

Abstract
Background
The complex life cycle of malaria parasites requires well-orchestrated stage specific gene expression. In the vertebrate host the parasites grow and multiply by schizogony in two different environments: within erythrocytes and within hepatocytes. Whereas erythrocytic parasites are well-studied in this respect, relatively little is known about the exo-erythrocytic stages.
Methods
In an attempt to fill this gap, genome wide RNA-seq analyses of various exo-erythrocytic stages of Plasmodium berghei including sporozoites, samples from a time-course of liver stage development and detached cells were performed. These latter contain infectious merozoites and represent the final step in exo-erythrocytic development.
Results
The analysis represents the complete transcriptome of the entire life cycle of P. berghei parasites with temporal detailed analysis of the liver stage allowing comparison of gene expression across the progression of the life cycle. These RNA-seq data from different developmental stages were used to cluster genes with similar expression profiles, in order to infer their functions. A comparison with published data from other parasite stages confirmed stage-specific gene expression and revealed numerous genes that are expressed differentially in blood and exo-erythrocytic stages. One of the most exo-erythrocytic stage-specific genes was PBANKA_1003900, which has previously been annotated as a “gametocyte specific protein”. The promoter of this gene drove high GFP expression in exo-erythrocytic stages, confirming its expression profile seen by RNA-seq.
Conclusions
The comparative analysis of the genome wide mRNA expression profiles of erythrocytic and different exo-erythrocytic stages could be used to improve the understanding of gene regulation in Plasmodium parasites and can be used to model exo-erythrocytic stage metabolic networks toward the identification of differences in metabolic processes during schizogony in erythrocytes and hepatocytes.
Keywords: Malaria, Life cycle, Plasmodium liver stage, Transcriptome, RNA-seq, Merosome
Background
Malaria is a devastating disease caused by apicomplexan parasites of the genus Plasmodium. Almost half of the world’s population is permanently at risk of malaria resulting in over 200 Million malaria cases worldwide mostly in African countries. There were more than 400,000 deaths in 2017 [1], and the majority of them were children under the age of five.
The life cycle of Plasmodium parasites involves the injection of sporozoites into the vertebrate host during a blood meal of an infected female mosquito. For the rodent parasite Plasmodium berghei it has been shown that a proportion of injected sporozoites actively invade blood vessels and then are passively transported to the liver [2]. After crossing the blood vessel endothelia in the liver to reach the parenchyma, the parasite transmigrates through several hepatocytes before it settles in one. Upon entry into the ultimate host cell, the host plasma membrane invaginates forming a parasitophorous vacuole (PV) in which the parasite resides, develops and multiplies by exo-erythrocytic schizogony. The intracellular parasite extensively remodels the parasitophorous vacuole membrane (PVM), in particular by excluding or removing host cell proteins and by incorporating parasite proteins [3].
Each exo-erythrocytic stage parasite (EEF: exo-erythrocytic form) generates tens of thousands of nuclei by the process of exo-erythrocytic schizogony. This rapid nuclear division is accompanied by growth and replication of organelles including the Golgi apparatus, endoplasmic reticulum, mitochondrion and apicoplast and a vast expansion of the plasma membrane [4–6]. Nuclei and organelles are eventually segregated into individual merozoites. Once EEF merozoites have completed their development, the PVM ruptures. This process requires an orchestrated action of multiple Plasmodium proteins such as lipases (e.g. PbPL) [7], proteases (e.g. SUB1) [8, 9] and possibly perforins as shown for erythrocytic stage parasites (EF: erythrocytic form) [10–12]. Upon rupture of the PVM, EEF merozoites disperse in the host cell cytoplasm and the host cell actin cytoskeleton collapses [13]. The final developmental stage of the EEF in in vitro cultures, is the formation of detached cells (DCs) and merosomes, host cell plasma membrane enclosed merozoites [14, 15]. In the in vivo mouse model, infected cells become excluded from the liver tissue upon PVM rupture and merosomes are formed and pushed into the lumen of adjacent blood vessels. At tissue sites with small capillaries, merosomes rupture to release merozoites into the blood [16]. Liberated merozoites immediately invade red blood cells (RBC), where they undergo repeated asexual reproduction cycles. In contrast to the tens of thousands merozoites generated by a single EEF, within erythrocytes the parasites produce only a limited number (12 to 32) merozoites by erythrocytic schizogony. Another difference between erythrocytic and exo-erythrocytic schizogony is that after rupture of the PVM the EEF merozoites can reside in the host cell cytoplasm for up to several hours, whereas EF merozoites are liberated from the PVM and the host cell plasma membrane almost simultaneously [17]. Some EF will differentiate into male and female gametocytes. When these are ingested by a mosquito during a blood meal, they mature into macrogametes and microgametes and are liberated from the RBC. These sexual forms fuse to form zygotes and transform into motile ookinetes, which are able to cross the midgut epithelium of the mosquito to develop into oocysts, in which thousands of sporozoites are formed. After about 9–16 days (depending on the parasite species and the environmental temperature), sporozoites are liberated and invade the salivary glands of the mosquito (reviewed in [18]), whereupon they are then ready to be injected into a host during the next blood meal.
Studying the entire life cycle using human parasites under live or laboratory conditions is difficult due to ethical and safety reasons. However, the use of model organisms, such as the rodent malaria parasite P. berghei is experimentally tractable and the genetic manipulation of this parasite is relatively easy and well established [19–21]. This model allows investigating the EEF development and filling gaps of knowledge that might also be relevant for human Plasmodium species. Many transcriptomic studies have been undertaken for different Plasmodium species, either by microarrays or RNA-seq. Table 1 lists the transcriptomic data integrated in PlasmoDB [22].
Table 1.
Genome wide transcriptome data from studies integrated in PlasmoDB release43; http://www.plasmodb.org (np: not published)
| Method | Plasmodium sp. | Life cycle stages | Refs. |
|---|---|---|---|
| RNA-seq | P. berghei ANKA | 5 asexual and sexual stage transcriptomes | [49] |
| Female and male gametocyte | [85] | ||
| P. chabaudi chabaudi | Trophozoite transcriptomes after mosquito transmission or direct injection into mice | [86] | |
| P. yoelii yoelii 17X | Salivary gland sporozoite transcriptomes: WT vs. Puf2-KO | [87] | |
| P. falciparum 3D7 | NSR-seq transcript profiling of malaria-infected pregnant women and children | [88] | |
| Polysomal and steady-P. berghei state asexual stage transcriptomes | [89] | ||
| Blood stage transcriptome (3D7) | [90] | ||
| Transcriptomes of 7 sexual and asexual life stages | [91] | ||
| Intraerythrocytic cycle transcriptome (3D7) | np | ||
| Strand specific transcriptomes of 4 life cycle stages | [91] | ||
| Transcriptome during intraerythrocytic development | [92] | ||
| Strand specific transcriptome of the intraerythrocytic developmental cycle | [93] | ||
| Ribosome and steady state mRNA sequencing of asexual cell cycle stages | [94] | ||
| Mosquito or cultured sporozoites and blood stage transcriptome (NF54) | np | ||
| Female and male gametocyte transcriptomes | [50] | ||
| Ring, oocyst and sporozoite transcriptomes | [95] | ||
| P. vivax P01 | Hypnozoite RNAseq | [29] | |
| Transcription profile of intraerythrocytic cycle | [96] | ||
| P. cynomolgi strain M | P. cynomolgi transcriptome of whole blood and bone marrow collected during 100-day infection of M. mulatta | [97] | |
| Hypnozoite, schizont and blood stage transcriptomes (laser microdissection) | [98] | ||
| Liver stage hypnozoite vs schizont transcriptomes (primary culture) | [30] | ||
| DNA micro-array | P. berghei ANKA | DOZI mutant transcript profile | [51] |
| Transcript profiling of developmental stages—high producer (HP/HPE) | [99] | ||
| AP2-G2 knock out and WT expression profiles | [100] | ||
| P. yoelii yoelii 17X | Liver, mosquito and blood stage expression profiles | [28] | |
| Life cycle stages | [101] | ||
| P. falciparum 3D7 | Pfal3D7 real-time transcription and decay | np | |
| Invasion pathway knockouts | [102] | ||
| Three isogenic lines w/CQ treatment: expression profiles | [103] | ||
| Life cycle expression data (3D7) | [104] | ||
| Sexually vs asexually committed schizont transcriptional profiles | [105] | ||
| Gametocyte stages I-V transcriptomes | [106] | ||
| Two Sir2 KO lines expression profiling | [107] | ||
| Asexual blood stage transcriptomes of clonal strains | [108] | ||
| Erythrocytic expression time series (3D7, DD2, HB3) | [109, 110] | ||
| Microarray expression from patient samples | [111] | ||
| mRNA half life | [112] | ||
| P. vivax P01 | Intraerythrocytic developmental cycle of three isolates | [113] | |
| Sporozoite expression profiles | [114] | ||
| P. knowlesi strain H | Intraerythrocytic cycle expression profile: in vitro and ex vivo (Pkno PK1(A+)) | [115] |
Recently, single cell transcriptomic profiles have also been published for EF of Plasmodium falciparum, Plasmodium knowlesi and P. berghei [23–25]. One of these studies (the Malaria Cell Atlas) also analysed the liver stage, but only a single time point (44hpi) [25]. Two further RNA-seq studies focused on the host cell transcriptome and did not attempt to group parasite genes in a systematic manner and did not include data from later stage EEFs (beyond 48hpi) [26, 27]. Three further studies analysed the transcriptome of Plasmodium EEFs, one of which was done using the rodent parasite Plasmodium yoelii [28] and the other two using Plasmodium vivax with an emphasis on the dormant parasite stage, the hypnozoite [29, 30]. Since P. berghei is a widely used model in malaria research and the so far published data for its EEF stages are incomplete, genome wide RNA-seq analyses of EEF development in a time course fashion were performed and expression data were compared to already published data of gene-expression of other life cycle stages of P. berghei. In particular, EEF merozoites originating from DCs/merosomes were compared to EF merozoites (from in vitro cultivated schizonts) and it was found that their transcription profiles differ substantially. Remarkably, differences were identified that are predicted to have an impact on metabolic processes during schizogony in erythrocytes and hepatocytes despite the fact, that both types of merozoites infect RBC.
Methods
Mice, parasites, infections
BALB/c mice used for mosquito infections were between 6 and 10 weeks of age and were purchased from the Central animal facility at University of Bern, Harlan (Horst, the Netherlands) or Janvier Labs (Le Genest Saint Isle, France). For RNA work: mice were infected by intraperitoneal injection of blood stabilates of marker-free P. berghei strain ANKA expressing mCherry under the control of hsp70 regulatory sequences (PbmCherryhsp70) [7]. At parasitaemia of ~ 4%, the mouse was bled and 40 μl of infected blood was injected intravenously into phenylhydrazine treated mice (200 μl of 6 mg/ml in PBS, 2–3 days before). At day 3 to 4 after infection, each mouse with at least 7% parasitaemia was anaesthetized with Ketasol/Xylasol and exposed to ~ 150 female Anopheles stephensi mosquitoes (which were sugar starved for 5 h). Mosquitoes were kept at 20.5 °C and 80% humidity. From day 16–26 post infection salivary glands of infected mosquitos (sorted by fluorescence stereomicroscope Olympus SZX10/U-HGLGPS) were dissected into serum free IMDM (Iscove’s Modified Dulbecco’s Medium, Sigma-Aldrich). Sporozoites were liberated from the glands and were used to infect confluent HeLa cells (per time point 10 wells of 96 well plates were seeded with 40,000 cells/well the day before). Each well was infected with ~ 20,000 PbmCherryhsp70 sporozoites for 6 h. The cells were detached with accutase (Innovative Cell Technology), pooled and the equivalent of 10 wells were seeded in 25 cm2 cell culture flask. 1/6th of the cells was washed once with PBS and pelleted by centrifugation (2 min 100 g). The pellet was loosened by flicking and the cells were resuspended with 250 μl of RNAlater and stored at 4 °C till all time points were harvested. It has been reported that RFP in contrast to GFP is preserved in RNAlater treated cells [31].
Media of the cultured cells were changed at 24 hpi and 48 hpi. At the respective time points the cells were detached from the surface with accutase, washed once with PBS and then resuspended in 250 μl RNAlater and stored at 4 °C. The use of RNAlater shortened the parasite’s time in an unnatural state not being in the incubator in adequate environment and medium down to 10 min compared to 1–2 h in case of sorting fresh cells.
To generate transgenic parasites expressing gfp under control of the promoter of PBANKA_1003900 (PBANKA_1003900GFP), an episomal PbGFPcon vector was used [32]. First, the PBANKA_1003900 promoter (1.7 kb) was amplified using primers GCTCTACCAATTTTGTGTCAC and GGATCCTTAAAAATTAATTTTGTATAAAATCG and cloned into a pCR2.1-TOPO vector (Invitrogen) and sequenced. Then the P. berghei elongation factor-1α promoter of PbGFPcon was exchanged for the PBANKA_1003900 promoter (EcoRV from pCR2.1-TOPO vector/BamHI) and the gfp gene was re-introduced in the correct orientation as a BamHI fragment. Finally the construct (Additional file 2: Fig. S1) was used to transfect the reference wild type P. berghei ANKA parasite line (cl15cy1 (ANKAwt)) [19] to generate line 300 (PBANKA_1003900GFP). Transfection with the episomal construct and positive selection of transfected parasites with pyrimethamine was performed as described previously [19].
For Microscopy work: confluent HeLa cells in 96-well plate (40,000 cells seeded the day before) were infected with ~ 20,000 PBANKA_1003900GFP sporozoites. Cells were washed and detached 2 h post-infection using accutase and seeded on glass covers in 24 wells. At the indicated time points cells were fixed with 4% PFA in PBS for 10 min, washed with PBS and kept at 4 °C. Nuclei were stained with 1 μM Hoechst 33342 for 20 min, embedded with Dako-mounting medium. Fluorescent microscopy pictures were taken on a Leica DM5500B. Signals were photographed using same exposure settings.
For live cell imaging, infected cells were seeded onto glass bottom dishes (35-20-1.5-N, Cellvis, Mountain View). Live cell microscopy was performed with a Leica DMI6000B epifluorescence microscope equipped with a SOLA-SE-II light source starting at 30hpi.
FACS sorting
Cells kept in RNAlater were FACS sorted on a BD FACSARIA III, FACSflow was used as sheath fluid. A 561 nm laser was used in combination of 610/20 nm filter detect the infected cells (see Additional file 2: Fig. S2). To obtain maximal purity the sorting was performed using the 4-way-purity mode with 100 microns nozzle. The sorted cells were collected into RNAlater (500 μl in Eppedorf tube). 100,000 non-infected cells at each time point were sorted as negative controls.
RNA isolation, library preparation, sequencing
Prior to isolation of RNA by ReliaPrep™ RNA Cell Miniprep System RNAlater was removed from the cells by adding an equal volume of ddH2O to the cells. The cells were then centrifuged (200 g, 2 min). The RNA from the pelleted cells was extracted according to the manufacturer’s protocol and kept at − 80 °C. RNA extraction and Illumina m RNA-sequencing were performed in duplicates. Following RNA isolation, total RNA was quantified with a Qubit Fluorometer (Life Technologies). Quality of the extracted RNA was checked by the RNA integrity number (RIN), measured using an Agilent 2100 BioAnalyser (Agilent Technologies). The SMARTer™ Ultra Low RNA kit from Clontech was used for the reverse transcription and cDNA amplification according to manufacturer’s specifications, starting with 10 ng of total RNA as input. The Nextera XT kit (Illumina, San Diego, CA, USA) was used for cDNA libraries preparation using 200 pg of cDNA. Library molarity and quality was assessed with the Qubit and Tapestation using a DNA High sensitivity chip (Agilent Technologies). The cDNA libraries were pooled and loaded at 12.5 pM, multiplexed on 4 lanes of HiSeq Rapid PE v2 Flow cells for generating paired reads of 100 bases on an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA, USA).
Data processing
Short reads generated in this study were deposited at the European Nucleotide Archive “(http://www.ebi.ac.uk/ena/) and are accessible through the accession number PRJEB23770 (Secondary study accession number: ERP105548)”. Publicly available data was obtained from SRA (SRP027529, ERS092084 and ERS092085). All reads were quality-checked with FastQC (bioinformatics.babraham.ac.uk/projects/fastqc). For the publicly available data, illumina adaptor sequences and low-quality reads were removed with TrimGalore (version 0.4.1 with the parameter-illumina), http://www.bioinformatics.babraham.ac.uk/projects/trim_galore). For the data generated in this study, Nextera transposase sequences and low quality reads were removed with Trimmomatic (version 0.33 with the parameters ILLUMINACLIP:adapters/NexteraPE-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:5:30 MINLEN:50 [33]. Low complexity reads were removed with fqtrim (version 0.9.4) [34]. For paired-end reads, if only one end was removed, the remaining read end was treated as single-end read. To remove potential contamination with host RNA, all reads were aligned to the human genome (ensembl82) with Bowtie2 (version, 2.2.5) [35]. Single-end reads and read pairs with none of the ends aligning to the human genome were kept and aligned to the P. berghei ANKA reference genome (PlasmoDB Release 33) with Subread (i.e. subjunc, version 1.4.6-p5) [36] allowing up to 10 alignments per read (options: -n 20 -m 5 -B 10 -H –all Junctions, always in single-end mode, i.e., ignoring the reverse read-end of paired-end reads). Count tables were generated with Rcount [37] with an allocation distance of 100 bp for calculating the weights of the reads with multiple alignments and a minimal number of 5 hits. Count tables are available in Additional file 1: Table S2. Multireads were included to avoid underestimation of expression of genes with similar sequences (see for example [38, 39]). To ensure that each multiread contributes only once to the final expression values, alignments of multireads were weighted according to the number of unique read alignments within 100 bp of a multiread alignment site (for details, see [37]).
Differential expression
Variation in gene expression was analysed with a general linear model in R with the package DESeq 2 (version 1.16.1) [40] according to a design with a single factor comprising all different experimental groups. Specific groups were compared with linear contrasts and P-values were adjusted for multiple testing [41], (i.e., false discovery rate). Genes with an adjusted P value (FDR) below 0.01 and a minimal logFC of 2 were considered to be differentially expressed. Normalized gene expression data for plotting and clustering was likewise obtained with DESeq 2 (version 1.16.1) [40].
Gene co-expression network
To identify groups of genes with similar expression patterns across the life cycle of P. berghei, a gene co-expression network (GCN) was constructed. Therefore an adjacency matrix with pairwise Pearson correlation coefficients was calculated, Fisher’s z-transformation was applied and each pairwise correlation coefficient for being significantly bigger than zero was tested (as described in [42]). P-values were adjusted for multiple testing [41] and correlation coefficients with an adjusted P-value below 0.001 were identified as significant. The significant pairwise correlation coefficients were then used to construct the GCN. To resolve the community structure of the GCN, a modularity optimization algorithm [43] implemented by the function “cluster_louvain” in the R package “igraph” (version 1.0.1) was used [44]. Communities with less than 11 genes were collapsed into a single “mixed” community (70 communities with a total of 197 genes). The network was visualized with Cytoscape (version 3.5.1, “prefuse force directed layout”) [45]. The GeneIDs per community are listed in Additional file 1: Table S4. It should be considered that the GCN analysis was partly based on already published data and for that reason could not be corrected for possible batch effects. The focus of the current study was the liver stage including the sporozoite stage and detached cells as a final point of liver stage development.
Gene ontology enrichment
To functionally characterize the network communities or genes found to be differentially expressed, enrichment of gene ontology (GO) terms was tested with topGO (version 3.4.1) [46] in conjunction with the GO annotation available from PlasmoDB [22]. Analysis was based on gene counts (genes in the set of interest compared to all annotated genes) using the “weight” algorithm with Fisher’s exact test (both implemented in topGO). A term was identified as significant if the P-value was below 0.05.
Enrichment of selected gene groups
To test for enrichment of a specific group of genes (e.g., “merozoite invasion genes” from [47]) within a gene set of interest compared to all genes annotated with any of the tested groups, we used Fisher’s exact test (two-by-two contingency table). P-values were adjusted for multiple testing [41] and groups with an adjusted P-value (FDR) below 0.05 were identified as significant.
Results and discussion
High fidelity exo-erythrocytic stage RNA-seq data and sample selection criteria
HeLa cells were infected with P. berghei sporozoites that express mCherry under the control of a constitutive Hsp70 promoter, allowing the detection of fluorescent parasites in all developmental stages [7]. Exo-erythrocytic form (EEF) parasites were isolated at different time-points of infection by FACS sorting (6 h, 24 h, 48 h, 54 h and 60 h). At 69 h detached cells and merosomes (DCs/merosomes) were collected from the culture medium supernatant. To preserve RNA integrity during FACS sorting of infected cells, cells were treated with RNAlater [31, 48]. In addition, sporozoite samples were generated by processing infected salivary glands of mosquitoes at day 20 post feeding. For each sample, independent biological duplicates were collected. Following the isolation of total RNAs from infected HeLa cells and from infected salivary glands, libraries were sequenced with an Illumina HiSeq 2500 resulting in 34 to 61 million paired-end reads per sample (Additional file 1: Table S1). After removal of low quality sequences, sequencing adapters and sequences arising from host RNA, reads were aligned to the P. berghei ANKA reference genome, resulting in around 0.23 to 21.4 million weighted alignments (hits) within genic regions (Additional file 1: Table S1, raw counts are provided in Table S2). Samples collected 6 h after infection were excluded from further analysis as only low amounts of hits were recovered (22,154 and 35,312 hits) in both biological replicates. The reason is most likely that, at this time-point of infection, parasite transcripts represent only a small fraction compared to host cell transcripts.
4475 transcribed genes were identified (≥ 80 normalized read counts, corresponding on average to 20 RPKMs in at least one developmental stage of the EEF). In a previously reported transcriptome analysis of P. yoelii EEF stages using microarrays, about 2000 genes were detected [28]. This exemplifies the higher sensitivity of the Next Generation Sequencing (NGS) compared to array technology [39].
A hierarchical clustering of the different EEF samples was performed together with RNA-seq data of EF (rings, trophozoites and schizonts harvested 4, 16 and 22 h after infection of RBC), as well as with RNA-seq data of gametocytes and ookinetes [49]. The replicates exhibited a high Spearman’s correlation to each other and the different stages grouped well according to the host environment (exo-erythrocytic, erythrocytic) (Fig. 1).
Fig. 1.

Clustering of samples of different life cycle stages based on genes with the highest overall high variance (90th percentile, Spearman correlation and hierarchical clustering). Stages are sporozoites, exo-erythrocytic (EEF) stages (DCs/merosomes are detached cells and merosomes), erythrocytic (EF) stages (rings, trophozoites, schizonts, gametocytes) and ookinetes. _A, _B: Biological replicates. Heatmap was generated using normalized and log2(x + 1)-transformed gene expression values [40]. Heatmap drawn with the R-package gplots [116]
The analysis revealed that the transcripts profiles of the extracellular ookinetes and sporozoites cluster together, which might be due to the fact, that both are motile stages that traverse mosquito host cells. Ookinetes and sporozoites are, however, markedly different from the profiles of EEF and asexual EF stages. Notably, the expression profile of gametocytes is different from both EEF and asexual EF stages but shows similarities to the ookinetes. This is not surprising as in the Plasmodium life cycle gametocytes differentiate into ookinetes (via gamete and zygote stages). In female gametocytes various mRNAs are already produced but translationally repressed and only used during development of the zygote and ookinete [50–52]. Among the EEF stages, the highest similarities of gene expression profiles were observed as expected for adjacent stages/timepoints (e.g., 48 h and 54 h). However, the early stages/timepoints (24 h, 48 h and 54 h) were fairly distinct from the later time-points (60 h and detached cells). The asexual EF stages showed a high degree of similarity among them. In fact, the gene expression profile of detached cells, containing the EEF merozoites, was rather distinct from the profile of late stage EF schizonts, containing EF merozoites, although both need to be prepared to invade RBC. It is noteworthy that proteomics data of P. yoelii, revealed that 90% of the proteins of late EEF were also detected in the early EF [28].
To verify the RNA-seq data, the expression pattern of selected genes was compared to previously reported patterns. The expression pattern of the housekeeping proteins (GAPDH, actin 1 and alpha-tubulin 1; Additional file 2: Fig. S3), putative proteases (SERA 1 to 5; Additional file 2: Fig. S4), PVM proteins (EXP1, Exp2, UIS3 and UIS4; Additional file 2: Fig. S5), sporozoite surface proteins (CSP and TRAP; Additional file 2: Fig. S6), fatty acid biosynthesis enzymes (FabB/F, FabI, FabZ and FabG, Additional file 2: Fig. S7) as well as merozoite surface proteins (MSP 1, 4/5, 7, 8, 9, 10; Additional file 2: Fig. S8) were very similar to the already published data confirming the quality of the here presented RNA-seq data. Further information about the selected genes presented in Additional file 2: Figs. S3 to S8 is detailed in the supplementary information section.
Importantly, the expression profiles deduced from the RNA-seq analysis provide valuable information for the choice of promoters to drive expression of stage-specific transgenes, such as fluorescent or luminescent reporter proteins. Previously, the promoters of the housekeeping genes heat shock protein (hsp70) and eukaryotic elongation factor 1α (eef1α) have been used to drive expression of fluorescent reporters [7, 32, 53]. According to the here presented RNA-seq analysis, the hsp70 promoter is a better choice for driving constitutive expression of reporters as hsp70 mRNA exhibits a more uniform expression profile compared to eef1α mRNA (Additional file 2: Fig. S9).
Next, a more detailed computational analysis of the exo-erythrocytic stage transcriptome and a comparison with other developmental stages was performed.
Gene co-expression network
To further explore the complexity of the parasite transcriptome, in particular the gene expression similarities among the different developmental stages, a gene co-expression network (GCN) was computed [42] and genes with similar expression patterns (“communities”) [43] were extracted and visualized (Fig. 2). This analysis allowed to gain insight into the sets of similarly expressed genes in the different EEF and EF stages and into characteristic expression patterns within the entire transcriptome. These analyses were used to find functionally related genes based on similar expression patterns.
Fig. 2.

Gene co-expression network based on RNA-seq data of samples of different life cycle stages. Stages are sporozoites, exo-erythrocytic (EEF) stages (DCs/merosomes correspond to detached cells and merosomes), erythrocytic (EF) stages (rings, trophozoites, schizonts, gametocytes) and ookinetes. Each node represents a gene and each edge depicts a significant pairwise correlation. The network was visualized with Cytoscape [45] using the “prefuse force directed layout”. Nodes/genes are colored according to their membership in 14 communities and a ‘mixed’ (M) community (pool of communities with less than 11 genes per community), identified with a modularity optimization algorithm [43]. For each community, a heatmap summarizes the expression patterns of all genes within the community. Expression values in the heatmaps correspond to gene-wise Z-scored of normalized and log2(x + 1)-transformed count data averaged across the replicates
The GCN analysis revealed 14 different communities comprised of 11 to 818 genes and a “mixed” community with 197 genes (a pool of communities with 10 or less genes per community) (Fig. 2, Additional file 1: Table S4). Of a total of 5104 genes, 4675 genes are represented in the GCN. 429 genes did not meet the GCN criteria, as the expression pattern of each of them did not significantly correlate to the expression pattern of another gene throughout all stages. Interestingly, the GCN analysis indicated marked differences between EEF and EF. Unexpectedly, even the transcriptome of EEF-derived and EF-derived merozoites (i.e., detached cells and late blood schizonts) were found to differ substantially. The 486 transcribed genes of community 1 were strongly expressed in DCs/merosomes, which contain EEF-derived merozoites and extended into the ring stage (initial phase of EF development). The 596 transcribed genes of community 2 were as well enriched in DCs/merosomes, but expression of these genes persisted longer during the EF (into trophozoite and partly into schizont stage) whereas their expression was strongly reduced in the sporozoites. To functionally characterize the communities defined in the GCN, a gene ontology (GO) term enrichment analysis from the domain “Biological Process (BP)” was performed (Fig. 3, Additional file 1: Table S3).
Fig. 3.

GO terms in GCN communities (expressed as − log10(P-value)). The colors refer to the different communities in Fig. 1b. Only GO-terms with P-values < 0.01 were included. Communities containing less than 25 genes were ignored because of potential false significance (following recommendations in GeneSetEnrichmentAnalysis from the Broad Institute)
The first two communities in Fig. 2 contain genes that are expressed specifically in DCs/merosomes and EF stages. The majority of the corresponding gene products were predicted to be involved in gene expression, ribosome biogenesis and transcription (Community 1), or in mRNA processing and translation (Community 2) (Fig. 3). All these functions are involved in DNA/RNA biology, particularly in gene expression and regulation. This is not surprising as invasive forms like the merozoites in DCs/merosomes prepare for the next growth phase after invasion and most likely need stored transcripts for a rapid protein synthesis after invasion of erythrocytes, comparable to the storage of repressed transcripts in mature gametocytes and sporozoites [50–52, 54–58]. On the other hand, the 160 genes of community 3 were highly specific to the EF stages (Fig. 3). Conspicuously, this community has almost no GO-term annotations for biological processes (only 3 out of 160 genes were annotated). However, this community was highly enriched for small nucleolar RNAs (snoRNAs), PIR pseudogenes (Plasmodium interspersed repeat pseudo genes) and genes of the three large multigene families in rodent parasites, coding for PIR proteins, fam-a proteins and fam-b proteins. SnoRNAs are important components of ribosome biogenesis that consist in non-coding RNAs with a diversity of function like pseudo-uridylation and 2′-O-methylation of RNAs or synthesis of telomeric DNA [59]. PIR, fam-a and fam-b proteins are exported by EF stages into the cytoplasm of the host erythrocyte. Recently it has been shown that a subset of PIR, fam-a and fam-b proteins are also expressed in EEF stages [60], however the function of most of these proteins remains unknown.
Community 6, consisting of 776 genes, was enriched for genes expressed in sporozoites, but also frequently expressed at elevated levels in gametocytes, ookinetes and schizonts. Not surprisingly, genes preferentially expressed in community 6 were involved in host cell entry, host cell exit and parasite motility (Fig. 3). In addition, genes that are involved in transport of subcellular components and DNA repair were present. Genes whose expression was found to be more specific to sporozoites, with persisting expression during the early EEF stages, but almost complete absence in DCs/merosomes (community 5), were involved in DNA synthesis and metabolic processes, consistent with the high multiplication observed following hepatocyte invasion by the sporozoites (Fig. 3).
Communities 7, 8 and 9 contain 45 genes in total, the expression of which were mostly specific to the developing EEF stages. According to guidelines of the Broad Institute on GeneSetEnrichmentAnalysis, small size communities should not be interpreted.
The expression of the 31 genes of community 10 was enriched in EF schizonts and in gametocytes, but these genes were also found well expressed in late EEF stages. Although the GO term ‘DNA metabolic process’ is listed for this community, it should be assessed with caution due to the reason mentioned above.
The 176 genes of community 11 were expressed during the developing EEF and EF stages, with a slight bias towards the developing EEF stages (85% of all genes in the community were on average expressed at a higher level in the EEF stages). In this community, 3 out of 9 genes coding for enzymes of fatty acid biosynthesis have been identified as hits. This is in agreement with the high fatty acid usage of EEF stages to generate various parasite membranes [61]. Apart from genes involved in fatty acid biosynthesis, it is very likely that genes identified in this community are involved in schizogony and merozoite development in both EEF and EF stages.
The remaining communities were mostly defined by genes with almost complete absence of expression in sporozoites (community 12; 499 genes), in sporozoites and DCs/merosomes (community 13; 480 genes) or in sporozoites, 24 h EEF stage and partly DCs/merosomes (community 14; 818 genes). However, whereas genes of community 12 and 13 were generally expressed throughout the EEF and the EF stages, genes of community 14 were more specific to gametocytes and ookinetes (Fig. 2). Sporozoites are not growing or proliferating and therefore it can be expected that in sporozoites, expression of genes involved in several metabolic processes, protein lipidation, phosphorylation and signal peptide processing is less pronounced than in other stages.
Altogether, the GCN analysis allowed to identify 14 clearly defined communities and a pool of small communities (mix) with totally 4675 genes attributed (see Additional file 1: Table S4 for GeneIDs of the members of the communities and for genes excluded during the GCN analysis).
The generated GCN provides a first comprehensive overview of gene regulation in a Plasmodium parasite throughout EEF and EF development including several life cycle stages in the mosquito vector (ookinetes, sporozoites). The identification of clear communities of genes with comparable expression profiles may help identifying common signatures in the untranslated promotor regions that may be involved in regulation of gene expression.
Differences in gene expression between developing exo-erythrocytic and erythrocytic parasites
To better elucidate the differences between EEF and EF stage parasites, differential expression analyses were conducted. A limitation of this approach was that blood stage gene transcription was based on already published data, which cannot be corrected for batch effects or contaminations with sexual stage parasites. Known confounders are the contamination of the 4 h EF ring stage with about 3% mature gametocytes and the 22 h EF schizont cultures with about 10% immature gametocytes [49]. In addition, the transition between ring forms and young trophozoites is not exactly defined by morphological or molecular means. Apart from these reasonable limitations, a comparison of the highly enriched and homogeneous stages during blood and liver stages revealed very interesting differences.
In order to compare EF and EEF parasites, averaged gene expression of developing EEF stages (24 h, 48 h, 54 h and 60 h) and of developing EF stages (ring 4 h, trophozoite 16 h) was compared, despite that such an averaging bears a potential bias towards earlier or late stages. In this comparison, 299 genes were significantly upregulated in the EEF stages (LogFC ≥ 2, adjP ≤ 0.01) and 392 genes were significantly upregulated in the EF stages (Fig. 4; Additional file 1: Table S5). GO-term enrichment (summarized in Table 2) revealed that genes preferentially expressed in the EEF stages were enriched for the fatty acid biosynthesis pathway (e.g. fabB/fabF in Fig. 4), entry into the host cell, leading strand elongation, tetrapyrrol biosynthesis and DNA replication. Considering that a single P. berghei EEF parasite generates more than 10,000 merozoites and an individual EF stage parasite only 12–18, an enrichment of expression in genes associated with fatty acid biosynthesis and DNA replication is expected and has already been confirmed in other studies [62, 63] and reviewed in [64]. In contrast, genes preferentially expressed in the EF stages were enriched for the GO terms for cell motility and intracellular, organellar transport (summarized as “movement of cell or subcellular components”), protein export into host cell cytoplasm, exit from host cell and pathogenesis. Enrichment of gene expression related to translocation of proteins (e.g. membrane associated histidine-rich protein 1: mahrp1a and b in Fig. 4) across the PVM is also required for parasite remodeling of the host RBC [65], including proteins transported to the surface of the RBC involved in RBC sequestration [65–67]. Many P. berghei proteins are known to be transported into the host red blood cell [60, 67] whereas only a few proteins have been identified to be exported into the host hepatocyte, for example, CSP [68, 69] and LISP2 [70].
Fig. 4.

Volcano plot of differently expressed genes of developing exo-erythrocytic (EEF) stages (EEF_24 h–60 h) compared to developing erythrocytic stages (EF_ring, EF trophozoites). In this analysis the DCs/merosomes and schizonts stages are not included. The graph shows LogFC values relative to FDR (− Log10(adjusted P-value). Positive LogFC values represent preferentially EEF stage expressed genes. Negative LogFC values represent preferentially EF stage genes. Genes in green and red show highest expression in liver stages compared to all other stages (including sporozoites, gametocytes and ookinetes)
Table 2.
Gene ontology (GO) term annotation of genes preferentially expressed in (i) EEF stages and (ii) asexual EF stages
| Biological process ID | Process description | Annotated | Observed | P value |
|---|---|---|---|---|
| (i) Preferentially expressed in exo-erythrocytic stage (24 h, 48 h, 54 h and 60 h) | ||||
| GO:0006633 | Fatty acid biosynthetic process | 9 | 6 | 4e−06 |
| GO:0044409 | Entry into host | 33 | 7 | 0.0036 |
| GO:0006272 | Leading strand elongation | 2 | 2 | 0.0040 |
| GO:0033014 | Tetrapyrrole biosynthetic process | 9 | 4 | 0.0042 |
| GO:0006260 | DNA replication | 39 | 10 | 0.0110 |
| GO:0055114 | Oxidation–reduction process | 83 | 12 | 0.0215 |
| GO:0006270 | DNA replication initiation | 4 | 2 | 0.0219 |
| GO:0006334 | Nucleosome assembly | 4 | 2 | 0.0219 |
| (ii) Preferentially expressed in erythrocytic stage (ring 4 h, trophozoite 16 h) | ||||
| GO:0006928 | Movement of cell or subcellular component | 34 | 6 | 0.00036 |
| GO:0044053 | Translocation of peptides or proteins into host cell cytoplasm | 3 | 2 | 0.00256 |
| GO:0040011 | Locomotion | 22 | 5 | 0.00644 |
| GO:0035891 | Exit from host cell | 8 | 2 | 0.02169 |
| GO:0009405 | Pathogenesis | 8 | 2 | 0.02169 |
| GO:0030833 | Regulation of actin filament polymerization | 1 | 1 | 0.02978 |
| GO:0006166 | Purine ribonucleoside salvage | 1 | 1 | 0.02978 |
| GO:0019510 | S-Adenosylhomocysteine catabolic process | 1 | 1 | 0.02978 |
| GO:0010323 | Negative regulation of isopentenyl diphosphate biosynthetic process, methylerythritol 4-phosphate pathway | 1 | 1 | 0.02978 |
| GO:0020035 | Cytoadherence to microvasculature, mediated by symbiont protein | 1 | 1 | 0.02978 |
| GO:0007050 | Cell cycle arrest | 1 | 1 | 0.02978 |
The number of annotated genes (number genes per GO term present in the entire RNA-seq study) and observed genes (number of genes per GO term preferentially expressed in…) per Biological Process are listed. Only GO terms with P-values < 0.05 are shown
Exo-erythrocytic and erythrocytic merozoites: same but different
In contrast to the limited differences in gene expression observed during parasite development in hepatocytes and RBC, the differences between first generation merozoites generated at the end of the liver stage and subsequent generation merozoites generated during the repeated rounds of blood stage development, were much more pronounced (i.e. see communities 1, 2, 4, 5, 10, 13 in Fig. 2) even though the 22 h EF schizont sample was not entirely pure but also contained some immature schizonts and immature gametocytes as stated in the corresponding publication [49]. Still, 880 and 1275 genes were preferentially expressed in DC s/merosomes and in 22 h schizonts, respectively (Fig. 5 and Additional file 1: Table S6). GO-term enrichment analysis (Table 3) indicated clear differences between these sets. Genes preferentially expressed in first generation merozoites (DCs/merosomes) were found enriched for the GO-terms: gene expression, ribosome biogenesis, amide biosynthetic process, RNA biosynthetic process and mRNA splicing. In contrast, genes preferentially expressed in subsequent generation merozoites (22 h EF schizonts) were enriched for the GO-terms: small GTPase-mediated signal transduction, DNA replication and recombination, protein localization and modification, and signal peptide processing. At first glance, it is rather surprising that merozoites derived from EEF and EF stage differ so markedly. However, it might reflect the fact that the mechanism of egress from their respective host cells differ markedly. EF stage-derived merozoites almost simultaneously rupture the PVM and the plasma membrane of the RBC [17]. On the other hand, EEF stage-derived merozoites are initially freed from the PVM but can then stay for several hours in the host cell until they are extruded inside merosomes into a blood vessel [7, 14] and are eventually released from merosomes in the fine capillaries of the lungs [16]. Also, DCs/merosomes do not form entirely synchronously. In vitro generation of detached cells starts as early as 54 h, peaks at 65 h but continues until 69 h and even later. Since for the current study DCs/merosomes were collected at 69 h to increase the yield, some merozoites might be at different developmental and activation stages and thus might express different sets of genes. However, the vast majority of 22 h EF schizonts and 69 h DCs/merosomes contain fully matured and infectious merozoites and thus are certainly the best defined stages to compare between blood and liver stages. The next section focuses in more detail on genes involved in the egress of EEF stage-derived merozoites in comparison to EF stage-derived merozoites.
Fig. 5.

Volcano plot of differently expressed genes of detached cells and merosomes compared to EF schizonts (sample 22 h). Positive LogFC values represent preferentially DCs/merosomes expressed genes. Negative LogFC values represent preferentially EF schizont expressed genes. The graph shows LogFC values relative to FDR (− Log10(adjusted P-value)
Table 3.
Gene ontology (GO) term annotation of genes preferentially expressed in (i) EEF merozoites and (ii) EF merozoites
| Biological process ID | Process description | Annotated | Observed | P value |
|---|---|---|---|---|
| (i) Preferentially expressed in exo-erythrocytic stage merozoites (DCs/merosomes) | ||||
| GO:0010467 | Gene expression | 462 | 181 | 3.0e−22 |
| GO:0042254 | Ribosome biogenesis | 63 | 47 | 4.6e−20 |
| GO:0043604 | Amide biosynthetic process | 219 | 98 | 1.1e−17 |
| GO:0032774 | RNA biosynthetic process | 103 | 35 | 0.00017 |
| GO:0017183 | Peptidyl-diphthamide biosynthetic process from peptidyl-histidine | 5 | 4 | 0.00700 |
| GO:0000398 | mRNA splicing, via spliceosome | 43 | 15 | 0.01718 |
| GO:0008295 | Spermidine biosynthetic process | 2 | 2 | 0.04105 |
| GO:0008612 | Peptidyl-lysine modification to peptidyl-hypusine | 2 | 2 | 0.04105 |
| (ii) Preferentially expressed in erythrocytic stage merozoites (22 h schizonts) | ||||
| GO:0007264 | Small gtpase mediated signal transduction | 26 | 14 | 0.0014 |
| GO:0006260 | DNA replication | 39 | 20 | 0.0049 |
| GO:0045184 | Establishment of protein localization | 93 | 34 | 0.0060 |
| GO:0007017 | Microtubule-based process | 35 | 15 | 0.0075 |
| GO:0000041 | Transition metal ion transport | 7 | 6 | 0.0151 |
| GO:0006465 | Signal peptide processing | 5 | 4 | 0.0153 |
| GO:0006848 | Pyruvate transport | 3 | 3 | 0.0154 |
| GO:0006497 | Protein lipidation | 21 | 10 | 0.0196 |
| GO:0006511 | Ubiquitin-dependent protein catabolic process | 38 | 16 | 0.0248 |
| GO:0043248 | Proteasome assembly | 8 | 5 | 0.0267 |
| GO:0006310 | DNA recombination | 9 | 6 | 0.0366 |
| GO:0006468 | Protein phosphorylation | 82 | 28 | 0.0437 |
The number of annotated genes (number genes per GO term present in the entire RNA-seq study) and observed genes (number of genes per GO term preferentially expressed in…) per Biological Process are listed. Only GO terms with P-values < 0.05 are shown
Comparison of mRNA expression patterns at the end of exo-erythrocytic and erythrocytic stage development
Given that EF merozoites egress within minutes upon PVM rupture, whereas EEF merozoites remain in the hepatocyte cytoplasm for up to several hours, genes were searched that might be (i) necessary for the rapid egress specifically from RBC, (ii) required for survival in the hepatocyte cytoplasm and (iii) specific for both developmental stages (i.e. necessary for PVM rupture in RBC and hepatocytes). Interestingly, 74 genes were upregulated in 22 h EF schizonts in comparison to EEF samples (LogFC ≥ 2, adjP ≤ 0.01, Additional file 1: Table S7), but only four of these were upregulated when compared to all other stages (Additional file 1: Table S8). These four genes code for the following proteins: (i) the high mobility group protein B1 (PBANKA_0601900) of which the orthologue in P. falciparum has been shown to potently activate pro-inflammatory cytokines, suggesting a role in triggering host inflammatory immune responses [71]; (ii) a protein of the PIR multigene family (PBANKA_0500781); the function of PIRs is not known but these proteins are believed to be involved in antigenic variation and evasion from host immune responses [72]; (iii) a conserved Plasmodium protein of unknown function (PBANKA_0915200); (iv) MSP9 (Merozoite surface protein 9, PBANKA_1443300). The orthologue of MSP9 in P. falciparum has been shown to bind to erythrocyte band 3 protein and to form a complex with MSP1 [73, 74]. Plasmodium falciparum merozoites are able to infect RBC via two different invasion mechanisms: one is sialic acid-dependent, involving MSP9 and the other one is MSP9 and sialic acid-independent. Interestingly msp9 is barely transcribed in DCs/merosomes and also hardly in other developmental EEF stages, which might be indicative that EEF stage-derived merozoites do not invade in the sialic acid-dependent manner. Attempts to knock out P. berghei MSP9 were not successful [75]. In P. yoelii, MSP9 has been found in the erythrocyte cytoplasm [76] and might, in addition to invasion, also be involved in egress of merozoites from RBC.
Thereafter, 293 genes were identified that are specifically upregulated in DCs/merosomes when compared to all other stages (Additional file 1: Table S9). It is conceivable that most of them are involved in the EEF stage-specific egress, in parasite survival in the dying host hepatocyte or in early RBC remodeling. As discussed earlier, it is plausible that transcripts are translationally suppressed in merozoites until the next stage in the life cycle, like has been described for mature gametocytes and sporozoites [51, 77]. Among the highly upregulated transcripts, sbp1 (skeleton binding protein 1) (PBANKA_1101300), mahrp1a and mahrp1b (PBANKA_1145800 and PBANKA_1145900) were identified. These gene products are all involved in the trafficking of exported proteins from the parasite to the surface of the RBC [65]. Knock out of MAHRP1a or SBP1 reduced the sequestration of infected cells [65]. It is plausible that EEF stage-derived merozoites express high levels of these mRNAs in order to sufficiently express these proteins immediately after infection of RBC. Plausibly, this may allow the infected RBC to be efficiently sequestered in the periphery to avoid immediate clearance by the spleen. Why expression of sbp1 and mahrp1a and mahrp1b appears to be lower in EF stage merozoites than in EEF stage merozoites is unknown. One reason might be, that in course of EF stage infection, the spleen is heavily remodeled [78–80] and allows for passage of infected RBC, making an efficient sequestration and thus a pronounced expression of sequestration ligands on the surface of the infected RBC less necessary [65]. The parasite might thus be less dependent on intense trafficking of sequestration ligands. Another reason might be that mahrp1a and sbp1 are already expressed during ring and trophozoite stage and thus less transcript would be needed to fulfill the same function as in EEF.
Genes predominantly expressed in exo-erythrocytic stages
One of the goals of this study was to identify EEF stage-specific expressed genes. The EEF data were therefore compared with data from all other stages. The comparison revealed 5 highly specifically expressed transcripts for EEF stages with a LogFC > 6 and adjP < 0.01 (Fig. 6). The genes lisp1 (PBANKA_1024600) and lisp2 (PBANKA_1003000) have been previously reported to be expressed exclusively during EEF stage development [80–82]. This is clearly reflected by the here presented RNA-seq analysis where substantial lisp1 and lisp2 mRNA levels were only found in EEF stages, from 24 hpi to DCs/merosomes. Along with lisp1 and lisp2 two conserved Plasmodium genes (PBANKA_0518900 and PBANKA_0519500) were identified, one of which is annotated as membrane protein, although there is no transmembrane domain other than the signal peptide (PBANKA_0518900). The fifth gene in this EEF-specific group of genes is PBANKA_1003900. An averaged logFC of PBANKA_1003900 from later stages compared to non-EEF stages was similarly high as for lisp2 and lisp1 (Table 4).
Fig. 6.

Expression profiles of the top 5 genes predominantly expressed in exo-erythrocytic stages compared to all other stages. Gene expression values corresponding to normalized and log2(x + 1)-transformed read counts. The data were normalized with DESeq 2 (with default parameters) [40]
Table 4.
LogFC and FDR (adjP) values of EEF-specific genes
| 48 h to DCs/merosomes compared to other stages | ||||
|---|---|---|---|---|
| Mean LogFC | Min adjP | Max adjP | ||
| Liver specific protein 2 (lisp2) | PBANKA_1003000 | 9.946 | 2.26E−228 | 1.22E−18 |
| Liver specific protein 3 (lisp3) | PBANKA_1003900 | 8.730 | 9.44E−133 | 2.17E−10 |
| Conserved Plasmodium membrane protein, unknown function | PBANKA_0518900 | 8.697 | 3.36E−175 | 9.55E−17 |
| Liver specific protein 1 (lisp1) | PBANKA_1024600 | 8.475 | 1.21E−189 | 1.24E−38 |
| Conserved Plasmodium protein, unknown function | PBANKA_0519500 | 7.035 | 7.81E−64 | 2.28E−21 |
LogFC is indicated as mean of EEF late stages by single comparisons to each other stage. FDR (adjP) values are presented as min and max values of the different comparisons
PBANKA_1003900 is a syntenic ortholog of P. falciparum sexual stage-specific protein precursor (Pfs16; PF3D7_0406200) which is expressed early during development of P. falciparum gametocytes [83]. Plasmodium berghei parasites expressing an mCherry-tagged PBANKA_1003900 provided experimental evidence that this gene is also expressed in gametocytes and was, therefore, annotated as a gametocyte specific protein [84]. However, substantial PBANKA_1003900 transcript levels were only detected in EEF stages. A transgenic parasite line expressing GFP under the control of the promoter of PBANKA_1003900 (PBANKA_1003900GFP) was generated and analysed for GFP expression by fluorescence microscopy in the different EEF and EF stages. GFP was not detectable in any of the EF stages, including gametocytes. In contrast, GFP expression was detected by fluorescence microscopy of infected HeLa cells fixed at different time points (Fig. 7; 24 h, 48 h, 54 h, 60 h). At 24 h of EEF stage development no or only very weak GFP-fluorescence was detectable. At 48 h of EEF stage development, the signal was readily visible and at 54 h and 60 h post infection the fluorescent signal was profoundly intense. When performing live cell imaging, the first signals were observed at 30 h post infection (Additional file 3: Movie S1, starting from 30 hpi). From 45 h onwards the protein was substantially expressed confirming the results obtained from fixed cells. These fluorescence patterns (fixed and live) nicely confirmed the RNA-seq data during EEF stage development. Interestingly, analyses of gene-deletion mutants lacking PBANKA_1003900 demonstrated that the gene is not essential at any developmental stage throughout the complete parasite life cycle ([84] and own unpublished data). According to the expression profile deduced from the RNA-seq analysis and also confirmed by the promoter analyses, it is appropriate to revise the annotation as “gametocyte specific protein” and to rename it as “liver specific protein 3 (LISP3)”.
Fig. 7.
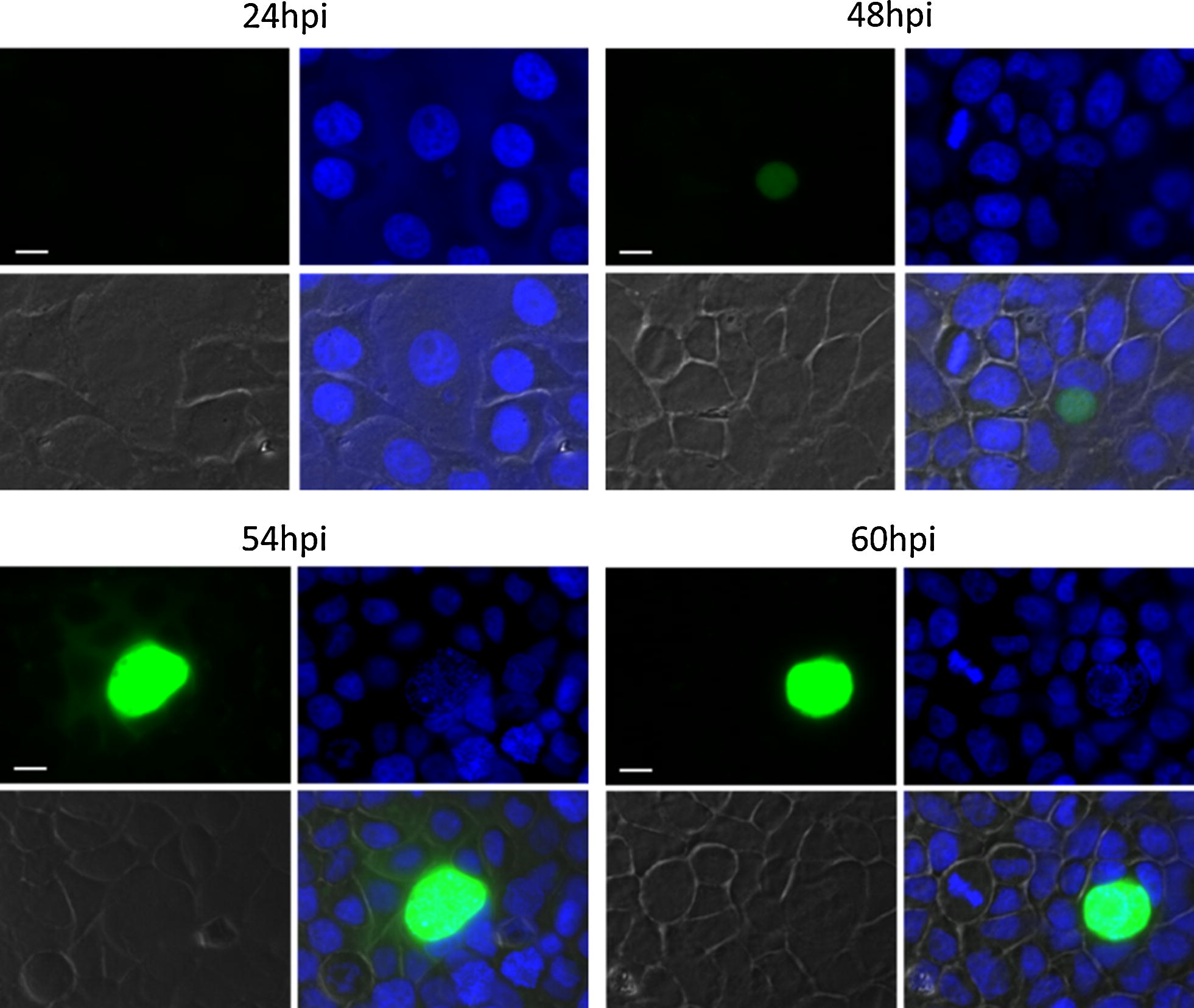
GFP expression during EEF stage development of the transgenic line 300 expressing GFP under control of the promoter region of PBANKA_1003900 (PBANKA_1003900GFP). Infected HeLa cells were fixed with 4% PFA/PBS at indicated times post infection with sporozoites. Nuclei were stained with Hoechst. Microscopy-settings (e.g. exposure time) were kept the same for all samples (bar 10 µm)
Conclusion
This study presents a time-course transcriptomic assessment of P. berghei liver stage development with emphasis on late stage EEF stages and an extended comparative gene transcription analysis including published transcriptomic data from erythrocytic stages (sexual and asexual), as well as the ookinete stage. This offers a comprehensive overview of gene transcription throughout most of the life cycle and allows a better understanding of gene regulation in different life cycle stages. In particular, the transcriptome of these different life cycle stages provides an invaluable tool for systems biology approaches to model metabolic pathways that are essential in different steps of the Plasmodium life cycle. In a first attempt to analyse transcription profiles during the Plasmodium life cycle, almost 4700 genes were segemented into 14 distinct communities based on their different expression profile and attributed gene ontology terms to the individual communities. A more detailed analysis of the promoter regions of the genes in the communities with similar expression profiles could help identifying common DNA domains that might support the further understanding of regulation of gene expression in Plasmodium. The RNA-seq data provided here are of considerable interest for fundamental research questions with respect to the parasite biology as well as for applied research at the identification of new protein targets for vaccine and drug development.
Supplementary information
Additional file 1: Table S1. RNA-seq data of different life cycle stages of P. berghei used in this study. Table S2. RNA-seq analysis: Raw sequencing counts per gene in the different life cycle stages. Table S3. Gene ontology (GO) term annotation of genes of the individual communities in the GCN. Table S4. P. berghei geneIDs of genes of the different communities in the GCN. Table S5. Genes preferentially expressed in developing EEF stages compared to developing EF stages. Table S6. Genes preferentially expressed in detached cells (DCs/merosomes) compared to erythrocytic schizonts. Table S7. Genes preferentially expressed in blood schizonts (22 h) compared to EEF stages. Table S8. Genes preferentially expressed in blood schizonts (22 h) compared to all other stages. Table S9. Genes preferentially expressed in detached cells (DCs/merosomes) compared to all other stages.
Additional file 2: Figure S1. Generation and genotyping of parasites expressing gfp under control of the promoter of PBANKA_1003900 (PBANKA_1003900GFP). Figure S2. Fluorescence-activated cell sorting of infected HeLa cells preserved in RNAlater. Figure S3. RNA expression profiles of 3 housekeeping genes (gapdh, actinI, tubulin1). Figure S4. RNA expression profiles of 5 genes encoding serine-repeat antigens, serine-type proteases (SERA1-5). Figure S5. RNA expression profiles of 5 genes encoding proteins of the parasitophorous vacuole membrane (Exported protein 1, Exported protein 2, UIS3, UIS4). Figure S6. RNA expression profiles of genes encoding 2 sporozoite surface proteins (CSP and TRAP). Figure S7. RNA expression profiles of 4 genes encoding enzymes involved in fatty acid biosynthesis (FabB/F, FabI, FabZ, FabG). Figure S8. RNA expression profiles of 6 genes encoding merozoite surface proteins (MSP). Figure S9. RNA expression profiles of 3 genes whose promoter regions have been used to drive expression of fluorescent/luminescent reporter proteins (HSP70, two genes for EF1α).
Additional file 3. Time laps: GFP expression under the control of PBANKA_1003900 promoter during EEF stage development.
Acknowledgements
FACS sorting was performed at the Cytometry Laboratory Core facility of the University of Bern; we are thankful for the kind support by Stefan Müller. RNA-sequencing was performed at the iGE3 genomics platform of the University of Geneva.
Authors’ contributions
VH and DSF conceived and coordinated the study, VH and RC designed the experiments; RC, SD performed the experiments; MWS, RC and SD conducted data analysis; BFF and CJJ generated reporter parasite lines; RC and MWS prepared figures; VH and RC wrote the original draft; DSF, CJJ, VH, RC, BFF, MWS, SD reviewed and edited the paper. All authors read and approved the final manuscript.
Funding
DSF and VH were supported by the RTD Grant MalarX (Grant Number: 51RTPO_151032), within SystemsX.ch (the Swiss Initiative for Systems Biology http://www.systemsx.ch/projects/research-technology-and-development-projects/malarx/).
Availability of data
Short reads generated in this study were deposited at the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) and are accessible through the accession number PRJEB23770 (Secondary study accession number: ERP105548).
Ethics approval and consent to participate
Experiments were performed in accordance with the guidelines of the Swiss Act on animal protection (TSchG) and approved by the animal experimentation commission of the canton Bern (authorization numbers BE109/13 and BE132/16). The generation of the parasite line expressing GFP under the promoter of PBANKA_1003900 was approved by the Animal Experiments Committee of the Leiden University Medical Center (DEC 12042). The Dutch Experiments on Animal Act is established under European guidelines (EU directive no. 86/609/EEC regarding the Protection of Animals used for Experimental and Other Scientific Purposes).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Reto Caldelari, Email: reto.caldelari@izb.unibe.ch.
Volker Heussler, Email: volker.heussler@izb.unibe.ch.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12936-019-2968-7.
References
- 1.WHO . World malaria report 2016. Geneva: World Health Organization; 2017. [Google Scholar]
- 2.Douglas RG, Amino R, Sinnis P, Frischknecht F. Active migration and passive transport of malaria parasites. Trends Parasitol. 2015;31:357–362. doi: 10.1016/j.pt.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spielmann T, Montagna GN, Hecht L, Matuschewski K. Molecular make-up of the Plasmodium parasitophorous vacuolar membrane. Int J Med Microbiol. 2012;302:179–186. doi: 10.1016/j.ijmm.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Kaiser G, De Niz M, Zuber B, Burda P-C, Kornmann B, Heussler VT, et al. High resolution microscopy reveals an unusual architecture of the Plasmodium berghei endoplasmic reticulum. Mol Microbiol. 2016;102:775–791. doi: 10.1111/mmi.13490. [DOI] [PubMed] [Google Scholar]
- 5.Stanway RR, Mueller N, Zobiak B, Graewe S, Froehlke U, Zessin PJM, et al. Organelle segregation into Plasmodium liver stage merozoites. Cell Microbiol. 2011;13:1768–1782. doi: 10.1111/j.1462-5822.2011.01657.x. [DOI] [PubMed] [Google Scholar]
- 6.Burda PC, Schaffner M, Kaiser G, Roques M, Zuber B, Heussler VT. A Plasmodium plasma membrane reporter reveals membrane dynamics by live-cell microscopy. Sci Rep. 2017;7:9740. doi: 10.1038/s41598-017-09569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burda P-C, Roelli MA, Schaffner M, Khan SM, Janse CJ, Heussler VT. A Plasmodium phospholipase is involved in disruption of the liver stage parasitophorous vacuole membrane. PLoS Pathog. 2015;11:e1004760. doi: 10.1371/journal.ppat.1004760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tawk L, Lacroix C, Gueirard P, Kent R, Gorgette O, Thiberge S, et al. A key role for Plasmodium subtilisin-like SUB1 protease in egress of malaria parasites from host hepatocytes. J Biol Chem. 2013;288:33336–33346. doi: 10.1074/jbc.M113.513234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suarez C, Volkmann K, Gomes AR, Billker O, Blackman MJ. The malarial serine protease SUB1 plays an essential role in parasite liver stage development. PLoS Pathog. 2013;9:e1003811. doi: 10.1371/journal.ppat.1003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deligianni E, Morgan RN, Bertuccini L, Wirth CC, de Monerri NC, Spanos L, et al. A perforin-like protein mediates disruption of the erythrocyte membrane during egress of Plasmodium berghei male gametocytes. Cell Microbiol. 2013;15:1438–1455. doi: 10.1111/cmi.12131. [DOI] [PubMed] [Google Scholar]
- 11.Garg S, Agarwal S, Kumar S, Shams Yazdani S, Chitnis CE, Singh S. Calcium-dependent permeabilization of erythrocytes by a perforin-like protein during egress of malaria parasites. Nat Commun. 2013;4:1736. doi: 10.1038/ncomms2725. [DOI] [PubMed] [Google Scholar]
- 12.Wirth CC, Glushakova S, Scheuermayer M, Repnik U, Garg S, Schaack D, et al. Perforin-like protein PPLP2 permeabilizes the red blood cell membrane during egress of Plasmodium falciparum gametocytes. Cell Microbiol. 2014;16:709–733. doi: 10.1111/cmi.12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burda P-C, Caldelari R, Heussler VT. Manipulation of the host cell membrane during Plasmodium liver stage egress. MBio. 2017;8:e00139. doi: 10.1128/mBio.00139-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, Rennenberg A, et al. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science. 2006;313:1287–1290. doi: 10.1126/science.1129720. [DOI] [PubMed] [Google Scholar]
- 15.Graewe S, Stanway RR, Rennenberg A, Heussler VT. Chronicle of a death foretold: Plasmodium liver stage parasites decide on the fate of the host cell. FEMS Microbiol Rev. 2012;36:111–130. doi: 10.1111/j.1574-6976.2011.00297.x. [DOI] [PubMed] [Google Scholar]
- 16.Baer K, Klotz C, Kappe SHI, Schnieder T, Frevert U. Release of hepatic Plasmodium yoelii merozoites into the pulmonary microvasculature. PLoS Pathog. 2007;3:e171. doi: 10.1371/journal.ppat.0030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glushakova S, Yin D, Li T, Zimmerberg J. Membrane transformation during malaria parasite release from human red blood cells. Curr Biol. 2005;15:1645–1650. doi: 10.1016/j.cub.2005.07.067. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh AK, Jacobs-Lorena M. Plasmodium sporozoite invasion of the mosquito salivary gland. Curr Opin Microbiol. 2009;12:394–400. doi: 10.1016/j.mib.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janse CJ, Ramesar J, Waters AP. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc. 2006;1:346–356. doi: 10.1038/nprot.2006.53. [DOI] [PubMed] [Google Scholar]
- 20.Philip N, Orr R, Waters AP. Transfection of rodent malaria parasites. Methods Mol Biol. 2013;923:99–125. doi: 10.1007/978-1-62703-026-7_7. [DOI] [PubMed] [Google Scholar]
- 21.Kaiser G, De Niz M, Burda P-C, Niklaus L, Stanway RL, Heussler V. Generation of transgenic rodent malaria parasites by transfection of cell culture-derived merozoites. Malar J. 2017;16:305. doi: 10.1186/s12936-017-1949-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37:D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poran A, Nötzel C, Aly O, Mencia-Trinchant N, Harris CT, Guzman ML, et al. Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature. 2017;551:95–99. doi: 10.1038/nature24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reid AJ, Talman AM, Bennett HM, Gomes AR, Sanders MJ, Illingworth CJR, et al. Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites. Elife. 2018;7:e33105. doi: 10.7554/eLife.33105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howick VM, Russell A, Andrews T, Heaton H, Reid AJ, Natarajan KN, et al. The Malaria Cell Atlas: a comprehensive reference of single parasite transcriptomes across the complete Plasmodium life cycle. bioRxiv. 2019;527:556. doi: 10.1126/science.aaw2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Posfai D, Sylvester K, Reddy A, Ganley JG, Wirth J, Cullen QE, et al. Plasmodium parasite exploits host aquaporin-3 during liver stage malaria infection. PLoS Pathog. 2018;14:e1007057. doi: 10.1371/journal.ppat.1007057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaMonte GM, Orjuela-Sanchez P, Calla J, Wang LT, Li S, Swann J, et al. Dual RNA-seq identifies human mucosal immunity protein Mucin-13 as a hallmark of Plasmodium exoerythrocytic infection. Nat Commun. 2019;10:488. doi: 10.1038/s41467-019-08349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tarun AS, Peng X, Dumpit RF, Ogata Y, Silva-Rivera H, Camargo N, et al. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc Natl Acad Sci USA. 2008;105:305–310. doi: 10.1073/pnas.0710780104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gural N, Mancio-Silva L, Miller AB, Galstian A, Butty VL, Levine SS, et al. In vitro culture, drug sensitivity, and transcriptome of Plasmodium vivax hypnozoites. Cell Host Microbe. 2018;23:395–406. doi: 10.1016/j.chom.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voorberg-van der Wel A, Roma G, Gupta DK, Schuierer S, Nigsch F, Carbone W, et al. A comparative transcriptomic analysis of replicating and dormant liver stages of the relapsing malaria parasite Plasmodium cynomolgi. Elife. 2017;6:e29605. doi: 10.7554/eLife.29605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaitoun I, Erickson CS, Schell K, Epstein ML. Use of RNAlater in fluorescence-activated cell sorting (FACS) reduces the fluorescence from GFP but not from DsRed. BMC Res Notes. 2010;3:328. doi: 10.1186/1756-0500-3-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franke-Fayard B, Trueman H, Ramesar J, Mendoza J, van der Keur M, van der Linden R, et al. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pertea G. fqtrim: v0.9.4 release. 2015.
- 35.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid MW, Grossniklaus U. Rcount: simple and flexible RNA-Seq read counting. Bioinformatics. 2015;31:436–437. doi: 10.1093/bioinformatics/btu680. [DOI] [PubMed] [Google Scholar]
- 38.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 39.Schmid MW, Schmidt A, Klostermeier UC, Barann M, Rosenstiel P, Grossniklaus U. A powerful method for transcriptional profiling of specific cell types in eukaryotes: laser-assisted microdissection and RNA sequencing. PLoS ONE. 2012;7:e29685. doi: 10.1371/journal.pone.0029685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 42.Coman D, Rütimann P, Gruissem W. A flexible protocol for targeted gene co-expression network analysis. Methods Mol Biol. 2014;1153:285–299. doi: 10.1007/978-1-4939-0606-2_21. [DOI] [PubMed] [Google Scholar]
- 43.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech Theory Exp. 2008. p. 10008.
- 44.Csardi G, Nepusz T. The Igraph software package for complex network research. InterJournal Complex Systems. 2006;1695:1–9. [Google Scholar]
- 45.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 47.Bozdech Z, Llinás M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:e5. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishimoto KP, Newkirk D, Hou S, Fruehauf J, Nelson EL. Fluorescence activated cell sorting (FACS) using RNAlater to minimize RNA degradation and perturbation of mRNA expression from cells involved in initial host microbe interactions. J Microbiol Methods. 2007;70:205–208. doi: 10.1016/j.mimet.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 49.Otto TD, Böhme U, Jackson AP, Hunt M, Franke-Fayard B, Hoeijmakers WAM, et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol. 2014;12:86. doi: 10.1186/s12915-014-0086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lasonder E, Rijpma SR, van Schaijk BCL, Hoeijmakers WAMAM, Kensche PR, Gresnigt MS, et al. Integrated transcriptomic and proteomic analyses of P. falciparum gametocytes: molecular insight into sex-specific processes and translational repression. Nucleic Acids Res. 2016;44:6087–6101. doi: 10.1093/nar/gkw536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mair GR, Braks JAM, Garver LS, Dimopoulos G, Hall N, Wiegant JCAG, et al. Translational repression is essential for Plasmodium sexual development and mediated by a DDX6-type RNA helicase. Science. 2006;313:667–669. doi: 10.1126/science.1125129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mair GR, Lasonder E, Garver LS, Franke-Fayard BMD, Carret CK, Wiegant JCAG, et al. Universal features of post-transcriptional gene regulation are critical for Plasmodium zygote development. PLoS Pathog. 2010;6:e1000767. doi: 10.1371/journal.ppat.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hliscs M, Nahar C, Frischknecht F, Matuschewski K. Expression profiling of Plasmodium berghei HSP70 genes for generation of bright red fluorescent parasites. PLoS ONE. 2013;8:e72771. doi: 10.1371/journal.pone.0072771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saeed S, Carter V, Tremp AZ, Dessens JT. Translational repression controls temporal expression of the Plasmodium berghei LCCL protein complex. Mol Biochem Parasitol. 2013;189:38–42. doi: 10.1016/j.molbiopara.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao PN, Santos JM, Pain A, Templeton TJ, Mair GR. Translational repression of the cpw-wpc gene family in the malaria parasite Plasmodium. Parasitol Int. 2016;65:463–471. doi: 10.1016/j.parint.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 56.Zhang M, Fennell C, Ranford-Cartwright L, Sakthivel R, Gueirard P, Meister S, et al. The Plasmodium eukaryotic initiation factor-2α kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. J Exp Med. 2010;207:1465–1474. doi: 10.1084/jem.20091975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Silvie O, Briquet S, Müller K, Manzoni G, Matuschewski K. Post-transcriptional silencing of UIS4 in Plasmodium berghei sporozoites is important for host switch. Mol Microbiol. 2014;91:1200–1213. doi: 10.1111/mmi.12528. [DOI] [PubMed] [Google Scholar]
- 58.Zhang M, Joyce BR, Sullivan WJ, Nussenzweig V. Translational control in Plasmodium and Toxoplasma parasites. Eukaryot Cell. 2013;12:161–167. doi: 10.1128/EC.00296-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109:145–148. doi: 10.1016/S0092-8674(02)00718-3. [DOI] [PubMed] [Google Scholar]
- 60.Fougère A, Jackson AP, Paraskevi Bechtsi D, Braks JAM, Annoura T, Fonager J, et al. Variant exported blood-stage proteins encoded by Plasmodium multigene families are expressed in liver stages where they are exported into the parasitophorous vacuole. PLoS Pathog. 2016;12:1–37. doi: 10.1371/journal.ppat.1005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarun AS, Vaughan AM, Kappe SHI. Redefining the role of de novo fatty acid synthesis in Plasmodium parasites. Trends Parasitol. 2009;25:545–550. doi: 10.1016/j.pt.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 62.Yu M, Kumar TRS, Nkrumah LJ, Coppi A, Retzlaff S, Li CD, et al. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe. 2008;4:567–578. doi: 10.1016/j.chom.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vaughan AM, O’Neill MT, Tarun AS, Camargo N, Phuong TM, Aly ASI, et al. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol. 2009;11:506–520. doi: 10.1111/j.1462-5822.2008.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shears MJ, Botté CY, McFadden GI. Fatty acid metabolism in the Plasmodium apicoplast: drugs, doubts and knockouts. Mol Biochem Parasitol. 2015;199:34–50. doi: 10.1016/j.molbiopara.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 65.De Niz M, Ullrich A-K, Heiber A, Soares AB, Pick C, Lyck R, et al. The machinery underlying malaria parasite virulence is conserved between rodent and human malaria parasites. Nat Commun. 2016;7:11659. doi: 10.1038/ncomms11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fonager J, Pasini EM, Braks JAM, Klop O, Ramesar J, Remarque EJ, et al. Reduced CD36-dependent tissue sequestration of Plasmodium-infected erythrocytes is detrimental to malaria parasite growth in vivo. J Exp Med. 2012;209:93–107. doi: 10.1084/jem.20110762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pasini EM, Braks JA, Fonager J, Klop O, Aime E, Spaccapelo R, et al. Proteomic and genetic analyses demonstrate that Plasmodium berghei blood stages export a large and diverse repertoire of proteins. Mol Cell Proteomics. 2013;12:426–448. doi: 10.1074/mcp.M112.021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hügel FU, Pradel G, Frevert U. Release of malaria circumsporozoite protein into the host cell cytoplasm and interaction with ribosomes. Mol Biochem Parasitol. 1996;81:151–170. doi: 10.1016/0166-6851(96)02701-6. [DOI] [PubMed] [Google Scholar]
- 69.Hollingdale MR, Leland P, Leef JL, Leef MF, Beaudoin RL. Serological reactivity of in vitro cultured exoerythrocytic stages of Plasmodium berghei in indirect immunofluorescent or immunoperoxidase antibody tests. Am J Trop Med Hyg. 1983;32:24–30. doi: 10.4269/ajtmh.1983.32.24. [DOI] [PubMed] [Google Scholar]
- 70.Orito Y, Ishino T, Iwanaga S, Kaneko I, Kato T, Menard R, et al. Liver-specific protein 2: a Plasmodium protein exported to the hepatocyte cytoplasm and required for merozoite formation. Mol Microbiol. 2013;87:66–79. doi: 10.1111/mmi.12083. [DOI] [PubMed] [Google Scholar]
- 71.Kumar K, Singal A, Rizvi MMA, Chauhan VS. High mobility group box (HMGB) proteins of Plasmodium falciparum: DNA binding proteins with pro-inflammatory activity. Parasitol Int. 2008;57:150–157. doi: 10.1016/j.parint.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 72.Cunningham D, Lawton J, Jarra W, Preiser P, Langhorne J. The pir multigene family of Plasmodium: antigenic variation and beyond. Mol Biochem Parasitol. 2010;170:65–73. doi: 10.1016/j.molbiopara.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 73.Li X, Chen H, Oo TH, Daly TM, Bergman LW, Liu S-C, et al. A co-ligand complex anchors Plasmodium falciparum merozoites to the erythrocyte invasion receptor band 3. J Biol Chem. 2004;279:5765–5771. doi: 10.1074/jbc.M308716200. [DOI] [PubMed] [Google Scholar]
- 74.Kariuki MM, Li X, Yamodo I, Chishti AH, Oh SS. Two Plasmodium falciparum merozoite proteins binding to erythrocyte band 3 form a direct complex. Biochem Biophys Res Commun. 2005;338:1690–1695. doi: 10.1016/j.bbrc.2005.10.154. [DOI] [PubMed] [Google Scholar]
- 75.Bushell E, Gomes AR, Sanderson T, Anar B, Girling G, Herd C, et al. Functional profiling of a Plasmodium genome reveals an abundance of essential genes. Cell. 2017;170(260–272):e8. doi: 10.1016/j.cell.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Siau A, Huang X, Weng M, Sze SK, Preiser PR. Proteome mapping of Plasmodium: identification of the P. yoelii remodellome. Sci Rep. 2016;6:31055. doi: 10.1038/srep31055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cui L, Lindner S, Miao J. Translational regulation during stage transitions in malaria parasites. Ann N Y Acad Sci. 2015;1342:1–9. doi: 10.1111/nyas.12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin-Jaular L, Ferrer M, Calvo M, Rosanas-Urgell A, Kalko S, Graewe S, et al. Strain-specific spleen remodelling in Plasmodium yoelii infections in Balb/c mice facilitates adherence and spleen macrophage-clearance escape. Cell Microbiol. 2011;13:109–122. doi: 10.1111/j.1462-5822.2010.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.del Portillo HA, Ferrer M, Brugat T, Martin-Jaular L, Langhorne J, Lacerda MVG. The role of the spleen in malaria. Cell Microbiol. 2012;14:343–355. doi: 10.1111/j.1462-5822.2011.01741.x. [DOI] [PubMed] [Google Scholar]
- 80.De Niz M, Helm S, Horstmann S, Annoura T, Del Portillo HA, Khan SM, et al. In vivo and in vitro characterization of a Plasmodium liver stage-specific promoter. PLoS ONE. 2015;10:e0123473. doi: 10.1371/journal.pone.0123473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishino T, Boisson B, Orito Y, Lacroix C, Bischoff E, Loussert C, et al. LISP1 is important for the egress of Plasmodium berghei parasites from liver cells. Cell Microbiol. 2009;11:1329–1339. doi: 10.1111/j.1462-5822.2009.01333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Helm S, Lehmann C, Nagel A, Stanway RR, Horstmann S, Llinas M, et al. Identification and characterization of a liver stage-specific promoter region of the malaria parasite Plasmodium. PLoS ONE. 2010;5:e13653. doi: 10.1371/journal.pone.0013653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dechering KJ, Thompson J, Dodemont HJ, Eling W, Konings RN. Developmentally regulated expression of pfs16, a marker for sexual differentiation of the human malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 1997;89:235–244. doi: 10.1016/S0166-6851(97)00123-0. [DOI] [PubMed] [Google Scholar]
- 84.Deligianni E, Andreadaki M, Koutsouris K, Siden-Kiamos I. Sequence and functional divergence of gametocyte-specific parasitophorous vacuole membrane proteins in Plasmodium parasites. Mol Biochem Parasitol. 2018;220:15–18. doi: 10.1016/j.molbiopara.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 85.Yeoh LM, Goodman CD, Mollard V, McFadden GI, Ralph SA. Comparative transcriptomics of female and male gametocytes in Plasmodium berghei and the evolution of sex in alveolates. BMC Genomics. 2017;18:734. doi: 10.1186/s12864-017-4100-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Spence PJ, Jarra W, Lévy P, Reid AJ, Chappell L, Brugat T, et al. Vector transmission regulates immune control of Plasmodium virulence. Nature. 2013;498:228–231. doi: 10.1038/nature12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lindner SE, Mikolajczak SA, Vaughan AM, Moon W, Joyce BR, Sullivan WJ, et al. Perturbations of Plasmodium Puf2 expression and RNA-seq of Puf2-deficient sporozoites reveal a critical role in maintaining RNA homeostasis and parasite transmissibility. Cell Microbiol. 2013;15:1266. doi: 10.1111/cmi.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vignali M, Armour CD, Chen J, Morrison R, Castle JC, Biery MC, et al. NSR-seq transcriptional profiling enables identification of a gene signature of Plasmodium falciparum parasites infecting children. J Clin Invest. 2011;121:1119–1129. doi: 10.1172/JCI43457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bunnik EM, Chung D-WD, Hamilton M, Ponts N, Saraf A, Prudhomme J, et al. Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum. Genome Biol. 2013;14:R128. doi: 10.1186/gb-2013-14-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Otto TD, Wilinski D, Assefa S, Keane TM, Sarry LR, Böhme U, et al. New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol Microbiol. 2010;76:12–24. doi: 10.1111/j.1365-2958.2009.07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.López-Barragán MJ, Lemieux J, Quiñones M, Williamson KC, Molina-Cruz A, Cui K, et al. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics. 2011;12:587. doi: 10.1186/1471-2164-12-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bártfai R, Hoeijmakers WA, Salcedo-Amaya AM, Smits AH, Janssen-Megens E, Kaan A, et al. H2A.Z demarcates intergenic regions of the Plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLoS Pathog. 2010;6:e1001223. doi: 10.1371/journal.ppat.1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Siegel T, Hon C-C, Zhang Q, Lopez-Rubio J-J, Scheidig-Benatar C, Martins RM, et al. Strand-specific RNA-Seq reveals widespread and developmentally regulated transcription of natural antisense transcripts in Plasmodium falciparum. BMC Genomics. 2014;15:150. doi: 10.1186/1471-2164-15-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Caro F, Ahyong V, Betegon M, DeRisi JL. Genome-wide regulatory dynamics of translation in the Plasmodium falciparum asexual blood stages. Elife. 2014;3:04106. doi: 10.7554/eLife.04106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zanghì G, Vembar SS, Baumgarten S, Ding S, Guizetti J, Bryant JM, et al. A specific PfEMP1 is expressed in P. falciparum sporozoites and plays a role in hepatocyte infection. Cell Rep. 2018;22:2951–2963. doi: 10.1016/j.celrep.2018.02.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu L, Mok S, Imwong M, Jaidee A, Russell B, Nosten F, et al. New insights into the Plasmodium vivax transcriptome using RNA-Seq. Sci Rep. 2016;6:20498. doi: 10.1038/srep20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Joyner C, Moreno A, Meyer EVS, Cabrera-Mora M, Kissinger JC, Barnwell JW, et al. Plasmodium cynomolgi infections in rhesus macaques display clinical and parasitological features pertinent to modelling vivax malaria pathology and relapse infections. Malar J. 2016;15:451. doi: 10.1186/s12936-016-1480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cubi R, Vembar SS, Biton A, Franetich J-F, Bordessoulles M, Sossau D, et al. Laser capture microdissection enables transcriptomic analysis of dividing and quiescent liver stages of Plasmodium relapsing species. Cell Microbiol. 2017;19:e12735. doi: 10.1111/cmi.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hall N, Karras M, Raine JD, Carlton JM, Kooij TWA, Berriman M, et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005;307:82–86. doi: 10.1126/science.1103717. [DOI] [PubMed] [Google Scholar]
- 100.Yuda M, Iwanaga S, Kaneko I, Kato T. Global transcriptional repression: an initial and essential step for Plasmodium sexual development. Proc Natl Acad Sci USA. 2015;112:12824–12829. doi: 10.1073/pnas.1504389112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhou Y, Ramachandran V, Kumar KA, Westenberger S, Refour P, Zhou B, et al. Evidence-based annotation of the malaria parasite’s genome using comparative expression profiling. PLoS One. 2008;3:e1570. doi: 10.1371/journal.pone.0001570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stubbs J, Simpson KM, Triglia T, Plouffe D, Tonkin CJ, Duraisingh MT, et al. Molecular mechanism for switching of P. falciparum invasion pathways into human erythrocytes. Science. 2005;309:1384–1387. doi: 10.1126/science.1115257. [DOI] [PubMed] [Google Scholar]
- 103.Jiang H, Patel JJ, Yi M, Mu J, Ding J, Stephens R, et al. Genome-wide compensatory changes accompany drug- selected mutations in the Plasmodium falciparum crt gene. PLoS One. 2008;3:e2484. doi: 10.1371/journal.pone.0002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Le Roch KG, Zhou Y, Blair PL, Grainger M, Moch JK, Haynes JD, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301:1503–1508. doi: 10.1126/science.1087025. [DOI] [PubMed] [Google Scholar]
- 105.Pelle KG, Oh K, Buchholz K, Narasimhan V, Joice R, Milner DA, et al. Transcriptional profiling defines dynamics of parasite tissue sequestration during malaria infection. Genome Med. 2015;7:19. doi: 10.1186/s13073-015-0133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Young JA, Fivelman QL, Blair PL, de la Vega P, Le Roch KG, Zhou Y, et al. The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology-based pattern identification. Mol Biochem Parasitol. 2005;143:67–79. doi: 10.1016/j.molbiopara.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 107.Tonkin CJ, Carret CK, Duraisingh MT, Voss TS, Ralph SA, Hommel M, et al. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009;7:e84. doi: 10.1371/journal.pbio.1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rovira-Graells N, Gupta AP, Planet E, Crowley VM, Mok S, Ribas de Pouplana L, et al. Transcriptional variation in the malaria parasite Plasmodium falciparum. Genome Res. 2012;22:925–938. doi: 10.1101/gr.129692.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bozdech Z, Llinás M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003;1:85–100. doi: 10.1371/journal.pbio.0000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Llinás M, Bozdech Z, Wong ED, Adai AT, DeRisi JL. Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Res. 2006;34:1166–1173. doi: 10.1093/nar/gkj517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lemieux JE, Gomez-Escobar N, Feller A, Carret C, Amambua-Ngwa A, Pinches R, et al. Statistical estimation of cell-cycle progression and lineage commitment in Plasmodium falciparum reveals a homogeneous pattern of transcription in ex vivo culture. Proc Natl Acad Sci USA. 2009;106:7559–7564. doi: 10.1073/pnas.0811829106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shock JL, Fischer KF, DeRisi JL. Whole-genome analysis of mRNA decay in Plasmodium falciparum reveals a global lengthening of mRNA half-life during the intra-erythrocytic development cycle. Genome Biol. 2007;8:R134. doi: 10.1186/gb-2007-8-7-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bozdech Z, Mok S, Hu G, Imwong M, Jaidee A, Russell B, et al. The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc Natl Acad Sci USA. 2008;105:16290–16295. doi: 10.1073/pnas.0807404105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Westenberger SJ, McClean CM, Chattopadhyay R, Dharia NV, Carlton JM, Barnwell JW, et al. A systems-based analysis of Plasmodium vivax life cycle transcription from human to mosquito. PLoS Negl Trop Dis. 2010;4:e653. doi: 10.1371/journal.pntd.0000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lapp SA, Mok S, Zhu L, Wu H, Preiser PR, Bozdech Z, et al. Plasmodium knowlesi gene expression differs in ex vivo compared to in vitro blood-stage cultures. Malar J. 2015;14:110. doi: 10.1186/s12936-015-0612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, et al. gplots: various R programming tools for plotting data. Version 3.0.1. 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. RNA-seq data of different life cycle stages of P. berghei used in this study. Table S2. RNA-seq analysis: Raw sequencing counts per gene in the different life cycle stages. Table S3. Gene ontology (GO) term annotation of genes of the individual communities in the GCN. Table S4. P. berghei geneIDs of genes of the different communities in the GCN. Table S5. Genes preferentially expressed in developing EEF stages compared to developing EF stages. Table S6. Genes preferentially expressed in detached cells (DCs/merosomes) compared to erythrocytic schizonts. Table S7. Genes preferentially expressed in blood schizonts (22 h) compared to EEF stages. Table S8. Genes preferentially expressed in blood schizonts (22 h) compared to all other stages. Table S9. Genes preferentially expressed in detached cells (DCs/merosomes) compared to all other stages.
Additional file 2: Figure S1. Generation and genotyping of parasites expressing gfp under control of the promoter of PBANKA_1003900 (PBANKA_1003900GFP). Figure S2. Fluorescence-activated cell sorting of infected HeLa cells preserved in RNAlater. Figure S3. RNA expression profiles of 3 housekeeping genes (gapdh, actinI, tubulin1). Figure S4. RNA expression profiles of 5 genes encoding serine-repeat antigens, serine-type proteases (SERA1-5). Figure S5. RNA expression profiles of 5 genes encoding proteins of the parasitophorous vacuole membrane (Exported protein 1, Exported protein 2, UIS3, UIS4). Figure S6. RNA expression profiles of genes encoding 2 sporozoite surface proteins (CSP and TRAP). Figure S7. RNA expression profiles of 4 genes encoding enzymes involved in fatty acid biosynthesis (FabB/F, FabI, FabZ, FabG). Figure S8. RNA expression profiles of 6 genes encoding merozoite surface proteins (MSP). Figure S9. RNA expression profiles of 3 genes whose promoter regions have been used to drive expression of fluorescent/luminescent reporter proteins (HSP70, two genes for EF1α).
Additional file 3. Time laps: GFP expression under the control of PBANKA_1003900 promoter during EEF stage development.
Data Availability Statement
Short reads generated in this study were deposited at the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) and are accessible through the accession number PRJEB23770 (Secondary study accession number: ERP105548).