbial products can influence host cell physiology, host factors such as chromogranins and secretogranins appear to influence gut microbial composition83. Overall, large cohort analyses have provided a detailed characterization and indicate substantial changes of gut microbiota composition in IBD based on metagenomics data. Integrating functional microbial measurements, such as metatranscriptomics and metabolomics data, and measurements of host responses will be crucial to generate testable hypothesis and gain mechanistic insights into the role of the microbiome in disease.
Microbial strains implicated in IBD
As highlighted by the example of R. gnavus in the previous section, strain-level variation within a species can be important. One of the difficulties in drawing conclusions from amplicon sequencing data is that diversity within a bacterial species is not fully captured. Organisms of the same species are defined to have 70% DNA–DNA hybridization or 95% average nucleotide identity84; therefore, strains of the same species can possess important functional differences, including antibiotic resistance or pathogenicity. Most E. coli strains are harmless and part of a healthy gut microbiota, but some are pathogenic and can cause serious and fatal diseases by producing, for example, Shiga toxin. Furthermore, specific E. coli strains such as adhesive invasive E. coli (AIEC) are implicated in IBD. AIEC is a pathobiont and its role in IBD was recently reviewed in detail85. Briefly, AIEC is able to evade the immune system and to adhere and invade intestinal epithelial cells and macrophages in genetically susceptible hosts, including IBD patients. In addition to taking advantage of IBD host genetics that impair autophagy mechanisms, AIEC can suppress autophagic processes enabling it to survive and thrive inside cells. First, bacterial translocation allows AIEC to gain access to the lamina propria, where it is engulfed by macrophages but escapes autophagy. Continuous replication within macrophages results in the secretion of high levels of TNFα without inducing host cell death, leading to gut inflammation and AIEC over-colonization. Hyper-motility and increased acetate-consumption are associated with strains of E. coli from CD patients compared to those from healthy individuals, suggesting AIEC adaptation within the host86. Various strategies for targeting AIEC strains and/or inhibiting AIEC adhesion, including bacteriophages and small molecule drugs, are being investigated as potential therapies for IBD87.
In the absence of metagenomic sequencing, studies relied on culture-based methods to distinguish pathogenic strains. Culture-based methods were instrumental in identifying the ability of human oral Klebsiella strains to drive inflammation in the colon of genetically susceptible mice (that is, Il10−/−). Although some oral isolates of Klebsiella species from both controls and IBD patients induced murine colon inflammation, this was not consistent across the tested human, mouse, and environmental strains88. In particular, the Klebsiella pneumoniae 2H7 strain strongly induced accumulation of interferon-γ+ CD4+ Th1 cells in the intestinal lamina propria, however, only in genetically susceptible mice. Strain-specific genes associated with Th1 cell induction are predicted to encode hemolysin-coregulated protein and enzymes involved in fructose-, galactitol-, mannose-, and long-chain fatty acid-related uptake and metabolic pathways. Furthermore, the ability of pathobionts to outgrow other bacteria in the inflamed gut is supported by sequencing data, in which Klebsiella species and other bacteria associated with the oral cavity are more abundant in the guts of IBD patients compared to controls28,88,89. Klebsiella species are also more abundant in mouse models and human cohort studies of other gut diseases90,91. The specific mechanisms involved in bacterial translocation and inflammatory responses, however, remain to be elucidated.
Beneficial species that elicit positive effects and provide protection for the host can also be diverse as evidenced by F. prausnitzii, which contains numerous subspecies or phylogroups. Targeted qPCR studies suggested that the presence or absence of F. prausnitzii phylogroups may be indicative of disease92. The implication of the presence or absence of different F. prausnitzii phylogroups on disease is currently unclear as the full diversity of this group is not apparent in the short hypervariable regions sequenced in most amplicon-based studies. Even when the full 16S rRNA gene is sequenced, many F. prausnitzii strains do not fall into any of the identified phylogroups92.
Studies of strain-specific phenotypes, such as those outlined here, will likely become more common and will enable a deeper understanding of the role of microorganisms in IBD pathogenesis. As the number of genomes associated with pathogenesis-promoting strains increases, fewer genes will be shared exclusively among these disease-causing strains. This will eventually facilitate the identification of virulence factors (either presence or absence) and enable the comparison of potential virulence genes (including SNP identification) across species to identify commonalities.
Insights from metatranscriptomics
Metagenomics is limited to revealing the functional potential of microorganisms, rather than the actual functional activity. The presence of a gene or pathway does not necessarily mean that it is expressed. In addition to isolating DNA for metagenomics, RNA can be extracted from a sample, reversed transcribed into cDNA and sequenced for metatranscriptomics that measures actual microbial gene expression. Few studies have investigated the functional activity of the gut microbiome to date. One study established the feasibility of metatranscriptomics for fecal samples and found that metatranscriptional profiles varied more between individuals than metagenome functional profiles, highlighting the importance of measuring actual gene expression in addition to functional potential inferred from gene presence for understanding disease-related microbiota changes. This subject-specific, whole-community regulation suggests that bacteria interact with their host in a very specific, individualistic manner93.
Another study further showed that directly measuring functional activity reveals important insight into gut microbial community dynamics, including IBD-specific transcriptional activity that was either more pronounced or only detectable on the RNA level94. The authors observed that the distribution of microbial species encoding a pathway can remain fairly constant in a patient over time while pathway transcription can change. For example, in the context of the methylerythritol phosphate pathway, increases in disease severity for a patient co-occurred with A. putredinis domination of pathway transcription while no DNA-level changes were observed. Species-specific transcriptional biases in metabolic pathways were also observed. Transcription of many pathways such as the dTDP-L-rhamnose biosynthesis I pathway that produces deoxysugar β-l-rhamnopyranose — a building block of surface glycans that are often targets of the immune system — was dominated by F. prausnitzii.
Microbial transcriptional programs can respond rapidly to environmental cues such as changes in inflammation and oxygen levels, which may not necessarily be reflected on the DNA level. However, some caveats apply to fecal metatranscriptomics, such as variation due to subject-specific transit times, and that it only captures extractable, non-degraded RNA restricted to organisms that are present in stool. Additionally, owing to a high ratio of host to microbial DNA and RNA, performing metagenomics or metatranscriptomics on biopsy samples is not yet cost effective. In the absence of metatranscriptomic data, the ratio of reads located near the chromosomal origin of replication compared to reads near the terminus for a given bacteria can be used to estimate growth dynamics from metagenomic data95. Although it does not answer questions about gene expression, this approach cleverly enables one to determine which bacteria are actively dividing and presumably transcribing their genes.
Metabolites associated with health and IBD
Several classes of small molecules produced or modified by the gut microbiota can modulate immune and epithelial cell function (Box 3)96,97. Comparisons between conventional and germ-free or SPF mice show drastic differences in serum and tissue metabolites, underscoring the importance of the microbiome for host metabolism system-wide (reviewed in Ref. 98). In mouse models, intestinal inflammation and clinical response to dextran sulfate sodium (DSS)-induced colitis can be altered by postbiotically modulating levels of the microbial metabolites taurine, histamine, and spermine, highlighting the potential clinical relevance of microbial metabolites99. A combination of microbial and diet-derived metabolites likely contributes to inflammatory diseases such as IBD96.
Box 3. Microbial immunomodulatory molecules.
Microbial metabolites are critical for host-microbiota interactions and regulate host immune responses.
Short-chain fatty acids (SCFAs), including butyrate, propionate, and acetate, are byproducts of bacterial breakdown of dietary fibers. SCFAs can affect gene expression and cell proliferation during immune responses by modulating histone deacetylases148. Butyrate and butyrate-producing bacteria are less abundant in the stool of inflammatory bowel disease (IBD) patients21.
Tryptophan can be converted into bioactive indole-containing metabolites by gut bacteria. Significant differences in tryptophan metabolite levels have been observed in the serum of germ-free mice compared to conventional mice149. Indole derivatives can affect the host by activating aryl hydrocarbon receptor, which regulates inflammation150. Further, indoleacrylic acid, a specific indole derivative produced by the mucus-utilizing bacterium Peptostreptococcus, induces mucin gene expression and activates the NRF2 pathway47. This further affects host tryptophan metabolism by decreasing tryptophan availability, which reduces host metabolism products such as serotonin151. In IBD patients, bacterial metabolism of tryptophan is reduced.
Bile acid metabolites are generated by bacteria from host-produced bile acids. These secondary bile acids are sensed by host receptors including FXR and TGR5 and can regulate genes involved in immune cell maturation, cytokine release, and microbial defense152. Production of secondary bile acids is reduced in IBD patients. Taurine, which is cleaved from certain classes of primary bile acids, is an inflammatory metabolite that activates IL-18 expression99. Additionally, conjugated bile acids cause CD4+ T effector cell-mediated upregulation of the xenobiotic transporter, Mdr1, to maintain homeostasis. Mdr1 function in IBD patients is reduced relative to controls153.
Succinate is produced by Bacteroides species through the breakdown of dietary fiber, which in turn is metabolized by Clostridium species to produce butyrate and ATP154. Succinate increases IL-1β production by stabilizing hypoxia-inducible factor-1α in macrophages and stimulating dendritic cells via succinate receptor 1155. Serum succinate levels are increased in hypertension, ischemic heart disease, type 2 diabetes, and obesity156. Murine colonic succinate levels are affected by dietary fiber concentrations, antibiotics, and chemically induced intestinal motility disturbances157,158.
Sphingolipids, including sphingosine, ceramide, and sphingomyelin, are a class of plasma membrane-associated lipids that are produced by both the host and specific bacteria. Sphingolipids, most prominently ceramide, are closely tied to metabolic, apoptotic, and inflammatory pathways in host cells. In IBD, the cellular levels and distribution of different sphingolipids is significantly different between inflamed and non-inflamed intestinal tissue53,159.
Several human studies identified metabolite differences in stool79,100,101, serum101,102, or mucosa of IBD patients compared to controls. Taurine and cadaverine levels are increased in UC, and carnosine, ribose, and choline levels correlate with inflammation as measured by fecal calprotectin101. In a twin-pair study of healthy individuals and IBD patients, increases in tryptophan, bile acids, and unsaturated fatty acids were linked to ileal CD103. Strong associations between disease-associated microorganisms and metabolites were found by pairing metabolomics with microbial taxonomic analyses, showing increased levels of bile acids, sphingolipids and tryptophan79,100,103. IBD patients with inactive disease displayed similar microbiomes and metabotypes to their healthy first-degree relatives100. Although the mechanistic relationship between host disease, microorganisms, and metabolites is becoming clearer, key questions about disease-associated metabolites remain to be answered — whether they are bacterially produced or metabolized, affect bacteria directly or indirectly by altering host physiology, or a combination of these possibilities.
Studies often perform a combination of targeted (known) and untargeted (unknown) metabolomics, but subsequent analyses, as those described above, have focused on the small subset of known molecules. The majority of the gut metabolome is uncharacterized, and untargeted metabolomics has great potential to identify novel disease-associated molecules. Computational approaches will have a crucial role in prioritizing microorganism–metabolite associations for experimental validations. This knowledge can be further used to develop therapeutic approaches that either inhibit disease-associated microbial metabolism and the corresponding microorganisms (for example, tungstate treatment) or augment beneficial metabolites and their respective species (for example, probiotic and postbiotic treatments; Figure 3).
Figure 3. Microbiome-based therapies for inflammatory bowel disease.
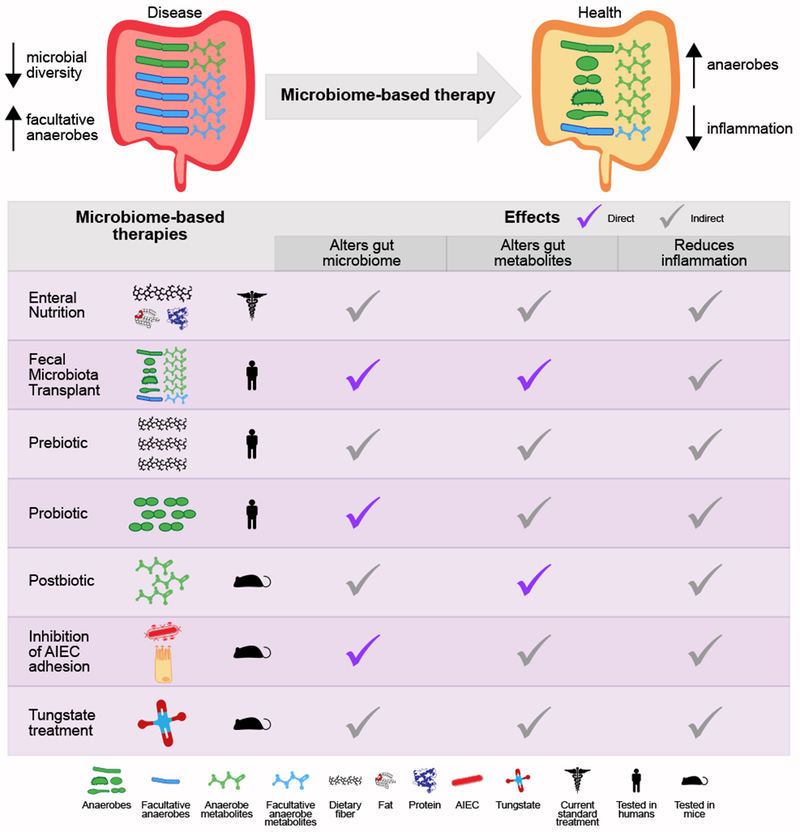
Microbiome-based therapies for inflammatory bowel disease aim to restore the gut microbial balance, which includes increasing microbial diversity, in particular anaerobic bacteria, reducing facultative anaerobes, and reducing gut inflammation (part a). Current and developing treatments either alter nutrition, administer microbial organisms and/or metabolites, or directly target microorganisms and/or pathways (part b). The effects of these treatments, including reduction of gut inflammation and alterations of the gut microbial communities and metabolites, are either direct or indirect. AIEC, adhesive invasive Escherichia coli.
Fungi and viruses in IBD
Most efforts to date have focused on bacterial alterations of the IBD gut microbiome, in part due to the popularity of 16S rRNA amplicon sequencing. However, the gut also harbors a diverse community of fungi and viruses that may have roles in IBD pathogenesis, affecting the host directly or indirectly by affecting bacterial members of the gut microbiota (for example, bacteriophages). Although relatively few studies have investigated IBD-associated fungi, ASCA is elevated in IBD, suggesting a role for fungi in disease pathogenesis104 (Box 2). One study observed fungal taxonomic shifts in IBD with an increased Basidiomycota species to Ascomycota species ratio, a decreased proportion of S. cerevisiae, and an increased proportion of Candida albicans compared to healthy controls105. Similarly, another study found that disease severity was positively correlated with fungal representation80 and hypothesized that the CD-specific gut environment may favor fungi at the expense of bacteria or that antibiotic treatment creates a niche for fungal expansion. Recent studies have started to investigate the interactions between intestinal fungi and immune cells; for example, CX3CR1+ mononuclear phagocytes are essential for the initiation of immune responses to intestinal fungi, and a missense mutation in the gene encoding CX3CR1 was associated with impaired antifungal responses in CD patients106. Furthermore, S. cerevisiae enhanced host purine metabolism in murine colitis models, leading to an increase in uric acid production. Treatment with uric acid in turn increased gut permeability in mice.107 In addition, Malassezia restricta, a fungus typically found on the human skin, is enriched in the colonic mucosa of CD patients with a disease-linked polymorphism in CARD9, a signaling adaptor important for anti-fungal defense. In mouse models, M. restricta exacerbated colitis through mechanisms requiring Card9108.
Enteric viruses are known to trigger disease onset in genetically susceptible mice. An interaction between murine norovirus infection and a mutation in the autophagy gene Atg16L1, a known CD susceptibility allele, induces abnormalities in granule packaging and unique patterns of gene expression in Paneth cells, albeit Paneth cells are not infected by the virus109. CD-like pathologies were observed in response to DSS-induced colitis but were dependent on this virus-plus-susceptibility gene interaction, as mice carrying either alone did not exhibit similar pathologies. Furthermore, pathologies were TNFα-dependent and IFNγ-dependent, and preventable with broad spectrum antibiotics, implicating the commensal gut microbiota. Thus, a combination of environmental factors, commensal bacteria, and a virus-plus-susceptibility gene interaction led to an IBD phenotype. Other studies linked FUT2 mutations, which lead to asymptomatic norovirus infections, with the pathogenesis of ileal CD110. Recent findings revealed that noroviruses target tuft cells within the intestinal epithelium111, and tuft cell-specific gene expression in the colon decreases with broad-spectrum antibiotic treatment. In antibiotic-treated mice, IL-4 and IL-25 stimulation induce tuft cell hyperplasia in the ileum but not in the colon, indicating that both intestinal bacteria and type 2 cytokines regulate tuft cells in a tissue-specific manner.
Several studies have recently investigated the role of the enteric virome in human IBD using virus-like particle enrichment112,113. A significant expansion of Caudovirales bacteriophages was detected in CD and UC patients; however, this expansion was cohort-specific and highlights the need for further studies. Analysis of viral sequencing data is currently challenging as the large majority of reads are of unknown origin and cannot be assigned to reference genomes. Viral classification is further complicated by integration of many virus genomes into the host genome. As more tools are developed and viral reference genomes are sequenced, we will be better able to determine the role of the virome in IBD.
Microbiome-based IBD therapies
Several treatments for IBD are available; however, most of these treatments have remission rates of less than 50%. In some cases, knowledge of a patient’s microbiome composition can predict response to specific IBD treatments. For example, the response to anti-integrin treatment114, anti-TNFα therapy115, or ustekinumab therapy can be predicted based on a combination of the gut microbiome and other clinical factors116. Another study examined treatment-naive pediatric patients, whose treatment was not standardized in the study protocol, and was able to predict a patient treatment response with 76.5% accuracy based on the abundance of six bacterial genera in pre-treatment samples: Faecalibacterium, Veillonella, Fusobacterium, Coprococcus, Akkermansia, and Adlercreutzia117. Although additional replications of these results are required, they highlight the therapeutic potential of the gut microbiome in choosing optimal treatment strategies for IBD patients.
IBD treatments targeting host factors and microorganisms (including microbial physiology and metabolites) that influence disease are in various stages of development (Figures 1 and 3). Current standard treatments suppress the immune system and alter diet, with dietary modification being one of the most common behavioral interventions for IBD. This includes prebiotic effects that shift the microbial composition as a result of changes in nutrient availability; however, the scientific evidence for many dietary modifications is lacking (reviewed in Ref. 118). To date, the strongest clinical evidence was observed with enteral nutrition treatment for CD. Although not effective for UC patients, enteral nutrition has comparable efficacy to corticosteroids in pediatric CD patients. The effect of enteral nutrition is less compelling in adults, which may in part be due to poor compliance80,119,120.
Diet has a strong influence on gut microbial communities121 and dietary changes have rapid effects on gut microbiota composition independent of inflammation and antibiotics80. Moreover, dietary patterns have been associated with IBD risk (recently reviewed in Ref. 122). Many challenges arise when evaluating dietary effects on disease, including accuracy of information on dietary intake, complex interactions between foods consumed, and differences in food metabolism among individuals122. Despite these challenges, clinical studies have shown that certain dietary components can promote or prevent intestinal inflammation and influence IBD risk. A prospective study following 170,776 women monitored long-term intake of dietary fiber. Intake of the highest quintile (median of 24.3 g/day) was associated with a 40% reduction of CD risk compared to the lowest quintile (median of 12.7 g/day)123. The protective effect of dietary fiber on CD risk may be mediated through gut microorganisms123 that metabolize fiber into SCFAs, which leads to an increased mucosal immune tolerance through the activation of GPCRs and the subsequent activation of Tregs121. In addition, interactions between the gut microbiota and dietary concentrations of proteins and fiber can change intestinal permeability and severity of intestinal inflammation in mice124. Reduced consumption of red and processed meat, however, did not decrease the rate of CD flares in a separate study125. In the future, engineered diets that restrict deleterious components but supplement beneficial nutrients may be used alone or in combination with other therapies to maintain or prevent disease.
Fecal microbiota transplantation (FMT), in which the stool of a healthy donor is transferred to the intestinal tract of a patient, has been highly effective in treating Clostridium difficile infections and has been assessed for the treatment of IBD126. Many FMT studies have been limited by small sample number and have employed different methods for administering FMT, making results across studies difficult to compare. Despite these challenges, evidence supports that FMT induces clinical remission in UC, particularly when patients received multiple, lower gastrointestinal infusions126. Clinical remission was achieved for 28% of UC patients across four randomized controlled trials127. Variable response rates to FMT in UC are likely due to the heterogeneity of the disease. The emerging consensus in the field, however, is that FMT has potential as a treatment for UC if 1) antibiotics are administered prior to FMT treatment; 2) inflammation can be controlled, which would likely result in higher efficacy rates; and 3) a designed cocktail is used that replaces missing and boosts beneficial organisms. Encouragingly, engraftment of species after FMT can be predicted based on the abundance and phylogeny of the bacteria in the donor sample and pre-FMT patient sample, and donor strains engraft in an all-or-nothing manner128. Although there is evidence that FMT may induce clinical remission in CD, fewer studies of FMT in CD have been conducted and the confidence interval is broad126. Only one study reported endoscopic outcomes, which may not correlate with clinical outcomes in CD described in other studies. In this study, no patient achieved endoscopic remission126,129. The long-term effects of FMT and the ability to use FMT as a maintenance therapy have not yet been examined in IBD.
The microbiome alters host immune function and provides a number of therapeutic leads and targets34,41,47,51,57–59,61,62,130,131. Little evidence supports the efficacy of prebiotic or probiotic treatment for IBD; however, the possibility remains that the most efficacious probiotic bacterial strains have yet to be identified. The adaptive immune response could be modulated by administrating microbially-derived metabolites or enzymes in postbiotic therapies. For example, nitrogen scavenging pathways of Proteobacteria are a potential therapeutic target, as Proteobacteria gain a growth advantage from host-derived nitrogen, which exacerbates colitis in mouse models132,133. Host nitrogen sources include nitric oxide produced by immune cells or urea produced as a by-product of host metabolism. In fact, one study showed that tungstate treatment, which specifically targets the molybdenum cofactor-requiring enzymes needed to use nitric oxide for anaerobic respiration, can inhibit Proteobacteria replication and ameliorate colitis in mice134. These types of therapies are still in early stages of development; however, increasing our knowledge of mechanisms by which specific microorganisms interact with the immune system will increase our ability to develop directed microbiome-based therapies.
Conclusions and future perspectives
IBD is a complex disease involving host, microbial, and environmental factors. Adopting multi-disciplinary approaches that connect genetic risk factors, microorganisms, and microbial metabolites with altered immune responses and epithelial cell functions will be essential to fully understand the underlying mechanisms of host–microbiota interactions in disease135,136. Gut commensals contribute important functions for human health that include developing and maintaining a robust immune system, providing colonization resistance against pathogens, maintaining the intestinal mucosal barrier, and regulating host immunity. There is no single causative organism and IBD is a polymicrobial disease with more severe disease linked to reduced gut microbial diversity and blooms of bacteria such as R. gnavus and E. coli. At the same time, the gut microbiome is required for disease onset, as mouse models in germ-free conditions rarely develop IBD-like phenotypes, and broad-spectrum antibiotics can prevent disease onset in mice. In humans, antibiotics can lead to remission in severe cases, and enteral nutrition reliably induces remission in pediatric CD, albeit reducing gut diversity. Neither of these approaches, however, result in lasting remission or curing the disease. This paradox highlights that in order to develop effective therapies and eventually cure IBD we need to understand the mechanisms underlying aberrant interactions between the host immune system and the gut microbiome.
For future research in the field it will be particularly important to develop an understanding of how microorganisms and microbial products affect the immune status of the host in both health and disease. Functional differences between strains (for example, R. gnavus and inflammation-inducing oral Klebsiella species) can provide important insights and opportunities for mechanistic studies. Multi-omics technologies have potential to predict disease-relevant host–microbial mechanisms by identifying transcriptional alterations and metabolic changes in IBD. However, the large proportion of microbial genes with unknown function and microorganisms without a sequenced genome need to be taken into account to fully characterize the impact of the microbiome. Fungal and viral communities in the gut have also been implicated in IBD, but are currently largely unexplored. The ability of norovirus to trigger IBD-like phenotypes and the tropism of norovirus for tuft cells suggest that tuft cells may have a yet-unknown role in IBD. Furthermore, because gut microbial composition is highly individualized, we must focus clinical studies on longitudinal tracking of unique patient subsets to understand how normal host–microbiota interactions are shifted in IBD and in response to treatments. Investigating the initial microbiome state of treatment-naive patients and identifying changes over time implicated in disease progression, such as the development of complications135, disease course and treatment efficacy28, will be particularly illuminating. Finally, rather than identifying microorganisms and microbial products that cause inflammation in mice and exploring their relevance in humans, it will be important for future functional studies to focus on human disease-relevant microbial factors and test their effects in mouse models137.
All of these research avenues have high clinical value and will reveal important factors implicated in IBD development. One of the biggest limitations of the current standard of care is that it treats disease symptoms, such as chronic inflammation, rather than restores the host–microbial balance that leads to IBD. Microbiome-based therapeutic interventions could augment beneficial bacteria, target pathogenic organisms, use synthetic organisms or leverage microbial bioactive metabolites to reverse specific defects in IBD by restoring community structure and promoting barrier restitution, immune tolerance and tissue healing. Importantly, IBD is a heterogeneous disease with distinct clinical manifestations. As findings are translated to the clinic, significant microbiome-related differences between disease subtypes will be crucial to consider in order to develop targeted therapies and improve treatment efficacy rates.
Supplementary Material
Acknowledgements
We thank Theresa Reimels for editorial assistance and for help with figure design. R.J.X. received funding from the National Institutes of Health (P30 DK043351 and R01 AT009708), the Crohn’s and Colitis Foundation of America, and the Center for Microbiome Informatics and Therapeutics at MIT. RJX is a consultant to Nestle and Novartis.
Glossary
- 16S rRNA gene
A gene conserved among bacteria often used for taxonomic classification
- Anti-TNFα therapy
Drugs that target TNFα to decrease inflammation are often used to treat autoimmune diseases and IBD
- Atypical perinuclear anti-neutrophil cytoplasmic antibody (pANCA)
Anti-neutrophil cytoplasmic antibody (ANCA) is classified based on staining patterns. Cytoplasmic ANCA (cANCA) refers to staining of the entire cytoplasm, and perinuclear ANCA (pANCA) refers to staining of the area around the nucleus. pANCAs have been implicated in IBD; however, their target antigens are unknown, and they are therefore described as atypical pANCA
- Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy
Autoimmune disease characterized by destruction of endocrine tissues, chronic mucocutaneous candidiasis and ectodermal disorders
- Barrier function
Epithelial cell-cell junctions plus mucosal layer that permit nutrients and prevent luminal contents from accessing the rest of the body
- Chitobioside
A building block of the N-acetylglucosamine-based glycan chitin and a component of the cell walls of microorganisms
- Chromogranin/Secretogranin
A family of water-soluble acidic glycoproteins that are mainly produced by endocrine cells such as the enteroendocrine cells of the gut. Also known as granins, they are precursors of biologically active peptides involved in inflammation
- Colectomy
A surgical procedure removing all or part of the colon
- Culturomics
The process of using classical microbiological techniques to culture and identify unknown bacteria that inhabit a given environment
- Glycemic response
The glycemic response to food describes its effect on blood glucose levels after consumption
- Hygiene hypothesis
According to the hygiene hypothesis, a lack of early childhood microbial exposure affects the development of the immune system. This has been ascribed to the increase of allergic and autoimmune diseases in Western countries
- Indole metabolites
Indole metabolites derive from microbial metabolism of tryptophan and can be recognized by several host receptors that regulate host-microbial homeostasis
- Irritable bowel syndrome (IBS)
A chronic condition, which affects the large intestine and causes abdominal pain, cramping, and shifts in bowel movement patterns. In contrast to inflammatory bowel disease, IBS is not associated with mucosal inflammation, ulcers or other damage to the bowel
- Keystone taxa
Species with high connectivity in microbial networks (built based on statistical associations), suggesting that they are a key component of the ecosystem and their removal would result in drastic changes to the microbial ecosystem
- Laminaribioside
A glucose disaccharide building block of laminarin and a component of the cell walls of fungi and algae
- Mannan
A mannose polymer and component of fungal and plant cell walls
- Mannobioside
A disaccharide of mannose
- Metagenome
All of the DNA in an environment
- Metatranscriptome
All of the RNA in an environment
- Metabolome
All of the metabolites in an environment
- Microbiome
The genes, genomes and products of the microbiota
- Microbiota
The collection of microorganisms in a particular environment
- Non-caseating granulomas
Granulomas are clusters of immune cells that form during infection, inflammation, or in the presence of a foreign substance to prevent a systemic spread. The absence of necrosis is a defining feature of non-caseating granulomas. In Crohn’s disease non-caseating granulomas are formed during inflammation without an obvious infectious trigger
- Pre-biotics
Certain foods or food components that confer a beneficial effect by providing a competitive advantage to beneficial commensal bacteria capable of metabolizing these substrates or by augmenting the production of metabolic products that result from their fermentation
- Pro-biotics
An organism or multiple organisms that confer beneficial effects to the host
- Post-biotics
Bacterial metabolic products that mediate benefits to the host
- RORγt+ Tregs
Regulatory T cells that express the transcription factor RORγt, a nuclear hormone receptor and critical regulator of anti-microbial immunity
- Strain
The classical microbiological definition of strain is a single bacterial isolate. In the context of metagenomic data, it refers to a combination of single nucleotide polymorphisms that are computationally predicted to be linked and originating from an individual strain genome
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41579-019-0213-6.
References
- 1.Ijssennagger N et al. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc Natl Acad Sci U S A 112, 10038–10043, doi: 10.1073/pnas.1507645112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yano JM et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 161, 264–276, doi: 10.1016/j.cell.2015.02.047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reinhardt C et al. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 483, 627–631, doi: 10.1038/nature10893 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho I et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626, doi: 10.1038/nature11400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee WJ & Hase K Gut microbiota-generated metabolites in animal health and disease. Nat Chem Biol 10, 416–424, doi: 10.1038/nchembio.1535 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Fischbach MA & Segre JA Signaling in Host-Associated Microbial Communities. Cell 164, 1288–1300, doi: 10.1016/j.cell.2016.02.037 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh PJ et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484, doi: 10.1038/nature07540 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Chatelier E et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546, doi: 10.1038/nature12506 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Qin J et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60, doi: 10.1038/nature11450 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Karlsson FH et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103, doi: 10.1038/nature12198 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Wang Z et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63, doi: 10.1038/nature09922 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koeth RA et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19, 576–585, doi: 10.1038/nm.3145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang WH et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368, 1575–1584, doi: 10.1056/NEJMoa1109400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsiao EY et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463, doi: 10.1016/j.cell.2013.11.024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimura KE et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med 22, 1187–1191, doi: 10.1038/nm.4176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arrieta MC et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med 7, 307ra152, doi: 10.1126/scitranslmed.aab2271 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Vatanen T et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 165, 1551, doi: 10.1016/j.cell.2016.05.056 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Zhao G et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc Natl Acad Sci U S A 114, E6166–E6175, doi: 10.1073/pnas.1706359114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang H et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547, 173–178, doi: 10.1038/nature22969 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamas B et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 22, 598–605, doi: 10.1038/nm.4102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imhann F et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 67, 108–119, doi: 10.1136/gutjnl-2016-312135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng SC et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778, doi: 10.1016/S0140-6736(17)32448-0 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Zhernakova A et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569, doi: 10.1126/science.aad3369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothschild D et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215, doi: 10.1038/nature25973 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Kronman MP, Zaoutis TE, Haynes K, Feng R & Coffin SE Antibiotic exposure and IBD development among children: a population-based cohort study. Pediatrics 130, e794–803, doi: 10.1542/peds.2011-3886 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrante M et al. New serological markers in inflammatory bowel disease are associated with complicated disease behaviour. Gut 56, 1394–1403, doi: 10.1136/gut.2006.108043 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dotan I et al. Antibodies against laminaribioside and chitobioside are novel serologic markers in Crohn’s disease. Gastroenterology 131, 366–378, doi: 10.1053/j.gastro.2006.04.030 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Schirmer M et al. Compositional and Temporal Changes in the Gut Microbiome of Pediatric Ulcerative Colitis Patients Are Linked to Disease Course. Cell Host Microbe 24, 600–610 e604, doi: 10.1016/j.chom.2018.09.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaubeck M et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 65, 225–237, doi: 10.1136/gutjnl-2015-309333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Couturier-Maillard A et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 123, 700–711, doi: 10.1172/JCI62236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elinav E et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757, doi: 10.1016/j.cell.2011.04.022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garrett WS et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45, doi: 10.1016/j.cell.2007.08.017 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henao-Mejia J et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185, doi: 10.1038/nature10809 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivanov II et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498, doi: 10.1016/j.cell.2009.09.033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sellon RK et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 66, 5224–5231 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schultz M et al. IL-2-deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am J Physiol 276, G1461–1472 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Rath HC, Wilson KH & Sartor RB Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with Bacteroides vulgatus or Escherichia coli. Infect Immun 67, 2969–2974 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xavier RJ & Podolsky DK Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434, doi: 10.1038/nature06005 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Bunker JJ & Bendelac A IgA Responses to Microbiota. Immunity 49, 211–224, doi: 10.1016/j.immuni.2018.08.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mkaddem SB et al. IgA, IgA receptors, and their anti-inflammatory properties. Curr Top Microbiol Immunol 382, 221–235, doi: 10.1007/978-3-319-07911-0_10 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Moon C et al. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521, 90–93, doi: 10.1038/nature14139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohanan V et al. C1orf106 is a colitis risk gene that regulates stability of epithelial adherens junctions. Science, doi: 10.1126/science.aan0814 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaudier E et al. Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am J Physiol Gastrointest Liver Physiol 287, G1168–1174, doi: 10.1152/ajpgi.00219.2004 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Furusawa Y et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450, doi: 10.1038/nature12721 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Kim MH, Kang SG, Park JH, Yanagisawa M & Kim CH Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 145, 396–406 e391–310, doi: 10.1053/j.gastro.2013.04.056 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Venkatesh M et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 41, 296–310, doi: 10.1016/j.immuni.2014.06.014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wlodarska M et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 22, 25–37 e26, doi: 10.1016/j.chom.2017.06.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work identified a tryptophan derivative produced by gut bacteria that increases mucus production and decreases inflammatory cytokine production.
- 48.Dominguez-Bello MG et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 107, 11971–11975, doi: 10.1073/pnas.1002601107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Backhed F et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 690–703, doi: 10.1016/j.chom.2015.04.004 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Jakobsson HE et al. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut 63, 559–566, doi: 10.1136/gutjnl-2012-303249 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Atarashi K et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341, doi: 10.1126/science.1198469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olszak T et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493, doi: 10.1126/science.1219328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.An D et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156, 123–133, doi: 10.1016/j.cell.2013.11.042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper showed sphingolipids produced by intestinal bacteria play a role in colonic invariant natural killer T cell homeostasis.
- 54.Lopez-Serrano P et al. Environmental risk factors in inflammatory bowel diseases. Investigating the hygiene hypothesis: a Spanish case-control study. Scand J Gastroenterol 45, 1464–1471, doi: 10.3109/00365521.2010.510575 (2010). [DOI] [PubMed] [Google Scholar]
- 55.von Mutius E Allergies, infections and the hygiene hypothesis--the epidemiological evidence. Immunobiology 212, 433–439, doi: 10.1016/j.imbio.2007.03.002 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Britton GJ et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORgammat(+) Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 50, 212–224 e214, doi: 10.1016/j.immuni.2018.12.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gaboriau-Routhiau V et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31, 677–689, doi: 10.1016/j.immuni.2009.08.020 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Mao K et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 554, 255–259, doi: 10.1038/nature25437 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Atarashi K et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163, 367–380, doi: 10.1016/j.cell.2015.08.058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobes J et al. Gastrointestinal Autoimmunity Associated With Loss of Central Tolerance to Enteric alpha-Defensins. Gastroenterology 149, 139–150, doi: 10.1053/j.gastro.2015.05.009 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Round JL & Mazmanian SK Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 107, 12204–12209, doi: 10.1073/pnas.0909122107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atarashi K et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236, doi: 10.1038/nature12331 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Gevers D et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392, doi: 10.1016/j.chom.2014.02.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sokol H, Lay C, Seksik P & Tannock GW Analysis of bacterial bowel communities of IBD patients: what has it revealed? Inflamm Bowel Dis 14, 858–867, doi: 10.1002/ibd.20392 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Scanlan PD, Shanahan F, O’Mahony C & Marchesi JR Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J Clin Microbiol 44, 3980–3988, doi: 10.1128/JCM.00312-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ott SJ et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 53, 685–693 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gophna U, Sommerfeld K, Gophna S, Doolittle WF & Veldhuyzen van Zanten SJ Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol 44, 4136–4141, doi: 10.1128/JCM.01004-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frank DN et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104, 13780–13785, doi: 10.1073/pnas.0706625104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Swidsinski A, Weber J, Loening-Baucke V, Hale LP & Lochs H Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43, 3380–3389, doi: 10.1128/JCM.43.7.3380-3389.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Swidsinski A et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 122, 44–54 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Kotlowski R, Bernstein CN, Sepehri S & Krause DO High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut 56, 669–675, doi: 10.1136/gut.2006.099796 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mylonaki M, Rayment NB, Rampton DS, Hudspith BN & Brostoff J Molecular characterization of rectal mucosa-associated bacterial flora in inflammatory bowel disease. Inflamm Bowel Dis 11, 481–487 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Conte MP et al. Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut 55, 1760–1767, doi: 10.1136/gut.2005.078824 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schultsz C, Van Den Berg FM, Ten Kate FW, Tytgat GN & Dankert J The intestinal mucus layer from patients with inflammatory bowel disease harbors high numbers of bacteria compared with controls. Gastroenterology 117, 1089–1097 (1999). [DOI] [PubMed] [Google Scholar]
- 75.Prindiville T, Cantrell M & Wilson KH Ribosomal DNA sequence analysis of mucosa-associated bacteria in Crohn’s disease. Inflamm Bowel Dis 10, 824–833 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Yilmaz B et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med, doi: 10.1038/s41591-018-0308-z (2019). [DOI] [PubMed] [Google Scholar]
- 77.Morgan XC et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79, doi: 10.1186/gb-2012-13-9-r79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vich Vila A et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci Transl Med 10, doi: 10.1126/scitranslmed.aap8914 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Franzosa EA et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4, 293–305, doi: 10.1038/s41564-018-0306-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work pairs metagenomic and metabolomic data from a cross-sectional IBD cohort to identify associations between gut bacteria and metabolites that are predictive of IBD status.
- 80.Lewis JD et al. Inflammation, Antibiotics, and Diet as Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s Disease. Cell Host Microbe 18, 489–500, doi: 10.1016/j.chom.2015.09.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that microbial dysbiosis results from independent effects of inflammation, diet, and antibiotics and is an important factor in the context of enteral nutrition and anti-TNFα treatment in pediatric Crohn’s disease patients.
- 81.Hall AB et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med 9, 103, doi: 10.1186/s13073-017-0490-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Human Microbiome Project C Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, doi: 10.1038/nature11234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sundin J et al. Fecal chromogranins and secretogranins are linked to the fecal and mucosal intestinal bacterial composition of IBS patients and healthy subjects. Sci Rep 8, 16821, doi: 10.1038/s41598-018-35241-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Konstantinidis KT, Rossello-Mora R & Amann R Uncultivated microbes in need of their own taxonomy. ISME J 11, 2399–2406, doi: 10.1038/ismej.2017.113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martinez-Medina M & Garcia-Gil LJ Escherichia coli in chronic inflammatory bowel diseases: An update on adherent invasive Escherichia coli pathogenicity. World J Gastrointest Pathophysiol 5, 213–227, doi: 10.4291/wjgp.v5.i3.213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elhenawy W, Tsai CN & Coombes BK Host-Specific Adaptive Diversification of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Cell Host Microbe 25, 301–312 e305, doi: 10.1016/j.chom.2018.12.010 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Palmela C et al. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 67, 574–587, doi: 10.1136/gutjnl-2017-314903 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Atarashi K et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 358, 359–365, doi: 10.1126/science.aan4526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study links specific strains of oral Klebsiella species that are increased in IBD with Th1 cell induction and inflammation.
- 89.Rashid T, Ebringer A & Wilson C The role of Klebsiella in Crohn’s disease with a potential for the use of antimicrobial measures. Int J Rheumatol 2013, 610393, doi: 10.1155/2013/610393 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Garrett WS et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300, doi: 10.1016/j.chom.2010.08.004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carvalho FA et al. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med 206, 2179–2189, doi: 10.1084/jem.20090741 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lopez-Siles M, Duncan SH, Garcia-Gil LJ & Martinez-Medina M Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J 11, 841–852, doi: 10.1038/ismej.2016.176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Franzosa EA et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci U S A 111, E2329–2338, doi: 10.1073/pnas.1319284111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schirmer M et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol 3, 337–346, doi: 10.1038/s41564-017-0089-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that metatranscriptomic profiling can reveal species-specific biases in transcriptional activity of gut bacteria, including IBD-specific microbial characteristics providing new insight into the potential mechanisms of host-microbial dysbiosis in disease.
- 95.Korem T et al. Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 349, 1101–1106, doi: 10.1126/science.aac4812 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thorburn AN, Macia L & Mackay CR Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 40, 833–842, doi: 10.1016/j.immuni.2014.05.014 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Donia MS & Fischbach MA HUMAN MICROBIOTA. Small molecules from the human microbiota. Science 349, 1254766, doi: 10.1126/science.1254766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ursell LK et al. The intestinal metabolome: an intersection between microbiota and host. Gastroenterology 146, 1470–1476, doi: 10.1053/j.gastro.2014.03.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Levy M et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443, doi: 10.1016/j.cell.2015.10.048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jacobs JP et al. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. Cell Mol Gastroenterol Hepatol 2, 750–766, doi: 10.1016/j.jcmgh.2016.06.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kolho KL, Pessia A, Jaakkola T, de Vos WM & Velagapudi V Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J Crohns Colitis 11, 321–334, doi: 10.1093/ecco-jcc/jjw158 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Scoville EA et al. Alterations in Lipid, Amino Acid, and Energy Metabolism Distinguish Crohn’s Disease from Ulcerative Colitis and Control Subjects by Serum Metabolomic Profiling. Metabolomics 14, doi: 10.1007/s11306-017-1311-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jansson J et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One 4, e6386, doi: 10.1371/journal.pone.0006386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mitsuyama K et al. Antibody markers in the diagnosis of inflammatory bowel disease. World J Gastroenterol 22, 1304–1310, doi: 10.3748/wjg.v22.i3.1304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sokol H et al. Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048, doi: 10.1136/gutjnl-2015-310746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leonardi I et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236, doi: 10.1126/science.aao1503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chiaro TR et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci Transl Med 9, doi: 10.1126/scitranslmed.aaf9044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Limon JJ et al. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe 25, 377–388 e376, doi: 10.1016/j.chom.2019.01.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cadwell K et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145, doi: 10.1016/j.cell.2010.05.009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McGovern DP et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet 19, 3468–3476, doi: 10.1093/hmg/ddq248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wilen CB et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 360, 204–208, doi: 10.1126/science.aar3799 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Norman JM et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460, doi: 10.1016/j.cell.2015.01.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lopetuso LR, Ianiro G, Scaldaferri F, Cammarota G & Gasbarrini A Gut Virome and Inflammatory Bowel Disease. Inflamm Bowel Dis 22, 1708–1712, doi: 10.1097/MIB.0000000000000807 (2016). [DOI] [PubMed] [Google Scholar]
- 114.Ananthakrishnan AN et al. Gut Microbiome Function Predicts Response to Anti-integrin Biologic Therapy in Inflammatory Bowel Diseases. Cell Host Microbe 21, 603–610 e603, doi: 10.1016/j.chom.2017.04.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kolho KL et al. Fecal Microbiota in Pediatric Inflammatory Bowel Disease and Its Relation to Inflammation. Am J Gastroenterol 110, 921–930, doi: 10.1038/ajg.2015.149 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Doherty MK et al. Fecal Microbiota Signatures Are Associated with Response to Ustekinumab Therapy among Crohn’s Disease Patients. MBio 9, doi: 10.1128/mBio.02120-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shaw KA et al. Dysbiosis, inflammation, and response to treatment: a longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med 8, 75, doi: 10.1186/s13073-016-0331-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Halmos EP & Gibson PR Dietary management of IBD--insights and advice. Nat Rev Gastroenterol Hepatol 12, 133–146, doi: 10.1038/nrgastro.2015.11 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Narula N et al. Enteral nutritional therapy for induction of remission in Crohn’s disease. Cochrane Database Syst Rev 4, CD000542, doi: 10.1002/14651858.CD000542.pub3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dziechciarz P, Horvath A, Shamir R & Szajewska H Meta-analysis: enteral nutrition in active Crohn’s disease in children. Aliment Pharmacol Ther 26, 795–806, doi: 10.1111/j.1365-2036.2007.03431.x (2007). [DOI] [PubMed] [Google Scholar]
- 121.Albenberg LG & Wu GD Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology 146, 1564–1572, doi: 10.1053/j.gastro.2014.01.058 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee D et al. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology 148, 1087–1106, doi: 10.1053/j.gastro.2015.01.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ananthakrishnan AN et al. A prospective study of long-term intake of dietary fiber and risk of Crohn’s disease and ulcerative colitis. Gastroenterology 145, 970–977, doi: 10.1053/j.gastro.2013.07.050 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Llewellyn SR et al. Interactions Between Diet and the Intestinal Microbiota Alter Intestinal Permeability and Colitis Severity in Mice. Gastroenterology 154, 1037–1046 e1032, doi: 10.1053/j.gastro.2017.11.030 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Albenberg L et al. A Diet Low in Red and Processed Meat Does Not Reduce Rate of Crohn’s Disease Flares. Gastroenterology, doi: 10.1053/j.gastro.2019.03.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Paramsothy S et al. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. J Crohns Colitis 11, 1180–1199, doi: 10.1093/ecco-jcc/jjx063 (2017). [DOI] [PubMed] [Google Scholar]
- 127.Levy AN & Allegretti JR Insights into the role of fecal microbiota transplantation for the treatment of inflammatory bowel disease. Therap Adv Gastroenterol 12, 1756284819836893, doi: 10.1177/1756284819836893 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Smillie CS et al. Strain Tracking Reveals the Determinants of Bacterial Engraftment in the Human Gut Following Fecal Microbiota Transplantation. Cell Host Microbe 23, 229–240 e225, doi: 10.1016/j.chom.2018.01.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vermeire S et al. Donor Species Richness Determines Faecal Microbiota Transplantation Success in Inflammatory Bowel Disease. J Crohns Colitis 10, 387–394, doi: 10.1093/ecco-jcc/jjv203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Palm NW et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010, doi: 10.1016/j.cell.2014.08.006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study pioneered the use of IgA coating to identify inflammatory members of the IBD gut microbiota
- 131.Larmonier CB, Shehab KW, Ghishan FK & Kiela PR T Lymphocyte Dynamics in Inflammatory Bowel Diseases: Role of the Microbiome. Biomed Res Int 2015, 504638, doi: 10.1155/2015/504638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ni J et al. A role for bacterial urease in gut dysbiosis and Crohn’s disease. Sci Transl Med 9, doi: 10.1126/scitranslmed.aah6888 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Winter SE et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711, doi: 10.1126/science.1232467 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhu W et al. Precision editing of the gut microbiota ameliorates colitis. Nature 553, 208–211, doi: 10.1038/nature25172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that tungstate treatment inhibits molybdenum-cofactor-dependent microbial respiratory pathways in the mammalian gut and prevents the dysbiotic expansion of Enterobacteriaceae during gut inflammation.
- 135.Kugathasan S et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 389, 1710–1718, doi: 10.1016/S0140-6736(17)30317-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hyams JS et al. Clinical and biological predictors of response to standardised paediatric colitis therapy (PROTECT): a multicentre inception cohort study. Lancet, doi: 10.1016/S0140-6736(18)32592-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tanoue T et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605, doi: 10.1038/s41586-019-0878-z (2019). [DOI] [PubMed] [Google Scholar]
- 138.Podolsky DK Inflammatory bowel disease. N Engl J Med 347, 417–429, doi: 10.1056/NEJMra020831 (2002). [DOI] [PubMed] [Google Scholar]
- 139.Hering NA, Fromm M & Schulzke JD Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J Physiol 590, 1035–1044, doi: 10.1113/jphysiol.2011.224568 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Salzman NH et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11, 76–83, doi: 10.1038/ni.1825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.VanDussen KL et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology 146, 200–209, doi: 10.1053/j.gastro.2013.09.048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Adolph TE et al. Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276, doi: 10.1038/nature12599 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Surawicz CM, Haggitt RC, Husseman M & McFarland LV Mucosal biopsy diagnosis of colitis: acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology 107, 755–763 (1994). [DOI] [PubMed] [Google Scholar]
- 144.Van der Sluis M et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129, doi: 10.1053/j.gastro.2006.04.020 (2006). [DOI] [PubMed] [Google Scholar]
- 145.McDole JR et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483, 345–349, doi: 10.1038/nature10863 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Desai D, Faubion WA & Sandborn WJ Review article: biological activity markers in inflammatory bowel disease. Aliment Pharmacol Ther 25, 247–255, doi: 10.1111/j.1365-2036.2006.03184.x (2007). [DOI] [PubMed] [Google Scholar]
- 147.Francescone R, Hou V & Grivennikov SI Cytokines, IBD, and colitis-associated cancer. Inflamm Bowel Dis 21, 409–418, doi: 10.1097/MIB.0000000000000236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fernando MR, Saxena A, Reyes JL & McKay DM Butyrate enhances antibacterial effects while suppressing other features of alternative activation in IL-4-induced macrophages. Am J Physiol Gastrointest Liver Physiol 310, G822–831, doi: 10.1152/ajpgi.00440.2015 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Wikoff WR et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A 106, 3698–3703, doi: 10.1073/pnas.0812874106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zelante T et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385, doi: 10.1016/j.immuni.2013.08.003 (2013). [DOI] [PubMed] [Google Scholar]
- 151.Williams BB et al. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 16, 495–503, doi: 10.1016/j.chom.2014.09.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Devlin AS & Fischbach MA A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol 11, 685–690, doi: 10.1038/nchembio.1864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cao W et al. The Xenobiotic Transporter Mdr1 Enforces T Cell Homeostasis in the Presence of Intestinal Bile Acids. Immunity 47, 1182–1196 e1110, doi: 10.1016/j.immuni.2017.11.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fischbach MA & Sonnenburg JL Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe 10, 336–347, doi: 10.1016/j.chom.2011.10.002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tannahill GM et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242, doi: 10.1038/nature11986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Serena C et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J, doi: 10.1038/s41396-018-0068-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ferreyra JA et al. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16, 770–777, doi: 10.1016/j.chom.2014.11.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.De Vadder F et al. Microbiota-Produced Succinate Improves Glucose Homeostasis via Intestinal Gluconeogenesis. Cell Metab 24, 151–157, doi: 10.1016/j.cmet.2016.06.013 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Abdel Hadi L, Di Vito C & Riboni L Fostering Inflammatory Bowel Disease: Sphingolipid Strategies to Join Forces. Mediators Inflamm 2016, 3827684, doi: 10.1155/2016/3827684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Segata N GraPhlAn. https://bitbucket.org/nsegata/graphlan/wiki/Home (2014).
- 161.Vaughn BP et al. Increased Intestinal Microbial Diversity Following Fecal Microbiota Transplant for Active Crohn’s Disease. Inflamm Bowel Dis 22, 2182–2190, doi: 10.1097/MIB.0000000000000893 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.