

Abstract

Here we describe a facile, tandem synthetic route for indolo[3,2-c]quinolinones, a class of natural alkaloid analogues of high biological significance. A Ugi four-component reaction with indole-2-carboxylic acid and an aniline followed by a Pd-catalyzed cyclization yields tetracyclic indoloquinolines in good to moderate yields. Commercially available building blocks yield highly diverse analogues in just two simple steps.
Exploring better synthetic strategies to obtain natural products, and analogues/skeletons thereof, are at the very heart of synthetic organic chemistry and medicinal chemistry.1 Approaches achieving atom economy are highly sought after, offering various advantages, minimization of waste, time, and resources. Multicomponent reactions (MCRs) are such an advanced class of organic reactions which, opposite to classical organic reactions, allow for the easy, fast, and efficient generation of chemical diversity in just one assembly step.2 The scaffold diversity of MCRs and the window in chemical space have been undoubtedly recognized by the synthetic community in industry and academia as a great tool to design and discover a variety-oriented series of building blocks with potentially interesting biological activities.2a
Indole fused polyheterocycles, as constituents of diverse natural alkaloids and pharmaceutical agents, have drawn much attention from organic and bioorganic chemists during the past several decades.3 Among these, 2,3-fused indoles are of particular interest as they are a part of a large number of natural products of biological interest such as reserpine (reuptake inhibitor),4a fumitremorgin C (BCRP-specific inhibitor),4b evodiamine (anticancer),4c and terpendole E (KSP inhibitor) (Figure 1).4d
Figure 1.

Some bioactive 2,3-fused indole compounds.
Indoloquinolinones are very important in the fused indole family due to their wide occurrence in numerous bioactive natural products.5 Natural products containing the indoloquinolinone scaffold show diverse biological and pharmacological activities, such as effective DNA intercalators5a and inhibition of Plasmodium falciparum cyclin-dependent protein kinase as potential antimalarial agents.5b This tetracyclic structure can also be utilized as useful building blocks for the synthesis of natural products such as isocryptolepine6 and many other potential antineoplastics.5a
The ubiquity of the indoloquinolinone scaffold in compounds that exhibit promising biological and pharmacological properties inspire research into developing efficient methods for their construction.7 The Wang group disclosed an efficient synthesis of indolo[3,2-c]quinolinones from N-(o-bromophenyl)-3-indolecarboxamide using a Pd-catalyst (Scheme 1, cutoff a). The bromo group on the N-aryl moiety was crucial for the success of this reaction.7a The skeleton can also be assembled from 3-oxo-3-phenylpropanoate derivatives and substituted anilines that involves Pd/Cu catalyzed C–C bond formation and I(III)-mediated oxidative C–N bond formation which was reported by Zhang in 2013 (cutoff b).7b Doyle and co-workers synthesized the skeleton from an indole-3-carboxylate derivative via intramolecular lactamization (cutoff c).7c The indoloquinolinone skeleton can also be constructed through a microwave-assisted thermal electrocyclization of a phenyl isocyanate (cutoff d).7d Furthermore, the Xu group reported a base-free process to access the skeleton via a palladium-catalyzed intramolecular cross dehydrogenative coupling (CDC) reaction (cutoff e).7e The indoloquinolinone skeleton can also be constructed through an intramolecular displacement reaction involving an aromatic fluorine (cutoff f).7f The CuI-catalyzed photochemical or thermal reaction of 3-(2-azidobenzylidene)-lactams can also provide the desired indoloquinolinone skeleton (cutoff g).7g While these methods are useful for the synthesis of valuable indolo[3,2-c]quinolinones, they mainly rely on the manipulation of prefunctionalized substrates and overall require multistep transformations. Moreover, most of them suffer from a limited substrate scope and poor functional group compatibility and require protection of the indole NH. From the perspective of atom economy and step efficiency, an ideal synthesis that could overcome these shortcomings is still highly desired.
Scheme 1. Synthetic Routes of Indolo[3,2-c]quinolinones through Different Cutoffs.

Recently, Lingkai and his colleagues developed an efficient method to construct indolo[3,2-c]quinolinones starting from indole-2-carboxamides in the presence of a Pd-catalyst.8 From our point of view, the methodology is ideally suitable for a multicomponent reaction. Therefore, our aim is to use easily accessible starting materials in a Ugi four-component reaction followed by a tandem/sequential Pd(OAc)2-catalyzed dual C(sp2)–H functionalization of the Ugi products toward indolo[3,2-c]quinolinone analogues, involving a 1,2-acyl migration. To the best of our knowledge, this is the first study on the use of MCR in the synthesis of an indolo[3,2-c]quinolinone library.
Isocyanide-based multicomponent reactions (IMCRs) have attracted much attention, due to the fact that versatile functional groups can be introduced in the MCR adducts, which can undergo further condensations or cyclization reactions leading to an array of structurally diverse scaffolds.9 In this study, starting from the Ugi-4CR of aniline 1a, benzaldehyde 2a, indole-2-carboxylic acid 3a, and tert-butyl isocyanide 4a in methanol at room temperature resulted in the corresponding Ugi adduct 5a in a good yield of 83% after 12 h. With compound 5a in hand, we were keen to investigate the C–H functionalization and optimize the reaction conditions (Table 1). When the reaction was carried out in the presence of 10 mol % of Pd(OAc)2 using 3.0 equiv of Cu(OAc)2 as the oxidant in DMF at 140 °C under N2 for 9 h, the desired product 6a was obtained in 49% yield (entry 1). Although Pd(TFA)2 (51% yield, entry 2) was slightly superior to Pd(OAc)2, we chose Pd(OAc)2 as the catalyst for further scope and limitation studies from the point of view of economy. Reducing the amount of solvent resulted in higher yields (entries 3–4). To our delight, the desired product 6a was formed in 71% yield with the addition of 4.0 equiv of pivalic acid (PivOH) (entry 5). Decreasing the amount of PivOH to 2.0 equiv afforded 6a in 67% yield (entry 6). However, further increasing the amount of PivOH to 6.0 equiv improved the yield of 6a to 78% (entry 7). Increasing the reaction time did not help improve the outcome of the product (entry 8). Other oxidants such as CuBr2 and Cu(NO3)2 failed to give the desired product 6a (entries 9–10). Reducing the amount of Pd(OAc)2 to 5 mol % gave a lower yield (67%) of 6a (entry 11). When the amount of Cu(OAc)2 was reduced to 2.0 or 1.0 equiv, the yield of 6a was decreased to 73% and 58%, respectively (entries 12 and 13). The yield of 6a dramatically decreased to 32% at a lower temperature of 120 °C (entry 14). We also tested a higher temperature of 160 °C but without any increase of the yield (entry 15). Other solvents were also examined such as acetonitrile, 1,4-dioxane, and DMAc (N,N-dimethylacetamide). It was found that DMF was the best solvent for this reaction among the selected solvents (entry 7 vs entries 16–18). Notably, no desired product was obtained in the absence of Pd(OAc)2 or Cu(OAc)2 (entries 19 and 20). Finally, the optimized reaction conditions were concluded to be 10 mol % Pd(OAc)2, 3.0 equiv of Cu(OAc)2, and 6.0 equiv of PivOH in DMF (1 mL) at 140 °C under N2 (entry 7).
Table 1. Optimization Studies for the Formation of 6aa,b.

| entry | catalyst (mol %) | [O] (equiv) | additive (equiv) | solvent | T (°C) | product yield (%) 6a |
|---|---|---|---|---|---|---|
| 1c | Pd(OAc)2 (10) | Cu(OAc)2 (3) | – | DMF | 140 | 49 |
| 2c | Pd(TFA)2 (10) | Cu(OAc)2 (3) | – | DMF | 140 | 51 |
| 3d | Pd(OAc)2 (10) | Cu(OAc)2 (3) | – | DMF | 140 | 55 |
| 4e | Pd(OAc)2 (10) | Cu(OAc)2 (3) | – | DMF | 140 | 62 |
| 5 | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (4) | DMF | 140 | 71 |
| 6 | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (2) | DMF | 140 | 67 |
| 7 | Pd(OAc)2(10) | Cu(OAc)2(3) | PivOH (6) | DMF | 140 | 78 |
| 8f | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (6) | DMF | 140 | 76 |
| 9 | Pd(OAc)2 (10) | CuBr2 (3) | PivOH (6) | DMF | 140 | trace |
| 10 | Pd(OAc)2 (10) | Cu(NO3)2 (3) | PivOH (6) | DMF | 140 | trace |
| 11 | Pd(OAc)2 (5) | Cu(OAc)2 (3) | PivOH (6) | DMF | 140 | 67 |
| 12 | Pd(OAc)2 (10) | Cu(OAc)2 (2) | PivOH (6) | DMF | 140 | 73 |
| 13 | Pd(OAc)2 (10) | Cu(OAc)2 (1) | PivOH (6) | DMF | 140 | 58 |
| 14 | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (6) | DMF | 120 | 32 |
| 15 | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (6) | DMF | 160 | 75 |
| 16 | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (6) | CH3CN | 140 | trace |
| 17 | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (6) | 1,4-Dioxane | 140 | trace |
| 18 | Pd(OAc)2 (10) | Cu(OAc)2 (3) | PivOH (6) | DMAc | 140 | 46 |
| 19 | Pd(OAc)2 (10) | – | PivOH (6) | DMF | 140 | trace |
| 20 | – | Cu(OAc)2 (3) | PivOH (6) | DMF | 140 | ND |
The Ugi-reaction was carried out using 1a (1.0 mmol), 2a (1.0 mmol), 3a (1.05 mmol), and 4a (1.05 mmol) in MeOH (1 M) for 12 h at rt.
Reaction conditions: 5a (0.3 mmol), Pd(OAc)2 (10 mol %), [O] (0.9 mmol), PivOH (1.8 mmol), solvent (1 mL), 140 °C, N2, isolated yields.
Solvent (6 mL).
Solvent (3 mL).
Solvent (1 mL).
Reaction time: 16 h.
With the optimal conditions in hand, a series of Ugi products were synthesized in good to excellent yields and were examined to determine the scope of cyclization reaction by reacting substituted anilines with different aldehydes/ketones, isocyanides, and indole-2-carboxylic acids in methanol followed by Pd(OAc)2-catalyzed C(sp2)–H functionalization to furnish the corresponding library 6a–r (Scheme 2). All the substrates 1, 2, 3, and 4 led to the expected indolo[3,2-c]quinolinone products 6a–r in 35–78% yields in two steps. Substituted anilines with electron-donating groups such as p-methyl (1c), p-anisole (1e), 3,5-dimethyl (1f), p-NHBoc (1k), and p-methoxy (1l) reacted smoothly with 58%, 64%, 42%, 49%, and 72% yields, respectively. Electron-withdrawing substituents such as chloro and fluoro reacted nicely to give the cyclized products in 55% and 59% yields, respectively (6b, 6g). Notably, the bromo or iodo group was cleaved in the presence of a palladium catalyst to give the major product 6a when 4-bromoaniline or 4-iodoaniline was employed in the cyclization reactions. Besides, commercially available 5-chloro, 5-methoxy, and 6-methoxy substituted indole-2-carboxylic acid (3j, 3m, 3o) reacted to give the expected product in moderate to good yields. Surprisingly, N-methyl substituted indole-2-carboxylic acid (3r) also formed the polyheterocycle in 65% yield, which was not the case with the substrate of N,1-dimethyl-N-phenyl-1H-indole-2-carboxamide.8Scheme 2 clearly indicates that there are no electronic or steric effects on the outcome of the reaction.
Scheme 2. Synthesis of Indolo[3,2-c]quinolinones 6,,,

The amine, aldehyde, isocyanide, and acid components are depicted with pink, blue, red, and green color, respectively.
The Ugi-reaction was carried out using 1 (1.0 mmol), 2 (1.0 mmol), 3 (1.05 mmol), and 4 (1.05 mmol) in MeOH (1M) for 12 h at rt.
Reaction conditions: 5 (0.3 mmol), Pd(OAc)2 (10 mol %), Cu(OAc)2 (0.9 mmol), PivOH (1.8 mmol), DMF (1.0 mL), 140 °C, 9 h, isolated yields. Under nitrogen.
Yield refers to the purified products.
After successfully demonstrating the cyclization reactions with different anilines and indole-2-carboxylic acids, we then focused on different aldehydes/ketones and isocyanides. Paraformaldehyde was utilized in most cases and results in good yields. Benzaldehyde and p-nitrobenzaldehyde also reacted smoothly to achieve the cyclized products 6d, 6h in 40% and 35% yields, respectively. It is worth mentioning that the cyclic ketone reacted without any interruption to obtain a moderate yield (6i). Furthermore, the benzyl isocyanide (4k) and substituted benzyl isocyanides with electron-donating groups such as p-methoxy (4p), 2,3-dimethoxy (4o) reacted smoothly with 49%, 59%, and 65% yields, respectively. The Ugi adduct bearing a 1-methoxy-4-ethylbenzene substituent on the amide moiety also underwent the reaction, affording the highly strained polycyclic indole compound 6j in good yield. Similarly, aliphatic cyclic and branched isocyanides (6n, 6m, 6q) also furnished the different tetraheterocycles in good yields.
Several structures have been confirmed by X-ray single-crystal analyses (Figure 2 and Supporting Information). The following interesting motifs could be observed in the solid state: the scaffold in general is flat, and therefore, stocking interactions with neighboring molecules are always observed (Supporting Information, Figure S2).
Figure 2.
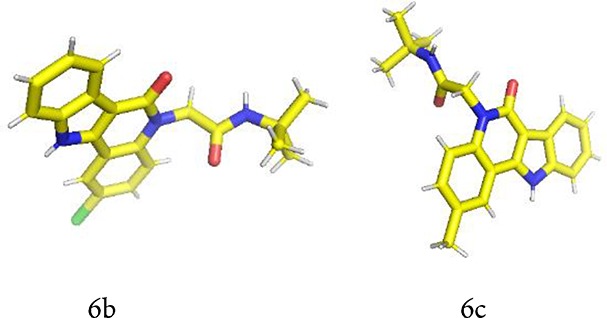
X-ray structures of selected products; crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos.: CCDC1912181 (6b) and CCDC1912182 (6c).
Furthermore, the scalability of this method was investigated (Scheme 3). A four-component reaction of 4-chloroaniline 1b, paraformaldehyde 2b, indole-2-carboxylic acid 3b, and tert-butyl isocyanide 4b was conducted in 10 mmol scale, while the product 6b could be obtained in 42% yield (1.6 g) via two steps. Therefore, this Ugi reaction of indole-2-carboxylic acid and the following Pd-catalyzed dual C(sp2)–H functionalization could be easily scaled up demonstrating it is a practical method.
Scheme 3. Gram-Scale Reaction.

To gain further insight into the reaction mechanism, a radical trapping experiment as a control experiment was examined (Scheme 4). The reaction was not inhibited by the addition of 3.0 equiv of TEMPO, and 6a was still obtained in 45% yield (Scheme 4a). The results suggested that a radical pathway was most likely not involved in this reaction. By changing the oxidant from copper acetate to oxygen, 6a could still be obtained in 28% yield (Scheme 4b). However, without palladium acetate, product 6a could not be observed (Scheme 4c). These results proved again that copper acetate might serve only as an oxidant.
Scheme 4. Control Experiments.

A plausible mechanism of the cyclization path was explained based on the previous report as shown in Scheme 5.8 After obtaining the Ugi adduct 5a, Pd(OAc)2 attacks the indole to give the iminium intermediate A, which undergoes a nucleophilic attack of the N-aryl to the iminium moiety which forms the intermediate B. Then B is further converted to the intermediate C via a nucleophilic addition process. Subsequently, the formed C undergoes 1,2-acyl migration to give intermediate D, which after protonation and oxidative aromatization gives the product 6a.
Scheme 5. Proposed Reaction Mechanism.

Moreover, we were also interested in potential biological applications of the synthesized compounds. For this aim, all the compounds were docked in the cocrystal structure of human proto-oncogene serine threonine kinase (PIM1) [PDB code 3CY2]. The original cocrystallized ligand binds in a non-ATP mimetic binding mode. In the docking poses, we noticed that our ligands fit the pocket nicely and there is a good overlap of the indole moieties (nonsubstituted or with Cl- or OMe- substituents) of the docking compounds with the original ligand. The pyridone moiety is reversed in the docked poses in most of the cases; however, this carbonyl group did not participate in hydrogen bonds in the original ligand either. Interestingly, we observed that the conformation of the piperidine moiety was mimicked in the 6a by the tert-butyl acetamide and the NH-moiety formed a hydrogen bond with the carbonyl group of Asn172, similar to one of the interactions of the original piperidine (Figure 3A). In the case of the ketone-derivated ligand 6i, the NH-moiety of the tert-butyl acetamide is able to form a hydrogen bond with Glu171, whereas the cyclopentane ring is filling a more hydrophobic part of the pocket, forming van der Waals interactions with Phe49 (Figure 3B). We hypothesize that these compounds could have potential as kinase inhibitors (Figure 3).
Figure 3.
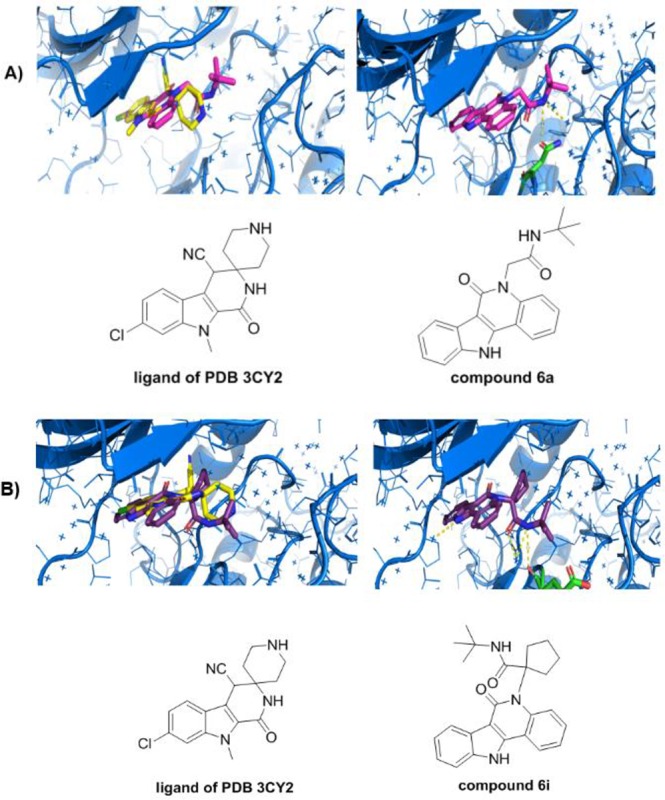
(A) Docking poses. Top left: Overlap of compound 6c (magenta sticks) with the ligand (yellow sticks) of PDB 3CY2. Top right: hydrogen bonds (yellow dots) of compound 6c (magenta sticks) with Asn172 (green sticks). (B) Bottom left: Overlap of compound 6i (purple sticks) with the ligand (yellow sticks) of PDB 3CY2. Bottom right: hydrogen bonds (black yellow dots) of compound 6i (purple sticks) with Glu171 (green sticks).
Conclusions
In summary, an indolo[3,2-c]quinolinone library was successfully established based on MCR starting from commercially available materials. Diversity can be achieved through the aniline, aldehyde/ketone, isocyanide, and indole-2-carboxylic acid, all four components. Regarding potential applications, docking studies indicate that these types of derivatives could be useful as kinase inhibitors, and biological work is ongoing and will be reported in due course.
Experimental Section
General Information
Nuclear magnetic resonance spectra were recorded on a Bruker Avance 500 spectrometer. Chemical shifts for 1H NMR were reported relative to TMS (δ 0 ppm) or internal solvent peak (CDCl3 δ 7.26 ppm, DMSO-d6 δ 2.50 ppm or CD3OD δ 3.31 ppm), and coupling constants were in hertz (Hz). The following abbreviations were used for spin multiplicity: s = singlet, d = doublet, t = triplet, dt = double triplet, ddd = doublet of double doublet, m = multiplet, and br = broad. Chemical shifts for 13C NMR reported in ppm relative to the solvent peak (CDCl3 δ 77.23 ppm, DMSO δ 39.52 ppm, CD3OD δ 49.00 ppm). Flash chromatography was performed on a Grace Reveleris X2 using Grace Reveleris Silica columns (12 g), and a gradient of petroleum ether/ethyl acetate (0–100%) or dichloromethane/methanol (0–20%) was applied. Thin layer chromatography was performed on Fluka precoated silica gel plates (0.20 mm thick, particle size 25 μm). Reagents were available from commercial suppliers and used without any purification unless otherwise noted. All isocyanides were made in house by performing the Ugi procedure. Other reagents were purchased from Sigma-Aldrich, ABCR, Acros, Fluorochem, and AK Scientific and were used without further purification. Mass spectra were measured on a Waters Investigator Supercritical Fluid Chromatograph with a 3100 MS Detector (ESI) using a solvent system of methanol and CO2 on a Viridis silica gel column (4.6 mm × 250 mm, 5 μm particle size) and reported as (m/z). High resolution mass spectra (HRMS) were recorded using an LTQ-Orbitrap-XL (Thermo Fisher Scientific; ESI pos. mode) at a resolution of 60000@m/z400. Electrospray ionization mass spectra (ESI-MS) were recorded on a Waters Investigator Semiprep 15 SFC-MS instrument. Melting points were obtained on a melting point apparatus and were uncorrected. Yields given refer to chromatographically purified and spectroscopically pure compounds unless otherwise stated.
General Experimental Procedure and Characterization
General Procedure A
A solution of aldehyde or ketone 1 (1.0 mmol) and amine 2 (1.0 mmol) in methanol (1 mL) was stirred at room temperature for 30 min. Subsequently, isocyanide 3 (1.05 mmol) and indole-2-carboylic acid 4 (1.05 mmol) were added and the reaction was stirred at room temperature overnight. Reaction progress was monitored via TLC and SFC-MS. Upon completion of the reaction, the mixture was concentrated in vacuo and purified by column chromatography to give the desired product 5.
General Procedure B
Ugi adduct 5 (0.3 mmol), Pd(OAc)2 (0.03 mmol), Cu(OAc)2 (0.9 mmol), PivOH (1.8 mmol), and DMF (1 mL) were placed in a flask under N2. After the completion of the addition, the reaction mixture was allowed to react at 140 °C in an oil bath for 9 h. Then, the reaction mixture was cooled to room temperature, treated with H2O, and then extracted with EA. The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After removal of the EA, the residue was purified by flash chromatography to afford the product 6.
Gram-Scale Synthesis of 6b
An oven-dried 50 mL flask equipped with a magnetic stirrer bar was charged with 4-chloroaniline (1.27 g, 10 mmol) and paraformaldehyde (300 mg, 10 mmol). 15 mL of MeOH were added, and the reaction was stirred for 30 min. Then indole-2-carboxylic acid (1.69 g, 10.5 mmol) was added followed by tert-butyl isocyanide (872 mg, 10.5 mmol). The mixture was stirred at rt for 24 h. Solvent was removed under vacuum, and the residue was purified by silica gel column chromatography using ethyl acetate/petroleum ether (v/v, 1:1) as eluent to give Ugi product 5b (3.14 g, 82%). Subsequently, Ugi adduct 5b (3.14 g, 8.2 mmol), Pd(OAc)2 (184 mg, 0.82 mmol), Cu(OAc)2 (4.5 g, 25 mmol), PivOH (5 g, 49 mmol), and DMF (20 mL) were placed in a 50 mL flask under N2. After the completion of the addition, the reaction mixture was allowed to react at 140 °C in an oil bath for 12 h. Then, the reaction mixture was cooled to room temperature, was treated with H2O, and then extracted with EA. The combined organic layers were washed with brine and dried over anhydrous Na2SO4. After removal of the EA, the residue was purified by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford the product 6b (1.6 g, 51%) as an off-white solid.
N-(2-(tert-Butylamino)-2-oxoethyl)-N-phenyl-1H-indole-2-carboxamide (5a)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 5a (287 mg, 82%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.66 (s, 1H), 7.58–7.48 (m, 3H), 7.48–7.35 (m, 4H), 7.31–7.22 (m, 1H), 7.05 (ddd, J = 7.9, 6.9, 0.9 Hz, 1H), 6.37 (s, 1H), 5.36 (d, J = 2.1 Hz, 1H), 4.45 (s, 2H), 1.38 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.6, 162.7, 142.9, 135. 6, 130.1, 129.0, 128.7, 128.4, 127.6, 125.0, 122.4, 120.4, 111.6, 108.2, 56.5, 51.4, 28.8 ppm.
N-(2-(tert-Butylamino)-2-oxoethyl)-N-(4-chlorophenyl)-1H-indole-2-carboxamide (5b)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 5b (326 mg, 85%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.52 (s, 1H), 7.50–7.44 (m, 3H), 7.43–7.36 (m, 3H), 7.32–7.24 (m, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.21 (s, 1H), 5.47 (d, J = 2.1 Hz, 1H), 4.39 (s, 2H), 1.28 (s, 9H). 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.6, 162.7, 142.9, 135.6, 130.1, 129.0, 128.7, 128.4, 127.6, 125.0, 122.4, 120.4, 111.6, 108.2, 56.5, 51.4, 28.8 ppm.
N-(2-(tert-Butylamino)-2-oxoethyl)-N-(p-tolyl)-1H-indole-2-carboxamide (5c)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 5c (265 mg, 73%) as a white solid. 1H NMR (500 MHz, Chloroform-d) δ 9.56 (s, 1H), 7.44–7.39 (m, 2H), 7.30–7.26 (m, 5H), 7.08–7.02 (m, 1H), 6.38 (s, 1H), 5.41 (d, J = 2.1 Hz, 1H), 4.42 (s, 2H), 2.47 (s, 3H), 1.39 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.9, 162.9, 140.2, 139.1, 137.2, 135.6, 130.7, 128.6, 128.0, 127.6, 125.0, 122.4, 120.4, 111.7, 109.9, 108.4, 56.6, 51.5, 28.8, 21.3 ppm.
N-(2-(tert-Butylamino)-2-oxo-1-phenylethyl)-N-phenyl-1H-indole-2-carboxamide (5d)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 5d (383 mg, 90%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.35 (s, 1H), 7.36 (q, J = 6.0, 4.8 Hz, 3H), 7.33 (s, 1H), 7.29 (s, 2H), 7.27–7.23 (m, 5H), 7.23–7.20 (m, 1H), 7.05–6.92 (m, 1H), 6.11 (d, J = 3.7 Hz, 1H), 5.79 (s, 1H), 5.16–5.01 (m, 1H), 3.52 (s, 1H), 1.38 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 168.6, 162.3, 140.1, 135.3, 134.5, 131.1, 130.4, 128.7, 128.5, 128.4, 127.7, 124.7, 122.4, 120.2, 111.4, 107.7, 67.3, 51.7, 28.7 ppm.
N-(2-(tert-Butylamino)-2-oxoethyl)-N-(4-phenoxyphenyl)-1H-indole-2-carboxamide (5e)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 5e (331 mg, 75%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.33 (s, 1H), 7.49–7.33 (m, 6H), 7.30 (d, J = 1.1 Hz, 1H), 7.23–7.18 (m, 1H), 7.16–7.06 (m, 5H), 6.32 (s, 1H), 5.45 (dd, J = 2.2, 1.0 Hz, 1H), 4.40 (s, 2H), 1.40 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.7, 162.6, 157.9, 156.4, 137.6, 135.4, 130.0, 129.9, 128.6, 127.7, 125.1, 124.1, 122.4, 120.6, 119.7, 119.4, 111.6, 108.1, 56.6, 51.4, 28.8 ppm.
N-(2-(tert-Butylamino)-2-oxoethyl)-N-(3,5-dimethylphenyl)-1H-indole-2-carboxamide (5f)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 35% ethyl acetate in petroleum ether) to afford 5f (287 mg, 76%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.55 (s, 1H), 7.52–7.34 (m, 2H), 7.27 (d, J = 11.5 Hz, 1H), 7.15 (d, J = 16.5 Hz, 1H), 7.03 (d, J = 30.8 Hz, 3H), 6.41 (s, 1H), 5.46 (s, 1H), 4.41 (s, 2H), 2.37 (s, 6H), 1.39 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.9, 162.6, 142.5, 139.9, 135.5, 130.7, 128.7, 127.7, 125.8, 124.9, 122.4, 120.4, 111.6, 108.2, 56.7, 51.3, 28.8, 21.3 ppm.
N-(2-(tert-Butylamino)-2-oxoethyl)-N-(4-fluorophenyl)-1H-indole-2-carboxamide (5g)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 5g (331 mg, 90%) as a white solid. 1H NMR (500 MHz, Chloroform-d) δ 9.47 (s, 1H), 7.46–7.38 (m, 4H), 7.30–7.25 (m, 1H), 7.20 (dd, J = 9.2, 7.9 Hz, 2H), 7.07 (ddd, J = 8.0, 6.9, 1.0 Hz, 1H), 6.23 (s, 1H), 5.36 (t, J = 1.3 Hz, 1H), 4.39 (s, 2H), 1.38 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.5, 163.5, 161.6, 138.9 (d, J = 3.4 Hz), 135.5, 130.4 (d, J = 8.7 Hz), 128.5, 127.6, 125.1, 122.5, 120.6, 117.1 (d, J = 22.6 Hz), 111.6, 108.1, 56.4, 51.5, 28.8 ppm.
N-(2-(tert-Butylamino)-1-(4-nitrophenyl)-2-oxoethyl)-N-phenyl-1H-indole-2-carboxamide (5h)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 35% ethyl acetate in petroleum ether) to afford 5h (400 mg, 85%) as yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.33 (s, 1H), 8.15–8.06 (m, 2H), 7.52–7.47 (m, 2H), 7.44 (ddd, J = 8.6, 4.9, 1.3 Hz, 1H), 7.41–7.32 (m, 4H), 7.29 (d, J = 1.2 Hz, 1H), 7.26 (ddt, J = 8.3, 7.0, 1.3 Hz, 1H), 7.03 (ddd, J = 8.0, 6.9, 1.1 Hz, 1H), 6.20 (d, J = 10.0 Hz, 2H), 5.15 (dd, J = 2.2, 1.0 Hz, 1H), 1.41 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.6, 162.6, 147.7, 141.6, 139.6, 135.5, 131.2, 130.7, 129.6, 129.4, 128.7, 127.6, 125.2, 123.4, 122.5, 120.5, 111.5, 108.2, 66.3, 52.0, 28.7 ppm.
N-(1-(tert-Butylcarbamoyl)cyclopentyl)-N-phenyl-1H-indole-2-carboxamide (5i)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 5i (262 mg, 65%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.31 (s, 1H), 7.60–7.51 (m, 3H), 7.47–7.42 (m, 2H), 7.35 (ddt, J = 7.3, 1.9, 1.0 Hz, 2H), 7.23 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 7.01 (ddd, J = 7.9, 6.8, 0.9 Hz, 1H), 6.36 (s, 1H), 4.92 (dd, J = 2.1, 1.0 Hz, 1H), 2.50–2.41 (m, 2H), 1.93–1.83 (m, 2H), 1.77–1.58 (m, 4H), 1.41 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 173.0, 162.6, 140.2, 135.1, 130.9, 130.1, 129.6, 129.3, 127.7, 124.7, 122.3, 120.2, 111.4, 107.0, 75.3, 51.0, 36.9, 28.7, 23.7 ppm.
5-Chloro-N-(2-((4-methoxyphenethyl)amino)-2-oxoethyl)-N-phenyl-1H-indole-2-carboxamide (5j)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 5j (406 mg, 88%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.38 (s, 1H), 7.54–7.42 (m, 3H), 7.39–7.32 (m, 2H), 7.25–7.16 (m, 3H), 7.13–7.08 (m, 2H), 6.74–6.66 (m, 2H), 6.42 (s, 1H), 5.18 (dd, J = 2.1, 1.1 Hz, 1H), 4.45 (s, 2H), 3.62 (d, J = 0.8 Hz, 3H), 3.58 (q, J = 6.4 Hz, 2H), 2.81 (t, J = 6.7 Hz, 2H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 168.2, 162.2, 158.3, 142.4, 133.7, 130.5, 130.1, 129.8, 129.7, 129.2, 128.5, 128.2, 126.1, 125.5, 121.6, 114.0, 112.7, 107.5, 55.6, 55.1, 40.6, 34.5 ppm.
tert-Butyl (4-(N-(2-(Benzylamino)-2-oxoethyl)-1H-indole-2-carboxamido)phenyl)carbamate (5k)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 5k (369 mg, 74%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.24 (s, 1H), 7.53–7.47 (m, 2H), 7.43 (dd, J = 8.1, 1.1 Hz, 1H), 7.38 (dt, J = 8.3, 1.1 Hz, 1H), 7.36–7.32 (m, 2H), 7.31–7.23 (m, 6H), 7.05 (ddd, J = 8.1, 6.9, 1.0 Hz, 1H), 6.78 (t, J = 5.9 Hz, 1H), 6.71 (s, 1H), 5.48 (dd, J = 2.2, 1.0 Hz, 1H), 4.54–4.50 (m, 4H), 1.58 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 168.5, 162.8, 152.5, 139.1, 138.0, 137.2, 135.5, 129.0, 128.7, 128.5, 127.7, 127.7, 127.5, 125.0, 122.6, 120.5, 119.3, 111.5, 108.4, 55.8, 43.6, 28.4 ppm.
N-(2-(tert-Butylamino)-2-oxoethyl)-6-methoxy-N-(4-methoxyphenyl)-1H-indole-2-carboxamide (5l)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 5l (377 mg, 92%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.58 (s, 1H), 7.35–7.25 (m, 2H), 7.05–6.97 (m, 2H), 6.83 (d, J = 2.2 Hz, 1H), 6.72 (dd, J = 8.8, 2.2 Hz, 1H), 6.44 (d, J = 3.7 Hz, 1H), 5.32 (s, 1H), 4.40 (d, J = 1.9 Hz, 2H), 3.90 (s, 3H), 3.83 (s, 3H), 1.36 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 168.0, 162.8, 159.8, 158.6, 135.7, 129.5, 127.7, 123.2, 122.1, 115.2, 112.0, 108.5, 93.3, 56.7, 55.6, 55.5, 51.3, 28.8 ppm.
N-(2-(Butylamino)-2-oxoethyl)-5-methoxy-N-(p-tolyl)-1H-indole-2-carboxamide (5m)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 85% ethyl acetate in petroleum ether) to afford 5m (350 mg, 89%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.53 (s, 1H), 7.28 (d, J = 3.8 Hz, 4H), 7.12–6.98 (m, 1H), 6.93 (dd, J = 9.0, 2.5 Hz, 1H), 6.80 (d, J = 2.5 Hz, 1H), 6.58 (t, J = 5.9 Hz, 1H), 5.34 (s, 1H), 4.50 (s, 2H), 3.78 (s, 3H), 3.31 (q, J = 6.8 Hz, 2H), 2.47 (s, 3H), 1.51 (p, J = 7.3 Hz, 2H), 1.39–1.33 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 168.7, 162.8, 154.4, 140.2, 139.1, 131.0, 130.7, 129.0, 128.0, 116.8, 112.6, 107.9, 102.2, 55.9, 55.6, 39.4, 31.6, 21.3, 20.1, 13.8 ppm.
N-(2-(Cyclohexylamino)-2-oxoethyl)-6-methoxy-N-phenyl-1H-indole-2-carboxamide (5n)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 80% ethyl acetate in petroleum ether) to afford 5n (361 mg, 85%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.21 (s, 1H), 7.50 (q, J = 2.9 Hz, 3H), 7.45–7.33 (m, 2H), 7.24 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 2.3 Hz, 1H), 6.72 (dd, J = 8.8, 2.2 Hz, 1H), 6.43 (d, J = 8.3 Hz, 1H), 5.25 (d, J = 2.2 Hz, 1H), 4.48 (s, 2H), 3.85 (s, 3H), 2.04–1.88 (m, 2H), 1.72 (dt, J = 13.4, 4.1 Hz, 3H), 1.66–1.53 (m, 1H), 1.39 (q, J = 12.5 Hz, 2H), 1.21 (qd, J = 10.4, 9.1, 3.8 Hz, 3H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.7, 162.7, 158.7, 142.8, 136.5, 130.1, 129.0, 128.4, 127.5, 123.3, 122.0, 112.1, 108.6, 93.2, 55.7, 55.5, 48.2, 33.0, 25.5, 24.7 ppm.
N-(2-((2,3-Dimethoxybenzyl)amino)-2-oxoethyl)-6-methoxy-N-(p-tolyl)-1H-indole-2-carboxamide (5o)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 70% ethyl acetate in petroleum ether) to afford 5o (419 mg, 86%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.18 (s, 1H), 7.28–7.21 (m, 4H), 7.01 (t, J = 7.9 Hz, 1H), 6.89 (ddd, J = 19.8, 7.7, 1.5 Hz, 4H), 6.79 (d, J = 2.3 Hz, 1H), 6.71 (dd, J = 8.8, 2.2 Hz, 1H), 5.34–5.29 (m, 1H), 4.53 (t, J = 5.3 Hz, 3H), 4.49 (s, 2H), 3.90 (d, J = 3.7 Hz, 2H), 3.86 (s, 6H), 3.84 (s, 3H), 2.46 (s, 3H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 168.5, 162.7, 158.6, 152.6, 147.2, 140.2, 138.9, 136.5, 131.7, 130.7, 128.1, 127.7, 124.2, 123.2, 121.3, 112.0, 112.0, 108.5, 93.3, 60.7, 55.8, 55.6, 55.5, 38.9, 21.3 ppm.
5-Chloro-N-(2-((1-(4-methoxyphenyl)ethyl)amino)-2-oxoethyl)-N-phenyl-1H-indole-2-carboxamide (5p)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 5p (379 mg, 82%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.57 (s, 1H), 7.56–7.42 (m, 3H), 7.39–7.29 (m, 4H), 7.26–7.21 (m, 2H), 7.19 (dd, J = 8.8, 2.0 Hz, 1H), 6.89–6.77 (m, 2H), 6.66 (d, J = 8.1 Hz, 1H), 5.22 (d, J = 2.1 Hz, 1H), 5.16–5.08 (m, 1H), 4.60–4.42 (m, 2H), 3.78 (s, 3H), 1.50 (d, J = 6.9 Hz, 3H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.3, 162.4, 158.8, 142.5, 135.1, 133.8, 130.1, 129.8, 129.2, 128.5, 128.4, 127.3, 126.0, 125.5, 121.5, 114.0, 112.8, 107.5, 55.6, 55.3, 48.4, 21.8 ppm.
N-(2-oxo-2-((2,4,4-trimethylpentan-2-yl)amino)ethyl)-N-(o-tolyl)-1H-indole-2-carboxamide (5q)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 30% ethyl acetate in petroleum ether) to afford 5q (332 mg, 79%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 9.87 (s, 1H), 7.49–7.42 (m, 2H), 7.42–7.34 (m, 4H), 7.30–7.24 (m, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.72 (d, J = 5.6 Hz, 1H), 5.24 (q, J = 2.2 Hz, 1H), 4.85 (dd, J = 14.3, 8.2 Hz, 1H), 3.90 (dd, J = 14.8, 5.0 Hz, 1H), 2.25 (d, J = 1.8 Hz, 3H), 1.77 (d, J = 3.4 Hz, 2H), 1.46 (d, J = 2.4 Hz, 6H), 1.03(s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.4, 162.8, 141.7, 136.0, 135.7, 131.9, 129.5, 129.1, 128.7, 127.9, 127.8, 125.0, 122.5, 120.4, 111.7, 107.3, 56.3, 55.4, 52.1, 31.6, 31.5, 29.0, 17.7 ppm.
N-(2-(tert-Butylamino)-2-oxoethyl)-1-methyl-N-phenyl-1H-indole-2-carboxamide (5r)
Synthesized according to procedure A in 1 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 5r (284 mg, 78%) as a yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 7.42 (dt, J = 7.9, 1.0 Hz, 1H), 7.36–7.29 (m, 2H), 7.29–7.21 (m, 4H), 7.06 (ddd, J = 7.9, 6.8, 1.1 Hz, 1H), 6.27 (s, 1H), 6.11 (d, J = 0.8 Hz, 1H), 4.46 (s, 2H), 3.97 (s, 3H), 1.41 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.7, 164.2, 143.8, 138.1, 131.2, 129.5, 127.5, 127.0, 126.1, 123.9, 122.0, 120.1, 109.8, 108.1, 55.7, 51.4, 31.7, 28.8 ppm.
N-(tert-Butyl)-2-(6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6a)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 6a (81 mg, 78%) as an off-white solid; mp: 364–366 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.61 (s, 1H), 8.40–8.17 (m, 2H), 7.99 (s, 1H), 7.67–7.57 (m, 2H), 7.42–7.28 (m, 4H), 5.03 (s, 2H), 1.30 (d, J = 1.7 Hz, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.1, 159.5, 140.4, 139.1, 138.3, 130.0, 125.1, 124.6, 123.1, 122.1, 121.6, 121.2, 115.9, 113.4, 112.1, 106.0, 50.9, 44.3, 28.9. HRMS (ESI)calcd for C21H22N3O2 [M + H]+, 348.1712; found, 348.1711.
N-(tert-Butyl)-2-(2-chloro-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6b)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 6b (63 mg, 55%) as an off-white solid; mp: 353–355 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.67 (s, 1H), 8.41 (d, J = 2.5 Hz, 1H), 8.22 (d, J = 7.9 Hz, 1H), 8.01 (s, 1H), 7.68–7.62 (m, 2H), 7.42 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.35 (d, J = 9.1 Hz, 1H), 7.33–7.29 (m, 1H), 5.02 (s, 2H), 1.29 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 166.8, 159.3, 139.1, 138.3, 137.8, 129.5 (d, J = 18.6 Hz), 126.4, 125.4–124.9 (m), 124.8, 122.3, 121.9, 121.3, 118.5–117.4 (m), 114.7, 112.4, 106.7, 50.9, 44.4, 29.0 ppm. HRMS (ESI) calcd for C21H21ClN3O2 [M + H]+, 382.1322; found, 382.1323.
N-(tert-Butyl)-2-(2-methyl-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6c)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 6c (63 mg, 58%) as a yellow solid; mp: 348–349 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.56 (s, 1H), 8.21 (d, J = 7.8 Hz, 1H), 8.13–8.08 (m, 1H), 7.96 (s, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.46–7.35 (m, 2H), 7.32–7.21 (m, 2H), 5.00 (s, 2H), 2.46 (s, 3H), 1.29 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.1, 159.5, 140.3, 138.3, 137.1, 131.2, 131.1–130.7 (m), 125.1, 124.6, 122.9, 121.6, 121.2, 115.8, 113.3, 112.2, 106.1, 50.9, 44.3, 29.0, 20.8 ppm. HRMS (ESI) calcd for C22H24N3O2 [M + H]+, 362.1868; found, 362.1867.
N-(tert-Butyl)-2-(6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)-2-phenylacetamide (6d)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 70% ethyl acetate in petroleum ether) to afford 6d (51 mg, 40%) as a yellow solid; mp: 323–325 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.67 (s, 1H), 8.26 (dd, J = 7.1, 2.3 Hz, 2H), 7.75 (s, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.41 (dddd, J = 11.7, 7.2, 5.0, 2.9 Hz, 3H), 7.35–7.27 (m, 5H), 7.27–7.18 (m, 3H), 1.28 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.7, 160.2, 140.8, 138.4, 138.3, 137.5, 128.8, 128.5, 128.0, 127.4, 125.2, 124.7, 122.9, 122.2, 121.7, 121.3, 119.7, 114.1, 112.3, 105.9, 61.8–56.6 (m), 51.3, 28.9 ppm. HRMS (ESI) calcd for C27H26N3O2 [M + H]+, 424.2025; found, 424.2019.
N-(tert-Butyl)-2-(6-oxo-2-phenoxy-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6e)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 6e (84 mg, 64%) as a yellow solid; mp: 333–335 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.55 (s, 1H), 8.23 (d, J = 7.9 Hz, 1H), 8.02 (d, J = 2.6 Hz, 1H), 7.99 (s, 1H), 7.60 (d, J = 8.1 Hz, 1H), 7.47–7.41 (m, 2H), 7.41–7.34 (m, 3H), 7.29 (t, J = 7.3 Hz, 1H), 7.18 (dd, J = 7.9, 6.7 Hz, 1H), 7.13–7.08 (m, 2H), 5.03 (s, 2H), 1.31 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.0, 159.3, 157.9, 151.2, 139.7, 138.3, 135.6, 130.6, 125.0, 124.8, 123.8, 121.8, 121.7, 121.4, 118.5, 117.9, 114.4, 112.6, 112.3, 106.5, 50.9, 44.5, 29.0 ppm. HRMS (ESI)calcd for C27H26N3O3 [M + H]+, 440.1974; found, 440.1969.
N-(tert-Butyl)-2-(1,3-dimethyl-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6f)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 6f (47 mg, 42%) as a yellow solid; mp: 338–340 °C. 1H NMR (500 MHz, DMSO-d6) δ 11.44 (s, 1H), 8.28 (d, J = 7.8 Hz, 1H), 8.01 (s, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.37 (td, J = 8.1, 7.6, 1.3 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.04 (d, J = 4.1 Hz, 2H), 5.77 (s, 1H), 5.03 (s, 2H), 2.95 (s, 3H), 2.41 (s, 3H), 1.30 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.2, 159.4, 140.0, 139.8, 138.8, 138.5, 134.4, 126.0, 124.4, 124.2, 121.7, 121.0, 120.9, 114.3, 112.9, 110.9, 106.3, 50.9, 44.7, 29.0, 23.4, 21.9 ppm. HRMS (ESI) calcd for C23H26N3O2 [M + H]+, 376.2025; found, 376.2019.
N-(tert-Butyl)-2-(2-fluoro-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6g)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 6g (65 mg, 59%) as an off-white solid; mp: 331–333 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.63 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 8.13 (dd, J = 9.1, 3.0 Hz, 1H), 8.00 (s, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.51 (ddd, J = 11.2, 8.4, 3.0 Hz, 1H), 7.42 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.37 (dd, J = 9.4, 4.5 Hz, 1H), 7.34–7.28 (m, 1H), 5.03 (s, 2H), 1.29 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 166.9, 1593, 158.4, 156.5, 139.5, 138.3, 135.9, 125.1, 124.9, 121.9, 121.4, 118.1, 114.2 (d, J = 9.0 Hz), 112.3, 108.6, 106.7, 50.8, 44.5, 29.0 ppm. HRMS (ESI) calcd for C21H21FN3O2 [M + H]+, 366.1618; found, 366.1619.
N-(tert-Butyl)-2-(4-nitrophenyl)-2-(6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6h)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 6h (49 mg, 35%) as a yellow solid; mp: 328–330 °C. 1H NMR (500 MHz, Chloroform-d) δ 10.84 (s, 1H), 8.08–8.02 (m, 2H), 7.60–7.54 (m, 2H), 7.46–7.41 (m, 2H), 7.41–7.33 (m, 3H), 7.33–7.30 (m, 1H), 7.25–7.18 (m, 3H), 6.22 (s, 1H), 1.28 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 166.6, 163.7, 147.7, 144.0, 142.4, 136.8, 132.1, 131.4, 129.8, 129.2, 127.6, 126.9, 126.2, 123.3, 122.0, 120.7, 113.9, 75.3, 52.4, 28.5 ppm. HRMS (ESI)calcd for C27H25N4O4 [M + H]+, 469.1876; found, 469.1866.
N-(tert-Butyl)-2-(4-nitrophenyl)-2-(6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6i)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 6i (66 mg, 55%) as a yellow solid; mp: 349–351 °C. 1H NMR (500 MHz, Chloroform-d) δ 9.71 (s, 1H), 8.41 (d, J = 7.7 Hz, 1H), 7.93 (dd, J = 7.8, 1.5 Hz, 1H), 7.89 (s, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.40 (tdd, J = 8.1, 3.7, 1.4 Hz, 2H), 7.37–7.33 (m, 1H), 7.26 (d, J = 7.6 Hz, 1H), 3.32 (dt, J = 12.5, 6.0 Hz, 2H), 1.70 (d, J = 57.9 Hz, 6H), 1.37 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 174.2, 164.4, 140.1, 139.9, 137.6, 127.8, 125.0, 124.7, 122.3, 122.1, 121.7, 121.3, 120.3, 114.9, 111.3, 109.3, 75.8, 51.1, 39.2, 28.6, 23.7 ppm. HRMS (ESI) calcd for C25H28N3O2 [M + H]+, 402.2182; found, 402.2180.
2-(8-Chloro-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)-N-(4-methoxyphenethyl)acetamide (6j)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 6j (106 mg, 77%) as a yellow solid; mp: 316–318 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.83 (s, 1H), 8.31–8.21 (m, 2H), 8.16 (d, J = 2.1 Hz, 1H), 7.66 (d, J = 8.6 Hz, 1H), 7.60 (ddd, J = 8.6, 7.2, 1.5 Hz, 1H), 7.41 (ddd, J = 7.5, 4.4, 2.2 Hz, 2H), 7.28 (d, J = 8.6 Hz, 1H), 7.15–7.09 (m, 2H), 6.87–6.82 (m, 2H), 5.02 (s, 2H), 3.72 (s, 3H), 3.32–3.26 (m, 2H), 2.66 (t, J = 7.2 Hz, 2H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.7, 159.3, 158.1, 141.6, 139.2, 136.8, 131.6, 130.1, 126.2, 126.2, 124.6, 123.3, 122.5, 120.2, 116.1, 114.2 (d, J = 9.9 Hz), 114.0, 113.8, 113.3, 105.6, 55.4 (d, J = 10.5 Hz), 44.4, 41.0, 34.7 ppm. HRMS (ESI) calcd for C26H23ClN3O3[M + H]+, 460.1428; found, 460.1406.
tert-Butyl(5-(2-(benzylamino)-2-oxoethyl)-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-2-yl)carbamate (6k)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 40% ethyl acetate in petroleum ether) to afford 6k (73 mg, 49%) as a brown solid; mp: 289–291 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.68 (s, 1H), 9.57 (s, 1H), 8.69 (t, J = 6.0 Hz, 1H), 8.57 (s, 1H), 8.22 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 8.1 Hz, 1H), 7.39 (ddd, J = 8.4, 5.4, 1.9 Hz, 2H), 7.37–7.29 (m, 4H), 7.29–7.26 (m, 3H), 5.10 (s, 2H), 4.32 (d, J = 6.0 Hz, 2H), 1.55 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 168.3, 159.3, 153.5, 140.3, 139.8, 138.5, 134.5, 134.4, 128.7, 127.7, 127.3, 125.1, 124.6, 121.6, 121.2, 116.5, 113.7, 112.5, 112.3, 106.4, 79.7, 44.5, 42.7, 28.7 (d, J = 13.6 Hz) ppm. HRMS (ESI) calcd for C29H29N4O4 [M + H]+, 497.2188; found, 497.2178.
N-(tert-Butyl)-2-(2,9-dimethoxy-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6l)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 6l (88 mg, 72%) as an off-white solid; mp: 341–343 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.40 (s, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.95 (s, 1H), 7.82 (d, J = 2.8 Hz, 1H), 7.26 (d, J = 9.3 Hz, 1H), 7.19 (dd, J = 9.2, 2.8 Hz, 1H), 7.10 (d, J = 2.2 Hz, 1H), 6.94 (dd, J = 8.6, 2.3 Hz, 1H), 4.98 (s, 2H), 3.90 (s, 3H), 3.88 (s, 3H), 1.29 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.2, 159.0, 157.9, 154.6, 139.5, 139.4, 133.1, 121.9, 119.0, 117.5, 117.2, 114.1, 111.1, 106.6, 105.3, 95.6, 56.1, 55.8, 50.9, 44.4, 29.0 ppm. HRMS (ESI) calcd for C23H26N3O4 [M + H]+, 408.1923; found, 408.1917.
N-Butyl-2-(9-methoxy-2-methyl-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6m)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 10% methanol in dichloromethane) to afford 6m (74 mg, 63%) as a yellow solid; mp: 325–327 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.43 (s, 1H), 8.15 (t, J = 5.7 Hz, 1H), 8.07 (s, 1H), 7.70 (d, J = 2.6 Hz, 1H), 7.52 (d, J = 8.7 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.23 (d, J = 8.6 Hz, 1H), 7.01 (dd, J = 8.8, 2.6 Hz, 1H), 5.00 (s, 2H), 3.84 (s, 3H), 3.09 (q, J = 6.6 Hz, 2H), 2.45 (s, 3H), 1.42–1.38 (m, 2H), 1.30–1.26 (m, 2H), 0.87 (t, J = 7.3 Hz, 3H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 167.8, 159.5, 155.2, 140.6, 137.0, 133.0, 131.2, 130.8, 125.8, 122.7, 115.9, 114.2, 113.6, 113.0, 106.1, 103.0 (d, J = 27.6 Hz), 55.8 (d, J = 25.5 Hz), 44.3, 38.8, 31.7, 20.9, 20.0, 14.1 ppm. HRMS (ESI) calcd for C23H26N3O3 [M + H]+, 392.1974; found, 392.1967.
N-Cyclohexyl-2-(9-methoxy-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6n)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 8% methanol in dichloromethane) to afford 6n (58 mg, 48%) as a yellow solid; mp: 307–309 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.47 (s, 1H), 8.24 (dd, J = 7.9, 1.6 Hz, 1H), 8.17 (d, J = 7.9 Hz, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.56 (ddd, J = 8.6, 7.1, 1.5 Hz, 1H), 7.39–7.30 (m, 2H), 7.10 (d, J = 2.3 Hz, 1H), 6.94 (dd, J = 8.6, 2.3 Hz, 1H), 5.04 (s, 2H), 3.88 (s, 3H), 1.80–1.67 (m, 4H), 1.56 (d, J = 12.4 Hz, 1H), 1.24 (q, J = 12.9 Hz, 6H) ppm. 13C{1H} NMR (126 MHz, DMSO) δ 166.9, 159.4, 157.9, 139.8, 139.4, 138.6, 129.3, 122.7, 122.1, 121.9, 118.9, 115.9, 113.6, 111.2, 106.3, 95.6, 55.9, 48.2, 44.2, 32.9, 25.6, 24.9 ppm. HRMS (ESI) calcd for C24H26N3O3 [M + H]+, 404.1974; found, 404.1971.
N-(2,3-Dimethoxybenzyl)-2-(9-methoxy-2-methyl-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6o)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 70% ethyl acetate in petroleum ether) to afford 6o (86 mg, 59%) as a yellow solid; mp: 283–285 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.44 (s, 1H), 8.58 (t, J = 5.9 Hz, 1H), 8.09–8.03 (m, 2H), 7.38 (dd, J = 8.7, 2.0 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 7.09 (d, J = 2.2 Hz, 1H), 7.04 (t, J = 7.9 Hz, 1H), 6.94 (ddd, J = 17.2, 8.4, 1.9 Hz, 2H), 6.84 (dd, J = 7.7, 1.6 Hz, 1H), 5.10 (s, 2H), 4.31 (d, J = 5.7 Hz, 2H), 3.87 (s, 3H), 3.80 (s, 3H), 3.73 (d, J = 6.5 Hz, 3H), 2.47 (s, 3H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 168.2, 159.3, 157.9, 152.7, 146.6, 139.7, 139.4, 136.6, 132.9, 131.2, 130.4, 124.3, 122.6, 121.8, 120.6, 119.0, 115.9, 113.6, 112.1, 111.1, 106.3, 95.6, 60.5, 56.2, 44.4, 37.5, 29.5, 20.9 ppm. HRMS (ESI) calcd for C28H28N3O5 [M + H]+, 486.2028; found, 486.2026.
2-(8-Chloro-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)-N-(1-(4-methoxyphenyl)ethyl)acetamide (6p)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 60% ethyl acetate in petroleum ether) to afford 6p (90 mg, 65%) as a yellow solid; mp: 326–328 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.82 (s, 1H), 8.71 (d, J = 8.0 Hz, 1H), 8.28 (dd, J = 7.9, 1.6 Hz, 1H), 8.15 (d, J = 2.2 Hz, 1H), 7.66 (d, J = 8.6 Hz, 1H), 7.60 (ddd, J = 8.7, 7.2, 1.5 Hz, 1H), 7.41 (dd, J = 8.5, 2.1 Hz, 2H), 7.35 (d, J = 8.6 Hz, 1H), 7.29–7.24 (m, 2H), 6.93–6.87 (m, 2H), 5.10 (d, J = 14.1 Hz, 2H), 4.97–4.86 (m, 1H), 3.75 (s, 3H), 1.38 (d, J = 7.0 Hz, 3H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 166.9, 159.3, 158.5, 141.6, 139.2, 136.8 (d, J = 6.2 Hz), 130.4 (d, J = 23.4 Hz), 127.6, 126.2, 124.6, 123.3, 122.4, 120.2, 116.2, 114.0, 114.0, 113.8, 113.2, 105.6, 55.6, 48.0 (d, J = 6.9 Hz), 44.2, 22.9 (d, J = 18.3 Hz) ppm. HRMS (ESI)calcd for C26H23ClN3O3 [M + H]+, 460.1428; found, 460.1423.
2-(4-Methyl-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)-N-(2,4,4-trimethylpentan-2-yl)acetamide (6q)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 6q (70 mg, 56%) as a yellow solid; mp: 326–328 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.47 (s, 1H), 8.22–8.10 (m, 2H), 7.65–7.58 (m, 2H), 7.43–7.35 (m, 2H), 7.28 (td, J = 7.4, 1.0 Hz, 2H), 5.03 (s, 2H), 2.70 (s, 3H), 1.71 (s, 2H), 1.34 (s, 6H), 0.99 (s, 9H) ppm. 13C{1H} NMR (126 MHz, DMSO-d6) δ 168.2, 161.3, 141.2, 140.1, 138.4, 135.0, 125.9, 124.9, 124.6, 122.5, 121.6, 121.3, 115.2, 112.2, 112.1, 105.8, 54.6, 50.8, 49.2, 31.7, 29.7, 29.6, 23.7 ppm. HRMS (ESI) calcd for C26H32N3O2[M + H]+, 418.2495; found, 418.2486.
N-(tert-Butyl)-2-(11-methyl-6-oxo-6,11-dihydro-5H-indolo[3,2-c]quinolin-5-yl)acetamide (6r)
Synthesized according to procedure B in 0.3 mmol scale, with purification of the crude product by column chromatography (silica gel; 50% ethyl acetate in petroleum ether) to afford 6r (74 mg, 68%) as a yellow solid; mp: 282–284 °C. 1H NMR (500 MHz, Chloroform-d) δ 8.52 (dd, J = 7.8, 1.6 Hz, 1H), 8.45 (d, J = 8.2 Hz, 1H), 7.65 (dd, J = 8.4, 1.2 Hz, 1H), 7.61–7.58 (m, 2H), 7.52 (ddd, J = 8.5, 7.1, 1.5 Hz, 1H), 7.48–7.40 (m, 2H), 7.29 (s, 1H), 5.01 (s, 2H), 4.41 (s, 3H), 1.30 (s, 9H) ppm. 13C{1H} NMR (126 MHz, Chloroform-d) δ 167.5, 157.1, 141.0, 135.6, 126.9, 126.5, 125.6, 123.8, 123.4, 122.9, 121.7, 121.3, 119.8, 119.6, 115.8, 110.7, 51.5, 48.4, 31.7, 28.6 ppm. HRMS (ESI) calcd for C22H24N3O2 [M + H]+, 362.1869; found, 362.1867.
Acknowledgments
The project leading to this application has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 713482 and No. 754425. Q.W. acknowledge the China Scholarship Council for supporting.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b01258.
The authors declare no competing financial interest.
Supplementary Material
References
- a Gaich T.; Baran P. S. Aiming for the Ideal Synthesis. J. Org. Chem. 2010, 75, 4657–4673. 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]; b Wender P. A. Toward the Ideal Synthesis and Molecular Function through Synthesis-informed Design. Nat. Prod. Rep. 2014, 31, 433–440. 10.1039/C4NP00013G. [DOI] [PubMed] [Google Scholar]; c Zarganes-Tzitzikas T.; Chandgude A. L.; Dömling A. Multicomponent Reactions, Union of MCRs and Beyond. Chem. Rec. 2015, 15, 981–996. 10.1002/tcr.201500201. [DOI] [PubMed] [Google Scholar]
- a Dömling A.; Wang W.; Wang K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dömling A. Recent Developments in IsocyanideBased Multicomponent Reactions in Applied Chemistry. Chem. Rev. 2006, 106, 17–89. 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; c Dömling A.; Ugi I. Multicomponent Reactions with Isocyanides. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. . [DOI] [PubMed] [Google Scholar]
- a Miller K. A.; Tsukamoto S.; Williams R. M. Asymmetric Total Syntheses of (+)-and (−)-Versicolamide B and Biosynthetic Implications. Nat. Chem. 2009, 1, 63–68. 10.1038/nchem.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Trost B. M.; Cramer N.; Bernsmann H. Concise Total Synthesis of (±)-Marcfortine B. J. Am. Chem. Soc. 2007, 129, 3086–3087. 10.1021/ja070142u. [DOI] [PubMed] [Google Scholar]; c Williams R. M.; Cox R. J. Paraherquamides, Brevianamides, and Asperparalines: Laboratory Synthesis and Biosynthesis. An Interim Report. Acc. Chem. Res. 2003, 36, 127–139. 10.1021/ar020229e. [DOI] [PubMed] [Google Scholar]; d Dalpozzo R.; Bartoli G.; Bencivenni G. Recent Advances in OrganocatalyticMethods for the Synthesis of Disubstituted 2-and 3-Indolinones. Chem. Soc. Rev. 2012, 41, 7247–7290. 10.1039/c2cs35100e. [DOI] [PubMed] [Google Scholar]
- a Erickson J. D.; Eiden L. E.; Hoffman B. Expression Cloning of a Reserpine-Sensitive Vesicular Monoamine Transporter. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 10993–10997. 10.1073/pnas.89.22.10993. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wu G.; Liu J.; Bi L.; Zhao M.; Wang C.; Baudy-Floc’h M.; Ju J.; Peng S. Toward Breast Cancer Resistance Protein (BCRP) Inhibitors: Design, Synthesis of ASeries of New Simplified Fumitremorgin C Analogues. Tetrahedron 2007, 63, 5510–5528. 10.1016/j.tet.2007.04.045. [DOI] [Google Scholar]; c Jiang J.; Hu C. Evodiamine: ANovel Anti-cancer Alkaloid from Evodiarutaecarpa. Molecules 2009, 14, 1852–1859. 10.3390/molecules14051852. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Oishi S.; Watanabe T.; Sawada J.; Asai A.; Ohno H.; Fujii N. Kinesin Spindle Protein (KSP) Inhibitors with 2,3-Fused Indole Scaffolds. J. Med. Chem. 2010, 53, 5054–5058. 10.1021/jm100476d. [DOI] [PubMed] [Google Scholar]
- a Chen Y.; Chung C.; Chen I.; Chen P.; Jeng H. Synthesis and Cytotoxic Activity Evaluation of Indolo-, Pyrrolo-, and Benzofuro-quinolin-2(1H)-ones and 6-Anilinoindoloquinoline Derivatives. Bioorg. Med. Chem. 2002, 10, 2705–2712. 10.1016/S0968-0896(02)00111-6. [DOI] [PubMed] [Google Scholar]; b Xiao Z.; Waters N. C.; Woodard C. L.; Li Z.; Li P. Design and Synthesis of PfmrkInhibitors as Potential Antimalarial Agents. Bioorg. Med. Chem. Lett. 2001, 11, 2875–2878. 10.1016/S0960-894X(01)00578-9. [DOI] [PubMed] [Google Scholar]
- Hayashi K.; Choshi T.; Chikaraishi K.; Oda A.; Yoshinaga R.; Hatae N.; Ishikura M.; Hibino S. A Novel Total Synthesis of IsocryptolepineBased on a Microwave-assisted Tandem CurtiusRearrangement and Aza-electrocyclicReaction. Tetrahedron 2012, 68, 4274–4279. 10.1016/j.tet.2012.03.055. [DOI] [Google Scholar]
- a Xu X.; Liu J.; Lu L.; Wang F.; Yin B. Pd-catalyzed RegioselectiveIntramolecular Direct Arylation of 3-Indolecarboxamides: Access to Spiro-indoline-3,3′-oxindoles and 5,11-Dihydro-6H-indolo[3,2-c]quinolin-6-ones. Chem. Commun. 2017, 53, 7796–7799. 10.1039/C7CC02256E. [DOI] [PubMed] [Google Scholar]; b Zhang X.; Zhang-Negrerie D.; Deng J.; Du Y.; Zhao K. Synthesis of Diversely Substituted Indoloquinolinones via Pd(II)/Cu(II)-mediated Oxidative C-C Bond Formation and I(III)-mediated C-N Bond Formation. J. Org. Chem. 2013, 78, 12750–12759. 10.1021/jo4023292. [DOI] [PubMed] [Google Scholar]; c Zhou L.; Doyle M. P. Lewis Acid Catalyzed Indole Synthesis via Intramolecular Nucleophilic Attack of Phenyldiazoacetates to Iminium Ions. J. Org. Chem. 2009, 74, 9222–9224. 10.1021/jo902089e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hayashi K.; Choshi T.; Chikaraishi K.; Oda A.; Yoshinaga R.; Hatae N.; Ishikura M.; Hibino S. A Novel Total Synthesis of IsocryptolepineBased on a Microwave-assisted Tandem CurtiusRearrangement and Aza-electrocyclicReaction. Tetrahedron 2012, 68, 4274–4279. 10.1016/j.tet.2012.03.055. [DOI] [Google Scholar]; e Cheng C.; Chen W. W.; Xu B.; Xu M. H. Access to Indole-fused Polyheterocycles via Pd-catalyzed Base-Free Intramolecular Cross DehydrogenativeCoupling. J. Org. Chem. 2016, 81, 11501–11507. 10.1021/acs.joc.6b02160. [DOI] [PubMed] [Google Scholar]; f Walser A.; Silverman G.; Flynn T.; Fryer R. I. Nucleophilic Displacement of Aromatic Fluorine, Part III†, Indoloquinolines and Benzofuranoquinolines. J. Heterocycl. Chem. 1975, 12, 351–358. 10.1002/jhet.5570120227. [DOI] [Google Scholar]; g Shi Z. J.; Ren Y. W.; Li B.; Lu S. C.; Zhang W. CuI-catalyzed Photochemical or Thermal Reactions of 3-(2-azidobenzylidene)lactams. Application to the Synthesis of Fused Indoles. Chem. Commun. 2010, 46, 3973–3975. 10.1039/c001715a. [DOI] [PubMed] [Google Scholar]
- Kong L.; Zheng Z.; Tang R.; Wang M.; Sun Y.; Li Y. Palladium-catalyzed Dual C(sp2)-H Functionalization of Indole-2-carboxamides Involving a 1,2-acyl Migration: a Synthesis of Indolo[3,2-c]quinolinones. Org. Lett. 2018, 20, 5696–5699. 10.1021/acs.orglett.8b02419. [DOI] [PubMed] [Google Scholar]
- Mahdavi M.; Hassanzadeh-Soureshjan R.; Saeedi M.; Ariafard A.; Ahmadi R. B.; Ranjbar P. R.; Shafiee A. Experimental and Computational Evidence for KOt-Bu-promoted Synthesis of Oxopyrazino[1,2-a]indoles. RSC Adv. 2015, 5, 101353–101361. 10.1039/C5RA17056G. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.