

Abstract
Dopamine (DA) is a well established modulator of prefrontal cortex (PFC) function, yet the cellular mechanisms by which DA exerts its effects in this region are controversial. A major point of contention is the consequence of D1 DA receptor activation. Several studies have argued that D1 receptors enhance the excitability of PFC pyramidal neurons by augmenting voltage-dependent Na+ currents, particularly persistent Na+ currents. However, this conjecture is based on indirect evidence. To provide a direct test of this hypothesis, we combined voltage-clamp studies of acutely isolated layer V–VI prefrontal pyramidal neurons with single-cell RT-PCR profiling. Contrary to prediction, the activation of D1 or D5 DA receptors consistently suppressed rapidly inactivating Na+ currents in identified corticostriatal pyramidal neurons. This modulation was attenuated by a D1/D5 receptor antagonist, mimicked by a cAMP analog, and blocked by a protein kinase A (PKA) inhibitor. In the same cells the persistent component of the Na+current was unaffected by D1/D5 receptor activation—suggesting that rapidly inactivating and persistent Na+ currents arise in part from different channels. Single-cell RT-PCR profiling showed that pyramidal neurons coexpressed three α-subunit mRNAs (Nav1.1, 1.2, and 1.6) that code for the Na+ channel pore. In neurons from Nav1.6 null mice the persistent Na+ currents were significantly smaller than in wild-type neurons. Moreover, the residual persistent currents in these mutant neurons—which are attributable to Nav1.1/1.2 channels—were reduced significantly by PKA activation. These results argue that D1/D5 DA receptor activation reduces the rapidly inactivating component of Na+ current in PFC pyramidal neurons arising from Nav1.1/1.2 Na+ channels but does not modulate effectively the persistent component of the Na+current that is attributable to Nav1.6 Na+channels.
Keywords: voltage-clamp, scRT-PCR, neuromodulation, monoamine, Na+ channel, molecular biology, protein kinase A, DA receptor, corticostriatal
Disordered dopaminergic signaling in the prefrontal cortex (PFC) has been implicated in a number of neuropsychiatric disorders, including schizophrenia. Despite its importance, the mechanisms by which dopamine (DA) modulates the activity of PFC neurons are controversial. In vivo studies suggest that spontaneous firing of PFC neurons is decreased either by activation of the mesocortical dopaminergic pathway or by application of iontophoretic DA (Bernardi et al., 1982; Mantz et al., 1988; Pirot et al., 1992). DA also has been shown to inhibit evoked activity in the PFC (Ferron et al., 1984). A number of in vitro studies are consistent with the conclusion that DA suppresses the excitability of layer V pyramidal neurons (Geijo-Barrientos and Pastore, 1995; Gulledge and Jaffe, 1998). However, other in vitro studies suggest that DA enhances the excitability of these neurons (Penit-Soria et al., 1987; Yang and Seamans, 1996; Shi et al., 1997; Gorelova and Yang, 2000).
The excitatory effects of DA in the PFC have been attributed to the activation of D1 class dopaminergic receptors. It has been argued that D1 receptor activation enhances Na+ currents, particularly persistent Na+ currents, in PFC pyramidal neurons (Yang and Seamans, 1996; Gorelova and Yang, 2000). However, this conclusion is based solely on recordings from neurons in slices in which it is virtually impossible to control adequately the electrotonically remote regenerative Na+currents. In studies in which these distal dendritic regions have been eliminated to achieve adequate voltage control, the consequences of D1 receptor stimulation appear to be qualitatively different. In both striatal and hippocampal neurons D1 receptors have been shown to suppress rapidly inactivating Na+ currents via the activation of protein kinase A (PKA) (Surmeier et al., 1992; Surmeier and Kitai, 1993; Schiffmann et al., 1995; Cantrell et al., 1997, 1999). These studies are consistent with work in heterologous expression systems showing that PKA phosphorylation of Nav1.1 or Nav1.2 Na+ channels reduces rapidly inactivating Na+ currents (Gershon et al., 1992; Li et al., 1992, 1993; Smith and Goldin, 1996, 1998).
In contrast to rapidly inactivating currents, the modulation of persistent Na+ currents by PKA has not been explored as thoroughly. This component of the Na+ current has been attributed to a “persistent” gating mode of channels underlying the rapidly inactivating current (Patlak, 1991; Alzheimer et al., 1993; Crill, 1996). Although both Nav1.1 and Nav1.6 channels produce currents with prominent persistent components in heterologous systems (Goldin, 1999), studies of brain neurons have implicated primarily the Nav1.6 channels in the generation of persistent currents (de Miera et al., 1997; Raman et al., 1997). It is unclear at this point how PKA phosphorylation of the Nav1.6 channel modulates its gating. This uncertainty makes it possible that, if Nav1.6 channels are major determinants of persistent Na+ currents in PFC pyramidal neurons, D1 receptor activation of PKA alters channel gating in a qualitatively different way than in previously studied cell types.
Our aim was to answer three questions. First, how does the activation of D1 receptors modulate rapidly inactivating Na+ currents in PFC pyramidal neurons in a preparation in which these currents can be voltage-clamped adequately? Second, are persistent Na+ currents in PFC pyramidal neurons modulated by D1 receptor activation? Third, if persistent Na+currents respond in a qualitatively different way to D1 receptor activation, is this difference attributable to channel heterogeneity? To answer these questions, we studied acutely isolated rodent layer V–VI PFC pyramidal neurons with voltage-clamp and single-cell RT-PCR techniques.
MATERIALS AND METHODS
Acute dissociation procedure. Prelimbic and infralimbic PFC pyramidal cells were obtained from young adult (3–4 weeks) Sprague Dawley rats (Harland, Indianapolis, IN). In some experiments Nav1.6 null mutant (Scn8amed) and congenic control mice (3 weeks) were used (Burgess et al., 1995; Kohrman et al., 1996). In both cases the neurons were acutely dissociated by using procedures similar to those previously described (Surmeier et al., 1995). Briefly, animals were anesthetized with isoflurane and decapitated; then brains were removed quickly, iced, and blocked for slicing. Sagittal slices (350 μm) were cut with a Leica VT1000S slicer (Leitz, Nussloch, Germany) while being bathed in a high-sucrose solution [containing (in mm): 250 sucrose, 11 glucose, 15 HEPES, 4 MgSO4, 1 Na2HPO4, 2.5 KCl, 1 kynurenic acid, 1 N-nitro-l-arginine, and 0.1 glutathione, pH 7.4, 300–305 mOsm/l]. Then the slices were incubated for 1–6 hr at room temperature (20–22°C) in a NaHCO3-buffered saline solution bubbled with 95% O2/5% CO2 [containing (in mm): 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 1 pyruvic acid, 0.2 ascorbic acid, 0.1NG-nitro-l-arginine, 1 kynurenic acid, and 10 glucose, pH 7.4, 300–305 mOsm/l]. All reagents were obtained from Sigma (St. Louis, MO). Next the slices were removed into low Ca2+ HEPES-buffered saline [containing (in mm): 140 Na-isothionate, 2 KCl, 4 MgCl2, 0.1 CaCl2, 23 glucose, and 15 HEPES, pH 7.4, 300–305 mOsm/l]; with the aid of a dissecting microscope the deep layers of the PFC were dissected and placed in an oxygenated Cell-Stir chamber (Wheaton, Millville, NJ) containing Pronase (1–2 mg/ml, Sigma protease type XIV) in HEPES-buffered HBSS at 37°C. In some experiments papain (20 U/ml, Worthington Biochemical, Lakewood, NJ) was used instead of Pronase. After 35–40 min of enzyme digestion the tissue was rinsed three times in zero Ca2+ buffer and dissociated mechanically with a graded series of fire-polished Pasteur pipettes. Then the cell suspension was plated into a 35 mm Petri dish (Nalge Nunc, Naperville, IL) containing saline; next, the dish was placed on the stage of an inverted microscope.
Retrograde labeling. In some cases PFC pyramidal cells were identified as projecting to the nucleus accumbens by retrograde labeling. For this purpose, 2–3 d before recording the animals were anesthetized with a mixture of ketamine (100 mg/kg; Fort Dodge Animal Health, Fort Dodge, IA) and xylazine (100 mg/kg; Phoenix Scientific, St. Joseph, MO); Fluoro-Gold (3% w/v; Fluorochrome, Denver, CO) was injected stereotaxically into the core of the nucleus accumbens with a microsyringe needle. Retrogradely labeled neurons, obtained by following the procedures described above, were observed with a fluorescence microscope, using a wide-band ultraviolet excitation filter.
Pharmacological agents. All of the stock solutions were prepared in water. Stock solutions of cBIMPS and Rp-cAMPS (Biolog Life Science Institute, Bremen, Germany) were aliquoted and frozen. Stock solutions of SKF 81297 and SCH 23390 (Sigma) were prepared freshly and protected from ambient light. Each of the stocks was diluted to the appropriate concentrations in the external recording solution immediately before the experiment.
Whole-cell recordings. Electrodes were pulled from Corning (Corning, NY) 7052 glass, coated with Sylgard (Dow Corning, Midland, MI), and fire-polished before use. The internal solution consisted of (in mm) 130N-methyl-d-glucamine, 20 HEPES, 20 CsCl, 2 MgCl2, 12 phosphocreatine, 2 Mg-ATP, 0.7 Na2GTP, and 0.1 leupeptin, pH 7.2, with CsOH/H2SO4 (osmolarity, 260–270 mOsm/l). The external solution consisted of (in mm) 15 NaCl, 110 tetraethylammonium chloride (TEA-Cl), 10 HEPES, 1 MgCl2, 2 BaCl2, and 0.3 CdCl2, pH 7.4, with CsOH (osmolarity, 300–305 mOsm/l). The persistent Na+ current was recorded by using an external solution containing (in mm) 115 NaCl, 45 TEA-Cl, 10 HEPES, 10 CsCl, 1 MgCl2, 2 BaCl2, and 0.3 CdCl2, pH 7.4, with CsOH (osmolarity, 300–305 mOsm/l). Solutions were applied by a gravity-fed sewer pipe system. An array of application capillaries (∼400 μm inner diameter) was positioned a few hundred micrometers from the cell under study. Solution changes were effected by altering the position of the array with a DC drive system controlled by a microprocessor-based controller (Newport, Irvine, CA). Solutions changes were complete within <1 sec. Recording were obtained with anAxon Instruments 200 patch-clamp amplifier (Foster City, CA) and controlled and monitored with a Macintosh computer running Pulse software (version 8.3, HEKA Elektronik, Lambrecht, Germany) with an ITC-Computer interface (Instrutech, Great Neck, NY). Electrode resistance was typically 1.5–3 MΩ in the bath. After formation of the gigaohm seal and subsequent cell rupture, series resistance was compensated (80–85%) and monitored periodically.
Data analyses. These were performed with IgorPro (WaveMetrics, Lake Oswego, OR) and Systat 5 running on a Macintosh computer (Systat, Evanston, IL). Curve fitting was done with IgorPro, using a least-squares criterion. Sample statistics are given as means or medians and ranges. Small nonmatched samples were analyzed with Kruskal–Wallis ANOVA. Box plots were used for graphic presentation of the data because of the small sample sizes (Tukey, 1977). The box plot represents the distribution as a box, with the median as a central line and the hinges as the edges of the box (the hinges divide the upper and lower halves of the distributions in two). The inner fences (shown as a line originating from the edges of the box) run to the limits of the distribution, excluding outliers [defined as points that are >1.5 times the interquartile range beyond the inner fence (Tukey, 1977)]. The outliers are shown as circles.
Single-cell reverse transcription-PCR procedures. In some experiments the neurons were aspirated after a recording in the whole-cell configuration. In these cases the recording solution was made RNase-free, and the total volume was kept to ∼5 μl in the electrode. The capillary glass used for the electrodes was autoclaved to 200°C for 1 hr. Sterile gloves were worn during all of the procedures to minimize RNase contamination. After aspiration the electrode was removed from the holder, and the content was ejected into a 0.5 ml Eppendorf tube (Madison, WI) containing 2.9 μl of diethyl pyrocarbonate-treated water, 0.7 μl of RNasin (28,000 U/ml), 0.7 μl of BSA (0.14 μg/μl), and 0.7 μl of oligo-dT (0.5 μg/μl). The mixture was heated to 70°C for 10 min and incubated on ice for 1 min. Single-strand cDNA was synthesized from the cellular mRNAs by the addition of SuperScript II RT (1 μl, 200 U/μl), 10× PCR buffer, MgCl2 (2 μl, 25 mm), DTT (2 μl, 0.1 m), and mixed dNTPs (1 μl, 10 mm), followed by incubation at 42°C for 50 min. The reaction was terminated by heating the mixture to 70°C for 15 min and then cooling it to 0°C. The RNA strand in the RNA–DNA hybrid was removed by adding 1 μl of RNase H (2 U/ml) and was incubated for 20 min at 37°C. All reagents except RNasin (Promega, Madison, WI) were obtained from Life Technologies (Grand Island, NY). The cDNA from the reverse transcription (RT) of RNA from a single PFC neuron was subjected to PCR to detect the expression of various mRNAs. Conventional PCR was performed with a thermal cycler (MJ Research, Watertown, MA). PCR primers were developed from GenBank with the commercially available software OLIGO (National Biosciences, Plymouth, MN). Primers for D1 and D5 DA receptors and for calmodulin kinase II (CaMKII) have been described previously (Yan et al., 1997; Vysokanov et al., 1998). Nav1.1 mRNA (GenBank accession number X03638) was detected with a pair of primers 5′-GAC CGG GTG ACA AAG CCA ATC (position 6210) and 5′-CCC TTT ACG CTG GTC CCT ACA GTC T (position 6538), which give a PCR product of 353 base pairs (bp). Nav1.2 mRNA (GenBank accession number X03639) was detected with a pair of primers 5′-CCT TCC ACA ACT TCT CCA CCT TCC TA (position 6109) and 5′-ATA TGG CAG GTG TGG CAG TTA AAA CA (position 6614), which give a PCR product of 531 bp. Nav1.5 mRNA (GenBank accession number M27902) was detected with a pair of primers 5′-TCT CCA GAT AGG GAC CGA GAG TCT (position 6225) and 5′-GGG TTA AGG AGA GGC AGT GTG AAC (position 6643), which give a PCR product of 442 bp. Nav1.6 mRNA (GenBank accession number L39018) was detected with a pair of primers 5′-AGA GGT CAG GGA GTC CAA GTG CTA (position 5907) and 5′-CGT CTG CCC AAG CGA TAG GAG (position 6142), which give a PCR product of 256 bp.
PCR techniques were performed by following procedures designed to minimize the chance of cross-contamination. Negative controls for contamination from extraneous and genomic DNA were run for every batch of neurons. To ensure that genomic DNA did not contribute to the PCR products, we aspirated and processed neurons in the normal manner, except that the reverse transcriptase was omitted. Contamination from extraneous sources was checked by replacing the cellular template with water. Both controls were consistently negative in these experiments.
RESULTS
PFC neurons from layers V–VI were acutely isolated for study, because previous studies have focused on the deeper cortical layers. Pyramidal neurons from these layers had a characteristic pyramidal shape with one apical dendrite (Fig.1A) and expressed CaMKII mRNA (see Figs. 3A, 4C) (Benson et al., 1992). To provide an additional level of certainty, we retrogradely labeled PFC pyramidal neurons projecting to the nucleus accumbens with Fluoro-Gold (Fig. 1A). The functional measures taken from these retrogradely labeled neurons were very similar to those derived from the majority of unlabeled neurons.
Fig. 1.
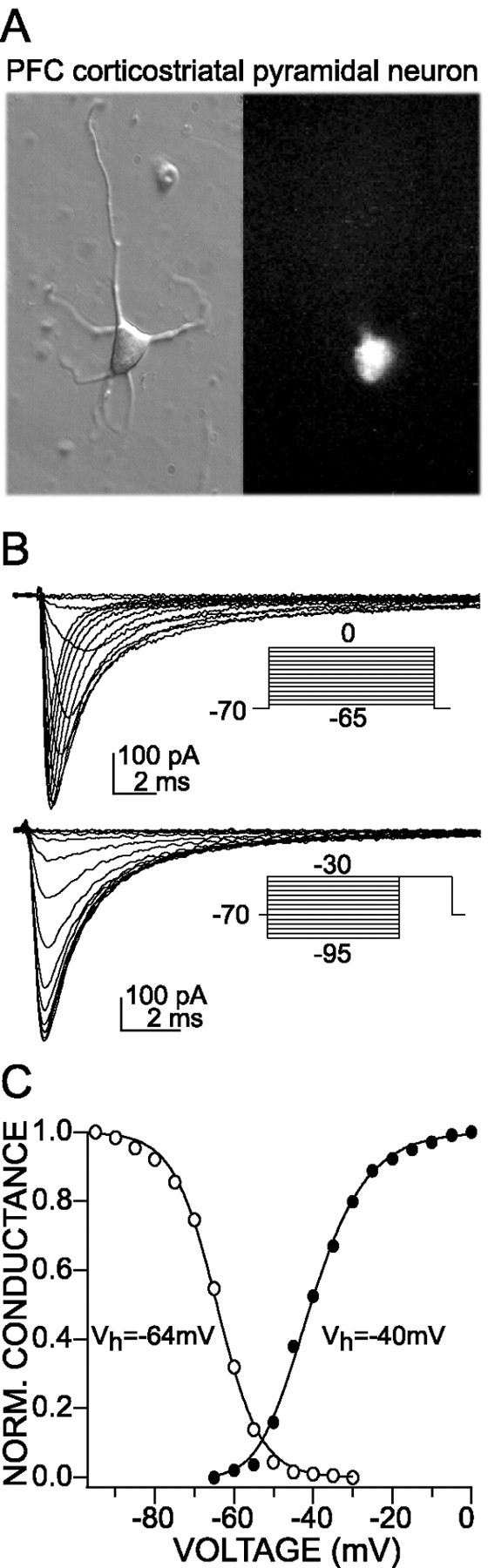
Steady-state activation and inactivation characteristics of transient Na+ currents in rat PFC neurons projecting to nucleus accumbens. A, A PFC pyramidal neuron that was retrogradely labeled from the nucleus accumbens with Fluoro-Gold (3%). Left, A bright-field photomicrograph of the acutely isolated neuron. Right, The epifluorescent view of the same field. B, Representative protocols and current traces (TTX-subtracted) that were used to study the voltage dependence of activation (top) and steady-state inactivation (bottom).C, Plot of the peak conductance as a function of the test pulse voltage (filled circles) and of the postpulse currents as a function of prepulse voltage (open circles). The lines are the best fit to the Boltzmann equation (activation: Vh = −40 mV, Vc = 6.7 mV; inactivation:Vh = −64 mV,Vc = 5.5 mV).
Fig. 3.

D1/D5 receptor activation reduces the rapidly inactivating Na+current. A, The scRT-PCR revealed that D1and/or D5 mRNA was expressed in PFC neurons. Shown are photographs of gels derived from two pyramidal neurons expressing CaMKII mRNA. One had detectable levels of D1 receptor mRNA; the other had detectable levels of D5 receptor mRNA. The sizing ladder is in the left-most lane of both gels.B, Plot of peak Na+ current evoked by a step from a holding potential of −80 to −30 mV as function of time. D1/D5 receptor agonist SKF 81297 (1 μm) reversibly suppresses the peak current.Inset, Representative currents used to constructB. C, Plot of peak Na+current evoked by a step from a holding potential of −80 to −35 mV as function of time. Dopamine application (50 μm) also reversibly suppresses the peak current. Inset, Representative currents used to construct C.D, Box plot summary of the modulation of Na+ transient current. SCH 23390 (1 μm) blocked the effect of SKF 81297 (1 μm;n = 7). This effect was mimicked by cBIMPS (50 μm; n = 7), a PKA activator, and was blocked by Rp-cAMPS (10 μm; n = 4), a PKA inhibitor, indicating the involvement of the PKA in the response that was observed. E, A representative steady-state inactivation plot derived from a PFC pyramidal neuron. SKF 81297 (1 μm) reduced the maximum current evoked by a step to −30 mV and shifted the voltage dependence of steady-state inactivation to slightly more negative potentials (Control:Vh = −62 mV,Vc = 5 mV; SKF:Vh = −65 mV,Vc = 4.9 mV). F, Current traces and protocols used to construct the steady-state inactivation plot shown in D.
Fig. 4.

The persistent Na+ current is not modulated by D1/D5 receptor stimulation. A, SKF 81297 (1 μm) failed to modulate the persistent Na+ current but decreased the fast transient Na+ current (shown ininset) in the same cell (protocol for theinset is the same as in Fig. 3B). The persistent Na+ current traces are TTX-subtracted and fit with a polynomial function. B, In cells in which the peak of the rapidly inactivating current was reduced by SKF 81297 (1 μm), the modulation was not seen 10–15 msec into the step pulse where only the persistent current would remain.Inset, Box plot summary of the percentage of modulation induced by SKF 81297 (1 μm) in the rapidly inactivating current at the peak (peak) and at 20 msec into the step pulse (late) in 15 mmNa+ (n = 6) and in persistent (n = 6) Na+ current measured at −25 mV, using the ramp protocol in 115 mmNa+. C, Single-cell RT-PCR profile of a PFC pyramidal neuron showing a coexpression of Nav1.1, 1.2, and 1.6 Na+ channel α-subunit mRNAs. D, Bar graph summarizing Nav1.1, 1.2, 1.5, and 1.6 mRNA detection in 40 PFC pyramidal neurons (including 21 retrogradely labeled corticoaccumbal neurons); the total length of each bar codes the percentage of the sample in which a particular mRNA was detected.
PFC pyramidal neurons display both rapidly inactivating and persistent Na+ currents
Both rapidly inactivating and slowly inactivating/persistent Na+ currents could be evoked in acutely isolated PFC pyramidal neurons. Rapidly inactivating currents were examined in an external solution containing a low Na+ concentration (15 mm) to reduce current amplitudes and to gain adequate control of the transmembrane voltage (the Na+concentration of the recording internal solution was ∼3 mm). At this external Na+concentration the properties of the currents evoked by depolarization were very similar to those in which the Na+ gradient was reversed, making the currents nonregenerative. Currents were evoked by standard activation and inactivation protocols (Fig. 1B), and steady-state plots were constructed by subtracting TTX-insensitive leak currents. Activation data were fit with a third-order Boltzmann function, whereas inactivation data were fit with single first-order Boltzmann function (Fig. 1C). The voltage dependence of activation (half-activation voltage,Vh = −38.4 ± 1.7 mV; slope factor, Vc = 5.8 ± 0.4 mV;n = 10) and inactivation (half-inactivation voltage,Vh = −66.2 ± 0.9 mV; slope factor, Vc = 6.2 ± 0.3 mV;n = 10) were similar to those found in a variety of other brain neurons (Surmeier et al., 1992; Cantrell et al., 1997).
All of the rat PFC pyramidal neurons that were tested (n = 31) exhibited a prominent persistent Na+ current (Crill, 1996). This component of the whole-cell Na+ current was defined operationally as the TTX-sensitive current, which was evoked by slowly ramping the membrane voltage from −80 to 0 mV (40 mV/sec; Fig.2A) in physiological concentrations of Na+ (115 mm). As shown in Figure 2A, this component of the Na+ current began to activate near −70 mV, peaked near −50 mV, and remained active at depolarized potentials. These properties are very similar to those described for persistent Na+ currents in cerebellar Purkinje cells, entorhinal cortex pyramidal neurons, and somatosensory cortex pyramidal neurons (Alzheimer et al., 1993; Kay et al., 1998; Magistretti and Alonso, 1999).
Fig. 2.

The persistent Na+ current evoked in PFC neurons is not solely attributable to a window current.A, Persistent Na+ currents evoked by slow voltage ramps (35 mV/sec, from −70 to 0 mV) in high external Na+ concentration (115 mm). The current was blocked completely by the application of TTX (300 nm).Inset, Leak-subtracted trace. B, In the same cell, in recording conditions (external Na+concentration, 15 mm) allowing for an adequate control of the transient Na+ currents, the steady-state activation and inactivation curves overlapped, generating a window current prominent between −65 and −35 mV. The window current has been increased 85-fold. C, D, The voltage dependence of the conductance underlying the persistent Na+ current is compared with the conductance resulting from the window current in two different cells. The predicted window current is represented by a dashedline, the conductance underlying the observed persistent current is represented by a thicksolid line, and the difference current showing the real persistent current is represented by a thin solid line. InC, the window current contributes to 45% of the persistent current when this participation is 25% in the cell shown inD. In both cells there is a clear difference between the window current and the persistent current.
In several neurons there was a prominent “hump” in the current–voltage relationship near −50 mV (see Fig.2A,C). These are precisely the position and shape expected of a window current (Hodgkin and Huxley, 1990). In this narrow voltage range there is a nonzero probability that a channel will be activated, but not inactivated, in the steady state. To estimate the contribution of this current to the measured persistent currents, we computed window currents by taking the product of the Boltzmann functions fit to the rapidly inactivating steady-state activation and inactivation plots in the same cell. An example of the estimated window current is shown on a normalized scale in Figure 2B. The window was significant between approximately −65 and −35 mV in most of the neurons that were examined (n = 10). The maximum window conductance was typically near 1% of the peak conductance of the rapidly inactivating current. The maximum conductance of the window current was estimated from that of the rapidly inactivating conductance and compared with that of the persistent current. At the peak of the persistent current (near −55 mV) the median contribution of the window current was estimated to be 25% of the total (n = 9). The maximum persistent conductance was 410 ± 86 pS, whereas the maximum potential contribution from a window conductance was 110 ± 49 pS (n = 9) in the same cells. Two examples are shown in Figure 2, C and D, in which the persistent estimated window and difference currents are plotted. Although the window current appears to be capable of making a larger contribution to the persistent current in PFC pyramidal neurons than in Purkinje neurons (Kay et al., 1998), it always has been a relatively small component of the total current. Moreover, at more positive membrane potentials (more than −35 mV) the window current is very small, but the persistent current is substantial. Together, these data argue that window currents make, at best, a small contribution to the persistent Na+ currents in PFC pyramidal neurons.
D1/D5 receptor activation decreases rapidly inactivating Na+ currents in PFC pyramidal neurons
In agreement with previous studies (Bergson et al., 1995; Gaspar et al., 1995), single-cell reverse transcription (scRT-PCR) analysis consistently revealed that PFC pyramidal neurons (n = 6) had detectable levels of D1 and/or D5 receptor mRNAs (Fig.3A). In most of the PFC pyramidal cells that were tested (84%, 16 of 19 neurons), the application of the D1/D5receptor agonist SKF 81297 (1 μm) reversibly suppressed the rapidly inactivating Na+currents evoked by a step from the holding potential (−70 mV) to −30 mV (average peak suppression, 27.6 ± 3.6%; n = 16; Fig. 3B,D). This D1/D5 receptor modulation also was observed in PFC pyramidal neurons retrogradely labeled from the nucleus accumbens (n = 5). Application of the D1/D5 receptor antagonist SCH 23390 (1 μm) significantly reduced the effect of equimolar SKF 81297 (average peak suppression, 10.7 ± 2.1%; n = 7; p < 0.05, Kruskal–Wallis; Fig. 3D), confirming that the observed effect resulted from D1/D5receptor activation. Dopamine (50 μm) also reversibly suppressed the rapidly inactivating Na+ currents in these neurons (average peak suppression, 37.7 ± 2.8%; n = 3; Fig.3C,D). A mixture of dopamine (50 μm) and the D2 receptor antagonist sulpiride (5 μm) had a similar effect (data not shown).
The best-characterized consequence of D1 class receptor stimulation is the activation of adenylyl cyclase, formation of cAMP, and activation of PKA (Stoof and Kebabian, 1981). As in other cell types (Surmeier and Kitai, 1993; Cantrell et al., 1997), D1/D5 receptor activation was mimicked by the membrane-permeant cAMP analog cBIMPS, a PKA activator (50 μm; average peak suppression, 23.4 ± 5.5%; n = 7; Fig. 3D). Conversely, the inclusion of the PKA inhibitor Rp-cAMPS (10 μm) in the patch pipette significantly reduced the effect of SKF 81297 (average peak suppression, 4.7 ± 1.2%; n = 4;p < 0.05, Kruskal–Wallis; Fig. 3D). These results argue that D1/D5receptor activation in layer V–VI PFC pyramidal neurons decreases rapidly inactivating Na+ currents via a PKA-dependent mechanism.
The D1/D5 receptor-mediated modulation of Na+ currents was produced primarily by a reduction in the peak open probability. There was also a consistent negative shift (Kruskal–Wallis, 0.1 >p > 0.05) in the voltage dependence of steady-state inactivation [Vh (control) = −66.5 ± 1.2 mV; Vh(D1/D5 agonist) = −70.1 ± 1.4 mV; n = 7; Figure 3E,F]. There was no detectable change in the voltage dependence of activation (data not shown). This pattern is very similar to that reported previously for striatal and hippocampal neurons (Surmeier et al., 1992;Cantrell et al., 1997).
D1/D5 receptor activation does not modulate the persistent Na+ current efficiently
SKF 81297 (1 μm) failed to alter significantly the persistent Na+ current in any of the rat pyramidal neurons that were studied (n = 21). An example drawn from one of these experiments is shown in Figure4A. To provide a positive control for the efficacy of the receptor activation, we examined the ability of SKF 81297 to modulate the rapidly inactivating Na+ current in the same neuron after testing the persistent current. As shown in the inset of Figure4A, the D1/D5 receptor agonist consistently reduced the rapidly inactivating currents in this paradigm. In four neurons that were tested in this way, the peak of the rapidly inactivating current was reduced by 31 ± 5%, whereas the persistent current was not altered significantly (Fig.4A). In those cells in which the peak of the rapidly inactivating current was reduced substantially by SKF 81297, it was difficult to discern the modulation 10–15 msec into the step response (Fig. 4B; median modulation, 0%; n = 5), providing additional evidence for a differential modulation of the persistent currents.
PFC pyramidal neurons coexpress Na+ channel α-subunit mRNAs
The simplest interpretation of the differential sensitivity of rapidly inactivating and persistent Na+currents to D1/D5 receptor activation is that they are attributable to different channel types. The pore-forming α-subunit of the Na+channel is the principal target of PKA (Costa et al., 1982; Costa and Catterall, 1984; Rossie and Catterall, 1987, 1989; Murphy et al., 1993;Cantrell et al., 1997). If the pattern of modulation is attributable to different channel types, then pyramidal neurons should express more than one α-subunit mRNA. To test this hypothesis, we profiled 40 PFC pyramidal neurons by using scRT-PCR techniques for Nav1.1, 1.2, 1.5, and 1.6 mRNAs—the four α-subunit mRNAs known to be expressed in the adult brain (Goldin, 1999; Hartmann et al., 1999). Twenty-one of these were retrogradely labeled corticoaccumbal neurons. Nav1.1, 1.2, and 1.6 mRNAs were seen consistently, with the most common pattern being the codetection of all three (Fig. 4C). A summary of the detection frequencies for each of these mRNAs is shown in Figure4D. There were no significant differences in the expression of Nav1.1, 1.2, and 1.6 mRNAs in corticoaccumbal and unlabeled deep layer PFC neurons. Nav1.5 mRNA was not detected in layer V–VI pyramidal neurons but was seen in pooled mRNA from either the PFC or the septum, in agreement with in situ hybridization experiments (Hartmann et al., 1999). The scRT-PCR profiling of septal neurons readily detected Nav1.5 mRNA, indicating that the transcript was detectable with single-cell techniques.
Nav1.6 Na+ channels make a prominent contribution to persistent Na+ currents
As shown above, the persistent Na+current in pyramidal PFC neurons cannot be attributed to a window current alone. A similar conclusion has been drawn for other cell types (Kay et al., 1998; Magistretti and Alonso, 1999). A commonly held view is that this current reflects an alternative, persistent gating mode of channels underlying the rapidly inactivating currents (Patlak, 1991;Crill, 1996). Studies in heterologous expression systems have shown that Nav1.1, Nav1.2, and Nav1.6 Na+channels give rise to rapidly inactivating and persistent Na+ currents (Goldin, 1999). Nav1.1 and Nav1.6 channels appear to enter this gating mode more frequently and produce more persistent current than Nav1.2 channels. However, several studies have suggested that Nav1.1 and Nav1.6 channels do not contribute equally to persistent currents that are found in brain neurons. For example, in cerebellar Purkinje neurons Nav1.6 channels appear to dominate persistent Na+ currents that are seen with somatic electrodes (Raman et al., 1997).
To determine whether the persistent Na+currents in PFC pyramidal neurons are dominated by Nav1.6 channels also, we studied cells from a Nav1.6 null mutant (Scn8amed) mouse (Burgess et al., 1995; Kohrman et al., 1996). As shown in Figure5A–C, the rapidly inactivating Na+ currents in PFC pyramidal neurons from the null mutants were indistinguishable in steady-state activation (wild-type: Vh = −31.9 ± 1.3 mV, Vc = 6.8 ± 0.2 mV, n = 13; null:Vh = −35 ± 1.6 mV,Vc = 6.2 ± 0.3 mV,n = 8) and inactivation (wild-type:Vh = −63.4 ± 1.1 mV,Vc = 6.1 ± 0.3 mV,n = 13; null: Vh = −61.8 ± 0.5 mV, Vc = 5.3 ± 0.3 mV, n = 8) properties from wild-type neurons. As expected from this result, the predicted window currents in wild-type and null neurons also were indistinguishable (see inset, Fig. 5C). However, the amplitude of the persistent Na+ current was reduced dramatically in pyramidal neurons from null mice (Fig. 5D). As shown in Figure 5E, the median persistent current density at −25 mV (outside the window region) was reduced by a factor of 3 in the Nav1.6 null mice [median current density, 1.5 pA/pF (n = 7); wild-type median current density, 4.6 pA/pF (n = 9);p < 0.05, Kruskal–Wallis; Fig. 5E]. In contrast, the density of the rapidly inactivating Na+ current in neurons from Nav1.6 null mice was reduced less substantially (Fig. 5F;p > 0.05, Kruskal–Wallis). These data indicate that Nav1.6 channels are major determinants of the persistent Na+ current in wild-type PFC pyramidal neurons, as in cerebellar Purkinje neurons.
Fig. 5.

A significant proportion of the persistent Na+ current is attributable to Na+ channels containing the Nav1.6 α-subunit.A,B, Comparison of the basic characteristics of the steady-state activation and inactivation kinetics of the rapidly inactivating Na+ currents in the wild-type (A) and null Nav1.6 allele mice (B) indicates that they are similar.C, The window current (inset) resulting from the rapidly inactivating Na+ currents is not modified in null allele mice (solid lines) as compared with controls (dottedlines).D, The persistent Na+ current is smaller in the null allele mice as compared with controls. The current traces represent the average of four cells in each case, and thesolid lines are the best corresponding polynomial fit. E, Box plot summary of the density of persistent Na+ current in wild-type (n = 7) and null allele mice (n= 7). F, Box plot summary of the density of the rapidly inactivating Na+ current indicating that there is no decrease in the null allele mice (n = 8) as compared with the control (n = 12).
Nav1.6 channels are modulated inefficiently by PKA activation in PFC pyramidal neurons
As described above, activation of the D1/D5 receptor/PKA cascade in wild-type PFC pyramidal neurons efficiently modulated the rapidly inactivating Na+ currents but failed to alter persistent Na+ currents significantly. Our working hypothesis was that this differential modulation was attributable to inefficient modulation of the channels underlying the persistent current. An alternative hypothesis was that PKA phosphorylation of the Na+ channel α-subunit altered gating in the rapidly inactivating, but not the persistent, mode. If the first hypothesis is correct, then persistent Na+ currents in neurons from the Nav1.6 null mutant should be modulated more efficiently by the activation of PKA than in wild-type neurons. Why? In mutant neurons the residual persistent Na+ current is attributable to Nav1.1/1.2 channels that have entered the alternative gating mode. As noted above, these channels are modulated efficiently by PKA phosphorylation. On the other hand, if the alternative hypothesis is correct, there should be no difference in the modulation of the persistent current in wild-type and null neurons.
As shown in Figure 6, activation of PKA with cBIMPS (50 μm) in neurons from Nav1.6 null mutants produced a robust reduction of persistent Na+ currents (mean reduction at −25 mV, 26.2 ± 5.5%; n = 5). In contrast, in neurons from wild-type mice, cBIMPS only modestly reduced persistent currents (mean reduction at −25 mV, 11.5 ± 2.7%; n = 10;p < 0.05, Kruskal–Wallis). The reduction of the persistent current seen in wild-type neurons is close to that expected from a modulation of Nav1.1/1.2 channels alone. On the basis of the change in persistent current density in the Nav1.6 null neurons, ∼32% of the persistent current is attributable to Nav1.1/1.2 channels. If PKA phosphorylation results in a 26% decline in the persistent (and rapidly inactivating) currents of Nav1.1/1.2 channels, then in wild-type neurons there should be an 8% decline (26 × 32%) in the mixed persistent Na+currents—if Nav1.6 channels are unaffected completely. This is not significantly different from what was observed (11%). The modulation of the rapidly inactivating Na+ currents by cBIMPS was indistinguishable in wild-type (n = 4) and mutant neurons (n = 3) (p > 0.05, Kruskal–Wallis). These data argue that the Nav1.6 channels that are mainly responsible for the persistent Na+ currents in PFC pyramidal neurons are not modulated by PKA activation.
Fig. 6.
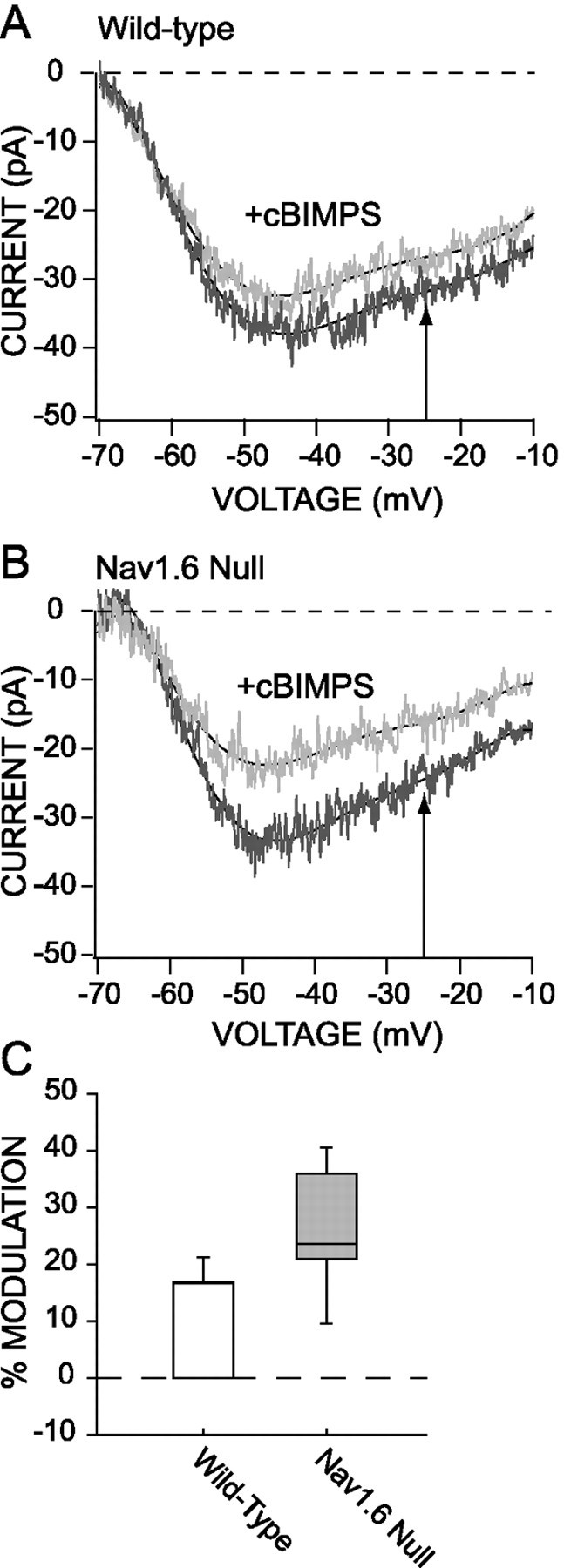
PKA activator decreases the persistent Na+ current in the Nav1.6 null mice.A, Application of cBIMPS (50 μm) produced only a small decrease of the persistent Na+ current in the control mice. B, Conversely, the application of cBIMPS in the Nav1.6 null mice induced a significant reduction of the persistent Na+ current. C, Box plot summarizes the reduction of the Na+ current in the wild-type mice (n = 10) and in the Nav1.6 null mice (n = 5).
DISCUSSION
Rapidly inactivating Na+ currents are suppressed by the activation of D1/D5 receptors
D1/D5 DA receptor activation suppresses rapidly inactivating Na+ currents in layer V–VI PFC pyramidal neurons. The modulation was similar in all responsive pyramidal neurons, including those that were retrogradely labeled from the nucleus accumbens. The attribution of the modulation to activation of D1 and/or D5 receptors is based on the ability of the D1 class agonist SKF 81297 to elicit the response, the ability of SCH 23390 to antagonize it, and the expression of D1 and/or D5 receptor mRNAs in responsive neurons. Both D1 and D5 receptors are positively coupled to adenylyl cyclase (AC) and the production of cAMP (Stoof and Kebabian, 1981; Sibley, 1995). As expected, the membrane-permeant cAMP analog cBIMPS mimicked the response to SKF 81297. Furthermore, a competitive antagonist of cAMP, Rp-cAMPS, blocked receptor-triggered modulation of Na+currents. These results strongly suggest that PKA is an obligate component of the signaling linkage between D1/D5 receptors and Na+ channels. This view is consistent with the similarity in the biophysical signature of the modulation in PFC pyramidal neurons and that seen in other neurons after the activation of D1 class receptors or PKA (Surmeier et al., 1992; Surmeier and Kitai, 1993; Schiffmann et al., 1995; Cantrell et al., 1997). Studies in heterologous systems (Smith and Goldin, 1998;Catterall, 2000) have shown that PKA phosphorylation of Na+ channel Nav1.1 or Nav1.2 α-subunits produces a very similar modulation to that seen in native expression systems that follow the activation of D1 class receptors.
D1/D5 receptor activation inefficiently modulated persistent Na+ currents
In contrast, persistent Na+ currents were not affected noticeably by D1/D5 receptor activation. This was true in all of the PFC pyramidal neurons that were examined, including those in which the D1/D5 receptor agonist SKF 81297 clearly reduced the rapidly inactivating currents. Why was there a difference in the susceptibility of these two components of the Na+ current to modulation? This is an important question for a number of reasons. Previous studies have shown that this current is an important determinant of repetitive activity and synaptic integration in pyramidal neurons (Geijo-Barrientos and Pastore, 1995; Crill, 1996; Yang et al., 1996). It is also a key element in several models of dopaminergic regulation of PFC pyramidal neurons (Geijo-Barrientos and Pastore, 1995; Yang et al., 1999).
An important first step toward understanding the differential modulation is determining the origin of the persistent current itself. There are three commonly advanced hypotheses. Perhaps the oldest is that the persistent current is a “window” current (Hodgkin and Huxley, 1990). Within a narrow range of membrane voltage at the foot of steady-state activation and inactivation gating curves, there is a finite probability that there will be a steady-state or persistent Na+ current. Although this hypothesis has been dismissed in other cell types (Kay et al., 1998), window currents do appear to be capable of making a modest contribution to the persistent Na+ currents in PFC pyramidal neurons at potentials near −50 mV. However, at more depolarized potentials (approximately −25 mV) the contribution of the window current is vanishingly small, demonstrating that another mechanism must be responsible for the lion's share of the persistent current.
There are two other explanations of the persistent current that have been offered. One is that the persistent current reflects the entry of rapidly inactivating Na+ channels into an alternative, persistent gating mode (Patlak, 1991; Alzheimer et al., 1993; Crill, 1996). All of the Na+channels expressed at significant levels in PFC pyramidal neurons (Nav1.1, 1.2, and 1.6) produce currents with rapidly inactivating and persistent components in heterologous expression systems (Smith and Goldin, 1998; Goldin, 1999). In the presence of auxiliary β-subunits the persistent Na+ currents of Nav1.1 and Nav1.6 channels are 2–5% of the rapidly inactivating component—in reasonable agreement with the amplitude of the currents observed here. Nav1.2 channels exhibit significantly less (≤1%) persistent gating. The other explanation is that persistent currents arise from a distinct Na+ channel (de Miera et al., 1997; Raman et al., 1997; Kay et al., 1998; Magistretti et al., 1999). Raman et al. (1997) have demonstrated that Nav1.6 channels are major contributors to both persistent and resurgent Na+ currents in Purkinje neurons, based on studies of mice lacking Nav1.6 channels. Similarly, on the basis of mRNA distribution, de Miera et al. (1997)suggested that in cerebellar Purkinje neurons the rapidly inactivating Na+ current in cerebellar Purkinje neurons was attributable to Nav1.1 channels, whereas the persistent current was attributable to Nav1.6 channels.
Our results are consistent with view that Nav1.6 channels are key determinants of persistent Na+ currents in PFC pyramidal neurons. There are four pieces of evidence supporting this conclusion. First, scRT-PCR profiling found that these neurons express Nav1.6 α-subunit mRNA in addition to mRNAs for Nav1.1 and Nav1.2 subunits. Second, window currents cannot account for the persistent currents, particularly at depolarized membrane potentials. Third, in PFC pyramidal neurons from Nav1.6 null mice (Burgess et al., 1995; Kohrman et al., 1996) persistent Na+current density was reduced to ∼30% of that in wild-type neurons. Last, persistent Na+ currents were modulated inefficiently by the activation of PKA in wild-type neurons conversely to persistent Na+ currents in neurons from Nav1.6 null mice—in this case because of Nav1.1 and Nav1.2 Na+ channels. The fact that previous studies have shown that both Nav1.1 and Nav1.2 channels are readily modulated by PKA phosphorylation (Smith and Goldin, 1998;Goldin, 1999) suggests that much of the persistent Na+ current is ascribable to another channel type—implicating Nav1.6 channels.
However, our results are not consistent with the suggestion that Nav1.6 channels are solely responsible for the persistent Na+ current or that these channels do not contribute to rapidly inactivating currents. There were persistent currents in neurons from the Nav1.6 null mutant mouse that were attributable to Nav1.1/1.2 channels. As mentioned above, all three channels yield currents with both rapidly inactivating and persistent components in heterologous expression systems (Goldin, 1999). Although it failed to reach statistical significance with our small sample, the median density of rapidly inactivating currents was smaller in neurons from Nav1.6 mutants. Furthermore, all three channel proteins are positioned appropriately in the somatodendritic membrane of cortical pyramidal neurons (Westenbroek et al., 1989; Gong et al., 1999;Caldwell et al., 2000). The most parsimonious interpretation of our work is that, whereas all three channels may contribute to both components of the somatodendritic Na+currents, Nav1.6 channels make a disproportionately large contribution to the persistent Na+ current in PFC pyramidal neurons.
The special role of Nav1.6 channels in pyramidal neurons provides a foundation for explaining the differential modulation of rapidly inactivating and persistent Na+ currents by D1 receptor activation. As mentioned above, PKA phosphorylation of sites in the I/II linker region lead to robust modulation of Nav1.1 and Nav1.2 channels (Smith and Goldin, 1998;Catterall, 2000). Our results argue that both the rapidly inactivating and persistent gating modes of Nav1.1/1.2 channels are modulated in a similar manner by PKA phosphorylation. However, of the five PKA consensus sites in the I/II linker region of Nav1.2, only three are present in Nav1.6 (Plummer et al., 1998; Smith et al., 1998). Moreover, the Nav1.6 α-subunit appears to lack a site in this region that is necessary for the expression of the PKA-mediated modulation (A. Goldin, personal communication).
Relationship to previous studies and functional implications
Our findings are clearly at odds with previous studies suggesting that D1 class receptors enhance Na+ currents in PFC pyramidal neurons (Geijo-Barrientos and Pastore, 1995; Yang and Seamans, 1996; Gorelova and Yang, 2000). The discrepancy cannot be ascribed to sampling different neuronal populations. It is much more likely to stem from the reliance on recordings in tissue slices in which dendritic regions that were critical to the observed response were not controlled adequately. Poor dendritic voltage control can lead to a variety of anomalous results, including the appearance of pseudo-persistent Na+ currents (White et al., 1995).
Although our findings strongly argue against the proposed involvement of voltage-dependent Na+ channels in DA-mediated enhancement of PFC excitability, they do not call into question the basic integrative phenomena this conjecture sought to explain. How is it that the activation of D1class receptors can result in increased excitability in some situations and decreased excitability in others? A very similar situation exists in the literature bearing on the D1 receptor modulation of the excitability of striatal medium spiny neurons. What has emerged from this apparent paradox is the recognition that the neuromodulatory effects of D1 receptor activation are state-dependent. Striatal medium spiny neurons move between depolarized up states and hyperpolarized down states (Wilson and Kawaguchi, 1996). When in the down state, the activation of D1 receptors suppresses the response to excitatory input by augmenting inwardly rectifying K+ currents and suppressing voltage-dependent Na+ currents (like those described here) (Surmeier et al., 1992). However, in the depolarized up state, D1 receptor stimulation enhances excitability by augmenting L-type Ca2+currents and possibly suppressing depolarization-activated K+ currents (Surmeier and Kitai, 1993;Surmeier et al., 1995; Hernandez-Lopez et al., 1997). This model not only provides an explanation for seemingly contradictory excitatory and inhibitory effects of D1 receptor stimulation but provides fundamental new insights into how dopamine is shaping striatal function.
A very similar situation may exist in PFC pyramidal neurons. Pyramidal neurons, like striatal medium spiny neurons, move between hyperpolarized down states and depolarized up states in which neurons spike (Steriade et al., 1993; Cowan and Wilson, 1994). Coordination of these state transitions and their associated spike activity has been postulated to control similar transitions in the striatum (Wilson and Kawaguchi, 1996). The transitions between these states are triggered by excitatory synaptic input to dendritic regions. By leaving the persistent Na+ currents mainly untouched, by enhancing L-type Ca2+ currents (Surmeier et al., 1995; Yang and Seamans, 1996), and by suppressing dendritic K+ currents (Hoffman and Johnston, 1998, 1999), D1/D5 receptor activation in these regions (Goldman-Rakic, 1999) should promote the amplification of EPSPs at depolarized up-state potentials (Stafstrom et al., 1985;Deisz et al., 1991; Markram et al., 1995; Schwindt and Crill, 1995;Stuart and Sakmann, 1995; Jung et al., 1997). In contrast, at hyperpolarized down-state membrane potentials, D1/D5 receptor activation should limit amplification of transient excitatory synaptic inputs by rapidly inactivating dendritic Na+channels. In this way, D1/D5 receptor activation could suppress the response to weak, temporally incoherent excitatory inputs while augmenting the response to maintained, synchronized excitatory inputs.
Footnotes
This work was supported by National Institutes of Health Grant NS 34696 to D.J.S. and Grant NS 34509 to M.M. We thank Sasha Ulrich for her help in the scRT-PCR experiments, Dr. Leslie Sprunger for husbandry assistance with the Nav1.6 mutant mice, Dr. Alan Goldin for his advice, and Dr. Caroline Rick for her careful reading of this manuscript.
Correspondence should be addressed to Dr. D. James Surmeier, Department of Physiology/Institute for Neuroscience, Northwestern University Medical School, Searle Building 5-447, 320 East Superior Street, Chicago, IL 60611. E-mail: j-surmeier@northwestern.edu.
REFERENCES
- 1.Alzheimer C, Schwindt PC, Crill WE. Modal gating of Na+ channels as a mechanism of persistent Na+ current in pyramidal neurons from rat and cat sensorimotor cortex. J Neurosci. 1993;13:660–673. doi: 10.1523/JNEUROSCI.13-02-00660.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson DL, Isackson PJ, Gall CM, Jones EG. Contrasting patterns in the localization of glutamic acid decarboxylase and Ca2+/calmodulin protein kinase gene expression in the rat central nervous system. Neuroscience. 1992;46:825–849. doi: 10.1016/0306-4522(92)90188-8. [DOI] [PubMed] [Google Scholar]
- 3.Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS. Regional, cellular, and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci. 1995;15:7821–7836. doi: 10.1523/JNEUROSCI.15-12-07821.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernardi G, Cherubini E, Marciani MG, Mercuri N, Stanzione P. Responses of intracellularly recorded cortical neurons to the iontophoretic application of dopamine. Brain Res. 1982;245:267–274. doi: 10.1016/0006-8993(82)90809-5. [DOI] [PubMed] [Google Scholar]
- 5.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease.”. Nat Genet. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 6.Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc Natl Acad Sci USA. 2000;97:5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantrell AR, Smith RD, Goldin AL, Scheuer T, Catterall WA. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel α-subunit. J Neurosci. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantrell AR, Scheuer T, Catterall WA. Voltage-dependent neuromodulation of Na+ channels by D1-like dopamine receptors in rat hippocampal neurons. J Neurosci. 1999;19:5301–5310. doi: 10.1523/JNEUROSCI.19-13-05301.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 10.Costa MR, Catterall WA. Cyclic AMP-dependent phosphorylation of the α-subunit of the sodium channel in synaptic nerve ending particles. J Biol Chem. 1984;259:8210–8218. [PubMed] [Google Scholar]
- 11.Costa MR, Casnellie JE, Catterall WA. Selective phosphorylation of the α-subunit of the sodium channel by cAMP-dependent protein kinase. J Biol Chem. 1982;257:7918–7921. [PubMed] [Google Scholar]
- 12.Cowan RL, Wilson CJ. Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. J Neurophysiol. 1994;71:17–32. doi: 10.1152/jn.1994.71.1.17. [DOI] [PubMed] [Google Scholar]
- 13.Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- 14.Deisz RA, Fortin G, Zieglgansberger W. Voltage dependence of excitatory postsynaptic potentials of rat neocortical neurons. J Neurophysiol. 1991;65:371–382. doi: 10.1152/jn.1991.65.2.371. [DOI] [PubMed] [Google Scholar]
- 15.de Miera EVS, Rudy B, Sugimori M, Llin↔ s R. Molecular characterization of the sodium channel subunits expressed in mammalian cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1997;94:7059–7064. doi: 10.1073/pnas.94.13.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferron A, Thierry AM, Le Douarin C, Glowinski J. Inhibitory influence of the mesocortical dopaminergic system on spontaneous activity or excitatory response induced from the thalamic mediodorsal nucleus in the rat medial prefrontal cortex. Brain Res. 1984;302:257–265. doi: 10.1016/0006-8993(84)90238-5. [DOI] [PubMed] [Google Scholar]
- 17.Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 18.Geijo-Barrientos E, Pastore C. The effects of dopamine on the subthreshold electrophysiological responses of rat prefrontal cortex neurons in vitro. Eur J Neurosci. 1995;7:358–366. doi: 10.1111/j.1460-9568.1995.tb00331.x. [DOI] [PubMed] [Google Scholar]
- 19.Gershon E, Weigl L, Lotan I, Schreibmayer W, Dascal N. Protein kinase A reduces voltage-dependent Na+ current in Xenopus oocytes. J Neurosci. 1992;12:3743–3752. doi: 10.1523/JNEUROSCI.12-10-03743.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldin AL. Diversity of mammalian voltage-gated sodium channels. Ann NY Acad Sci. 1999;868:38–50. doi: 10.1111/j.1749-6632.1999.tb11272.x. [DOI] [PubMed] [Google Scholar]
- 21.Goldman-Rakic PS. The “psychic” neuron of the cerebral cortex. Ann NY Acad Sci. 1999;868:13–26. doi: 10.1111/j.1749-6632.1999.tb11270.x. [DOI] [PubMed] [Google Scholar]
- 22.Gong B, Rhodes KJ, Bekele-Arcuri Z, Trimmer JS. Type I and type II Na+ channel α-subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J Comp Neurol. 1999;412:342–352. [PubMed] [Google Scholar]
- 23.Gorelova NA, Yang CR. Dopamine D1/D5 receptor activation modulates a persistent sodium current in rat prefrontal cortical neurons in vitro. J Neurophysiol. 2000;84:75–87. doi: 10.1152/jn.2000.84.1.75. [DOI] [PubMed] [Google Scholar]
- 24.Gulledge AT, Jaffe DB. Dopamine decreases the excitability of layer V pyramidal cells in the rat prefrontal cortex. J Neurosci. 1998;18:9139–9151. doi: 10.1523/JNEUROSCI.18-21-09139.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartmann HA, Colom LV, Sutherland ML, Noebels JL. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain [letter]. Nat Neurosci. 1999;2:593–595. doi: 10.1038/10147. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Lopez S, Bargas J, Surmeier DJ, Reyes A, Galarraga E. D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. J Neurosci. 1997;17:3334–3342. doi: 10.1523/JNEUROSCI.17-09-03334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve [1952 classical article]. Bull Math Biol. 1990;52:25–71. doi: 10.1007/BF02459568. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffman DA, Johnston D. Neuromodulation of dendritic action potentials. J Neurophysiol. 1999;81:408–411. doi: 10.1152/jn.1999.81.1.408. [DOI] [PubMed] [Google Scholar]
- 30.Jung HY, Mickus T, Spruston N. Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. J Neurosci. 1997;17:6639–6646. doi: 10.1523/JNEUROSCI.17-17-06639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kay AR, Sugimori M, Llin↔ s R. Kinetic and stochastic properties of a persistent sodium current in mature guinea pig cerebellar Purkinje cells. J Neurophysiol. 1998;80:1167–1179. doi: 10.1152/jn.1998.80.3.1167. [DOI] [PubMed] [Google Scholar]
- 32.Kohrman DC, Harris JB, Meisler MH. Mutation detection in the med and medJ alleles of the sodium channel SCN8a. Unusual splicing due to a minor class AT–AC intron. J Biol Chem. 1996;271:17576–17581. doi: 10.1074/jbc.271.29.17576. [DOI] [PubMed] [Google Scholar]
- 33.Li M, West JW, Lai Y, Scheuer T, Catterall WA. Functional modulation of brain sodium channels by cAMP-dependent phosphorylation. Neuron. 1992;8:1151–1159. doi: 10.1016/0896-6273(92)90135-z. [DOI] [PubMed] [Google Scholar]
- 34.Li M, West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- 35.Magistretti J, Alonso A. Biophysical properties and slow voltage-dependent inactivation of a sustained sodium current in entorhinal cortex layer II principal neurons. A whole-cell and single-channel study. J Gen Physiol. 1999;114:491–509. doi: 10.1085/jgp.114.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magistretti J, Ragsdale DS, Alonso A. High conductance sustained single-channel activity responsible for the low-threshold persistent Na+ current in entorhinal cortex neurons. J Neurosci. 1999;19:7334–7341. doi: 10.1523/JNEUROSCI.19-17-07334.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mantz J, Milla C, Glowinski J, Thierry AM. Differential effects of ascending neurons containing dopamine and noradrenaline in the control of spontaneous activity and of evoked responses in the rat prefrontal cortex. Neuroscience. 1988;27:517–526. doi: 10.1016/0306-4522(88)90285-0. [DOI] [PubMed] [Google Scholar]
- 38.Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. J Physiol (Lond) 1995;485:1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy BJ, Rossie S, De Jongh KS, Catterall WA. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel α-subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. J Biol Chem. 1993;268:27355–27362. [PubMed] [Google Scholar]
- 40.Patlak J. Molecular kinetics of the voltage-dependent Na+ channel. Physiol Rev. 1991;71:1047–1080. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- 41.Penit-Soria J, Audinat E, Crepel F. Excitation of rat prefrontal cortical neurons by dopamine: an in vitro electrophysiological study. Brain Res. 1987;425:263–274. doi: 10.1016/0006-8993(87)90509-9. [DOI] [PubMed] [Google Scholar]
- 42.Pirot S, Godbout R, Mantz J, Tassin JP, Glowinski J, Thierry AM. Inhibitory effects of ventral tegmental area stimulation on the activity of prefrontal cortical neurons: evidence for the involvement of both dopaminergic and GABAergic components. Neuroscience. 1992;49:857–865. doi: 10.1016/0306-4522(92)90362-6. [DOI] [PubMed] [Google Scholar]
- 43.Plummer NW, Galt J, Jones JM, Burgess DL, Sprunger LK, Kohrman DC, Meisler MH. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene scn8a. Genomics. 1998;54:287–296. doi: 10.1006/geno.1998.5550. [DOI] [PubMed] [Google Scholar]
- 44.Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 45.Rossie S, Catterall WA. Cyclic-AMP-dependent phosphorylation of voltage-sensitive sodium channels in primary cultures of rat brain neurons. J Biol Chem. 1987;262:12735–12744. [PubMed] [Google Scholar]
- 46.Rossie S, Catterall WA. Phosphorylation of the α-subunit of rat brain sodium channels by cAMP-dependent protein kinase at a new site containing Ser686 and Ser687. J Biol Chem. 1989;264:14220–14224. [PubMed] [Google Scholar]
- 47.Schiffmann SN, Lledo PM, Vincent JD. Dopamine D1 receptor modulates the voltage-gated sodium current in rat striatal neurones through a protein kinase A. J Physiol (Lond) 1995;483:95–107. doi: 10.1113/jphysiol.1995.sp020570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwindt PC, Crill WE. Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurophysiol. 1995;74:2220–2224. doi: 10.1152/jn.1995.74.5.2220. [DOI] [PubMed] [Google Scholar]
- 49.Shi WX, Zheng P, Liang XF, Bunney BS. Characterization of dopamine-induced depolarization of prefrontal cortical neurons. Synapse. 1997;26:415–422. doi: 10.1002/(SICI)1098-2396(199708)26:4<415::AID-SYN9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 50.Sibley DR. Molecular biology of dopamine receptors. In: Ariano MA, Surmeier DJ, editors. Molecular and cellular mechanisms of neostriatal function. Landes; Austin, TX: 1995. pp. 255–272. [Google Scholar]
- 51.Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse SCN8a sodium channel. J Neurosci. 1998;18:6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith RD, Goldin AL. Phosphorylation of brain sodium channels in the I–II linker modulates channel function in Xenopus oocytes. J Neurosci. 1996;16:1965–1974. doi: 10.1523/JNEUROSCI.16-06-01965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith RD, Goldin AL. Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci. 1998;18:811–820. doi: 10.1523/JNEUROSCI.18-03-00811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stafstrom CE, Schwindt PC, Chubb MC, Crill WE. Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. J Neurophysiol. 1985;53:153–170. doi: 10.1152/jn.1985.53.1.153. [DOI] [PubMed] [Google Scholar]
- 55.Steriade M, Nunez A, Amzica F. Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci. 1993;13:3266–3283. doi: 10.1523/JNEUROSCI.13-08-03266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoof JC, Kebabian JW. Opposing roles for D1 and D2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature. 1981;294:366–368. doi: 10.1038/294366a0. [DOI] [PubMed] [Google Scholar]
- 57.Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- 58.Surmeier DJ, Kitai ST. D1 and D2 dopamine receptor modulation of sodium and potassium currents in rat neostriatal neurons. Prog Brain Res. 1993;99:309–324. doi: 10.1016/s0079-6123(08)61354-0. [DOI] [PubMed] [Google Scholar]
- 59.Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci USA. 1992;89:10178–10182. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 61.Tukey JW. Exploratory data analysis. Addison–Wesley; Menlo Park, CA: 1977. [Google Scholar]
- 62.Vysokanov A, Flores-Hernandez J, Surmeier DJ. mRNAs for clozapine-sensitive receptors colocalize in rat prefrontal cortex neurons. Neurosci Lett. 1998;258:179–182. doi: 10.1016/s0304-3940(98)00882-9. [DOI] [PubMed] [Google Scholar]
- 63.Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- 64.White JA, Sekar NS, Kay AR. Errors in persistent inward currents generated by space-clamp errors: a modeling study. J Neurophysiol. 1995;73:2369–2377. doi: 10.1152/jn.1995.73.6.2369. [DOI] [PubMed] [Google Scholar]
- 65.Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397–2410. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein kinase C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015. doi: 10.1152/jn.1997.77.2.1003. [DOI] [PubMed] [Google Scholar]
- 67.Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V–VI rat prefrontal cortex neurons in vitro: modulation of dendritic–somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang CR, Seamans JK, Gorelova N. Electrophysiological and morphological properties of layers V–VI principal pyramidal cells in rat prefrontal cortex in vitro. J Neurosci. 1996;16:1904–1921. doi: 10.1523/JNEUROSCI.16-05-01904.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang CR, Seamans JK, Gorelova N. Developing a neuronal model for the pathophysiology of schizophrenia based on the nature of electrophysiological actions of dopamine in the prefrontal cortex. Neuropsychopharmacology. 1999;21:161–194. doi: 10.1016/S0893-133X(98)00112-2. [DOI] [PubMed] [Google Scholar]