

Abstract
Long-term exposure to nerve growth factor (NGF) is well established to have neurotrophic effects on basal forebrain cholinergic neurons, but its potential actions as a fast-acting neuromodulator are not as well understood. We report that NGF (0.1–100 ng/ml) rapidly (<60 min) and robustly enhanced constitutive acetylcholine (ACh) release (148–384% of control) from basal forebrain cultures without immediate persistent increases in choline acetyltransferase activity. More ACh was released in response to NGF when exposure was coupled with a higher depolarization level, suggesting activity dependence. In a long-term potentiation-like manner, brief NGF exposure (10 ng/ml; 60 min) induced robust and prolonged increases in ACh release, a capacity that was shared with the other neurotrophins. K252a (10–100 nm), BAPTA-AM (25 μm), and Cd2+ (200 μm) prevented NGF enhancement of ACh release, suggesting the involvement of TrkA receptors, Ca2+, and voltage-gated Ca2+ channels, respectively. Forskolin (10 μm), a cAMP generator, enhanced constitutive ACh release but did not interact synergistically with NGF. Tetrodotoxin (1 μm) and cycloheximide (2 μm) did not prevent NGF-induced ACh release, indicative of action at the level of the cholinergic nerve terminal and that new protein synthesis is not required for this neurotransmitter-like effect, respectively. In contrast, after a 24 hr NGF treatment, distinct protein synthesis-dependent and independent effects on choline acetyltransferase activity and ACh release were observed. These results indicate that neuromodulator/neurotransmitter-like (protein synthesis-independent) and neurotrophic (translation-dependent) actions likely make distinct contributions to the enhancement of cholinergic activity by NGF.
Keywords: brain-derived neurotrophic factor, choline acetyltransferase, neurotransmitter release, neuromodulation, cholinergic, neurotrophin
Basal forebrain cholinergic neurons (BFCNs) innervate cortical and associated structures (Fibiger, 1982), are important for attention (Baxter and Chiba, 1999), and degenerate in Alzheimer's disease (Bartus, 2000). Rapid modulation of acetylcholine (ACh) release by physiological and pathological factors [e.g., neurotransmitters (Raiteri et al., 1984; Hersi et al., 1995), certain growth factors (Kar et al., 1997), β-amyloid (Auld et al., 1998), and interleukins (Hanisch et al., 1993)] is likely critical for the consequences of innervation.
Neurotrophins, including nerve growth factor (NGF), are crucial for the survival and function of certain neuronal populations (Levi-Montalcini, 1987). Regions of BFCN innervation (e.g., hippocampus, cortex) are enriched in NGF (Korsching et al., 1985; Large et al., 1986), and NGF is retrogradely transported by BFCNs (DiStefano et al., 1992), with these neurons expressing TrkA and p75NTR receptors (Koh and Loy, 1989;Holtzman et al., 1992). NGF and TrkA are important for BFCN development, maintenance, and function in vivo (Vantini et al., 1989; Li et al., 1995; Chen et al., 1997; Fagan et al., 1997;Molnar et al., 1998; Debeir et al., 1999; Ruberti et al., 2000), and NGF exposure (days to weeks) enhances cholinergic markers in BF cultures (Hartikka and Hefti, 1988; Takei et al., 1988, 1989;Svendsen et al., 1994; Nonner et al., 1996; Pongrac and Rylett, 1998;Oosawa et al., 1999; Auld et al., 2001). The neurotrophins brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4) also enhance cholinergic markers (Nonomura et al., 1995; Nonner et al., 1996; Auld et al., 2001). Interestingly, even a 30 min exposure to NGF, BDNF, NT-3, or NT-4 increases choline acetyltransferase (ChAT) activity 24 hr later (Nonner et al., 2000).
Neurotrophins rapidly increase intracellular Ca2+ in several neuronal phenotypes, including BFCNs (Wildering et al., 1995; Stoop and Poo, 1996; Jiang and Guroff, 1997; Li et al., 1998; Jia et al., 1999; Nonner et al., 2000), and acutely modulate neurotransmission (Lu and Chow, 1999; Schinder and Poo, 2000). BDNF and NT-3 rapidly and Ca2+-dependently enhance neurotransmitter release from Xenopus motor neurons (Lohof et al., 1993;Stoop and Poo, 1996; He et al., 2000). BDNF can enhance hippocampal neurotransmission (Lessmann et al., 1994; Kang and Schuman, 1995; Li et al., 1998), inhibit high-frequency stimulation-associated fatigue (Gottschalk et al., 1998; Pozzo-Miller et al., 1999), and facilitate long-term potentiation (LTP) induction (Korte et al., 1995;Figurov et al., 1996; Patterson et al., 1996; Chen et al., 1999; Xu et al., 2000), with presynaptic TrkB typically being involved. Also, NGF rapidly modulates stimulated ACh release from hippocampal (Knipper et al., 1994) and visual-cortex (Sala et al., 1998) synaptosomes and likely influences visual-cortex LTP by modulating ACh release (Pesavento et al., 2000).
Using embryonic BF cultures, we report that NGF rapidly and potently enhanced ACh release in activity- and Ca2+-dependent manners and that increases persisted after NGF removal. Dichotomous actions consisting of protein synthesis-dependent “neurotrophic” effects on ChAT activity and protein synthesis-independent “neuromodulator” increases of ACh release were identified. These distinct capacities may make complementary contributions to NGF enhancement of BFCN function.
MATERIALS AND METHODS
Culture. All experiments followed guidelines of the Canadian Council on Animal Care and McGill University policies. Cultures were prepared as described previously (Auld et al., 2000a). BF regions (septum, diagonal band of Broca, and substantia innominata) of day 17 rat embryos (Charles River, St. Constant, QC, Canada) were dissected in HBSS (Life Technologies, Burlington, ON, Canada) containing 0.65% d(+)-glucose (Sigma, St. Louis, MO), 15 mm HEPES, 10 U/ml penicillin, and 10 mg/ml streptomycin (Life Technologies). These were dissociated at 37°C with 0.08% trypsin (Life Technologies) and 0.1% DNase I (Sigma) for 18 min [terminated with 10% fetal bovine serum (FBS; Immunocorp, Montréal, QC, Canada)]. The dissociation was completed mechanically with a fire-polished Pasteur pipette. Cultures were plated at 700,000–750,000 cells per well [precoated with poly-l-ornithine (0.3 μg/ml); Life Technologies] in 500 μl of growth medium in four-well tissue culture plates (Nunc, Naperville, IL), and cultures were maintained at 37°C and 5% CO2. The medium consisted of DMEM (#11965; Life Technologies) supplemented with KCl [20 mm; total, 25 mm; similar high-K+ conditions are associated with improved viability in BF cultures (Nakamura et al., 1994)], sodium pyruvate (1 mm),d(+)-glucose (35 mm), HEPES (15 mm), and FBS (10%). Under similar culture conditions, which were optimized for the study of ACh release, both release and ChAT activity steadily increased between plating and 10 dayin vitro (DIV 10) (Auld et al., 2000a).
Acetylcholine release. In most experiments, on DIV 7, the medium was removed, and cells were rinsed with Krebs' buffer [125 mm NaCl, 4.8 mm KCl, 1.2 mmKH2PO4, 25 mm HEPES, 1.2 mmMgSO4, 2.2 mmCaCl2, 10 mm glucose, 10 μm choline, and 200 nmneostigmine (all from Sigma), pH adjusted to 7.4] containing 6 mm K+. Unless described otherwise in Results, after a 60 min equilibration period at 37°C and 5% CO2, this buffer was discarded and replaced for a 45 min period with fresh buffer containing rhβNGF (R & D Systems, Minneapolis, MN; lots HS178041 or HS189011) or vehicle, from which ACh release was measured. Other compounds were delivered during the 60 min equilibration period as well as simultaneously with NGF during the ACh release period [BAPTA-AM (RBI, Natick, MA), CdCl2 (Sigma), cycloheximide (Sigma), forskolin (Sigma), K252a (Calbiochem, La Jolla, CA) Rp-cAMPS (RBI), tetrodotoxin (TTX; Tocris, Ballwin, MO)]. For the rhβNGF, rhNT-3, rhNT-4, or rhBDNF (R & D Systems; lots NG059091, OU02805, and OD048111) pretreatments (see Figs. 6, 7), medium with FBS was replaced with medium supplemented with B27 (2%; Life Technologies) containing the neurotrophin. After the indicated exposure period, culture wells were washed four times with NGF/neurotrophin-free buffer, and constitutive ACh release was then collected for 60 min periods in the buffer described above. In some experiments, p75NTR-IgG fusion protein (R & D Systems) was administered either concurrently with NGF or after NGF removal. All samples were kept at −80°C until ACh or ChAT activity quantification (<2 weeks).
Fig. 6.
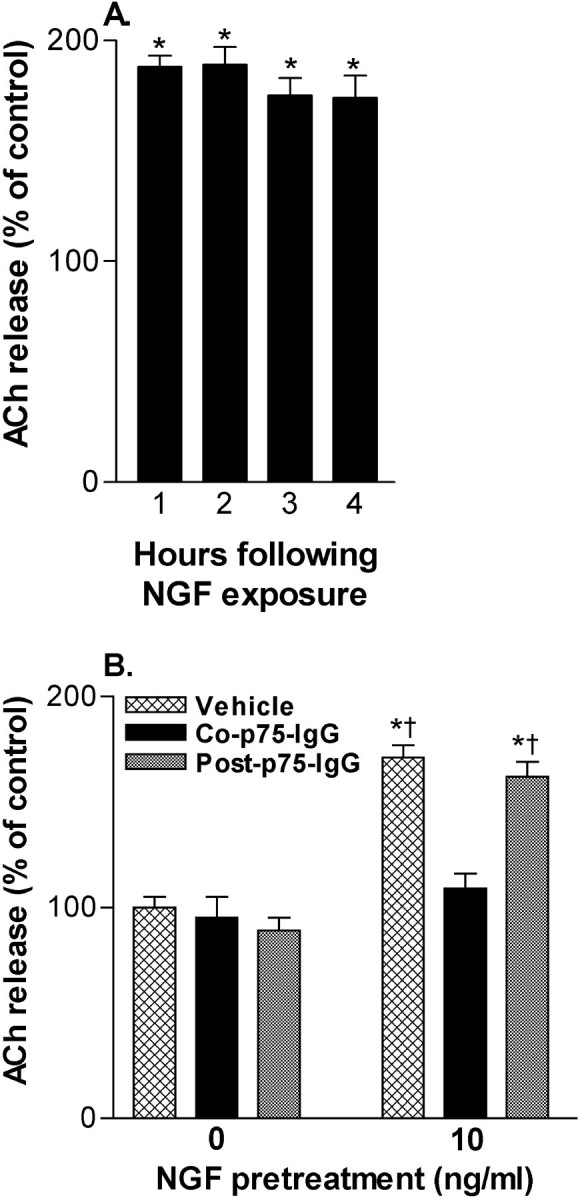
Brief exposure to NGF induces prolonged increase in ACh release. A, NGF (10 ng/ml for 60 min) treatment is associated with increased ACh release for at least 4 hr. Data are normalized according to release from control wells at the same hour and are expressed as mean ± SEM (n = 17–23). Statistical analysis was performed using repeated measure one-way ANOVA with Tukey's post-test; *p < 0.001 vs control.B, Treatment with NGF (10 ng/ml for 60 min) was associated with enhanced ACh release during the subsequent 60 min and was specific to availability during the defined exposure period, because a p75NTR-IgG fusion protein (5 μg/ml) only blocked the effect when coadministered with NGF. Data are normalized according to control wells and are expressed as mean ± SEM (n = 4–8). Statistical analysis was performed using a one-way ANOVA with Tukey's post-test: *p < 0.001 vs control,†p < 0.001 vs NGF/co-p75NTR-IgG fusion protein.
The percentage of cholinergic neurons in BF cultures maintained under similar conditions is low (∼1%) (Hartikka and Hefti, 1988; Svendsen et al., 1994). However, their unique ability to synthesize ACh makes quantification of supernatant ACh a reliable measure of their neurotransmitter release. To our knowledge, no report concerning an effect of NGF in BF cultures has indicated an indirect mechanism. BFCNs, but not GABAergic neurons, selectively express TrkA and p75NTR (Hartikka and Hefti, 1988; Koh and Loy, 1989; Holtzman et al., 1992;Svendsen et al., 1994). At a functional level, BFCNs, but not GABAergic neurons, respond to NGF and BDNF (Koliatsos et al., 1994). TrkB also appears to be selectively expressed on ChAT-immunoreactive somas in the BF (Molnar et al., 1998). Given that BFCNs selectively release ACh and respond to NGF, these cultures are an excellent model for studying their interactions.
Acetylcholine quantification. ACh was assayed by HPLC with electrochemical detection in conjunction with an enzyme reactor. Samples (100 μl) were injected manually via a 100 μl loop on a two-position valve (Valco, Houston, TX). ACh and choline, separated on a reverse-phase column (75 × 2.1 mm) pretreated with lauryl sulfate, passed through an enzyme reactor (10 × 2.1 mm) containing acetylcholinesterase (EC 3.1.1.7; Sigma, type VI-S) and choline oxidase (1.1.3.17; Sigma) covalently bound to glutaraldehyde-activated Lichrosorb NH2 (10 μm; Merck, Darmstadt, Germany). All column hardware and packing materials were from Chrompack (Raritan, NJ). The resultant hydrogen peroxide was electrochemically detected at a platinum electrode at a potential of +500 mV versus an Ag/AgCl reference electrode (Antec VT-03/Decade, Leiden, The Netherlands). The mobile phase, 0.2 maqueous potassium phosphate buffer, pH 8.0, with 1 mm tetramethylammonium hydroxide (Sigma), was delivered at 0.4–0.45 ml/min by a dual piston pump (ESA 580, Chelmsford, MA) connected to a degasser (CMA 260, Stockholm, Sweden). ACh eluted at ∼4 min.
Choline acetyltransferase activity. Cultures were homogenized in 200 μl ice-cold buffer (40 mmsodium phosphate buffer, pH 7.4, 200 mm NaCl, and 0.5% Triton X-100). Aliquots in duplicate were assayed for ChAT activity using [14C]-acetyl-CoA (New England Nuclear/DuPont, Markham, ON, Canada) and choline (Sigma) as substrate. After 60 min at 37°C, the reaction was stopped with ice-cold 10 mm sodium phosphate buffer, pH 7.4, containing 0.2 mm acetylcholine chloride (Sigma). Radioactive ACh was extracted using butyronitrile (Sigma) containing 15 mg/ml sodium tetraphenylborate (Sigma).
Statistical analysis. Data were statistically analyzed using either Student's t test (unpaired) or one- or two-way ANOVAs with Tukey's post hoc test, where appropriate. In all cases, p < 0.05 was considered statistically significant. The n represents individual culture wells evaluated in a given experiment, and unless indicated otherwise, data are expressed as mean ± SEM representing percentage of control wells receiving appropriate vehicle treatments.
RESULTS
NGF enhancement of ACh release is influenced by depolarization level
Exposure to NGF (100 ng/ml for 60 min in buffer with 6 mm K+) resulted in increased ACh release (∼192% of control) during an immediately subsequent 10 min period of K+ (25 mm) depolarization (control, 168 ± 22 fmol per well per minute; NGF, 322 ± 16; n = 4; p < 0.001). Because some aspects of synaptic plasticity are influenced by the level of neuronal electrical activity, including neurotrophin modulation of neurotransmission (Gottschalk et al., 1998; Boulanger and Poo, 1999a), we examined whether NGF-associated increased ACh release could be modified by activity level. An identical NGF treatment in sister culture wells was associated with a greater increase in the amount of ACh released when subsequently paired with increased depolarization. Indeed, the same NGF treatment resulted in a ∼2.4-fold greater increase of ACh release (femtomoles per well per minute) when followed by exposure to 25 mmK+ compared with 6 mm K+ (the increase in each K+ condition was calculated vs ACh release from the same depolarization conditions in the absence of NGF) (Fig. 1).
Fig. 1.
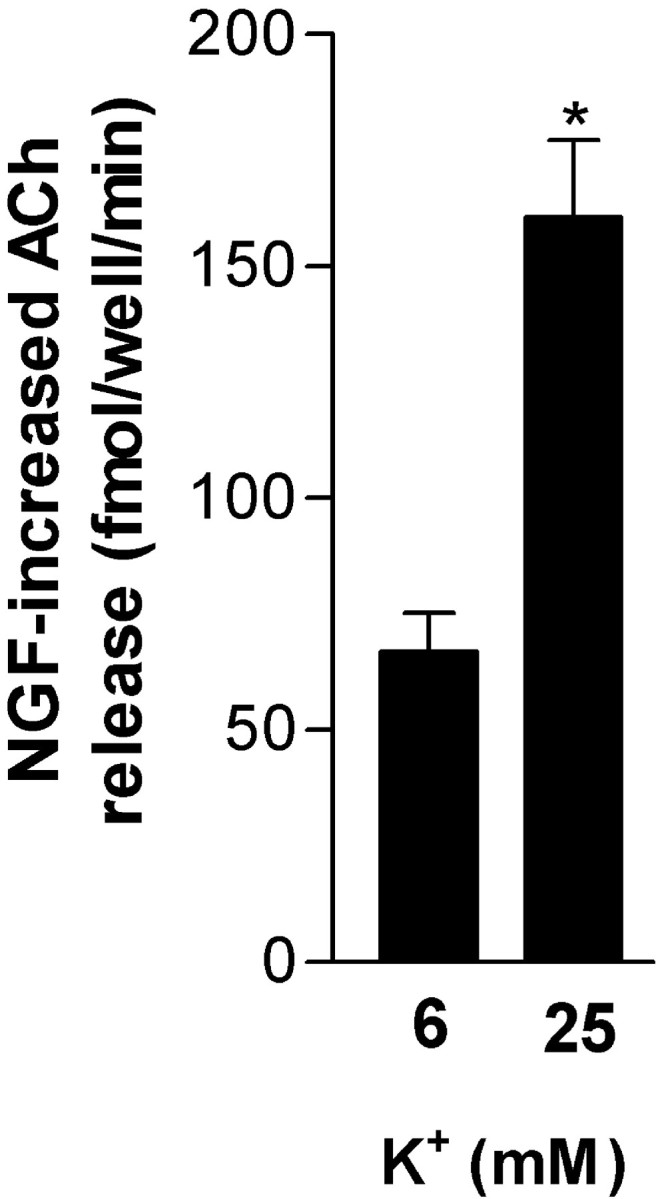
Activity-dependent enhancement of ACh release by NGF. Cultures were preexposed to NGF (10 ng/ml) for 60 min in low K+ (6 mm) buffer. ACh release was then evaluated from low-activity (6 mm K+) or high-depolarization (25 mm K+) conditions for a 15 min period. Columns represent increased ACh (femtomoles per well per minute ± SEM;n = 8) associated with NGF preexposure versus the same depolarizing conditions without NGF preexposure. Significance was determined using Student's t test (*p < 0.001 vs 6 mmK+).
We chose to further examine NGF enhancement of ACh release under conditions of constitutive ACh release associated with endogenous activity levels (in 6 mm K+buffer) because interpretation of mechanistic aspects of the NGF-associated increases would be complicated by the facts that K+ (25 mm) depolarization was associated with a large Ca2+-dependent induction of ACh release by itself, as well as the synergistic interaction between increased activity level and NGF action on ACh release. That most of the constitutive release was not sensitive to intracellular Ca2+ chelation (see below) enabled us to focus subsequent mechanistic studies directly on NGF-induced ACh release. It should be pointed out that although there was a large relative effect of NGF compared with this spontaneous release (possibly representing nonspecific leakage), NGF actually induced more ACh release when coupled with a depolarizing stimulation (see above).
NGF enhances constitutive ACh release: contribution of TrkA and calcium
NGF (0.1–100 ng/ml) robustly enhanced constitutive ACh release from embryonic BF cultures during a 45 min exposure period without inducing an immediate, persistent increase in ChAT activity (Fig.2). Furthermore, under these conditions, neither culture protein levels (control, 100 ± 1%,n = 21; NGF, 100 ng/ml, 103 ± 2%,n = 8) nor metabolic activity, indicated by MTT reduction (control, 100 ± 2%, n = 7; NGF 100 ng/ml, 99 ± 5%, n = 4), were altered; given the low percentage of cholinergic neurons in BF cultures, this was not unexpected (Hartikka and Hefti, 1988; Svendsen et al., 1994). The relative magnitude of NGF enhancement of ACh release was time dependent and during 15, 30, and 60 min exposure/release periods, NGF (1 ng/ml) enhanced constitutive ACh release to 127 ± 9% (n = 4; p < 0.05 vs control), 182 ± 11% (n = 6; p < 0.001), and 238 ± 25% (n = 6; p < 0.001) of control level, respectively. In the presence of TTX (1 μm), a 45 min exposure to NGF (1 ng/ml) still elicited robust ACh release (control, 100 ± 2%; TTX, 80 ± 6%; NGF, 287 ± 17%; NGF/TTX, 258 ± 27%;n = 6), suggesting an action at the level of the cholinergic nerve terminal.
Fig. 2.

NGF acutely enhances constitutive ACh release from embryonic basal forebrain neurons in a concentration-dependent manner during a short-term exposure but does not induce persistent ChAT activity changes. Data are expressed as a percentage of release or ChAT activity in the absence of NGF [mean ± SEM; 0 ng/ml (n = 59), 0.1 ng/ml (14), 0.5 ng/ml (14), 1 ng/ml (37), 10 ng/ml (27), and 100 ng/ml (19); control ACh release was ∼630 fmol/well for the 45 min exposure period, representing ∼14 fmol per well per minute]. Significance was determined using a one-way ANOVA with Tukey's post-test (*p < 0.001 vs control).
The tyrosine kinase inhibitor K252a blocked NGF (10 ng/ml)-induced increases in constitutive ACh release, suggesting the involvement of TrkA receptor signaling (Fig. 3). In the presence of 100 nm K252a, NGF did not increase ACh release beyond the control treated with K252a alone. Because cAMP signaling has been shown to enhance the effects of neurotrophins (Meyer-Franke et al., 1998; Boulanger and Poo, 1999b), we examined its actions on NGF-induced ACh release (Fig. 4). Forskolin, at a much higher concentration (10 μm), increased ACh release to a magnitude similar to NGF (1 ng/ml). The coapplication of NGF and forskolin, at these same concentrations, had only an additive effect without evidence of synergistic interaction. Furthermore, Rp-cAMPS (100 μm), a protein kinase A (PKA) antagonist, did not significantly decrease NGF (1 ng/ml)-induced increases in constitutive ACh release associated with a 45 min exposure (control, 100 ± 4%; Rp-cAMPS, 104 ± 1%; NGF, 329 ± 12%; NGF/Rp-cAMPS, 312 ± 12%;n = 4–9). Thus, the NGF increase of ACh release appears to not involve cAMP or PKA, at least under these low-K+ conditions.
Fig. 3.

K252a (10–100 nm) prevents NGF (10 ng/ml) enhancement of ACh release during a short-term exposure. Data are expressed as a percentage of release in the absence of NGF and K252a (mean ± SEM; n = 6–8). Statistical analysis was performed using a two-way ANOVA with Tukey's post-test (*p < 0.001 vs K252a only, at the same concentration; †p = 0.0627 vs cultures receiving neither K252a nor NGF).
Fig. 4.

Forskolin (10 μm) alone increases ACh release but does not synergistically enhance NGF (1 ng/ml)-induced ACh release during a short-term exposure in low K+(6 mm) conditions. Data are expressed as a percentage of release in the absence of forskolin and NGF (mean ± SEM;n = 6). Significance was determined using a one-way ANOVA with Tukey's post-test (*p < 0.001 vs control; †p < 0.001 vs both NGF and forskolin).
To determine whether Ca2+ was involved in the NGF-induced increases in constitutive ACh release, intracellular Ca2+ was chelated using BAPTA-AM (25 μm), and under these conditions NGF (1 ng/ml)-associated ACh release was prevented (Fig.5A). We next investigated the involvement of voltage-gated Ca2+ channels (VGCC) using Cd2+, a nonspecific antagonist. Cd2+ (200 μm) blocked NGF (1 ng/ml)-induced increases in ACh release, suggesting that voltage-gated Ca2+ channels were involved (Fig.5B). Together with inhibiting ACh release increases caused by K+ stimulation (25 mm; 10 min), Cd2+blocked NGF enhancement of release under these conditions as well (data not shown).
Fig. 5.
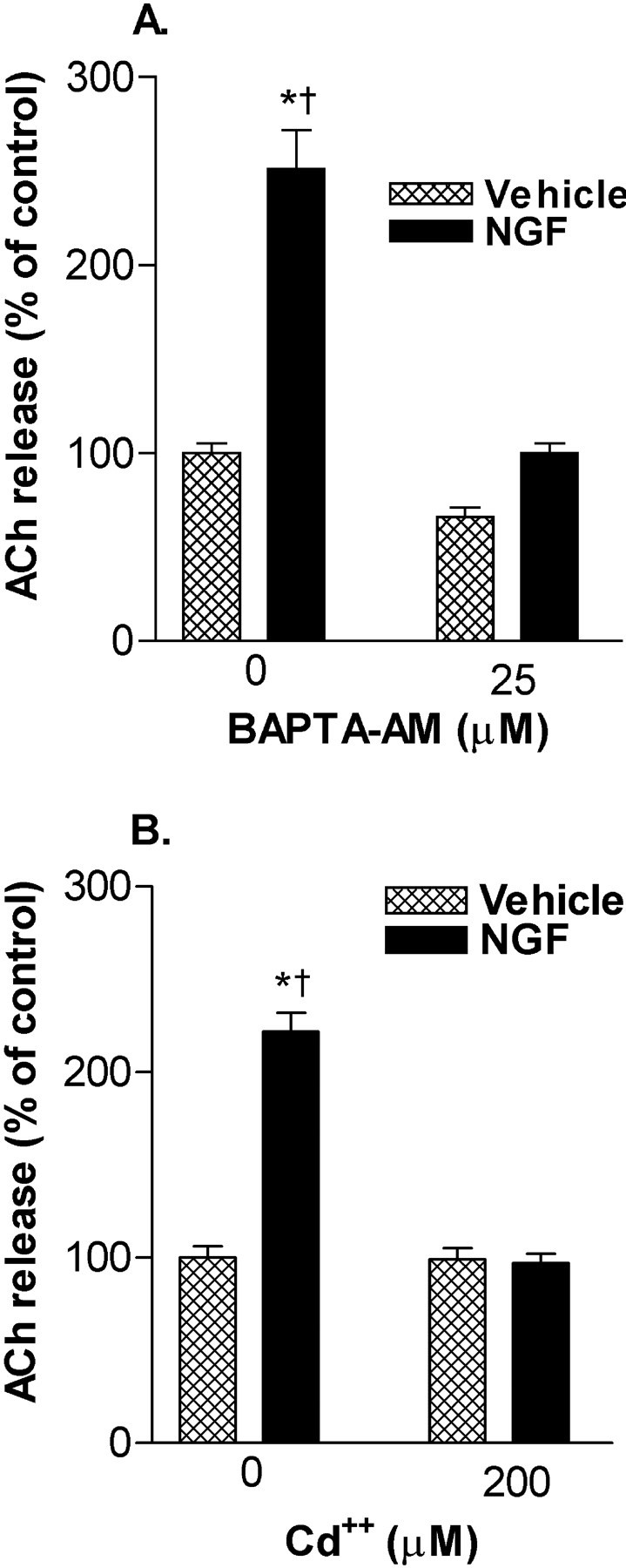
NGF (1 ng/ml)-enhanced ACh release involves Ca2+. A, The intracellular Ca2+ chelator BAPTA-AM (25 μm) inhibits NGF-induced ACh release during a short-term exposure. Data are expressed as a percentage of release in the absence of BAPTA-AM and NGF (mean ± SEM). Significance was determined using a one-way ANOVA with Tukey's post-test (*p < 0.001 vs control;† p < 0.001 vs NGF/BAPTA-AM).B, NGF-induced ACh release is inhibited by the voltage-gated Ca2+ channel antagonist Cd2+ (200 μm). Data are expressed as a percentage of release in the absence of Cd2+ and NGF (mean ± SEM; n = 6). Significance was determined using a one-way ANOVA with Tukey's post-test (*p < 0.001 vs control;†p < 0.001 vs NGF/Cd2+).
Brief exposure to neurotrophins induces prolonged ACh release
Treatment with NGF (10 ng/ml) for 60 min (followed by four rinses of culture plates) resulted in a robust increase of constitutive ACh release for at least the next 4 hr (Fig.6A). Interestingly, comparable to the 60 min treatment, a 5 min exposure to NGF (10 ng/ml) also resulted in increased ACh release during the hour-long period subsequent to NGF removal (289 ± 31% of control;n = 2; p < 0.05). Thus, it is likely that increased time after initial NGF exposure, rather than the duration of NGF exposure, was important for the time-dependent effect noted previously. The specificity of the prolonged influence of NGF on ACh release for the defined exposure period, as opposed to a possible influence of potential residual NGF left after rinse with NGF-free buffer, was indicated by the capacity of a p75NTR-IgG (5 μg/ml) fusion protein to block increases when coadministered with NGF (10 ng/ml), but not if given during the period of release determination immediately after NGF washout (Fig. 6B).
Considering that these neurons respond to NT-3, NT-4, and BDNF with increased ChAT activity (Nonomura et al., 1995; Nonner et al., 1996,2000), as well as retrogradely transport them from target regions (DiStefano et al., 1992), we sought to determine whether they acutely induced a persistent increase in ACh release. Under the same conditions as with NGF, a 60 min pretreatment with the other neurotrophins (10 ng/ml) also enhanced ACh release in the hour subsequent to their removal: NT-3, 153 ± 6% of control (n = 4;p < 0.001 vs control); NT-4, 161 ± 16% (n = 4; p < 0.01); and BDNF, 177 ± 19% (n = 4; p < 0.001). Furthermore, NGF was also associated with increased K+-stimulated ACh release that persisted after its removal (data not shown).
Differentiation between neurotrophic and neuromodulatory effects of NGF
Exposure to the protein synthesis inhibitor cycloheximide (2 μm) did not reduce NGF (1 ng/ml) enhancement of ACh release during a 45 min exposure period (control, 100 ± 3%; cycloheximide, 97 ± 5%; NGF, 329 ± 35%; NGF/cycloheximide, 362 ± 57%; n = 6–8), suggesting that new protein synthesis was not required for the rapid effect of NGF on ACh release. We next examined changes in ChAT activity and ACh release after 6–24 hr NGF (100 ng/ml) exposure and the contribution of protein synthesis to these effects (Fig.7). After 6 and 12 hr of NGF exposure, ChAT activity was not significantly increased versus vehicle-treated control. Regardless of treatment with cycloheximide (2 μm), there was increased release of ACh from NGF-treated groups. After 24 hr, ChAT activity was significantly increased in the NGF condition but was not increased in the NGF/cycloheximide-exposed condition. Remarkably, the enhancement of ACh release associated with NGF/cycloheximide treatment was slightly larger than that associated with the NGF-alone condition, despite the markedly lower level of ChAT activity in the NGF/cycloheximide-treated cultures after 24 hr exposures (Fig. 7). Our preliminary data suggest that after a 24-hr treatment, enhanced ACh release associated with the NGF/cycloheximide treatment decays faster on removal of NGF compared with conditions in which protein synthesis is not prevented, which are associated with increased ChAT activity (Auld et al., 2000b).
Fig. 7.

Evidence for protein synthesis-dependent and -independent effects of NGF on ChAT activity and ACh release, respectively. ChAT activity (A) and constitutive ACh release (B) were compared after 6, 12, and 24 hr exposures to NGF (100 ng/ml) and/or cycloheximide (CY; 2 μm). Data are normalized according to controls and are expressed as mean ± SEM (6 hr,n = 4; 12 hr, n = 4–8; 24 hr,n = 20–24). Statistical analysis was performed using two-way ANOVAs with Tukey's post-test: *p <0.05 vs vehicle-treated control and †p<0.05 vs NGF/CY (within ChAT and ACh, same hour);‡p < 0.05 vs NGF at 6 and 12 hr (within ChAT and ACh); §p< 0.05 vs NGF/CY at 6 and 12 hr (within ACh). Furthermore, at 6, 12, and 24 hr, the percentage changes in ACh release and ChAT activity were different within NGF and NGF/CY groups (p < 0.05).
DISCUSSION
We report that NGF influences BFCNs in a manner encompassing acute enhancement of ACh release in a neurotransmitter/neuromodulator-like manner. The concentrations of NGF enhancing ACh release indicate that it as one of the most potent ACh secretagogues ever recognized. More NGF-enhanced ACh release was associated with a greater depolarization, suggesting that this interaction could underlie some aspects of activity-dependent sculpting of BFCN synapses. Even brief exposures to NGF potentiated release for several hours after its removal, and this capacity was shared with BDNF, NT-4, and NT-3. After a 24 hr NGF treatment, distinct protein synthesis-dependent and -independent effects on ChAT activity and ACh release were observed. These findings imply that acute neurotransmitter-like as well as classical neurotrophic influences contribute to the effects of NGF on BFCN. These capabilities may make complementary contributions to the formation, maintenance, and activity of BFCN synapses.
The effective concentrations of NGF on ACh release suggest the involvement of TrkA receptors, which autophosphorylate at similarly low ligand concentrations (Kaplan et al., 1991). Inhibition of TrkA signaling with K252a prevented NGF-enhanced ACh release, consistent with K252a prevention of BDNF-enhanced neurotransmission in hippocampal cultures (Li et al., 1998). Considering that K252a (100 nm) increased ACh release to some extent in our model, it was not possible to reduce NGF-associated ACh release to vehicle-treated levels. Accordingly, a direct or modulatory role for p75NTR remains possible.
Although the steps linking TrkA to ACh release remain to be fully established, in our model, TTX-sensitive Na+ channels were not critical. However, NGF-enhanced ACh release was prevented by BAPTA-AM and Cd2+, suggesting that Ca2+ action after entry via VGCC was critical. In accord with these findings, NGF rapidly increases voltage-sensitive Ca2+ currents in molluscan neurons and PC12 cells (Wildering et al., 1995; Jia et al., 1999), as well as increasing intracellular Ca2+ in primary BF cultures (Nonner et al., 2000).
Depolarization augmented the quantity of ACh release associated with NGF exposure, implying a mechanism for preferentially maintaining more release at active synapses. This is in agreement with other reports of activity-dependent neurotrophin action on synaptic efficacy (Gottschalk et al., 1998; Boulanger and Poo, 1999a). Considering that synaptic fatigue may contribute to establishing ACh release levels during the high-K+ exposure period, it is interesting that fatigue accompanying high-frequency stimulation is prevented by BDNF (Gottschalk et al., 1998).
Our protocol involved a brief period of low activity (during and immediately before NGF exposure; 6 mmK+) after sustained high-activity levels (growth conditions; 25 mm K+). It is possible that depolarization during the maturation of BF cultures influenced NGF-associated signal transduction pathways and/or interacted with other BFCN characteristics to alter or facilitate NGF-induced ACh release. Indeed, short-term or multi-day K+ depolarization has been shown to modulate features of neurotrophin signaling pathways in central (Meyer-Franke et al., 1998) or peripheral neurons (Vaillant et al., 1999), respectively. Moreover, in Xenopus motor neurons, K+ depolarization rapidly increases the sensitivity of neurotransmission to enhancement by BDNF (Boulanger and Poo, 1999a). Nevertheless, our preliminary observations suggest that BF cultures grown under low-K+ conditions also respond acutely to NGF (10 ng/ml) with increased ACh release (data not shown). Thus, NGF-induced ACh release is not unique to cultures grown under high-K+ conditions, although more subtle differences could exist.
Activity upregulates synthesis and release of neurotrophins (Thoenen, 1995), with hippocampal expression and secretion of NGF being elevated by muscarinic and nicotinic receptor signaling (da Penha Berzaghi et al., 1993; Knipper et al., 1994; Blochl and Thoenen, 1995; French et al., 1999). The capacity of NGF to enhance ACh release and of ACh to increase NGF has been hypothesized to contribute to synaptic efficiency (Knipper et al., 1994). These characteristics suggest potential mechanisms for immediate (i.e., translation-independent secretagogue effects at the terminal level) and long-term (i.e., transcription/translation-dependent neurotrophic effects) strengthening of synaptic connectivity resulting from increased NGF availability. Moreover, the activity-dependent nature of NGF-enhanced ACh release implies that this feedback could be amplified at more active synapses.
TTX exposure did not prevent NGF-enhanced ACh release, and this is consistent with the selective expression of NGF receptors on BFCNs (Hartikka and Hefti, 1988; Koh and Loy, 1989; Holtzman et al., 1992;Svendsen et al., 1994). Because NGF is highly expressed in regions of BF innervation (Korsching et al., 1985; Large et al., 1986), action at the terminal agrees with the potential physiological modulation of ACh release by target-derived NGF. The TTX data also suggest that although increased depolarization is associated with greater NGF enhancement of ACh release, high levels of concurrent activity are not required. This may be significant for developing BFCNs first encountering target-derived NGF, which may increase ACh release from innervating fibers with low intrinsic activity, resulting in feedback between ACh release and NGF secretion and thereby increasing NGF available for retrograde transport. The translation-dependent actions of NGF likely include direct and indirect enhancement of action-potential generation probability. In an indirect neurotrophic manner, NGF increases BFCN excitability by altering properties of Ca2+ currents at the soma level (Levine et al., 1995) and increases excitability in other developing neurons by inducing expression of Na+, K+, and Ca2+channels (Lesser and Lo, 1995; Toledo-Aral et al., 1995; Hilborn et al., 1998). At the soma level, NGF increases BFCN firing under some conditions, indicating that it can directly induce action potentials (Palmer et al., 1993; Albeck et al., 1999). Thus, target-derived, retrogradely transported NGF is likely to facilitate generation of action potentials, and this would be amplified by the synapse-strengthening feedback between ACh release and NGF secretion at the terminal level. This could subsequently interact with the activity-dependent nature of the NGF secretagogue action to further promote synapse consolidation.
A 60 min NGF exposure increased ACh release for at least 4 hr after its removal. This was not observed when ACh release was stimulated with high K+ (without NGF), despite the ∼10-fold increase (Auld et al., 2000a). The long-term enhancement of ACh release was dependent on NGF availability only during the exposure period, suggesting that critical signal transduction/effector processes were initiated quickly and remained activated for several hours. Even a 5 min exposure was associated with prolonged enhancement of release. These time frames are similar to NGF-induced increases in Ca2+, TrkA phosphorylation, and downstream pathways in primary BF cultures (Knusel et al., 1992; Downen et al., 1993; Nonner et al., 2000). Thus, transitory target-derived NGF secretion may subsequently augment ACh release for several hours, greatly strengthening the synapse, although potential in vivo interactions with established circuitry could modify this response. Considering that NT-3, NT-4, and BDNF also induced prolonged ACh release, they could play a similar role during synaptic development and maintenance.
Complementary to our observations, a 30 min neurotrophin exposure increased ChAT activity measured 24 hr later in BFCNs (Nonner et al., 2000), and a 1 min exposure to NGF induced Na+ channel expression in PC12 cells (Toledo-Aral et al., 1995). Thus, even short exposures to neurotrophins can cause lasting effects via both neurotrophic and secretagogue mechanisms. Regarding prolonged responses of BFCNs to brief neurotrophin exposures, it seems likely that increases in ChAT activity (Nonner et al., 2000) and translation-independent ACh release (this report) depend on common (e.g., TrkA, Ca2+) and disparate (e.g., translation) mechanisms. Because Ca2+ is involved in NGF-induced ACh release, the mechanism(s) sustaining prolonged release likely involves Ca2+-dependent elements. Ca2+ activates kinases that regulate neurotransmission and vesicle trafficking, such as Ca2+/calmodulin-dependent kinase II (Llinas et al., 1985; Greengard et al., 1993). Interestingly, this kinase has been implicated in NT-3-induced neurotransmitter release from Xenopus motor neurons (He et al., 2000).
Rapid NGF enhancement of ACh release was not reduced by cycloheximide, suggesting that protein translation is not involved. In agreement with previous observations (Pongrac and Rylett, 1998), increased ChAT activity after 24 hr NGF exposure was dependent on new protein synthesis. Interestingly, even after 24 hr, and regardless of the lack of increase in ChAT activity, there was a large protein synthesis-independent enhancement of ACh release associated with the NGF/cycloheximide condition. Thus, NGF can enhance ACh release in a secretagogue manner after prolonged exposure, and this may have relevance for maintenance of ACh release levels at BFCN synapses that are exposed to target-derived NGF. It is also apparent that under some conditions, increased ChAT activity is not associated with—or required for—enhancement of ACh release after prolonged NGF exposure. Together, these data suggest that the translation-independent secretagogue action of NGF may contribute to a portion of increased ACh release, even after prolonged NGF exposures sufficient to induce transcription and translation-dependent increases in ACh synthesis capacity.
In summary, NGF rapidly enhanced ACh release from embryonic BF cultures. The NGF-associated increase was activity dependent and, unlike classical neurotransmitter modulation, persisted for several hours after NGF removal in a manner akin to LTP. The NGF-induced increase was dependent on TrkA signaling, Ca2+, and VGCCs, but not dependent on new protein synthesis. After a 24 hr treatment with NGF, distinct protein synthesis-dependent and -independent effects on ChAT activity and ACh release, respectively, were observed. Together, these data suggest that in addition to translation-dependent neurotrophic actions, NGF has strong influences on BFCN function via both rapid and prolonged modulation of ACh release in a neurotransmitter/neuromodulator-like manner.
Footnotes
This work was supported by the Medical Research Council of Canada (MRCC)/Canadian Institutes of Health Research (CIHR). D.S.A. holds a Doctoral Award from MRCC/CIHR. We thank Dr. Brian Collier and Dr. Freda Miller for critically reading this manuscript, and Dr. Joseph Rochford for advice concerning statistical analyses.
Correspondence should be addressed to Dr. Rémi Quirion, Scientific Director, Douglas Hospital Research Center, 6875 Boulevard Lasalle, Montréal, Québec, Canada H4H 1R3. E-mail:quirem@douglas.mcgill.ca.
D. S. Auld's present address: Centre de Recherche en Sciences Neurologiques, Département de Physiologie, Université de Montréal, P.O. Box 6128, Station Centre-ville, Montreal, Québec, Canada H3C 3J7.
REFERENCES
- 1.Albeck DS, Backman C, Veng L, Friden P, Rose GM, Granholm A. Acute application of NGF increases the firing rate of aged rat basal forebrain neurons. Eur J Neurosci. 1999;11:2291–2304. doi: 10.1046/j.1460-9568.1999.00644.x. [DOI] [PubMed] [Google Scholar]
- 2.Auld DS, Kar S, Quirion R. Beta-amyloid peptides as direct cholinergic neuromodulators: a missing link? Trends Neurosci. 1998;21:43–49. doi: 10.1016/s0166-2236(97)01144-2. [DOI] [PubMed] [Google Scholar]
- 3.Auld DS, Day JC, Mennicken F, Quirion R. Pharmacological characterization of endogenous acetylcholine release from primary septal cultures. J Pharmacol Exp Ther. 2000a;292:692–697. [PubMed] [Google Scholar]
- 4.Auld DS, Mennicken F, Quirion R. Differentiation between translation-independent neuromodulatory and translation-dependent neurotrophic influences of neurotrophins on basal forebrain cholinergic neurons. Soc Neurosci Abstr. 2000b;26:1869. [Google Scholar]
- 5.Auld DS, Mennicken F, Day JC, Quirion R. Neurotrophins differentially enhance acetylcholine release, acetylcholine content and choline acetyltransferase activity in basal forebrain neurons. J Neurochem. 2001;77:253–262. doi: 10.1046/j.1471-4159.2001.t01-1-00234.x. [DOI] [PubMed] [Google Scholar]
- 6.Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol. 2000;163:495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- 7.Baxter MG, Chiba AA. Cognitive functions of the basal forebrain. Curr Opin Neurobiol. 1999;9:178–183. doi: 10.1016/s0959-4388(99)80024-5. [DOI] [PubMed] [Google Scholar]
- 8.Blochl A, Thoenen H. Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur J Neurosci. 1995;7:1220–1228. doi: 10.1111/j.1460-9568.1995.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 9.Boulanger L, Poo M. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci. 1999a;2:346–351. doi: 10.1038/7258. [DOI] [PubMed] [Google Scholar]
- 10.Boulanger L, Poo M. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999b;284:1982–1984. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- 11.Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen KS, Nishimura MC, Armanini MP, Crowley C, Spencer SD, Phillips HS. Disruption of a single allele of the nerve growth factor gene results in atrophy of basal forebrain cholinergic neurons and memory deficits. J Neurosci. 1997;17:7288–7296. doi: 10.1523/JNEUROSCI.17-19-07288.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.da Penha Berzaghi M, Cooper J, Castren E, Zafra F, Sofroniew M, Thoenen H, Lindholm D. Cholinergic regulation of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) but not neurotrophin-3 (NT-3) mRNA levels in the developing rat hippocampus. J Neurosci. 1993;13:3818–3826. doi: 10.1523/JNEUROSCI.13-09-03818.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debeir T, Saragovi HU, Cuello AC. A nerve growth factor mimetic TrkA antagonist causes withdrawal of cortical cholinergic boutons in the adult rat. Proc Natl Acad Sci USA. 1999;96:4067–4072. doi: 10.1073/pnas.96.7.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiStefano PS, Friedman B, Radziejewski C, Alexander C, Boland P, Schick CM, Lindsay RM, Wiegand SJ. The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron. 1992;8:983–993. doi: 10.1016/0896-6273(92)90213-w. [DOI] [PubMed] [Google Scholar]
- 16.Downen M, Mudd L, Roback JD, Palfrey HC, Wainer BH. Early nerve growth factor-induced events in developing rat septal neurons. Dev Brain Res. 1993;74:1–13. doi: 10.1016/0165-3806(93)90077-n. [DOI] [PubMed] [Google Scholar]
- 17.Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM. A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci. 1997;17:7644–7654. doi: 10.1523/JNEUROSCI.17-20-07644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fibiger HC. The organization and some projections of the cholinergic neurons of the mammalian brain. Brain Res Rev. 1982;4:327–388. doi: 10.1016/0165-0173(82)90011-x. [DOI] [PubMed] [Google Scholar]
- 19.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 20.French SJ, Humby T, Horner CH, Sofroniew MV, Rattray M. Hippocampal neurotrophin and trk receptor mRNA levels are altered by local administration of nicotine, carbachol and pilocarpine. Mol Brain Res. 1999;67:124–136. doi: 10.1016/s0169-328x(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 21.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 23.Hanisch UK, Seto D, Quirion R. Modulation of hippocampal acetylcholine release: a potent central action of interleukin-2. J Neurosci. 1993;13:3368–3374. doi: 10.1523/JNEUROSCI.13-08-03368.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartikka J, Hefti F. Development of septal cholinergic neurons in culture: plating density and glial cells modulate effect of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J Neurosci. 1988;8:2967–2985. doi: 10.1523/JNEUROSCI.08-08-02967.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He X, Yang F, Xie Z, Lu B. Intracellular Ca(2+) and Ca(2+)/calmodulin-dependent kinase II mediate acute potentiation of neurotransmitter release by neurotrophin-3. J Cell Biol. 2000;149:783–792. doi: 10.1083/jcb.149.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hersi AI, Richard JW, Gaudreau P, Quirion R. Local modulation of hippocampal acetylcholine release by dopamine D1 receptors: a combined receptor autoradiography and in vivo dialysis study. J Neurosci. 1995;15:7150–7157. doi: 10.1523/JNEUROSCI.15-11-07150.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hilborn MD, Vaillancourt RR, Rane SG. Growth factor receptor tyrosine kinases acutely regulate neuronal sodium channels through the src signaling pathway. J Neurosci. 1998;18:590–600. doi: 10.1523/JNEUROSCI.18-02-00590.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holtzman DM, Li Y, Parada LF, Kinsman S, Chen CK, Valletta JS, Zhou J, Long JB, Mobley WC. p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron. 1992;9:465–478. doi: 10.1016/0896-6273(92)90184-f. [DOI] [PubMed] [Google Scholar]
- 29.Jia M, Li M, Liu XW, Jiang H, Nelson PG, Guroff G. Voltage-sensitive calcium currents are acutely increased by nerve growth factor in PC12 cells. J Neurophysiol. 1999;82:2847–2852. doi: 10.1152/jn.1999.82.6.2847. [DOI] [PubMed] [Google Scholar]
- 30.Jiang H, Guroff G. Actions of the neurotrophins on calcium uptake. J Neurosci Res. 1997;50:355–360. doi: 10.1002/(SICI)1097-4547(19971101)50:3<355::AID-JNR1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 31.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 33.Kar S, Seto D, Dore S, Hanisch U, Quirion R. Insulin-like growth factors-I and -II differentially regulate endogenous acetylcholine release from the rat hippocampal formation. Proc Natl Acad Sci USA. 1997;94:14054–14059. doi: 10.1073/pnas.94.25.14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knipper M, da Penha Berzaghi M, Blochl A, Breer H, Thoenen H, Lindholm D. Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur J Neurosci. 1994;6:668–671. doi: 10.1111/j.1460-9568.1994.tb00312.x. [DOI] [PubMed] [Google Scholar]
- 35.Knusel B, Rabin S, Widmer HR, Hefti F, Kaplan DR. Neurotrophin-induced trk receptor phosphorylation and cholinergic neuron response in primary cultures of embryonic rat brain neurons. NeuroReport. 1992;3:885–888. doi: 10.1097/00001756-199210000-00016. [DOI] [PubMed] [Google Scholar]
- 36.Koh S, Loy R. Localization and development of nerve growth factor-sensitive rat basal forebrain neurons and their afferent projections to hippocampus and neocortex. J Neurosci. 1989;9:2999–3018. doi: 10.1523/JNEUROSCI.09-09-02999.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koliatsos VE, Price DL, Gouras GK, Cayouette MH, Burton LE, Winslow JW. Highly selective effects of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 on intact and injured basal forebrain magnocellular neurons. J Comp Neurol. 1994;343:247–262. doi: 10.1002/cne.903430206. [DOI] [PubMed] [Google Scholar]
- 38.Korsching S, Auburger G, Heumann R, Scott J, Thoenen H. Levels of nerve growth factor and its mRNA in the central nervous system of the rat correlate with cholinergic innervation. EMBO J. 1985;4:1389–1393. doi: 10.1002/j.1460-2075.1985.tb03791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Large TH, Bodary SC, Clegg DO, Weskamp G, Otten U, Reichardt LF. Nerve growth factor gene expression in the developing rat brain. Science. 1986;234:352–355. doi: 10.1126/science.3764415. [DOI] [PubMed] [Google Scholar]
- 41.Lesser SS, Lo DC. Regulation of voltage-gated ion channels by NGF and ciliary neurotrophic factor in SK-N-SH neuroblastoma cells. J Neurosci. 1995;15:253–261. doi: 10.1523/JNEUROSCI.15-01-00253.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 43.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1161. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 44.Levine ES, Dreyfus CF, Black IB, Plummer MR. Differential effects of NGF and BDNF on voltage-gated calcium currents in embryonic basal forebrain neurons. J Neurosci. 1995;15:3084–3091. doi: 10.1523/JNEUROSCI.15-04-03084.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua-Couzens J, Clary DO, Knusel B, Mobley WC. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Llinas R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci USA. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 49.Lu B, Chow A. Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res. 1999;58:76–87. [PubMed] [Google Scholar]
- 50.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Jr, Reichardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molnar M, Tongiorgi E, Avignone E, Gonfloni S, Ruberti F, Domenici L, Cattaneo A. The effects of anti-nerve growth factor monoclonal antibodies on developing basal forebrain neurons are transient and reversible. Eur J Neurosci. 1998;10:3127–3140. doi: 10.1046/j.1460-9568.1998.00314.x. [DOI] [PubMed] [Google Scholar]
- 52.Nakamura M, Nishio C, Nonomura T, Hatanaka H. High potassium and cyclic AMP analog promote neuronal survival of basal forebrain cholinergic neurons in culture from postnatal 2-week-old rats. Dev Brain Res. 1994;81:218–229. doi: 10.1016/0165-3806(94)90308-5. [DOI] [PubMed] [Google Scholar]
- 53.Nonner D, Barrett EF, Barrett JN. Neurotrophin effects on survival and expression of cholinergic properties in cultured rat septal neurons under normal and stress conditions. J Neurosci. 1996;16:6665–6675. doi: 10.1523/JNEUROSCI.16-21-06665.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nonner D, Barrett EF, Barrett JN. Brief exposure to neurotrophins produces a calcium-dependent increase in choline acetyltransferase activity in cultured rat septal neurons. J Neurochem. 2000;74:988–999. doi: 10.1046/j.1471-4159.2000.0740988.x. [DOI] [PubMed] [Google Scholar]
- 55.Nonomura T, Nishio C, Lindsay RM, Hatanaka H. Cultured basal forebrain cholinergic neurons from postnatal rats show both overlapping and non-overlapping responses to the neurotrophins. Brain Res. 1995;683:129–139. doi: 10.1016/0006-8993(95)00357-v. [DOI] [PubMed] [Google Scholar]
- 56.Oosawa H, Fujii T, Kawashima K. Nerve growth factor increases the synthesis and release of acetylcholine and the expression of vesicular acetylcholine transporter in primary cultured rat embryonic septal cells. J Neurosci Res. 1999;57:381–387. [PubMed] [Google Scholar]
- 57.Palmer MR, Eriksdotter-Nilsson M, Henschen A, Ebendal T, Olson L. Nerve growth factor-induced excitation of selected neurons in the brain which is blocked by a low-affinity receptor antibody. Exp Brain Res. 1993;93:226–230. doi: 10.1007/BF00228389. [DOI] [PubMed] [Google Scholar]
- 58.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 59.Pesavento E, Margotti E, Righi M, Domenici L. Blocking the NGF-TrkA interaction rescues the developmental loss of LTP in the rat visual cortex: role for the cholinergic system. Neuron. 2000;25:165–175. doi: 10.1016/s0896-6273(00)80880-6. [DOI] [PubMed] [Google Scholar]
- 60.Pongrac JL, Rylett RJ. NGF-induction of the expression of ChAT mRNA in PC12 cells and primary cultures of embryonic rat basal forebrain. Mol Brain Res. 1998;62:25–34. doi: 10.1016/s0169-328x(98)00215-0. [DOI] [PubMed] [Google Scholar]
- 61.Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raiteri M, Leardi R, Marchi M. Heterogeneity of presynaptic muscarinic receptors regulating neurotransmitter release in the rat brain. J Pharmacol Exp Ther. 1984;228:209–214. [PubMed] [Google Scholar]
- 63.Ruberti F, Capsoni S, Comparini A, Di Daniel E, Franzot J, Gonfloni S, Rossi G, Berardi N, Cattaneo A. Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J Neurosci. 2000;20:2589–2601. doi: 10.1523/JNEUROSCI.20-07-02589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sala R, Viegi A, Rossi FM, Pizzorusso T, Bonanno G, Raiteri M, Maffei L. Nerve growth factor and brain-derived neurotrophic factor increase neurotransmitter release in the rat visual cortex. Eur J Neurosci. 1998;10:2185–2191. doi: 10.1046/j.1460-9568.1998.00227.x. [DOI] [PubMed] [Google Scholar]
- 65.Schinder AF, Poo MM. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23:639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- 66.Stoop R, Poo MM. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Svendsen CN, Kew JN, Staley K, Sofroniew MV. Death of developing septal cholinergic neurons following NGF withdrawal in vitro: protection by protein synthesis inhibition. J Neurosci. 1994;14:75–87. doi: 10.1523/JNEUROSCI.14-01-00075.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takei N, Tsukui H, Hatanaka H. Nerve growth factor increases the intracellular content of acetylcholine in cultured septal neurons from developing rats. J Neurochem. 1988;51:1118–1125. doi: 10.1111/j.1471-4159.1988.tb03076.x. [DOI] [PubMed] [Google Scholar]
- 69.Takei N, Tsukui H, Hatanaka H. Intracellular storage and evoked release of acetylcholine from postnatal rat basal forebrain cholinergic neurons in culture with nerve growth factor. J Neurochem. 1989;53:1405–1410. doi: 10.1111/j.1471-4159.1989.tb08531.x. [DOI] [PubMed] [Google Scholar]
- 70.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 71.Toledo-Aral JJ, Brehm P, Halegoua S, Mandel G. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron. 1995;14:607–611. doi: 10.1016/0896-6273(95)90317-8. [DOI] [PubMed] [Google Scholar]
- 72.Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol. 1999;146:955–966. doi: 10.1083/jcb.146.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vantini G, Schiavo N, Di Martino A, Polato P, Triban C, Callegaro L, Toffano G, Leon A. Evidence for a physiological role of nerve growth factor in the central nervous system of neonatal rats. Neuron. 1989;3:267–273. doi: 10.1016/0896-6273(89)90251-1. [DOI] [PubMed] [Google Scholar]
- 74.Wildering WC, Lodder JC, Kits KS, Bulloch AG. Nerve growth factor (NGF) acutely enhances high-voltage-activated calcium currents in molluscan neurons. J Neurophysiol. 1995;74:2778–2781. doi: 10.1152/jn.1995.74.6.2778. [DOI] [PubMed] [Google Scholar]
- 75.Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]