

Abstract
The exchange proteins directly activated by cAMP 1 and 2 (Epac1 and Epac2) are expressed in a cell specific manner in the liver, but their biological functions in this tissue are poorly understood. The current study was undertaken to begin to determine the potential roles of Epac1 and Epac2 in liver physiology and disease. Male C57BL/6J mice in which expression of Epac1 and/or Epac2 are deleted, were subjected to partial hepatectomy and the regenerating liver was analyzed with regard to lipid accumulation, cell replication and protein expression. In response to partial hepatectomy, deletion of Epac1 and/or Epac2 led to increased hepatocyte proliferation 36 h post surgery, and the transient steatosis observed in wild type mice was virtually absent in mice lacking both Epac1 and Epac2. The expression of the protein cytochrome P4504a14, which is implicated in hepatic steatosis and fibrosis, was substantially reduced upon deletion of Epac1/2, while a number of factors involved in lipid metabolism were significantly decreased. Moreover, the number of Küpffer cells was affected, and Epac2 expression was increased in the liver of wild type mice in response to partial hepatectomy, further supporting a role for these proteins in liver function. This study establishes hepatic phenotypic abnormalities in mice deleted for Epac1/2 for the first time, and introduces Epac1/2 as regulators of hepatocyte proliferation and lipid accumulation in the regenerative process.
Subject terms: Proteomics, Proteomics, Non-alcoholic fatty liver disease, Molecular medicine, Molecular medicine
Introduction
Diseases related to hepatic steatosis constitute an increasing health problem worldwide. A better understanding of the underlying molecular mechanisms in healthy and diseased liver tissue is required for understanding of etiology, and for the development of efficient treatment strategies. The second-messenger cAMP is a critical regulator of hepatic metabolic activity, and was in fact first described due to its effects on glycogenolysis in response to glucagon and epinephrine1. cAMP is also a central player in the control of liver lipogenesis2, and has been implicated in the transcriptional signaling responses that orchestrate liver regeneration in mammals in response to partial hepatectomy (PH)3. In general, the effects of cAMP in the liver have been ascribed to activation of the cAMP dependent protein kinase A (PKA), but numerous observations also link the exchange proteins directly activated by cAMP (Epac; also known as cAMP-guanine nucleotide exchange factor, cAMP-GEF) to hepatic cellular processes4.
The Epac proteins, Epac1 and Epac2 (hereafter collectively referred to as Epac1/2) are multi-domain proteins that function as cAMP-activated exchange factors for the small G-proteins Rap1 and Rap25,6. Epac1/2 are implicated in a wide range of biological processes including glucose stimulated insulin secretion, leptin signaling, neuronal growth and differentiation, and memory and learning (reviewed in7). Epac1 and Epac2 are encoded by Rapgef3 and Rapgef4, respectively, and differential splicing and alternative promoter usage in combination with epigenetic regulation of Rapgef4, give rise to three different Epac2 isoforms, Epac2A, Epac2B and Epac2C8–10. Whereas Epac1 is nearly ubiquitously expressed, Epac2 isoforms exhibit a strict tissue specific expression pattern. Of particular interest for the current study is that expression of the shortest isoform, Epac2C, is confined to hepatocytes10. Epac2C lacks the N-terminal cyclic nucleotide-binding domain (cNBD-A) found in Epac2A, which is required for insulin secretion11, and the Dishwelled-Egl-10-pleckstrin (DEP) domain found in Epac2A, Epac2B and in Epac112, that is essential for membrane association11. Overall, the Epac2C isoform has been sparsely studied. It is known that hepatic stellate cells (HSC) express Epac113, and cholangiocytes, both Epac1 and Epac2A14, and in vitro studies link Epac1/2 to hepatic cellular processes, such as formation of canalicular networks15, proliferation of cholangiocytes14 and glucagon-dependent secretion of the fibroblast growth factor 21 (Fgf21) from hepatocytes16. In HSC, Epac1 has been implicated in transforming growth factor β1 (TGFβ1) induced signaling13, which is believed to integrate pro- and anti-fibrotic signals13,17,18.
However, the functional roles of Epc1/2 in liver have not yet been studied in intact animals, and moreover, many of the in vitro studies undertaken to investigate the roles of Epac1/2 in hepatic cells have monitored the effects of the cAMP analogue 8-pCPT-2‘-O-Me-cAMP (8-pCPT). Although first described as a specific Epac1/2 agonist19, it is now clear that 8-pCPT and its derivatives have off target effects20–25, and that it is a rather poor activator of Epac226. Therefore, to delineate the roles of Epac1/2 in liver physiology we employed mouse models that are deleted for Epac1 (Epac1−/−), Epac2 (Epac2−/−; all isoforms), or a combined knockout model of both factors (Epac1/2−/−). The mice were subjected to partial hepatectomy (PH), which is widely used to gain insights into the mechanisms that control signaling, proliferation and growth in the liver27. We report here that PH provokes phenotypes in Epac1/2−/− mice that are related to cell proliferation and lipid turnover in the regenerating liver. Moreover, we found that Epac2C expression (mRNA and protein) were increased in response to PH, and comparison of the proteome of wild type (wt) and Epac1/2−/− mice demonstrated altered expression profiles of proteins related to hepatic lipid metabolism and disease development.
Results
Liver histology is normal in Epac1−/−, Epac2−/− and Epac1/2−/− mice
Histological analysis revealed no apparent abnormalities in liver morphology in Epac1−/−, Epac2−/− or Epac1/2−/− mice (Fig. 1a). Moreover, no differences in liver mass were observed in adult mice of the different genotypes, indicating that deletion of Epac1/2 does not affect post-natal liver growth (Supplementary Fig. S1). Since Epac1/2 signaling has been implicated in bile acid stimulated hepatocyte polarization15, we performed immunohistochemistry (IHC) analyses against multidrug resistance protein 1B (ABCB1) to label the apical hepatocyte membrane. The distribution of ABCB1 in wt and Epac1/2−/− mice was indistinguishable (Supplementary Fig. S2a), and canalicular length per cell was unaffected by Epac1/2 deletion both pre- and post-PH (Supplementary Fig. S2b). RT-PCR was performed to determine the expression of Epac1/2 in different hepatic cells (Fig. 1b). The primer pair used in the present study to amplify Epac2 transcripts recognizes all Epac2 isoforms, but we have demonstrated previously, both at mRNA and protein level, that the mouse liver only expresses the Epac2C isoform8. Epac1 transcripts were identified in sinusoidal endothelial cells, Küpffer (KC) cells and HSC, whereas Epac2C was found solely in hepatocytes (Fig. 1b). Thus, none of the cell types analyzed were found to co-express Epac1 and Epac2.
Figure 1.

Expression of Epac1/2 in liver cells. (a) H&E (20x) staining of liver sections from wt, Epac1−/−, Epac2−/− and Epac1/2−/− mice. Bar: 100 μm. (b) RT-PCR of Epac1 and Epac2 mRNA in hepatocytes (Hep), liver sinusoidal endothelial cells (LSEC), hepatic stellate cells (HSC), Küpffer cells (KC) and whole liver. −RT: without reverse transcriptase.
Deletion of Epac1/2 accelerates DNA replication after PH
To gain insight into the functional roles of Epac1/2 in the liver, wt, Epac1−/−, Epac2−/− and Epac1/2−/− mice were subjected to PH and euthanized at four post-operative time points (26 h, 36 h, 74 h and 168 h). Mice depleted for Epac1/2 displayed no adverse phenotypes in response to PH compared to wt mice (i.e. no change in post-operative hepatocellular necrosis or mortality), and the resected liver- to- body weight ratio was similar in all genotypes (Supplementary Fig. S3a), as was the restitution of hepatic index (Supplementary Fig. S3b). qPCR was performed to determine whether PH affects the expression of Epac1/2 in wt mice. As shown in Fig. 2a, Epac2c mRNA expression in total liver was increased at 26 h and 36 h post-PH, but then returned to basal levels. This finding correlates with increased Epac2C protein expression as determined by proteomic analyses (Supplementary file 2). In contrast, Epac1 mRNA levels decreased at 26 h post-PH (Fig. 2b). However, confounding the relevance of this finding is the very low level of Epac1 mRNA in total liver (see legend to Fig. 2 for ct-values), as also evident from undetectable levels of Epac1 in proteomic analyses of total liver (Supplementary file 2).
Figure 2.
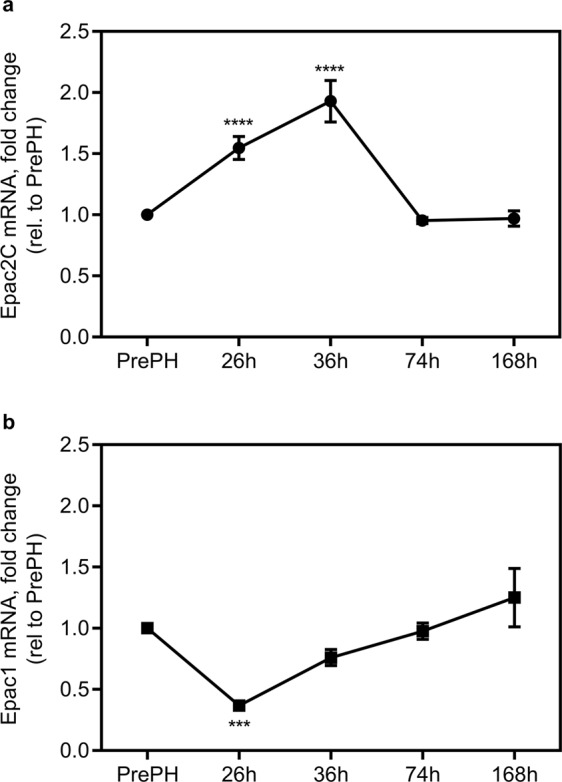
The expression of Epac2C mRNA is increased in liver after PH. qPCR analysis of (a) Epac2C and (b) Epac1 mRNA in liver pre-PH or 26 h, 36 h, 74 h and 168 h post-PH in wt mice. The CT values for resected tissue (4.39 ± 0.12 for Epac2C and 12.29 ± 0.38 for Epac1) were set to 1. Data are expressed as mean ± SD of three separate experiments performed in triplicates. One-way ANOVA with Dunnett’s adjustment for multiple comparisons was used to determine statistical differences. ***p < 0.001 and ****p < 0.0001: mRNA levels in wt livers pre-PH compared to mRNA levels in wt livers at the different postoperative time points (26 h, 36 h, 74 h or 168 h post-PH). F-statistics (a): F(4,9) = 75.57, p < 0.0001 and (b): F(4,10) = 24.50, p < 0.0001.
To determine whether the proliferative capacity after PH was affected as a consequence of deleting Epac1/2, BrdU incorporation in hepatocytes was analyzed to quantify DNA synthesis. In wt mice, BrdU-positive hepatocytes were detectable at 26 h, peaked at 36 h and returned to basal levels 74 h after PH, similar to previously reported28. Interestingly, the number of BrdU-positive hepatocytes was substantially increased in Epac1−/−, Epac2−/− and Epac1/2−/− mice compared to wt mice 36 h post-PH (Fig. 3a), demonstrating that more hepatocytes had entered the S-phase. The increased BrdU incorporation was accompanied by an increased number of DAPI stained nuclei per area 36 h post-PH (Fig. 3b). Since the hepatocyte size was similar in wt and Epac1/2−/− mice (Supplementary Fig. S3c), these results indicate increased hepatocyte proliferation in Epac1−/−, Epac2−/− and Epac1/2−/− mice, and not increased hepatocyte hypertrophy, which is often observed after PH29. The increased proliferation of hepatocytes in Epac1/2−/− mice was a result of PH, as DAPI staining revealed similar numbers of hepatocytes in resected tissue sections of all genotypes before PH, and because BrdU incorporation was equal in response to sham operation in all genotypes (Supplementary Fig. S3d).
Figure 3.
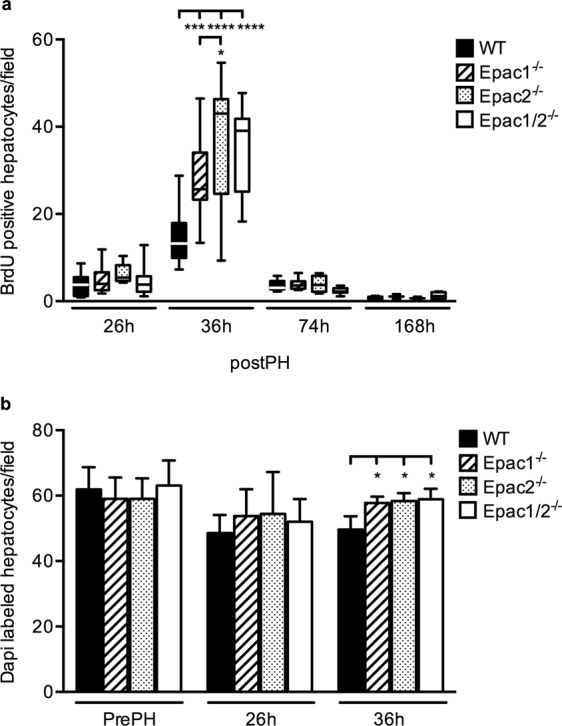
Increased BrdU-incorporation 36 h post-PH in Epac1−/−, Epac2−/− and Epac1/2−/− mice. (a) BrdU incorporation was analyzed in wt, Epac1−/−, Epac2−/− and Epac1/2−/− mice post-PH for the indicated time points. Data is shown as box and whiskers with median and min to max whiskers, n = 2–13 mice/group (6–13 mice/group at 26 h and 36 h). (b) Quantification of DAPI staining pre-PH, 26 h and 36 h post-PH. Data is shown as mean ± SD, n = 3–9 mice/group. Two-way ANOVA with Bonferroni’s adjustment for multiple comparisons was used to determine statistical differences between genotypes within each time point. *p < 0.05, ***p < 0.001 and ****p < 0.0001. F-statistics; (a) F(9,104) = 5.331, p < 0.0001 and (b) F(6,55) = 1.436, p = 0.2176. (a) 36 h post-PH (mean ± SD): wt: 15.04 ± 6.61; Epac1−/−: 27.97 ± 10.22; Epac2−/−: 36.57± 15.32; Epac1/2−/−: 34.86 ± 9.37. (b): 36 h post-PH (mean ± SD): wt: 49.6 ± 4.1; Epac1−/−: 57.8 ± 1.9; Epac2−/−: 58.4 ± 2.4; Epac1/2−/−: 58.9 ± 3.2.
Proteomic analyses indicated no changes in the expression of cyclin dependent kinases (Cdks) in Epac1/2−/− mice compared to wt mice (Supplementary dataset file 2). We were unable to detect other major cell cycle factors by this method, and therefore performed immunoblotting experiments. The results demonstrated similar levels of expression of Cyclin D1 and Cyclin E1 in the different genotypes (Supplementary Fig. S4a–l). In contrast, the expression of Interleukin 1 (IL-1), a suppressor of hepatocyte proliferation30, was moderately decreased in Epac1−/− mice 26 h post-PH (Supplementary Fig. S4a–d,m–p). Furthermore, the levels of Tumor Necrosis Factor alpha (TNFα), a potent pro inflammatory cytokine31, was increased in both Epac1−/− and Epac2−/− mice compared to wt mice pre-PH, with a more pronounced increase in Epac1−/− mice (Fig. S4a–d,q–p), and down-regulated in Epac1/2−/− mice 36 h post-PH (Fig. S4a–d,q–p) (see Supplementary Fig. S5 for the full- length blots).
The regenerating liver of Epac1/2−/− mice display early loss of fat vacuoles
Mice accumulate hepatic lipid droplets in response to PH, and this condition of steatosis is particularly evident 20–40 h post-PH32. Oil Red O (ORO) staining of the right and caudate lobes that remained after PH revealed extensive lipid accumulation in wt mice at 26 h and 36 h post-PH, followed by a return to basal levels at later time points (Fig. 4). Lipid droplets were also observed in Epac1−/−, Epac2−/− and Epac1/2−/− mice 26 h post-PH. The size, shape and distribution of the microvesicular fat vacuoles were comparable in all genotypes at this time point, although quantitative determination of the ORO staining revealed notable individual variations between animals within the same experimental group (Fig. 4b). This was especially evident in Epac1/2−/− mice where some individuals exhibited moderate steatosis 26 h post-PH, while others presented with a phenotype completely devoid of fat vacuoles (Fig. 4b). Of note, all Epac1/2−/− mice presented with a striking absence of fat vacuoles 36 h post-PH (Fig. 4a). Reduced fat accumulation was also evident in Epac2−/− mice at both time points (26 h and 36 h post-PH) compared to wt mice (Fig. 4).
Figure 4.

Epac1/2−/− mice are devoid of fat vacuoles 36 h post-PH. ORO staining of sections from livers from wt, Epac1−/−, Epac2−/− and Epac1/2−/− mice pre- and post-PH as indicated (20x). (a) Representative images of ORO staining. (b) Quantification of ORO-staining. The median and left lateral lobes resected during surgery were used as control tissue in these experiments. Data are shown as box and whiskers with median and min to max whiskers, n = 4–8 mice/group post-PH and n = 17–23 mice/group pre-PH. Two-way ANOVA with Bonferroni’s adjustment for multiple comparisons was used to determine statistical differences between genotypes within each time point. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. F-statistics: F(12,149) = 5.152, p < 0.0001.
Altered lipid profiles and protein expression in the absence of Epac1/2
Plasma lipid profiling revealed decreased basal (pre-PH) concentrations of circulating total cholesterol in Epac1−/−, Epac2−/− and Epac1/2−/− mice compared to wt mice (Table 1), as previously described in Epac1−/− mice33. The reduction in total cholesterol correlated with reduced levels of high-density lipoprotein (HDL) cholesterol, as neither low-density lipoprotein (LDL) cholesterol nor unesterified cholesterol levels were changed (Table 1). In accordance with the reduced level of HDL cholesterol, circulating phospholipid was also reduced in all Epac1/2 knockout models prior to PH. Moreover, the basal levels of triglycerides (TG) was elevated in plasma of Epac1/2−/− mice (Table 1), but the potential significance of this increase is currently unclear. The Epac1/2 knockout models exhibited reduced plasma levels of total cholesterol, HDL cholesterol, phospholipids (PL) and TGs, and elevated LDL cholesterol levels 36 h post-PH, compared to pre-PH conditions. The wt mice showed a similar profile, although the increase in LDL cholesterol was not significant (Table 1). Of note, Epac1/2−/− mice had a significantly higher level of plasma LDL cholesterol than wt and Epac2−/− mice post-PH, indicating increased LDL cholesterol secretion or decreased LDL cholesterol uptake in peripheral tissues. The concentration of circulating free fatty acids (FFA) was similar before and 36 h after PH in all genotypes (Table 1), demonstrating that the concentration of circulating FFA that peaks around 20 h post-PH in mice34, returns to normal also in the absence of Epac1/2.
Table 1.
Altered lipid profile in Epac−/− mice.
| WT | Epac1−/− | Epac2−/− | Epac1/2−/− | ||
|---|---|---|---|---|---|
| Control | |||||
| Plasma, mmol/l | C | 2.75 ± 0.09 | 2.26 ± 0.21aa | 2.25 ± 0.13aa | 2.37 ± 0.21aa |
| HDL-C | 2.38 ± 0.03 | 1.91 ± 0.37aaa | 1.99 ± 0.18aa | 1.90 ± 0.16aaaa | |
| LDL-C | 0.20 ± 0.05 | 0.21 ± 0.03 | 0.12 ± 0.04 | 0.23 ± 0.10 | |
| UC | 0.80 ± 0.08 | 0.75 ± 0.06 | 0.74 ± 0.04 | 0.78 ± 0.05 | |
| TG | 1.19 ± 0.33 | 1.34 ± 0.14 | 1.44 ± 0.20 | 1.54 ± 0.31a | |
| NEFA | 0.23 ± 0.14 | 0.017 ± 0.04 | 0.10 ± 0.13 | 0.11 ± 0.01 | |
| PL | 2.99 ± 0.12 | 2.60 ± 0.03a | 2.56 ± 0.16aa | 2.69 ± 0.16a | |
| Liver, μmol/g | C | 6.35 ± 0.43 | 7.77 ± 2.09 | 6.35 ± 0.51 | 6.69 ± 0.34 |
| TG | 5.25 ± 0.91 | 5.73 ± 0.46 | 8.86 ± 3.07 | 5.01 ± 0.50 | |
| NEFA | 0.61 ± 0.10 | 0.55 ± 0.08 | 0.55 ± 0.05 | 0.55 ± 0.06 | |
| PL | 21.36 ± 0.76 | 20.58 ± 0.86dd | 21.47 ± 0.69 | 22.54 ± 0.54 | |
| 36 h post PH | |||||
| Plasma, mmol/l | C | 1.62 ± 0.17bbbb | 1.68 ± 0.17bbb | 1.58 ± 0.23bbbb | 1.76 ± 0.22bbbb |
| HDL-C | 1.21 ± 0.19bbbb | 1.25 ± 0.13bbbb | 1.17 ± 0.16bbbb | 1.22 ± 0.18bbbb | |
| LDL-C | 0.29 ± 0.04ccc | 0.41 ± 0.09bb | 0.32 ± 0.13bbb, cccc | 0.54 ± 0.02bbbb | |
| UC | 0.75 ± 0.08 | 0.70 ± 0.05 | 0.73 ± 0.08 | 0.75 ± 0.09 | |
| TG | 0.68 ± 0.08bbbb | 0.65 ± 0.05bbbb | 0.57 ± 0.14bbbb | 0.54 ± 0.16bbbb | |
| NEFA | 0.16 ± 0.11 | 0.27 ± 0.15 | 0.23 ± 0.18 | 0.14 ± 0.07 | |
| PL | 1.77 ± 0.19bbbb | 1.74 ± 0.16bbbb | 1.68 ± 0.23bbbb | 1.74 ± 0.24bbbb | |
| Liver, μmol/g | C | 9.22 ± 0.82bbbb | 9.95 ± 0.90bb, c | 8.91 ± 1.01bbb | 7.81 ± 1.49 |
| TG | 78.78 ± 45.62bb | 39.47 ± 20.90 | 56.04 ± 46.97 | 7.68 ± 1.65 | |
| NEFA | 1.22 ± 0.41 | 0.94 ± 0.22 | 0.98 ± 0.41 | 0.66 ± 0.10 | |
| PL | 19.80 ± 1.10bb | 19.47 ± 0.91 | 20.38 ± 0.91 | 20.24 ± 0.80bbb | |
Enzymatic calorimetric lipid profiling of plasma and liver tissue isolated from wild type (wt), Epac1−/−, Epac2−/− and Epac1/2−/− control mice and PH operated mice 36 h post surgery. Data are presented as mean ± SD, n = 3–10 mice/group. Two-way ANOVA with Bonferroni’s adjustment for multiple comparisons or the non-parametric Kruskal-Wallis test with Dunn’s multiple comparison test was used to determine differences between genotypes and treatment groups for each factor. ap < 0.05, aap < 0.01, aaap < 0.001 and aaaap < 0.0001: control wt mice compared to control Epac1−/−, Epac2−/− and Epac1/2−/− mice. bbp < 0.01, bbbp < 0.001 and bbbbp < 0.0001: control mice compared to PH operated mice 36 h post surgery, same genotype. cp < 0.05, cccp < 0.001 and ccccp < 0.0001: PH operated Epac1/2−/− mice 36 h post surgery compared to PH operated wt, Epac1−/− and Epac2−/− mice 36 h post surgery. ddp < 0.01: control Epac1/2−/− mice compared to control wt, Epac1−/− and Epac2−/− mice. F-statistics for lipid profiling in plasma; C; F(3,42) = 5.597, p = 0.0025, HDL-C; F(3,41) = 5.777, p = 0.0022, LDL-C; F(3,40) = 4.116, p = 0.0123, UC; F(3,42) = 0.2445, p = 0.8647, TG; F(3,42) = 3.995, p = 0.0137, NEFA; F(3,40) = 3.423, p = 0.0261 and PL; F(3,41) = 2.368, p = 0.0847. F-statistics for lipid profiling in liver; C; F(3,42) = 1.583, p = 0.2078, TG; approximate p < 0.0001, Kruskal-Wallis statistic; 31.21, NEFA; approximate p = 0.0018, Kruskal-Wallis statistic; 22.85 and PL; F(3,42) = 1.210, p = 0.3177. C: Cholesterol, HDL-C: High-density lipoprotein-cholesterol, LDL-C: Low-density lipoprotein-cholesterol, UC: unesterified cholesterol, TG: Triglycerides, NEFA: Non-esterified fatty acids and PL: phospholipids.
The hepatic lipid profiles were nearly identical in the different genotypes in mice not subjected to PH (Table 1). The only variation found was that Epac1−/− mice had lower levels of phospholipids compared to Epac1/2−/−. However, taken together, these results indicate that under resting conditions, lipid metabolism is adequately maintained in the absence of Epac1/2. In response to PH, increased hepatic levels of TGs and cholesterol were observed in wt mice as reported35. Similarly, cholesterol was significantly increased in Epac1−/− ad Epac2−/− mice (Table 1). TG levels were also elevated in these genotypes, but due to large variations between individuals within the same genotype (Epac1−/− and Epac2−/−), the differences where not statistically significant. As also evident from the ORO staining (Fig. 4), Epac1/2−/− mice exhibited lower levels of TGs in the livers post-PH, but the large individual variations within each genotype resulted in non-significant statistical results. In contrast to the other genotypes, hepatic cholesterol was not increased in Epac1/2−/− mice after PH, also contributing to an overall reduced mass of neutral lipids in this group.
The dramatic effect on lipid accumulation in Epac1/2−/− mice 36 h after PH, but milder effects in mice deleted for either Epac1 or Epac2 (Fig. 4 and Table 1), provoked further analyses of the double knockout model. Proteomic analyses of liver tissue harvested pre-PH identified 113 proteins that were differentially expressed in Epac1/2−/− mice compared to wt mice (54 increased and 59 decreased). Gene ontology (GO) enrichment analyses of the proteins with increased expression pre-PH in Epac1/2−/− mice relative to wt mice indicated over-representation of proteins connected to sulfation and sulfotransferase activity, and biosynthetic processes related to, among other molecules, cholesterol biosynthesis and steroid metabolism (Fig. 5a; see Supplementary Table 1a for an overview of the proteins identified for each GO-term). The proteins with reduced expression in Epac1/2−/− mice compared to wt mice pre-PH were mainly associated with lipid metabolism, peroxisome biology and oxidation-reduction processes (Fig. 5b, and Supplementary Table 1b). Post-PH, again 113 proteins were differentially expressed in Epac1/2−/− mice compared to wt mice (38 increased and 75 decreased). GO enrichment analyses of the proteins with decreased expression post-PH predicted enrichment of similar or related pathways and processes as before PH, but interestingly, the statistical prediction was considerably strengthened for the terms related to peroxisome biology (Fig. 5c; see Supplementary Table 2 for an overview of the proteins identified for each GO-term). There were no significantly enriched GO-terms for the proteins that were increased in Epac1/2−/− mice compared to wt mice post-PH. In accordance with previous studies36, PH altered the hepatic protein composition in wt mice, resulting in enrichment of GO terms related to metabolic processes, mainly lipid metabolism, the inflammatory response and regeneration (Supplementary Fig. S6). The expression of proteins linked to lipid metabolism was also changed in Epac1/2−/− post-PH, and interestingly, the GO analyses predicted enrichment for the terms “Negative regulation of gluconeogenesis” and “Very-low density lipoprotein particle” in the absence of Epac1/2 (Supplementary Fig. S6).
Figure 5.

Functional annotation of differentially expressed proteins. GO enrichment analyses on proteins that were differentially expressed in wt and Epac1/2−/− mice (a/b) pre- or (c) 36 h post-PH. (a) Functional annotation of proteins with significantly higher expression pre-PH in Epac1/2−/− mice compared to wt. (b) Functional annotation of proteins with significantly lower expression pre-PH in Epac1/2−/− mice compared to wt. (c) Functional annotation of proteins with significantly lower expression post-PH in Epac1/2−/− mice compared to wt. Black bars indicate the respective p-values and grey bars indicate fold enrichment for the corresponding GO-term. The number of proteins annotated with each GO-term is shown behind each bar. BP: biological processes; MF: molecular function (MF); CC: cellular compartment.
Of particular interest is that the expression of cytochrome P4504A14 hydroxylase (Cyp4a14) was severely affected in Epac1/2−/− mice. This enzyme catalyzes omega-hydroxylation of medium-chain fatty acids, and is implicated in hepatic steatosis and fibrosis37. The proteomic analyses demonstrated a 10 and 14 fold repression of Cyp4a14 in Epac1/2−/− mice compared to wt mice before and after PH, respectively (Fig. 6a,b). IHC and immunoblotting analyses verified the decreased expression of Cyp4A14 in mice deleted for Epac1, Epac2 or Epac1/2 (Fig. 7 and Supplementary Fig. S7; see Supplementary Fig. S8 for full-length blots). The antibody used in IHC and immunoblotting recognizes Cyp4a10 and Cyp4a12, in addition to Cyp4a14. Thus, the IHC and immunoblotting results do not perfectly mimic the reduction in Cyp4A14 expression observed in the proteomic analyses. Cyp4a12 was downregulated 1.7 fold in Epac1/2−/− mice both before and after hepatectomy (Fig. 6), and Cyp4a10 was downregulated 2 and 4 fold in Epac1/2−/− mice pre- and post-PH, respectively (Fig. 6). The IHC analyses demonstrated that in wt mice, the highest expression of Cyp4A14 was in the centrilobular region38. Epac1−/−, Epac2−/− and Epac1/2−/− mice exhibited a distribution pattern with fewer centrilobular Cyp4A14 positive cells, a weaker staining intensity and a localization that included Cyp4A14 positive cells in the transition- and periportal regions pre-PH (Fig. 7).
Figure 6.

Differentially expressed proteins in Epac1/2−/− mice compared to wt. The proteins that were differently expressed in Epac1/2−/− mice compared to wt mice (a) pre- and (b) post-PH are listed. Blue bars indicate proteins that were decreased in Epac1/2−/− mice compared to wt mice, and red bars indicate proteins that were increased in Epac1/2−/− mice compared to wt mice. Statistics: two-tailed student’s two-sample t-test and Z-statistics for FC, only when p < 0.05 for both it was considered significant.
Figure 7.

Decreased expression of Cyp4A14 in Epac1/2−/− deficient mice. IHC analyses (DAB staining) to determine Cyp4a14 expression in liver sections from wt, Epac1−/−, Epac2−/− and Epac1/2−/− mice pre-PH (a–d) and 36 h post-PH (e–h). The antibody used also detects Cyp4a10 and Cyp4a12. 4–6 mice in each experimental group were analyzed (3 sections from each animal). Representative images are shown. Bar: 200 μm.
Cyp4a14 is a target gene for peroxisome proliferator-activated receptor alpha (PPARα)39, and it is notable that a considerable number of the proteins that were differentially expressed in Epac1/2−/− mice compared to wt mice (both pre- and post-PH) are known PPARα target genes. In particular, enzymes involved in fatty acid degradation were generally decreased in Epac1/2−/− mice as opposed to wt mice (Table 2). However, immunoblotting analyses demonstrated no effect on PPARα expression as a consequence of Epac1/2 deletion (Supplementary Fig. S9; see Supplementary Fig. S10 for full-length blots) and the proteomic analysis did not provide insight into a potential effect of PPARα expression in Epac1/2−/− mice, as we were unable to detect PPARα in this analysis. Cytochrome P4507α hydroxylase (Cyp7α1), was the protein found to be most significantly increased in Epac1/2−/− mice post-PH compared to before surgery (5-fold) (Fig. 6b). This was in sharp contrast to wt mice where the expression of CYP7α1 was decreased in response to PH, in accordance with the literature40 (Supplementary dataset file 2). Cyp7α1 is also a PPARα target gene39,41, and catalyzes the first reaction in the bile acid biosynthetic pathway42. The expression of other enzymes involved in this process (i.e. Akr1d1: Aldo-keto reductase family 1 member D1 and HSD17b4: Hydroxysteroid 17-beta dehydrogenase 4) was also altered in Epac1/2−/− mice. Of note is that the expression levels of the different PKA subunits (i.e. Prkar1a Prkar2a, Prkaca and Prkacb) were not changed in Epac1/2−/− mice (Supplementary dataset file 2), and neither were the expression of the different isoforms of the Epac1/2 downstream targets Ras-related protein Rap1/2 (Rap1a, Rap1b, Rap2a:Rap2c;Rap2b). Recently, several studied have shown that Epac1/2 is localized to the mitochondria43–46. In this regard, it is interesting to note that a substantial number of the proteins with decreased expression in Epac1/2−/− mice that are associated with lipid metabolism and oxidation-reduction processes, are mitochondrial proteins (Supplementary Table S3).
Table 2.
List of PPARα target genes that are less expressed in Epac1/2−/− mice.
| ID | Gene symbol | Protein name | prePH FC | postPH FC |
|---|---|---|---|---|
| P55096 | Abcd3 | ATP-binding cassette sub-family D member 3 | −1.23 | −1.19, ns |
| Q921H8 | Acaa1a | 3-ketoacyl-CoA thiolase A, peroxisomal | −1.51 | −1.58 |
| Q8VCH0 | Acaa1b | 3-ketoacyl-CoA thiolase B, peroxisomal | −1.64, ns | −2.25 |
| P51174 | Acadl | Long-chain specific acyl-CoA dehydrogenase, mitochondrial | −1.11, ns | −1.24 |
| P45952 | Acadm | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | −1.08, ns | −1.25 |
| Q9QYR9 | Acot2 | Acyl-coenzyme A thioesterase 2, mitochondrial | −1.61 | −4.18 |
| Q8BWN8 | Acot4 | Acyl-coenzyme A thioesterase 4 | −1.78, ns | −1.95 |
| Q9R0H0 | Acox1 | Peroxisomal acyl-coenzyme A oxidase 1 | −1.25 | −1.46 |
| P47740 | Aldh3a2 | Fatty aldehyde dehydrogenase | −1.48, ns | −2.75 |
| P52825 | Cpt2 | Carnitine O-palmitoyltransferase 2, mitochondrial | −1.02, ns | −1.24 |
| P47934 | Crat | Carnitine O-acetyltransferase | −1.59, ns | −1.92 |
| O88833 | Cyp4a10 | Cytochrome P450 4A10 | −2.01, ns | −3.93 |
| Q91WL5 | Cyp4a12a | Cytochrome P450 4A12A | −1.73 | −1.73 |
| O35728 | Cyp4a14 | Cytochrome P450 4A14 | −9.89 | −13.73 |
| Q9CQ62 | Decr1 | 2,4-dienoyl-CoA reductase, mitochondrial | −1.19, ns | −1.31 |
| O35459 | Ech1 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, mitochondrial | −1.31 | −1.35 |
| Q9DBM2 | Ehhadh | Peroxisomal bifunctional enzyme; Enoyl-CoA hydratase/3,2-trans-enoyl-CoA isomerase; 3-hydroxyacyl-CoA dehydrogenase | −1.18, ns | −1.59 |
| P51660 | Hsd17b4 | Peroxisomal multifunctional enzyme type 2;(3 R)-hydroxyacyl-CoA dehydrogenase; Enoyl-CoA hydratase 2 | −1.20 | −1.28 |
| Q9Z211 | Pex11a | Peroxisomal membrane protein 11A | −1.99 | −2.36 |
| Q9Z2Z6 | Slc25a20 | Mitochondrial carnitine/acylcarnitine carrier protein | −1.01, ns | −1.33 |
PrePH (WTprePH vs. Epac1/2−/−prePH), postPH (WTpostPH vs. Epac1/2−/−postPH), negative value: decreased protein expression in Epac1/2−/− mice relative to wt mice. ns: not significant. FC: fold change. Please see the Supplementary M&M section for details regarding statistical analysis.
The number of KCs is affected in Epac1−/− and Epac2−/− mice
It is intriguing that deletion of either Epac1 or Epac2 alone causes phenotypes in response to PH, and that some of the effects are reinforced when both factors are deleted. These findings suggest that also Epac1 expressing cells, and not only hepatocytes, are affected in the livers of the knockout models. To gain further insights into the potential effects on nonparenchymal cells, we quantified the relative numbers of KCs and HSCs by IHC. Both cell types are known to exert stimulatory and inhibitory effects on hepatocyte proliferation following PH47,48. We observed no significant differences between genotypes with regard to the number of platelet derived growth factor beta (PDGFβ) - positive HSCs (Fig. 8a). However, the number of F4/80-positive KCs was increased in Epac1−/− mice pre-PH, and decreased in Epac2−/− mice 36 h post-PH (Fig. 8b).
Figure 8.

Altered KC number in Epac1−/− and Epac2−/− mice. The number of (a) PDGFβ-positive stellate cells and (b) F4/80-positive KCs was determined in the livers of wt, Epac1−/−, Epac2−/− and Epac1/2−/− mice pre- and 36 h post-PH. Two-way ANOVA with Tukey’s adjustment for multiple comparisons was used to determine statistical differences between genotypes within each time point. n = 4–6 mice per group, *p < 0.05, **p < 0.01, and ****p < 0.0001. F-statistics: (a) F(3,16) = 2.670, p = 0.0836 and (b) F(3,15) = 6.630, p = 0.0045.
Discussion
The mammalian response to PH has been extensively studied, but the details of how the signaling cascades that control liver regeneration integrate, remain incompletely understood. It is known that cAMP and PKA are involved, and the production of cAMP is tightly controlled during the process49–52. Moreover, a delay in regeneration is observed in mice deleted for the cAMP-regulated transcription factor CREM (cAMP responsive element modulator)3. In the present study, we demonstrate for the first time that the cAMP binding proteins Epac1/2 are involved in regulating liver regeneration in an in vivo model. Deletion of Epac1 or Epac2, or both factors together, caused phenotypes manifested in increased lipid turnover and DNA synthesis early in the regenerative process. Moreover, we found that expression of Epac2C was upregulated in the regenerating liver, that the number of KCs cells was altered and that the liver proteome of Epac1/2−/− mice differed from wt mice, a disparity that was substantiated in response to PH. Taken together, it appears that loss of Epac1/2 is compatible with normal liver functions in mice living in a protected environment, but that these factors become important in the regulation of cellular responses in stressful situations. We used global knockout models in this study, and cannot disregard possible indirect effects of Epac1/2 deletion on liver regeneration, and thus, future analyses to decipher the molecular roles of Epac1 and Epac2 in different hepatic cell lines should include cell-specific knockout models. An example of a possible indirect effect is via blood platelets. It is clear that platelets and platelet-derived factors have central roles in liver regeneration after PH53–55, and in this regard it is interesting to note that we previously demonstrated that Epac1−/− mice (but not Epac2−/− mice) display a bleeding phenotype that is partly caused by altered platelet maturation and function56.
The fact that some of the observed phenotypic traits were substantiated in Epac1/2−/− mice compared to single knockouts, indicates that these proteins have distinct, but convergent roles in the regenerative process, and that lack of one factor (and both) causes defective cell communication and integration of signals. This notion is supported by the cell specific and mutual exclusive expression of Epac1 and Epac2 in the liver, and by the finding that the number of Epac1 expressing KCs is altered not only in Epac1−/− mice, but also in Epac2−/− mice post-PH. Epac1 exerts anti-inflammatory effects in different biological systems57, and at present, it is unclear whether the increased number of F4/80-positive KCs observed in Epac1−/− mice results from increased recruitment of circulating macrophages due to an increased inflammatory state in the absence of Epac1, or caused by intrinsic effects of the liver. Interestingly, the pronounced increase in TNFα that we observe in Epac1−/− mice before PH is in agreement with an enhanced recruitment of circulating monocyte-derived macrophages, which is known to secrete pro-inflammatory cytokines, whereas the pro-regenerative liver resident KCs secrete anti-inflammatory cytokines58.
The discovery that the number of KCs is changed in Epac2−/− mice 36 h post-PH might imply that the interaction between hepatocytes and KCs is disturbed. We have limited knowledge of how hepatocyte mediated signaling affects KC replenishment post-PH. However, it has been suggested that dysregulated secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) from hepatocytes interferes with, or delays KC replenishment post-PH59,60. This is interesting considering the suggested role for GM-CSF in hepatic lipid homeostasis61, and growing evidence demonstrating that KC renewal is affected by the metabolic state of the liver62. It is yet to be determined whether GM-CSF mediated signaling is affected in the absence of Epac2C.
Rap1 GTPase, the main downstream Epac1/2 effector, plays an important role in regulating cell proliferation. However, the roles of Epac1/2 in cell proliferation have remained elusive, as both pro-mitotic and anti-mitotic effects have been ascribed to these proteins depending on cellular context63. The increased BrdU incorporation observed in Epac1−/−, Epac2−/− and Epac1/2−/− mice at 36 h post-PH in the present study, indicates that the absence of Epac1/2 causes an accelerated first round of hepatocyte proliferation. In mice, coordinated waves of hepatocyte proliferation occur in response to PH, with the first round being the most robust64. The expression of cell cycle regulators are expressed within hours after PH, and interestingly, it has been demonstrated that the relative expression of the different cell cycle components is rather consistent throughout the liver regrowth, despite fluctuations in DNA synthesis during the process65. In line with this, although we detected increased proliferation at 36 h post-PH, we did not observe increased expression of Cyclin D1, Cyclin E1or cyclin dependent kinases (Cdks) in mice deleted for Epac1/2. However, we can not rule out that the levels of these factors differed between wt and Epac1/2 knockout models at time points earlier than 26 h and therefore escaped detection in our experimental set-up. We found that the expression of TNFα is lower in Epac1/2−/− mice, and partly in Epac2−/− mice compared to wt mice at the peak of DNA-synthesis (36 h post-PH). TNFα triggers the priming phase in hepatocyte proliferation after PH, whereas this factor is suppressed at later stages66. Thus, the observed decreased expression of TNFα in Epac1/2−/− mice substantiates the idea that hepatocyte proliferation occurs more rapidly in mice lacking Epac1/2. Taken together, we suggest that the increased number of BrdU-positive hepatocytes in Epac1−/−, Epac2−/− and Epac1/2−/− mice 36 h post-PH, demonstrate that the Epac proteins restrain hepatic cell proliferation under normal conditions.
A hallmark of the early stages of liver regeneration in response to PH is the accumulation of lipids, which contributes a greater portion of the energy substrates necessary to complete regrowth67. The finding that Epac1/2−/− mice are completely devoid of lipid droplets 36 h post-PH indicates regulatory roles for Epac1/2 in this process in wt mice. Despite large individual differences, the ORO-staining at 26 h points to the fact that Epac1/2−/− mice were capable of accumulating lipids at early time points after PH. We propose that ablation of Epac1/2 causes a shift in the regeneration process with a more rapid lipid turnover accompanied with increased hepatocyte proliferation. These irregularities did affect neither the overall regeneration process nor the survival of the animal, indicating that the role of Epac1/2 is to balance the regulatory mechanisms that control the process. This is consistent with several publications demonstrating that normal lipid accumulation in response to PH is not a prerequisite for normal regeneration34,68.
The proteomic analyses presented in this study disclosed that the expression of Cyp4A14 is dramatically decreased in Epac1/2−/− mice, both pre- and post-PH. This enzyme, and the human homolog (CYP4A hydroxylase), catalyze omega-hydroxylation of medium chain fatty acids and arachidonic acids69. Interestingly, Cyp4A14 expression is increased in murine models of spontaneous nonalcoholic fatty liver disease (NAFLD)70–72, as well as in livers of patients with NAFLD37. Moreover, overexpression of, or genetic ablation of Cyp4A14 in mice correlates with increased and reduced hepatic fat accumulation, respectively, in response to a high fat diet37. The result that Cyp4A14 is strongly repressed in livers from Epac1/2−/− mice both before and after hepatectomy provides a link between Epac1/2, Cyp4a14 and the regulation of hepatic lipid turnover. NAFLD is displayed as a range of diseases that can develop to non-alcoholic steatohepatitis and further to fibrosis73. Cyp4a14 also plays a role in the development of fibrosis, as diet induced portal and perisinusoidal fibrosis is attenuated in mice depleted for Cyp4A14 expression, as is the expression of fibrosis-related genes in HSC (activation of HSC is central in the development of hepatic fibrosis)37. In this context it is of interest that Epac1 has been proposed to exhibit anti-fibrotic functions, and that several studies point to Epac1 as an integrator of pro- and anti-fibrotic signals in HSC13,17,18. Whereas the pro-fibrogenic factor TGFβ1 decreases Epac1 expression in HSC, overexpression of Epac1 inhibits TGFβ1-induced production of collagen13. Expression of Epac1 is also repressed in fibrotic livers of mouse and human, and stimulation of Epac1 expression in HSC partly restores carbon tetrachloride induced fibrosis in mice17. The effects of high fat feeding in mice deleted for Epac1/2 is not yet clear, as conflicting results are obtained from different Epac knockout models23,33,74. A recent study describes increased obesity and fat accumulation in adipose tissue and liver in male mice deleted for Epac2A74. However, since the model used in this study74 expresses normal levels of Epac2C in hepatocytes, it does not provide information about the intrinsic roles for Epac2C in the liver. On the other hand, it does imply that Epac2A, acting outside of the liver, affects hepatic lipid accumulation indirectly, prompting future studies on hepatocyte specific Epac2C knockouts69.
Several mouse models that lack expression of Epac1/2 have been developed23,33,75–78. A common feature of these models is that deletion of either Epac1 or Epac2, or both, does not produce gross anatomical or physiological abnormalities, but that exposure to stress provokes phenotypes in a diversity of physiological processes, as we describe herein for the liver23,33,56,75,76,78–83. Collectively, these studies suggest that Epac1/2 may have evolved to balance cellular responses in challenging situations, and implicate Epac1/2 in a number of diseases. Epac1/2 are considered promising drug targets, and Epac2 has been suggested to be the target for both incretin-related drugs and sulfonylureas, which are widely used to treat type 2 diabetes84. Currently, considerable effort is invested in developing compounds that specifically act on either Epac1 or Epac2 to tailor treatment and reduce side effects26,85. The roles of Epac1/2 in liver physiology disclosed in the present study hold promise for that compounds acting via Epac1/2 may be used in the future to treat liver diseases.
Materials and Methods
Animals
Male mice (C57BL/6J background, 8–12 weeks) were used in this study. Epac1−/− and Epac2−/− mice were generated using the Cre-LoxP targeting strategy described elsewhere56,81–83. Of note, the targeting strategy used ablates the expression of all Epac2 isoforms (i.e. Epac2A, Epc2B and Epac2C). Mice deleted for Epac1 and Epac2 (Epac1/2−/−) were generated by crossing of Epac1−/− and Epac2−/− mice. Wt mice used in the study (originally from Taconic, Denmark) were littermates of Epac1−/− and Epac2−/− mice or, mice bred in the same room. The origin of the C57BL/6JBomTac mice is described at https://www.taconic.com/mouse-model/b6jbom. The mice were housed in groups of three to five and bred at the animal facility at Haukeland University Hospital and maintained under standard conditions with a temperature of 21 °C ± 0.5 °C, humidity of 55% ± 5%, lighting of 150 lux and 12 hours light/dark cycle (7 am/7 pm). Animals were housed in IVC II cages with food (RMI (E)) and water ab libtium. All animal protocols were approved by the Norwegian Animal Research Authority and performed according to the European Convention for the Protection of Vertebrates Used for Scientific Purposes. The Animal Care and Use Programs at University of Bergen are accredited by AAALAC international.
Hepatectomy
2/3 partial hepatectomy (PH) or sham laparotomy was performed as described86. At the selected post-operative endpoints (26 h, 36 h, 74 h and 168 h), the mice were euthanized by CO2- suffocation, with administration of BrdU (50 mg/kg IP) 2 h before sacrifice. Tissue removed at time of surgery (left lateral and median lobes) and regenerated tissue (right lateral and caudate lobes) removed at selected endpoints were flash-frozen on liquid nitrogen, or fixed in 4% paraformaldehyde (PFA; 10x w/v) for 18–24 h, embedded in paraffin and sectioned (4 μm). Trunk blood was obtained by cardiac puncture, collected into prechilled EDTA-tubes and blood plasma prepared. See Supplementary S1 Materials & Methods, for details on H&E, ORO staining, hepatocyte size quantification, lipid profiling, immunohistochemistry and immunoblotting.
BrDU analyses
Hepatocyte proliferation was assessed by monitoring BrdU-incorporation on paraffin sections according to manufacturer’s protocol (Anti-BrdU antibody (BU1/75 (ICR1), ab6326, Abcam)), with some modifications: Briefly; paraffin sections were dewaxed and DNA denaturized in HCl (2 N) for 30 min at 37 °C followed by neutralization in borate buffer (0.1 M) for 2 × 5 min at RT. Next, sections were rinsed 3 × 5 min in TBS, incubated in blocking buffer (10% normal donkey serum/2xTBS-T) for 60 min at RT and incubated with Anti-BrdU antibody (1:300 in 3% normal donkey serum/TBS-T) at 4 °C o/n with gentle agitation. Parallel sections used as negative controls were incubated with Rat IgG2a Isotype Control (1:300 in TBS-T) or in blocking buffer o/n at 4 °C. Next, sections were rinsed for 4 × 10 min in 2xTBS and incubated with Alexa Fluor® 488 Donkey Anti-Rat IgG (1:200 in 3% normal donkey serum/TBS-T) for 1.5 h at RT. Lastly, sections were rinsed 4 × 10 min in TBS, counterstained and mounted with Prolong® Gold Antifade Reagent with DAPI and coverslipped. DAPI-stained sections were not blocked, but directly counterstained and mounted. The stained sections were visualized with Zeiss Axioplan2 microscope and AxioVision version 4.5 Program (Zeiss), hepatocyte proliferation was determined by manually counting positive BrdU nuclear staining. For each mouse, ten 40x microscopic fields (in an area of 35.2 mm2) in three different sections were counted blindfolded by two different researchers. Likewise, DAPI staining was counted manually in ten 40x microscopic fields in one section from each mouse.
Real time (RT) and quantitative (q) PCR
Primary cell isolation and fluorescence-activated cell sorting
Hepatocytes and HSC were isolated as described previously8,87. Liver resident macrophages (KCs) were extracted as described previously88. In brief, mouse liver was perfused through the inferior vena cava with PBS. Livers were excised, minced and digested in solution I (0.8 mg/ml collagenase V, 0.625 mg/ml collagenase D, 1 mg/ml Dispase and 30 μg/ml DNase). The suspension was shaken on an orbital shaker at 37 °C for 25 min, with additional vigorous manual shaking every 5 min. Digest was strained through a 100 μm cell strainer. Following 2 × 5 min centrifugation at 300 g RBC lysis buffer was added for 3 min to lyse red blood cells. Cells were then centrifuged at 300 g for 5 min, resuspended in FACS buffer (2% FBS (Gibco) and 2 mM EDTA in Dulbecco’s phosphate-buffered saline no calcium, no magnesium (Gibco)) and filtered through a 35 μm strainer. Cells were blocked with 10% mouse serum (Sigma) containing 1% Purified anti-mouse CD16/32 (Biolegend) for 10 min. Cells were then incubated with fluorescent conjugated antibodies (FITC anti-mouse CD45.2, PE/Cy7 anti-mouse F4/80, APC/Cy7 anti-mouse/human CD11b, PE anti-mouse CD3, PE anti-mouse CD19, PE anti-mouse Ly-6G, APC anti-mouse CD31 and PE Rat Anti-Mouse Siglec-F) or isotype control at 4 °C for 30 min in the dark. Cells were washed, resuspended in FACS buffer and sorted using a FACS Aria II (BD Biosciences, USA). Following a positive CD45.2 gate and dump of CD3, CD19, Ly-6G, Siglec-F, and CD31 positive KCs were defined as the F4/80 high, CD11b intermediate population.
To isolate liver sinusoidal endothelial cells (LSEC), mouse liver was perfused through the inferior vena cava with PBS. Livers were excised, minced and digested in a solution containing 5 mg/ml collagenase I and 11μg/ml DNase. This cell suspension was shaken at 37 °C for 20 min. Digest was strained through a 70 μm cell strainer. Following 7 min centrifugation at 400 g, RBC Lysis buffer was added for 5 min. Cells were then centrifuged at 400 g for 7 min, resuspended in FACS buffer and filtered through a 35 μm strainer. Cells were blocked with 10% mouse serum and 1% CD16/32 for 10 min. Cells were then incubated with fluorescent conjugated antibodies (PE/Cy7 anti-mouse CD45 and APC anti-mouse CD31 clone. 390) or isotype control at 4 °C for 30 min in the dark. Cells were washed, resuspended in FACS buffer and sorted using a FACS Aria II. Endothelial cells were sorted from the CD31 positive, CD45 negative population.
RNA was extracted from the isolated cells using the RNeasy Micro kit (74004, Qiagen, Germany), and converted to cDNA using the SuperScript® III First-Strand Synthesis SuperMix for qRT-PCR kit (Invitrogen, Waltham, MA, USA). The RT-PCR reaction was carried out as described in8. For qPCR analyses, RNA was prepared from total liver and converted to cDNA using the GenElute Mammalian Total RNA miniprep Kit, and the iScript™ cDNA Synthesis kit (BioRad). qPCR was carried out on a LightCycler 480 II (Roche) using SYBR Green Supermix (BioRad) with following amendments in the thermal cycling protocol: 1 cycle for 300 sec at 95 °C and 40 cycles for 10 sec at 95 °C, 10 sec at 59 °C and 10 sec at 72 °C followed by a melting curve analysis. All samples were run in triplicates and repeated in three separate experiments. Expression levels of Epac1 and Epac2 Universal were normalized to the geometrical mean of SDHA and Ppib, and relative gene expression determined using the 2−ΔΔCt-method89–91. Primer sequences: Epac1 fwd: GAAAATGGCTGTGGGAACGTATCT, Epac1 rev: AGCTGCTCAGGGTGTGGGGT, Epac2 Universal fwd: ATCTACGAGACGAGCT CCTTCATATTAAA, Epac2 Universal rev: GACTACATTCACGGATCCTTTCAGA, β-Actin fwd: GGCCCAGAGCAAGAGAGGTATC, β-Actin rev: AGGCATACAGG GACAGCACAGC, Ppib fwd: GGAGATGGCACAGGAGGAAA, Ppib rev: CGTAG TGCTTCAGCTTGAAGTTCT, SDHA fwd: CATGCCAGGGAAGATTACAA and SDHA rev: GCACAGTCAGCCTCATTCAA.
Proteomic analyses
Frozen liver samples (10–12 µg) harvested pre- and 36 h post-PH were lysed in SDS lysis buffer (4% SDS in 0.1 M Tris-HCl, pH 7.6) and processed for LC-MS analysis according to the filter-aided sample preparation (FASP) procedure92, as described93. NanoLC mass spectrometry and proteomic data analyses were performed as described in Supplementary Materials & Methods. Proteins with significant differential expression were classified according to their biological processes using Gene ontology (GO) enrichment analysis with the online resource A.GO.TOOL software (University of Copenhagen, Denmark)94.
Statistics
Statistical analysis was performed using GraphPad Prism 6.0. The One-way ANOVA with Dunnett’s or Bonferroni’s adjustment for multiple comparisons (when applicable) or the Two-way ANOVA with Bonferroni’s adjustment for multiple comparisons (when applicable) was used to assess statistical differences between groups. The Brow-Forsynthe test was used to test the normality of the data. If the data did not meet the requirements for normal distribution, the non-parametric Kruskal-Wallis test with Dunn’s multiple comparison was used to assess statistical differences between groups. The data are shown as median with and min to max whiskers for box plots and mean ± SD for others. The degrees of freedom, F- and p-values for the different pair-wise comparisons are also provided in the figure legends. p < 0.05 was considered statistically significant. Proteomics: See Supplementary Materials & Methods.
Supplementary information
Acknowledgements
We thank Dr. Joël Maillet, BioTek Instruments GmbH, Kjellfrid Haukanes and Nina Lied Larsen (UiB) for excellent technical assistance. This work was supported by The Research Council of Norway (Grant no. 183451/S10; MB) and The Regional Health Authorities, Western Norway (Grant no. 911979; SOD).
Author Contributions
K.S.A., L.P. Planning and execution of experiments, preparation of manuscript; R.A., H.M., E.A., I.K.N.P., L.H., R.D., S.J., R.K.B., N.H., F.S. Planning and execution of experiments; S.O.D. Planning and financing study, preparing manuscript; M.B. Coordinating, planning and financing study, preparing manuscript. All authors read and approved the final manuscript.
Data Availability
The proteomics raw files and data were uploaded to the ProteomeXchange consortium (version 2.3.0) via the PRIDE partner repository, and with the dataset identifier PXD00616595,96. A detailed description of the LC-MS analysis is provided in the Supplementary Material & Methods section.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-50219-8.
References
- 1.Berthet J, Rall TW, Sutherland EW. The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. The Journal of biological chemistry. 1957;224:463–475. [PubMed] [Google Scholar]
- 2.Rui L. Energy metabolism in the liver. Comprehensive Physiology. 2014;4:177–197. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Servillo G, Della Fazia MA, Sassone-Corsi P. Transcription factor CREM coordinates the timing of hepatocyte proliferation in the regenerating liver. Genes Dev. 1998;12:3639–3643. doi: 10.1101/gad.12.23.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sequera C, Manzano S, Guerrero C, Porras A. How Rap and its GEFs control liver physiology and cancer development. C3G alterations in human hepatocarcinoma. Hepat Oncol. 2018;5:HEP05. doi: 10.2217/hep-2017-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Rooij J, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 6.Kawasaki H, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacological reviews. 2013;65:670–709. doi: 10.1124/pr.110.003707. [DOI] [PubMed] [Google Scholar]
- 8.Hoivik EA, et al. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PloS one. 2013;8:e67925. doi: 10.1371/journal.pone.0067925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niimura M, et al. Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. Journal of cellular physiology. 2009;219:652–658. doi: 10.1002/jcp.21709. [DOI] [PubMed] [Google Scholar]
- 10.Ueno H, et al. Characterization of the gene EPAC2: structure, chromosomal localization, tissue expression, and identification of the liver-specific isoform. Genomics. 2001;78:91–98. doi: 10.1006/geno.2001.6641. [DOI] [PubMed] [Google Scholar]
- 11.Alenkvist I, Gandasi NR, Barg S, Tengholm A. Recruitment of Epac2A to Insulin Granule Docking Sites Regulates Priming for Exocytosis. Diabetes. 2017;66:2610–2622. doi: 10.2337/db17-0050. [DOI] [PubMed] [Google Scholar]
- 12.Sugawara K, Shibasaki T, Takahashi H, Seino S. Structure and functional roles of Epac2 (Rapgef4) Gene. 2016;575:577–583. doi: 10.1016/j.gene.2015.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yokoyama U, et al. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc Natl Acad Sci USA. 2008;105:6386–6391. doi: 10.1073/pnas.0801490105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banales JM, et al. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49:160–174. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu D, Wakabayashi Y, Lippincott-Schwartz J, Arias IM. Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1403–1408. doi: 10.1073/pnas.1018376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cyphert HA, Alonge KM, Ippagunta SM, Hillgartner FB. Glucagon stimulates hepatic FGF21 secretion through a PKA- and EPAC-dependent posttranscriptional mechanism. PloS one. 2014;9:e94996. doi: 10.1371/journal.pone.0094996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schippers M, et al. Upregulation of Epac-1 in Hepatic Stellate Cells by Prostaglandin E2 in Liver Fibrosis Is Associated with Reduced Fibrogenesis. J Pharmacol Exp Ther. 2017;363:126–135. doi: 10.1124/jpet.117.241646. [DOI] [PubMed] [Google Scholar]
- 18.Insel PA, et al. cAMP and Epac in the regulation of tissue fibrosis. Br J Pharmacol. 2012;166:447–456. doi: 10.1111/j.1476-5381.2012.01847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enserink JM, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nature cell biology. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- 20.Herfindal L, et al. Off-target effect of the Epac agonist 8-pCPT-2′-O-Me-cAMP on P2Y12 receptors in blood platelets. Biochemical and biophysical research communications. 2013;437:603–608. doi: 10.1016/j.bbrc.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Herfindal L, et al. Introduction of aromatic ring-containing substituents in cyclic nucleotides is associated with inhibition of toxin uptake by the hepatocyte transporters OATP 1B1 and 1B3. PloS one. 2014;9:e94926. doi: 10.1371/journal.pone.0094926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poppe H, et al. Cyclic nucleotide analogs as probes of signaling pathways. Nature methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- 23.Yan J, et al. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Molecular and cellular biology. 2013;33:918–926. doi: 10.1128/MCB.01227-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almahariq M, Mei FC, Cheng X. Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends in endocrinology and metabolism: TEM. 2014;25:60–71. doi: 10.1016/j.tem.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vliem MJ, et al. 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. Chembiochem: a European journal of chemical biology. 2008;9:2052–2054. doi: 10.1002/cbic.200800216. [DOI] [PubMed] [Google Scholar]
- 26.Schwede F, et al. Structure-guided design of selective Epac1 and Epac2 agonists. PLoS Biol. 2015;13:e1002038. doi: 10.1371/journal.pbio.1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang J, Rudnick DA. Elucidating the metabolic regulation of liver regeneration. The American journal of pathology. 2014;184:309–321. doi: 10.1016/j.ajpath.2013.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudnick DA, Perlmutter DH, Muglia LJ. Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8885–8890. doi: 10.1073/pnas.151217998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyaoka Y, et al. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Current biology: CB. 2012;22:1166–1175. doi: 10.1016/j.cub.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 30.Liu M, Chen P. Proliferationinhibiting pathways in liver regeneration (Review) Mol Med Rep. 2017;16:23–35. doi: 10.3892/mmr.2017.6613. [DOI] [PubMed] [Google Scholar]
- 31.Tan Q, et al. The Role of IL-1 Family Members and Kupffer Cells in Liver Regeneration. Biomed Res Int. 2016;2016:6495793. doi: 10.1155/2016/6495793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shteyer E, Liao Y, Muglia LJ, Hruz PW, Rudnick DA. Disruption of hepatic adipogenesis is associated with impaired liver regeneration in mice. Hepatology. 2004;40:1322–1332. doi: 10.1002/hep.20462. [DOI] [PubMed] [Google Scholar]
- 33.Kai AK, et al. Exchange protein activated by cAMP 1 (Epac1)-deficient mice develop beta-cell dysfunction and metabolic syndrome. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:4122–4135. doi: 10.1096/fj.13-230433. [DOI] [PubMed] [Google Scholar]
- 34.Newberry EP, et al. Altered hepatic triglyceride content after partial hepatectomy without impaired liver regeneration in multiple murine genetic models. Hepatology. 2008;48:1097–1105. doi: 10.1002/hep.22473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng J, et al. Enhanced Liver Regeneration After Partial Hepatectomy in Sterol Regulatory Element-Binding Protein (SREBP)-1c-Null Mice is Associated with Increased Hepatocellular Cholesterol Availability. Cell Physiol Biochem. 2018;47:784–799. doi: 10.1159/000490030. [DOI] [PubMed] [Google Scholar]
- 36.Bracht T, et al. Proteome analysis of a hepatocyte-specific BIRC5 (survivin)-knockout mouse model during liver regeneration. Journal of proteome research. 2014;13:2771–2782. doi: 10.1021/pr401188r. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, et al. Ablation of cytochrome P450 omega-hydroxylase 4A14 gene attenuates hepatic steatosis and fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:3181–3185. doi: 10.1073/pnas.1700172114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jungermann K, Kietzmann T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31:255–260. doi: 10.1002/hep.510310201. [DOI] [PubMed] [Google Scholar]
- 39.Rakhshandehroo Maryam, Knoch Bianca, Müller Michael, Kersten Sander. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Research. 2010;2010:1–20. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang W, et al. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–236. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 41.Cheema SK, Agellon LB. The murine and human cholesterol 7alpha-hydroxylase gene promoters are differentially responsive to regulation by fatty acids mediated via peroxisome proliferator-activated receptor alpha. The Journal of biological chemistry. 2000;275:12530–12536. doi: 10.1074/jbc.275.17.12530. [DOI] [PubMed] [Google Scholar]
- 42.Kliewer SA, Mangelsdorf DJ. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig Dis. 2015;33:327–331. doi: 10.1159/000371670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valsecchi F, Ramos-Espiritu LS, Buck J, Levin LR, Manfredi G. cAMP and mitochondria. Physiology (Bethesda) 2013;28:199–209. doi: 10.1152/physiol.00004.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, et al. Identification of novel modulators of mitochondrial function by a genome-wide RNAi screen in Drosophila melanogaster. Genome Res. 2008;18:123–136. doi: 10.1101/gr.6940108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiao J, Mei FC, Popov VL, Vergara LA, Cheng X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J Biol Chem. 2002;277:26581–26586. doi: 10.1074/jbc.M203571200. [DOI] [PubMed] [Google Scholar]
- 46.DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci USA. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tao Y, Wang M, Chen E, Tang H. Liver Regeneration: Analysis of the Main Relevant Signaling Molecules. Mediators Inflamm. 2017;2017:4256352. doi: 10.1155/2017/4256352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123:1902–1910. doi: 10.1172/JCI66369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng Z, et al. Ten paths of PKA signaling pathway regulate hepatocyte proliferation in rat liver regeneration. Genes & Genomics. 2012;34:391–399. doi: 10.1007/s13258-011-0195-x. [DOI] [PubMed] [Google Scholar]
- 50.Diehl AM, Yang SQ, Wolfgang D, Wand G. Differential expression of guanine nucleotide-binding proteins enhances cAMP synthesis in regenerating rat liver. The Journal of clinical investigation. 1992;89:1706–1712. doi: 10.1172/JCI115771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Servillo G, Della Fazia MA, Sassone-Corsi P. Coupling cAMP signaling to transcription in the liver: pivotal role of CREB and CREM. Experimental cell research. 2002;275:143–154. doi: 10.1006/excr.2002.5491. [DOI] [PubMed] [Google Scholar]
- 52.Liu WH, et al. Hepatocyte proliferation during liver regeneration is impaired in mice with methionine diet-induced hyperhomocysteinemia. The American journal of pathology. 2010;177:2357–2365. doi: 10.2353/ajpath.2010.091131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Groeneveld D, et al. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood. 2019;133:1245–1256. doi: 10.1182/blood-2018-08-869057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirschbaum M, et al. Transient von Willebrand factor-mediated platelet influx stimulates liver regeneration after partial hepatectomy in mice. Liver Int. 2017;37:1731–1737. doi: 10.1111/liv.13386. [DOI] [PubMed] [Google Scholar]
- 55.Lesurtel M, et al. Platelet-derived serotonin mediates liver regeneration. Science. 2006;312:104–107. doi: 10.1126/science.1123842. [DOI] [PubMed] [Google Scholar]
- 56.Nygaard G, et al. Epac1-deficient mice have bleeding phenotype and thrombocytes with decreased GPIbbeta expression. Sci Rep. 2017;7:8725. doi: 10.1038/s41598-017-08975-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robichaux WG, 3rd, Cheng X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol Rev. 2018;98:919–1053. doi: 10.1152/physrev.00025.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elchaninov AV, et al. Dynamics of macrophage populations of the liver after subtotal hepatectomy in rats. BMC Immunol. 2018;19:23. doi: 10.1186/s12865-018-0260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eroglu A, et al. Effect of granulocyte-macrophage colony-stimulating factor on hepatic regeneration after 70% hepatectomy in normal and cirrhotic rats. HPB (Oxford) 2002;4:67–73. doi: 10.1080/136518202760378425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michalopoulos GK. Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology. 2017;65:1384–1392. doi: 10.1002/hep.28988. [DOI] [PubMed] [Google Scholar]
- 61.Hunt AN, et al. Hepatic Steatosis Accompanies Pulmonary Alveolar Proteinosis. Am J Respir Cell Mol Biol. 2017;57:448–458. doi: 10.1165/rcmb.2016-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. 2013;3:785–797. doi: 10.1002/cphy.c120026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Almahariq M, Mei FC, Cheng X. The pleiotropic role of exchange protein directly activated by cAMP 1 (EPAC1) in cancer: implications for therapeutic intervention. Acta biochimica et biophysica Sinica. 2016;48:75–81. doi: 10.1093/abbs/gmv115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zou Y, et al. Four waves of hepatocyte proliferation linked with three waves of hepatic fat accumulation during partial hepatectomy-induced liver regeneration. PLoS One. 2012;7:e30675. doi: 10.1371/journal.pone.0030675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rai RM, et al. Kupffer cell depletion abolishes induction of interleukin-10 and permits sustained overexpression of tumor necrosis factor alpha messenger RNA in the regenerating rat liver. Hepatology. 1997;25:889–895. doi: 10.1002/hep.510250417. [DOI] [PubMed] [Google Scholar]
- 67.Garcia-Arcos I, et al. Lipid analysis reveals quiescent and regenerating liver-specific populations of lipid droplets. Lipids. 2010;45:1101–1108. doi: 10.1007/s11745-010-3492-2. [DOI] [PubMed] [Google Scholar]
- 68.Pauta M, et al. Impaired liver regeneration in Ldlr-/- mice is associated with an altered hepatic profile of cytokines, growth factors, and lipids. Journal of hepatology. 2013;59:731–737. doi: 10.1016/j.jhep.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hardwick JP. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochemical pharmacology. 2008;75:2263–2275. doi: 10.1016/j.bcp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 70.Chatuphonprasert W, Nemoto N, Sakuma T, Jarukamjorn K. Modulations of cytochrome P450 expression in diabetic mice by berberine. Chem Biol Interact. 2012;196:23–29. doi: 10.1016/j.cbi.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 71.Yoshinari K, Takagi S, Sugatani J, Miwa M. Changes in the expression of cytochromes P450 and nuclear receptors in the liver of genetically diabetic db/db mice. Biol Pharm Bull. 2006;29:1634–1638. doi: 10.1248/bpb.29.1634. [DOI] [PubMed] [Google Scholar]
- 72.Enriquez A, Leclercq I, Farrell GC, Robertson G. Altered expression of hepatic CYP2E1 and CYP4A in obese, diabetic ob/ob mice, and fa/fa Zucker rats. Biochemical and biophysical research communications. 1999;255:300–306. doi: 10.1006/bbrc.1999.0202. [DOI] [PubMed] [Google Scholar]
- 73.Glen J, Floros L, Day C, Pryke R, Guideline Development G. Non-alcoholic fatty liver disease (NAFLD): summary of NICE guidance. Bmj. 2016;354:i4428. doi: 10.1136/bmj.i4428. [DOI] [PubMed] [Google Scholar]
- 74.Hwang M, et al. Epac2a-null mice exhibit obesity-prone nature more susceptible to leptin resistance. International journal of obesity. 2017;41:279–288. doi: 10.1038/ijo.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee K, et al. Involvement of cAMP-guanine nucleotide exchange factor II in hippocampal long-term depression and behavioral flexibility. Molecular brain. 2015;8:38. doi: 10.1186/s13041-015-0130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shibasaki T, et al. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suzuki S, et al. Differential roles of Epac in regulating cell death in neuronal and myocardial cells. The Journal of biological chemistry. 2010;285:24248–24259. doi: 10.1074/jbc.M109.094581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang Y, et al. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron. 2012;73:774–788. doi: 10.1016/j.neuron.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song WJ, Mondal P, Li Y, Lee SE, Hussain MA. Pancreatic beta-cell response to increased metabolic demand and to pharmacologic secretagogues requires EPAC2A. Diabetes. 2013;62:2796–2807. doi: 10.2337/db12-1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Okumura S, et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. The Journal of clinical investigation. 2014;124:2785–2801. doi: 10.1172/JCI64784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kopperud RK, et al. Increased microvascular permeability in mice lacking Epac1 (Rapgef3) Acta Physiol (Oxf) 2017;219:441–452. doi: 10.1111/apha.12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edland F, et al. Long-term consumption of an obesogenic high fat diet prior to ischemia-reperfusion mediates cardioprotection via Epac1-dependent signaling. Nutrition & metabolism. 2016;13:87. doi: 10.1186/s12986-016-0147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aesoy R, et al. Deletion of exchange proteins directly activated by cAMP (Epac) causes defects in hippocampal signaling in female mice. PloS one. 2018;13:e0200935. doi: 10.1371/journal.pone.0200935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takahashi H, et al. Role of Epac2A/Rap1 signaling in interplay between incretin and sulfonylurea in insulin secretion. Diabetes. 2015;64:1262–1272. doi: 10.2337/db14-0576. [DOI] [PubMed] [Google Scholar]
- 85.Barker Graeme, Parnell Euan, van Basten Boy, Buist Hanna, Adams David, Yarwood Stephen. The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation. Journal of Cardiovascular Development and Disease. 2017;4(4):22. doi: 10.3390/jcdd4040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nature protocols. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 87.Henderson NC, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5060–5065. doi: 10.1073/pnas.0511167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lynch RW, et al. An efficient method to isolate Kupffer cells eliminating endothelial cell contamination and selective bias. Journal of leukocyte biology. 2018;104:579–586. doi: 10.1002/JLB.1TA0517-169R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gubern C, et al. Validation of housekeeping genes for quantitative real-time PCR in in-vivo and in-vitro models of cerebral ischaemia. BMC molecular biology. 2009;10:57. doi: 10.1186/1471-2199-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kosir R, et al. Determination of reference genes for circadian studies in different tissues and mouse strains. BMC molecular biology. 2010;11:60. doi: 10.1186/1471-2199-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pernot F, Dorandeu F, Beaup C, Peinnequin A. Selection of reference genes for real-time quantitative reverse transcription-polymerase chain reaction in hippocampal structure in a murine model of temporal lobe epilepsy with focal seizures. Journal of neuroscience research. 2010;88:1000–1008. doi: 10.1002/jnr.22282. [DOI] [PubMed] [Google Scholar]
- 92.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 93.Hernandez-Valladares M, et al. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biological procedures online. 2016;18:13. doi: 10.1186/s12575-016-0043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scholz C, et al. Avoiding abundance bias in the functional annotation of post-translationally modified proteins. Nature methods. 2015;12:1003–1004. doi: 10.1038/nmeth.3621. [DOI] [PubMed] [Google Scholar]
- 95.Vizcaino JA, et al. 2016 update of the PRIDE database and its related tools. Nucleic acids research. 2016;44:D447–456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vizcaino JA, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature biotechnology. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics raw files and data were uploaded to the ProteomeXchange consortium (version 2.3.0) via the PRIDE partner repository, and with the dataset identifier PXD00616595,96. A detailed description of the LC-MS analysis is provided in the Supplementary Material & Methods section.