

Abstract
Alzheimer's Disease (AD) is characterized by cerebral accumulation of β-amyloid peptides (Aβ), which are proteolytically derived from β-amyloid precursor protein (βAPP). βAPP metabolism is highly regulated via various signal transduction systems, e.g., several serine/threonine kinases and phosphatases. Several growth factors known to act via receptor tyrosine kinases also have been demonstrated to regulate sβAPP secretion. Among these receptors, insulin and insulin-like growth factor-1 receptors are highly expressed in brain, especially in hippocampus and cortex. Emerging evidence indicates that insulin has important functions in brain regions involved in learning and memory. Here we present evidence that insulin significantly reduces intracellular accumulation of Aβ and that it does so by accelerating βAPP/Aβ trafficking from the trans-Golgi network, a major cellular site for Aβ generation, to the plasma membrane. Furthermore, insulin increases the extracellular level of Aβ both by promoting its secretion and by inhibiting its degradation via insulin-degrading enzyme. The action of insulin on βAPP metabolism is mediated via a receptor tyrosine kinase/mitogen-activated protein (MAP) kinase kinase pathway. The results suggest cell biological and signal transduction mechanisms by which insulin modulates βAPP and Aβ trafficking in neuronal cultures.
Keywords: β-amyloid, β-amyloid precursor protein, insulin, MAPK, Alzheimer's disease, diabetes mellitus, intracellular trafficking, endoplasmic reticulum, trans-Golgi network, plasma membrane
Neuropathological hallmarks of Alzheimer's Disease (AD) include deposition of β-amyloid (Aβ) plaques, neurofibrillary tangles, and neuronal cell loss in vulnerable brain regions. Plaques contain an aggregated population of heterogeneous Aβ peptides derived from β-amyloid precursor protein (βAPP). Full-length βAPP undergoes proteolytic β-secretase and γ-secretase activities to generate Aβ40 and Aβ42 peptides, the predominant Aβ variants. In addition to these amyloid-generating activities, full-length βAPP can undergo alternative processing by an enzymatic activity termed “α-secretase” that cleaves within the Aβ region. This activity releases a soluble fragment (sβAPPα) extracellularly and precludes Aβ formation. Several studies indicate that Aβ is toxic to neurons. Accumulation of Aβ peptides within the brain is believed to initiate the pathological cascade culminating in clinical AD, a hypothesis supported by the development of early-onset familial AD (FAD) within pedigrees harboring autosomal dominant gene mutations in βAPP that lead to the excessive generation of Aβ (for review, see Selkoe, 1998). Cell biological studies have demonstrated that both Aβ40 and Aβ42 are produced intracellularly (Cook et al., 1997; Xu et al., 1997; Lee et al., 1998; Skovronsky et al., 1998;Greenfield et al., 1999). Moreover, recent evidence raises the possibility that intracellular Aβ42 may play a direct pathogenic role in AD neuropathology (for review, see Wilson et al., 1999; Gouras et al., 2000).
βAPP metabolism is highly regulated via various signal transduction systems, e.g., various serine/threonine kinases and phosphatases (for review, see Mills and Reiner, 1999). Several growth factors known to act via receptors with intrinsic tyrosine kinase activity also have been demonstrated to regulate sβAPP secretion (Refolo et al., 1989;Schubert et al., 1989). Among these receptors, insulin and insulin-like growth factor-1 (IGF-1) receptors are highly expressed in brain, particularly in hippocampus and cortex (Werther et al., 1987). Recently, it has been demonstrated that insulin receptors are concentrated at the synaptic level and that they are a component of postsynaptic densities in cultured hippocampal neurons (Abbott et al., 1999). Moreover, insulin can recruit GABAAreceptors to the postsynaptic domain (Wan et al., 1997), suggesting a role for this hormone in synaptic plasticity. Emerging evidence indicates that insulin has important functions in brain regions involved in learning and memory (Wickelgren, 1998; Zhao et al., 1999). Recent findings demonstrated insulin receptor upregulation and reduced insulin receptor-mediated tyrosine kinase activity in AD brains (Frolich et al., 1998, 1999). Insulin and IGF-1 have been shown to regulate tau phosphorylation via GSK-3β, suggesting that neurofibrillary tangle formation in AD may be downstream of insulin signaling (Hong and Lee, 1997; Lesort et al., 1999; Lesort and Johnson, 2000). Recently, investigators have begun to address the potential role of insulin in βAPP metabolism. It was shown that insulin elevates sβAPP secretion in SH-SY5Y cells (Solano et al., 2000). Moreover, it was demonstrated that insulin-degrading enzyme (IDE) degrades extracellular Aβ in microglial and neuronal cultures and that insulin can prevent this degradation, thereby impairing the clearance of extracellular Aβ (Qiu et al., 1998; Vekrellis et al., 2000).
Here we report that insulin decreases intracellular levels and increases extracellular levels of both Aβ40 and Aβ42. These effects of insulin are associated with accelerated βAPP/Aβ trafficking from the Golgi/trans-Golgi network (TGN) to the plasma membrane and are prevented by inhibitors of tyrosine kinase and MAP kinase kinase.
MATERIALS AND METHODS
Cell cultures. N2a neuroblastoma cells transfected with human βAPP695 were maintained in medium containing 50% DMEM and 50% Opti-MEM, supplemented with 5% FBS, 200 μg/ml G418, and antibiotics (Life Technologies, Gaithersburg, MD). Primary neuronal cultures were derived from the cerebral cortices of day 17 (E17) embryos obtained from timed pregnant Sprague Dawley rats as described previously (Gouras et al., 1998). Neurons were used for experiments after 4–5 d in culture.
Pulse–chase experiments and insulin treatment. N2a cells (80% confluent in 10 cm dish) or primary neuronal cultures (107 cells/10 cm dish) were labeled for 20 min with 750 μCi/ml [35S]methionine (NEN-DuPont, Boston, MA) in methionine-free DMEM supplemented withl-glutamine (Life Technologies). Cells were chased at 37°C in serum-free DMEM (Life Technologies) or in serum- and glucose-free DMEM supplemented with 50 mm2-deoxy-d-glucose in the absence or presence of insulin (Sigma, St. Louis, MO). In some experiments 0.3–30 μmglucagon, 0.1–1 μm IGF-1, 2.5–25 ng/ml EGF, 25–250 ng/ml NGF, 1–10 ng/ml PDGF, 10–100 ng/ml aFGF, or 10–100 ng/ml bFGF (BD Transduction Laboratories, Franklin Lakes, NJ; gifts of Dr. J. Schlessinger, New York University Medical Center) was added during the chase. For continuous metabolic labeling the cells were incubated for 4 hr with 750 μCi/ml [35S]methionine in methionine-free DMEM supplemented with l-glutamine in the absence or presence of 1 μm insulin. For steady-state experiments the cells were treated for 4–16 hr with 0.3–1 μm insulin in the presence or absence of 10–25 μm tyrphostin-25, 500 nm wortmannin, 25 μmU73122, or 10 μm PD98059; the samples were analyzed by Western blot.
Aβ degradation assay. N2a cells expressing wild-type human βAPP695 were labeled continuously for 4 hr with 750 μCi/ml [35S]methionine, and medium was collected to serve as the source for labeled Aβ and sβAPPα proteins. Nonlabeled serum-free conditioned medium (CM) was collected after the incubation of primary neuronal cultures for 16 hr. This CM was mixed with the labeled Aβ/sβAPPα-containing medium and incubated further at 37°C for 16 hr in the absence or presence of 1 μm insulin and/or 1 mm 1,10-phenantroline. In some experiments IDE was eliminated from the cold CM by immunodepletion, using an anti-IDE monoclonal antibody (9B12) (Shii and Roth, 1986). The amounts of labeled Aβ or sβAPPα remaining in each sample after incubation were determined by immunoprecipitation with antibody 4G8 (for Aβ) or 6E10 (for sβAPPα) (Senetek PLC, St. Louis, MO), followed by 10–20% Tris/tricine (Aβ) or 4–12% Tris/glycine (sβAPPα) SDS-PAGE.
Immunoprecipitation and Western analysis. Media were collected, centrifuged briefly to remove cell debris, and sequentially immunoprecipitated first with 4G8 for Aβ and then with 6E10 for sβAPPα secreted from N2a cells or 22C11 (Roche Pharmaceuticals, Nutley, NJ) for sβAPP secreted from rat neurons. sβAPPβ secreted from N2a cells was determined by immunoprecipitation with 22C11 antibody after immunodepletion of sβAPPα with 6E10 antibody. Immunoprecipitation of IDE was performed with 28H1 monoclonal antibody (Shii and Roth, 1986). Cells (1–2 × 107) were scraped from plates in ice-cold PBS with a rubber policeman. After centrifugation the pellets were treated with 250 μl of 3% SDS in PBS containing 10 μl/ml of β-mercaptoethanol and subjected to vortexing and heating at 95°C for 10 min, followed by sonication and centrifugation at 100,000 × g for 10 min. The resultant supernatants were diluted 1:4; adjusted to a final concentration of 2% Triton X-100 and (in mm) 190 NaCl, 20 Tris-HCl, pH 8.8, and 2 EDTA; and subjected to immunoprecipitation and SDS-PAGE analysis that used 10–20% Tris/tricine gels (for Aβ) or 4–12% Tris/glycine gels (for sβAPPα detection). For Western blot the samples were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA or Bio-Rad, Hercules, CA), and the membranes were boiled in PBS for 5 min. Aβ and sβAPPα were detected by using 6E10 monoclonal antibodies. For Western blot analysis of full-length βAPP and presenilin-1 (PS-1) N-terminal fragment, the samples were analyzed on 12% SDS-PAGE gels, transferred to PVDF membrane (Millipore), and immunoblotted with antibody 369 (Buxbaum et al., 1990) and antibody Ab14 (Seeger et al., 1997), respectively. Anti-insulin receptor antibody was from Neomarkers (Fremont, CA). Intracellular IDE was detected by immunofluorescence as described previously (Greenfield et al., 1999) with 9B12 and 28H1 monoclonal antibodies (Shii and Roth, 1986).
Immunoprecipitation–mass spectrometry analysis. Media or cell lysates were immunoprecipitated with 4G8 antibody and protein A/G-agarose beads. The molecular masses and concentrations of immunoprecipitated Aβ species were measured by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis, as described previously (Xu et al., 1998). For analysis, Aβ12–28 and insulin internal standards were added to the samples.
Sucrose gradient fractionation. Fractionation by sucrose gradient was performed as described previously (Greenfield et al., 1999). After 16 hr of incubation in the absence or presence of 1 μm insulin, N2a cells were homogenized by using a stainless steel ball bearing homogenizer in 5 vol of 0.25 msucrose, 10 mm Tris-HCl, pH 7.4, 1 mmMgAc2, and a protease inhibitor mixture. The homogenate was loaded on top of a step gradient comprised of 1.5 ml of 2 m sucrose, 4 ml of 1.3 m sucrose, 3.0 ml of 1.16 m sucrose, and 2.0 ml of 0.8 m sucrose. All sucrose solutions contained 10 mm Tris-HCl, pH 7.4, and 1 mm MgAc2. The gradients were centrifuged for 2.5 hr at 100,000 × g in a Beckman SW41Ti rotor. Fractions were collected and assayed for total protein with BCA assay. Aβ and βAPP were assayed as described above. Proteins from each fraction were analyzed by Western blot with antibodies against calnexin, γ-adaptin (BD Transduction Laboratories), or ARF3 (Affinity BioReagents, Neshanic Station, NJ) to identify the fractions containing, respectively, endoplasmic reticulum (ER), TGN, and cytosol/post-TGN vesicles. To determine the fractions enriched in plasma membrane proteins, we fractionated N2a cells after biotinylation of surface proteins and detected biotinylated βAPP as described below.
Cell surface biotinylation. Biotinylation was performed on confluent monolayer N2a cells overexpressing βAPP695 by using sulfo-NHS-LC-biotin [sulfosuccinimidyl-6-(biotinamido)-hexanoate; Pierce, Rockford, IL]. The reagent was dissolved in PBS with calcium and magnesium, pH 7.2, at 0.5 mg/ml and added twice to the cultures for 20 min at 4°C. After thorough washing, the cells were lysed with 3%SDS as described above. βAPP was immunoprecipitated by using 369 antiserum and was analyzed by Western blot. Biotinylated βAPP was detected by using HRP-conjugated streptavidin and reaction with a chemiluminescent substrate (NEN-DuPont).
Endoglycosidase-H Digestion. To evaluate the effect of insulin on ER-to-Golgi trafficking of βAPP, we pulse-labeled N2a cells for 5 min with 750 μCi/ml [35S]methionine (NEN-DuPont) in methionine-free DMEM and chased the cells for 5–45 min in the absence or presence of 1 μm insulin as described above. [35S]-labeled βAPP was immunoprecipitated from the SDS-soluble lysates by 369 antiserum. The immune complexes were boiled for 5 min in a buffer containing 50 mm Tris-HCl, pH7.6, 0.5% SDS, and 1% β-mercaptoethanol to dissociate βAPP from the antibody; sodium citrate was added to a final concentration of 0.05 m. Then the samples were incubated in the presence or absence of 50 U of endoglycosidase-H (New England Biolabs, Beverly, MA) at 37°C for 16 hr. βAPP isoforms were separated on a 6% Tris/glycine gel, transferred to PVDF membrane, and detected by autoradiography.
Quantification and densitometry. Gels were exposed to x-ray film, and the films were scanned with an Agfa Arcus II scanner. Band intensities were analyzed and quantified by using NIH Image Quant software, version 1.52. Statistical analysis was performed with ANOVA, followed by a post hoc test.
RESULTS
Insulin increases extracellular levels of Aβ40 and Aβ42
To examine the effect of insulin on βAPP metabolism, we pulse-labeled neuroblastoma (N2a) cells overexpressing human βAPP695 or primary cultures of rat cortical neurons with [35S]methionine for 20 min and chased the cells in the absence or presence of insulin for 4 hr. Cells that were treated with insulin showed a three- to fourfold increase in extracellular levels of both 4 kDa Aβ1–40/42 and 3 kDa N-terminally truncated Aβx-40/42 peptides, mainly composed of Aβ11–40/42 (Gouras et al., 1998) (Fig.1a, top). The effect of insulin on extracellular levels of Aβ1–40/42 was concentration-dependent, with a minimal effective concentration of ∼50 nm and a half-maximal effect at ∼300 nm (Fig. 1b). Insulin caused a ∼1.4-fold increase in sβAPPα extracellular levels (Fig.1a, bottom, c), with a small increase occurring at the lowest concentration (0.2 nm) that was tested. Secretion of sβAPPβ was not altered by insulin treatment (data not shown). Immunoprecipitation–mass spectrometry (IP–MS) analysis revealed increases in extracellular Aβ1–40 in both primary neurons and N2a cells (Fig. 1d,e) and increases of Aβ1–42 in N2a cells (Fig. 1e). The effect of insulin on the extracellular levels of Aβ1–40/42 and Aβx-40/42 in cultures of N2a cells was evident at all incubation times from 30 min to 24 hr (Fig. 2a). Aβ secretion in the absence or presence of insulin peaked at 4 hr and decreased thereafter, indicating that Aβ is susceptible to degradation by proteases released from the cells. Extracellular sβAPPα was increased slightly by insulin at early, but not at late, incubation times (Fig. 2b) and reached a plateau level because of its resistance to the degrading activity of proteases. βAPP levels were not altered by insulin during pulse–chase experiments. Glucose-containing medium was required for the action of insulin on Aβ. Glucose concentrations higher than the standard growing concentration did not alter basal or insulin-stimulated Aβ levels (data not shown).
Fig. 1.

Effect of insulin on the extracellular levels of Aβ and sβAPPα in murine neuroblastoma cells (N2a) and primary rat cortical neuronal cultures. Cells were pulse-labeled for 20 min with [35S]methionine and incubated in serum-free medium in the absence or presence of various concentrations of insulin for 4 hr. a, Representative autoradiographic analysis of extracellular Aβ (top) and sβAPPα (bottom) after incubation with or without 300 nm insulin. b, Extracellular levels of Aβ from N2a cells as a function of insulin concentration.c, Extracellular levels of sβAPPα from N2a cells as a function of insulin concentration. For b andc the data represent means ± SD;n = 3. d, e, IP–MS analysis of extracellular Aβ from primary neurons (d) and N2a cells (e) after incubation in the absence or presence of 1 μm insulin for 4 hr.
Fig. 2.
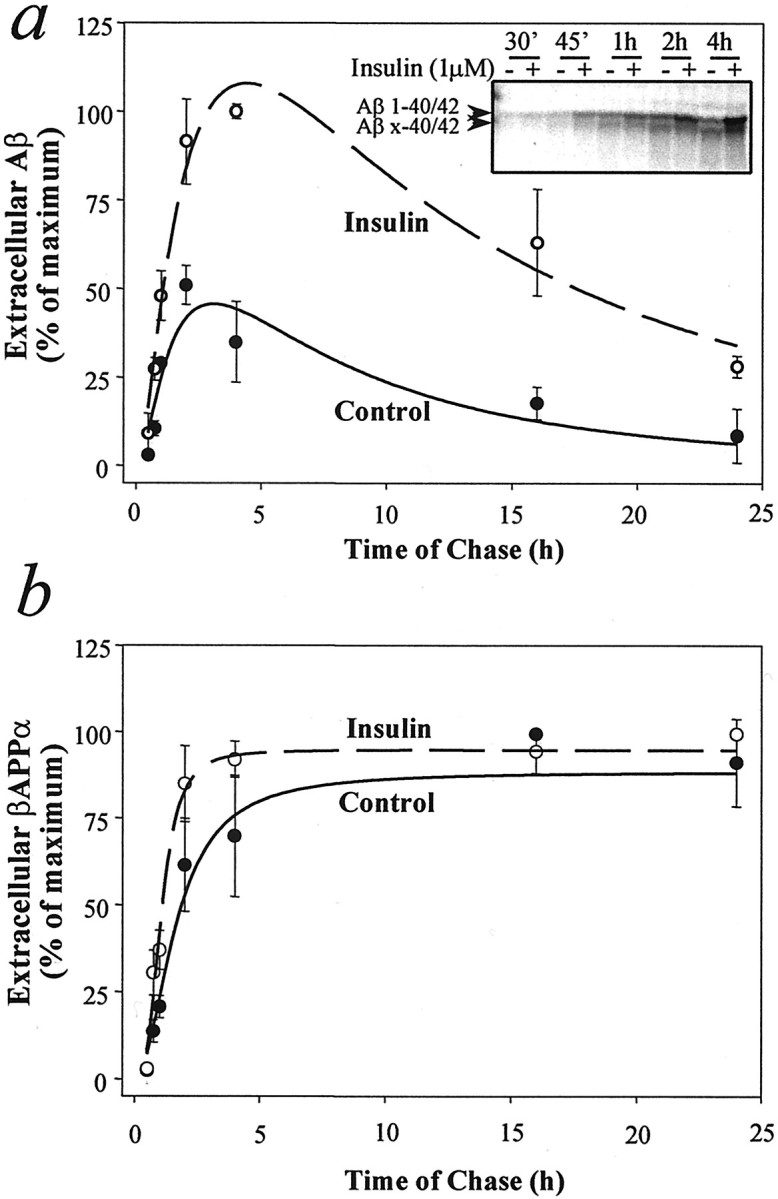
Time course of Aβ (a) and sβAPPα (b) extracellular levels from N2a cells in the absence or presence of insulin. Cells were pulse-labeled with [35S]methionine for 20 min and incubated in serum-free medium in the absence or presence of 1 μminsulin for different intervals. Data represent means ± SD;n = 3. Inset in ashows a representative autoradiographic analysis of extracellular Aβ after periods of chase from 30 min to 4 hr.
Insulin increases extracellular Aβ both by reducing IDE-mediated Aβ degradation and by stimulating Aβ secretion
To investigate whether the increased level of extracellular Aβ was attributable at least in part to the inhibition of extracellular degradation, we examined the effect of IDE on the levels of extracellular Aβ. IDE was found intracellularly by immunocytochemistry (data not shown), and pulse–chase experiments demonstrated a substantial amount of IDE in the medium of primary neuronal cultures after a 16 hr chase (Fig.3a). It was reported previously that IDE can cause the rapid degradation of Aβ1–40/42, but not Aβx-40/42, and that the enzymatic activity of IDE against Aβ1–40/42 could be prevented by insulin, phenantroline, or immunodepletion with an IDE-specific antibody (Qiu et al., 1998;Vekrellis et al., 2000). We have confirmed those earlier studies in the present investigation (Fig. 3b,c). Insulin and phenantroline each acted as a potent inhibitor of the degradation of Aβ by IDE in our in vitro setting (Fig. 3c). However, insulin, phenantroline, or immunodepletion of IDE from our system did not rescue Aβ completely from degradation, suggesting that other proteases may be involved in Aβ breakdown. To investigate whether insulin can stimulate the secretion of Aβ in addition to preventing its degradation, we treated N2a cells with or without insulin in the absence or presence of the IDE inhibitor 1,10-phenantroline for various time intervals (Fig. 3d). Phenantroline alone increased the extracellular levels of Aβ. However, the effect of phenantroline was less than that of insulin alone and was significant only at later intervals (4–16 hr). When phenantroline was present during the incubation period to inactivate IDE, insulin further enhanced the extracellular levels of Aβ. These results indicate that insulin can stimulate Aβ secretion in addition to inhibiting IDE-mediated Aβ degradation. In addition, the secretion of the 3 kDa N-terminally truncated Aβx-40/42 peptides, which are resistant to degradation by IDE (Qiu et al., 1998; Vekrellis et al., 2000) (Fig. 3b), was stimulated by insulin (see Figs. 1a, 2a).
Fig. 3.

Insulin inhibits Aβ degradation via IDE and stimulates the secretion of Aβ. a, IDE released in the medium from cultured neurons as a function of time. Primary cortical neurons were pulse-labeled for 20 min and chased for different intervals up to 16 hr. IDE was immunoprecipitated with 28H1 monoclonal anti-IDE antibody and analyzed on SDS-PAGE. b, c, N2a cells were incubated for 4 hr with [35S]methionine to produce labeled Aβ and sβAPPα. Serum-free conditioned medium was collected from cultured primary neurons, which had been incubated for 16 hr at 37°C. Then this medium was mixed with the medium containing labeled Aβ or sβAPPα and incubated for a further 16 hr in the absence or presence of the indicated substances (see Materials and Methods). b, Insulin inhibits Aβ degradation. Shown is an autoradiographic analysis of labeled Aβ (top) and sβAPPα (bottom). Media were collected before or after in vitro incubation in the absence or presence of 1 μm insulin, immunoprecipitated with anti-Aβ (4G8) or anti-sβAPP (22C11) antibodies, and analyzed on SDS-PAGE. Insulin caused a marked inhibition of Aβ1–40/42 degradation. c, IDE-mediated Aβ degradation was inhibited by 1 μm insulin (Ins), 1 mm 1,10-phenantroline (Phen), or immunodepletion of IDE with a monoclonal anti-IDE antibody (9B12; see Materials and Methods). Data represent means ± SD; n = 3. *p < 0.01 with respect to the sample with no incubation; **p < 0.01 with respect to control sample.d, Insulin stimulates Aβ secretion. N2a cells were pulse-labeled for 20 min and chased for various times in the absence or presence of 1 μm insulin, 1 mm1,10-phenantroline (Phen), or a combination of the two compounds. Data represent means ± SD; n = 3.
Insulin reduces intracellular Aβ40 and Aβ42
To examine further the role of insulin in βAPP metabolism, we examined the effect of insulin on intracellular Aβ, which recently has been hypothesized to be important in AD (Wild-Bode et al., 1997;Skovronsky et al., 1998; Chui et al., 1999; Wilson et al., 1999; Gouras et al., 2000; Mochizuki et al., 2000). N2a cells were treated with 1 μm insulin for 4 or 16 hr; Aβ was extracted with SDS and analyzed by immunoprecipitation and SDS-PAGE. Intracellular Aβ was reduced by 20% after 4 hr and by 45% after 16 hr of insulin treatment (Fig. 4a,b). Immunoprecipitation–mass spectrometry analysis revealed that intracellular Aβ40 and Aβ42 both were reduced after insulin treatment (Fig. 4c). No change was detected in the levels of full-length βAPP or PS-1 N-terminal fragment after insulin treatment for 4 hr (data not shown) or 16 hr (Fig. 4d,e).
Fig. 4.

Insulin reduces intracellular levels of Aβ in N2a cells. Cells were treated for 4 or 16 hr with or without 1 μm insulin and lysed in SDS. a, b, Intracellular Aβ was detected by immunoprecipitation with 4G8, followed by SDS-PAGE and Western blotting, using 6E10 monoclonal antibody, which recognizes only Aβ1–40/42. a, Representative autoradiographic analysis of intracellular Aβ after 16 hr of treatment in the absence or presence of 1 μminsulin. b, Quantitative analysis of intracellular Aβ after treatment with insulin for 4 or 16 hr. Data represent means ± SD; n = 5. *p < 0.05 versus control. c, IP–MS analysis of intracellular Aβ40/42 levels after 4 hr of treatment with 1 μm insulin.d, e, Western blot analysis for full-length βAPP (d) and the PS-1 N-terminal fragment (e) after 16 hr of treatment with 1 μm insulin.
Insulin reduces Aβ in the Golgi/TGN by accelerating βAPP/Aβ transport to the plasma membrane
βAPP resides predominantly in the Golgi/TGN, which is also the main site of Aβ generation (Cook et al., 1997; Hartmann et al., 1997;Xu et al., 1997; Lee et al., 1998; Skovronsky et al., 1998; Greenfield et al., 1999). Insulin is known to stimulate protein transport from TGN or post-TGN vesicles to the plasma membrane (Pessin et al., 1999). To define the subcellular compartments in which insulin exerts its action on βAPP and Aβ trafficking, we performed subcellular fractionation by using a well characterized sucrose gradient procedure (Greenfield et al., 1999) after 16 hr of incubation in the absence or presence of insulin. Insulin reduced Aβ in membrane fractions collected from the interfaces corresponding to secretory vesicles and Golgi/TGN, but not from those corresponding to heavy membranes such as the ER, plasma membranes, and lysosomes (Fig.5a,b). Moreover, after a 2 hr preincubation of cells at 20°C to accumulate newly synthesized [35S]-labeled βAPP in the TGN, insulin-dependent secretion of Aβ and sβAPPα was observed within 7.5–15 min of the addition of the hormone (data not shown), supporting the idea that insulin stimulates Aβ and sβAPPα trafficking from the TGN. After insulin treatment, full-length βAPP also is reduced in vesicle and Golgi-enriched fractions, whereas it is increased in the heavy membrane fractions (Fig. 5c,d). The total amount of βAPP was not changed overall by insulin treatment (see Fig.4d). Calnexin (ER), γ-adaptin (TGN), ARF3 (post-TGN vesicles, cytosol), and surface biotinylated βAPP (plasma membranes) were assayed to identify fractions that are enriched in these organelles (Fig. 5e). Our data indicate that insulin reduces intracellular Aβ and stimulates its secretion by increasing the βAPP/Aβ egress from the Golgi/TGN and from post-TGN vesicles. To determine whether the effect of insulin on trafficking was reflected in the number of βAPP molecules on the plasma membrane, we treated N2a cells for 4 hr with insulin and then labeled them with biotin. Insulin treatment resulted in an almost twofold increase in βAPP molecules at the cell surface (Fig. 5f).
Fig. 5.

Insulin influences Aβ and βAPP trafficking in N2a cells. Cells were treated for 16 hr in the absence or presence of 1 μm insulin, homogenized, and fractionated on an equilibrium flotation sucrose gradient (see Materials and Methods).a, Representative autoradiographic analysis and quantitative analysis (b) of Aβ subcellular distribution after insulin treatment. c, Representative autoradiographic analysis and quantitative analysis (d) of intracellular βAPP subcellular distribution after insulin treatment. e, Markers for subcellular compartments. Proteins from each fraction were precipitated by trichloroacetic acid and analyzed by Western blot, using the antibodies anti-calnexin (ER), anti-γ-adaptin (TGN), or anti-ARF3 (post-TGN vesicles, cytosol). Surface βAPP (plasma membrane) was determined as described (see Materials and Methods). Fraction 1 = 0.25 m sucrose solution (loading, cytosol). Fractions 2–5 correspond, respectively, to interfaces between 0.25/0.8 m (post-TGN vesicles), 0.8/1.16m (Golgi/TGN), 1.16/1.3 m, and 1.3m/2 m (heavy membranes such as ER, plasma membranes) sucrose solutions. f, N2a cells were treated for 4 hr in the absence or presence of 1 μminsulin. Surface proteins were labeled with biotin. Biotinylated βAPP was analyzed by immunoprecipitation with 369 antibody and Western blot with HRP-conjugated streptavidin.
Insulin does not affect ER-to-Golgi transport
It has been shown that insulin stimulates the export of leptin from the ER in rat adipocytes (Barr et al., 1997). However, an effect of insulin on ER-to-Golgi transport has not been reported for any other protein. To investigate whether insulin could affect βAPP trafficking between ER and Golgi, we pulse-labeled N2a cells for 5 min and chased them for up to 45 min in the absence or presence of insulin. Immunoprecipitated βAPP molecules were subjected to endoglycosidase-H (Endo-H) digestion, and the rate of appearance of Endo-H-resistant, N-linked oligosaccharide-modified isoforms of βAPP was evaluated. Newly synthesized βAPP (∼105 kDa) is sensitive to digestion by Endo-H (Fig. 6; t = 0 min). After 5–10 min of chase, a ∼115 kDa βAPP form appeared that was resistant to Endo-H digestion. Clearly, insulin treatment did not alter βAPP maturation significantly or the pattern of Endo-H resistance, an indicator of ER-to-Golgi trafficking, at any time of chase up to 45 min.
Fig. 6.

Lack of effect of insulin on βAPP trafficking from ER to Golgi. N2a cells were pulse-labeled for 5 min with [35S]methionine and chased in serum-free medium in the absence or presence of 1 μm insulin for 5–45 min. βAPP was immunoprecipitated by using 369 antibody; one-half of the sample was digested by endoglycosidase-H (Endo H). The arrowhead indicates the endoglycosidase-H-resistant βAPP species.
Insulin regulates βAPP processing via a receptor tyrosine kinase
Insulin receptors are present in both N2a and primary neuronal cultures (Fig. 7a). To determine whether the action of insulin involves a tyrosine kinase, we inhibited intrinsic tyrosine kinase activity with tyrphostin-25, a nonselective inhibitor of receptor and nonreceptor tyrosine kinases, and measured its effect on insulin-stimulated Aβ secretion in N2a cells. Tyrphostin-25 (25 μm) abolished the effect of insulin on Aβ secretion, which was accompanied by a small reduction in basal secretion (Fig. 7b). At a concentration of 10 μm, tyrphostin-25 partially inhibited the effect of insulin on Aβ levels without any effect on basal secretion (data not shown). These results indicate that tyrosine kinase activity is essential for the effect of insulin on Aβ trafficking. In addition, the effect of insulin on Aβ was mimicked by IGF-1, but not by glucagon, EGF, NGF, PDGF, aFGF, or bFGF (data not shown), indicating the specificity of the insulin effect. These various results indicate that the effect of insulin is mediated via interaction with the insulin/IGF-1 receptor.
Fig. 7.

The effect of insulin requires tyrosine kinase activity and is mediated via the MEK/MAP kinase cascade.a, Western blot analysis for insulin receptor (IR) in N2a cells and primary neurons. b, N2a cells were incubated for 16 hr in serum-free medium in the absence or presence of 1 μm insulin and/or 25 μmtyrphostin-25. Data represent means ± SD; n = 3. *p < 0.05 with respect to no addition; **p < 0.05 with respect to treatment with insulin alone; †Not significant with respect to tyrphostin-25 alone. c, N2a cells were incubated for 4 hr in serum-free medium in the absence or presence of 1 μminsulin, 25 μmU73122, 500 nm wortmannin, and/or 10 μm PD98059. Data represent means ± SD;n = 3. *p < 0.05 with respect to no addition; **p < 0.05 with respect to treatment with insulin alone.
Insulin regulates βAPP processing via MEK/MAP kinase cascade
To investigate the insulin pathway downstream of the insulin receptor, we studied the effect of selective inhibitors of three known insulin-activated signal transduction cascades. PD98059, a highly selective inhibitor of MAP kinase kinase activation (Alessi et al., 1995; Dudley et al., 1995), abolished the effect of insulin on Aβ secretion (Fig. 7c) and intracellular Aβ (data not shown) without altering basal levels of these parameters. In contrast, wortmannin, an inhibitor of PI-3 kinase, and U73122, an inhibitor of phospholipase C, caused a nonselective inhibition of both basal- and insulin-stimulated secretion of Aβ after incubation for 4 hr (Fig.7c).
DISCUSSION
The present observations, that intracellular Aβ decreases whereas extracellular Aβ increases in response to insulin, could be explained by a mechanism involving insulin-stimulated intracellular trafficking of βAPP and Aβ. This proposal is supported by substantial evidence showing that insulin selectively stimulates protein transport through the secretory pathway (Bogan and Lodish, 1999). In fact, we report here that insulin (1) accelerates βAPP/Aβ trafficking from the TGN, the main site of Aβ generation, to the plasma membrane; (2) increases the number of βAPP molecules on the plasma membrane; (3) increases the extracellular levels of Aβ even when IDE is inhibited by phenantroline; and (4) increases the secretion of 3 kDa Aβx-40/42 peptides, mainly composed of Aβ11–40/42 (Gouras et al., 1998), which are resistant to IDE degradation (Qiu et al., 1998; Vekrellis et al., 2000). βAPP levels and sβAPPβ secretion were not affected by insulin, suggesting that insulin may not regulate the β-cleavage of βAPP but only βAPP/Aβ trafficking. Elucidation of the potential contribution of the endosomal compartments to the effect of insulin on Aβ secretion awaits further investigation.
In agreement with previous studies (Qiu et al., 1998; Vekrellis et al., 2000), we have found in using both neuronal cell lines and primary neuronal cultures that IDE is a protease involved in Aβ degradation and that insulin inhibits Aβ degradation by competing for IDE. It was reported previously that IDE can be secreted by microglial cell cultures (Qiu et al., 1998), whereas it is cell-associated in primary neurons (Vekrellis et al., 2000). Although a significant amount of IDE is detectable in the medium of primary neurons after 16 hr, this is likely an experimental artifact of the cell culture setting, because IDE does not have the signal peptide required for targeting into the secretory pathway and therefore should not be secreted. The slow kinetics of IDE release further support this view. Evidence was presented recently that a neutral endopeptidase, but not IDE, was involved in Aβ degradation when radiolabeled Aβ was injected into rat brain (Iwata et al., 2000). Thus, IDE-mediated Aβ degradation may be of less physiological relevance in vivo. In addition, proteases other than IDE also may take part in Aβ degradation in neuronal cultures, as suggested by the incomplete inhibition of Aβ degradation by insulin, phenantroline, or immunodepletion of IDE in ourin vitro system (see Fig. 3c). This possibility is supported further by the observation that Aβ was still degraded when neuronal cultures were treated with phenantroline to inactivate IDE (see Fig. 3d).
We have performed a number of experiments on the signal transduction pathway by which insulin might stimulate the intracellular trafficking of βAPP and Aβ. The effect of insulin was abolished by tyrphostin-25 and was not mimicked by several other growth factors, indicating that the activation of the insulin/IGF-1 receptor tyrosine kinase is required for insulin-dependent Aβ secretion. Many of the present studies were performed by using a concentration of insulin of 1 μm. Although at this concentration a contribution of IGF-1 receptor could not be excluded, the fact that insulin is effective at a concentration of 50 nm indicates the specificity of its effect. A similar effect of IGF-1 on βAPP metabolism is to be expected because activation of IGF-1 receptors triggers the same downstream signaling molecules as those of insulin receptors (Lopaczynski, 1999). In an effort to elucidate the signaling pathway downstream of the insulin receptor, we studied the effect of selective inhibitors of the three known insulin-activated signal transduction cascades. The effect of insulin on Aβ secretion was abolished by PD98059, a highly selective inhibitor of the activation of the MAP kinase kinase (Alessi et al., 1995; Dudley et al., 1995), indicating that the effect of insulin is mediated by activation of the MAP kinase cascade. In contrast, wortmannin, an inhibitor of PI3-kinase, and U73122, a potent inhibitor of phospholipase C, nonselectively reduced basal- and insulin-stimulated secretion of Aβ.
Accumulation of Aβ plaques within the brain is widely believed to initiate the pathological cascade culminating in clinical AD. Generally, it is assumed that secreted Aβ, and particularly Aβ42, plays a crucial role in amyloid neuropathology. Recent evidence suggests the hypothesis that intracellular Aβ42 may play an important role in amyloid deposition and neuronal degeneration (for review, seeWilson et al., 1999; Gouras et al., 2000): high intracellular levels of Aβ42 are observed in cell lines expressing FAD mutant PS-1 (Wild-Bode et al., 1997); an insoluble pool of Aβ42 increases in a neuronal cell line during aging in culture (Skovronsky et al., 1998); intraneuronal accumulation of Aβ42 and extensive neuronal degeneration occur in the absence of Aβ plaques in transgenic mice expressing an FAD mutant form of PS-1 (Chui et al., 1999). Recent immunohistochemical studies have reported intraneuronal Aβ42 accumulation in early (Gouras et al., 2000) and late (Mochizuki et al., 2000) AD. Although direct neurotoxicity of intracellular Aβ has not been demonstrated, we cannot exclude that intracellular Aβ42 may play a direct role in initiating AD neuropathology.
Although basal insulin levels do not appear to be reduced in aging, insulin resistance and impaired insulin release in response to a glucose challenge are age-related phenomena (for review, see Lamberts et al., 1997; Perry, 1999). Several clinical studies suggest that insulin plays an important role in cognitive function and memory (for review, see Wickelgren, 1998; Lovestone, 1999). Recent findings demonstrated insulin receptor upregulation and reduced insulin receptor-mediated tyrosine kinase activity in AD brains (Frolich et al., 1998, 1999). Higher-fasting plasma insulin levels and reduced cerebrospinal fluid-to-plasma ratios of insulin, indicative of insulin resistance, have been described in patients with AD (Craft et al., 1998, 1999). Although recent cross-sectional and prospective population-based studies have indicated that diabetes mellitus is a risk factor for dementia and AD (Yoshitake et al., 1995; Ott et al., 1996; Leibson et al., 1997), the issue is still controversial (Landin et al., 1993; Mortel et al., 1993; Nielson et al., 1996;Heitner and Dickson, 1997). According to the present data, insulin dramatically reduces intracellular levels of Aβ40/42 and increases Aβ40/42 secretion in neuroblastoma cells and primary neuronal cultures by promoting βAPP trafficking via a known insulin-signaling pathway, suggesting that the insulin/IGF-1 pathway may play a role in AD pathogenesis. However, future studies must assess whether diabetes contributes to AD and, if so, whether such effects are mediated via the action of insulin on βAPP/Aβ metabolism.
Footnotes
This work was supported by National Institutes of Health Grant AG09464 to P.G., by the Alzheimer's Association (G.K.G., R.W., and H.X.), by the American Health Assistance Foundation (H.X.), and by the Ellison Medical Foundation (P.G. and H.X.). We are grateful to Dr. R. Roth (Stanford University) for the gift of 28H1 and 9B12 monoclonal antibodies; Dr. G. Thinakaran (University of Chicago) for providing human βAPP-transfected N2a cells; Drs. W. Netzer and H. Lin (Rockefeller University) for contributing to the optimization of Aβ detection techniques; Drs. D. Accili (Columbia University), M. Stoffel (Rockefeller University), and J. Schlessinger (New York University Medical Center) for helpful discussions; and Dr. J. Greenfield (Rockefeller University) for critical reading of this manuscript.
Correspondence should be addressed to Huaxi Xu, Laboratory of Molecular and Cellular Neuroscience, Fisher Center for Research on Alzheimer Disease, The Rockefeller University, 1230 York Avenue, Box 296, New York, NY 10021. E-mail: xuh@rockvax.rockefeller.edu.
Dr. Gasparini's present address: Nicox Research Institute, Milan, Italy.
REFERENCES
- 1.Abbott M-A, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci. 1999;19:7300–7308. doi: 10.1523/JNEUROSCI.19-17-07300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 3.Barr VA, Malide D, Zarnowski M, Taylor SI, Cushman SW. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology. 1997;138:4463–4472. doi: 10.1210/endo.138.10.5451. [DOI] [PubMed] [Google Scholar]
- 4.Bogan JS, Lodish HF. Two compartments for insulin-stimulated exocytosis in 3T3–L1 adipocytes defined by endogenous ACRP30 and GLUT4. J Cell Biol. 1999;146:609–620. doi: 10.1083/jcb.146.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadram TV, Uterbeck AJ, Greengard P. Processing of Alzheimer β/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chui DH, Tanahashi H, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin-1 accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 7.Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM-Y, Doms RW. Alzheimer's Aβ1–42 is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- 8.Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M, Porte D., Jr Cerebrospinal fluid and plasma insulin levels in Alzheimer's disease. Neurology. 1998;50:164–168. doi: 10.1212/wnl.50.1.164. [DOI] [PubMed] [Google Scholar]
- 9.Craft S, Asthana S, Schellenberg G, Cherrier M, Baker LD, Newcomer J, Plymate S, Latendresse S, Petrova A, Raskind M, Peskind E, Lofgreen C, Grimwood K. Insulin metabolism in Alzheimer's disease differs according to apolipoprotein E genotype and gender. Neuroendocrinology. 1999;70:146–152. doi: 10.1159/000054469. [DOI] [PubMed] [Google Scholar]
- 10.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frolich L, Blum-Degen D, Bernstein H-G, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P. Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm. 1998;105:423–438. doi: 10.1007/s007020050068. [DOI] [PubMed] [Google Scholar]
- 12.Frolich L, Blum-Degen D, Riederer P, Hoyer S. A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer's disease. Ann NY Acad Sci. 1999;893:290–293. doi: 10.1111/j.1749-6632.1999.tb07839.x. [DOI] [PubMed] [Google Scholar]
- 13.Gouras GK, Xu H, Jovanovic JN, Buxbaum JD, Wang R, Greengard P, Relkin NR, Gandy S. Generation and regulation of β-amyloid peptide variants by neurons. J Neurochem. 1998;71:1920–1925. doi: 10.1046/j.1471-4159.1998.71051920.x. [DOI] [PubMed] [Google Scholar]
- 14.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Aβ42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer β-amyloid peptides. Proc Natl Acad Sci USA. 1999;96:742–747. doi: 10.1073/pnas.96.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease Aβ40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 17.Heitner J, Dickson D. Diabetics do not have increased Alzheimer-type pathology compared with age-matched control subjects. A retrospective postmortem immunocytochemical and histofluorescent study. Neurology. 1997;49:1306–1311. doi: 10.1212/wnl.49.5.1306. [DOI] [PubMed] [Google Scholar]
- 18.Hong M, Lee VM-Y. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272:19547–19553. doi: 10.1074/jbc.272.31.19547. [DOI] [PubMed] [Google Scholar]
- 19.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee H-J, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 20.Lamberts SW, van den Beld AW, van der Lely AJ. The endocrinology of aging. Science. 1997;278:419–424. doi: 10.1126/science.278.5337.419. [DOI] [PubMed] [Google Scholar]
- 21.Landin K, Blennow K, Wallin A, Gottfries CG. Low blood pressure and blood glucose levels in Alzheimer's disease. Evidence for a hypometabolic disorder? J Intern Med. 1993;233:357–363. doi: 10.1111/j.1365-2796.1993.tb00684.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee S-J, Liyanage U, Bickel PE, Xia W, Lansbury PT, Kosik KS. A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 23.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, Palumbo PJ. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- 24.Lesort M, Johnson GVW. Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience. 2000;99:305–316. doi: 10.1016/s0306-4522(00)00200-1. [DOI] [PubMed] [Google Scholar]
- 25.Lesort M, Jope RS, Johnson GVW. Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3β and Fyn tyrosine kinase. J Neurochem. 1999;72:576–584. doi: 10.1046/j.1471-4159.1999.0720576.x. [DOI] [PubMed] [Google Scholar]
- 26.Lopaczynski W. Differential regulation of signaling pathways for insulin and insulin-like growth factor-1. Acta Biochim Pol. 1999;46:51–60. [PubMed] [Google Scholar]
- 27.Lovestone S. Diabetes and dementia. Neurology. 1999;53:1907–1909. doi: 10.1212/wnl.53.9.1907. [DOI] [PubMed] [Google Scholar]
- 28.Mills J, Reiner PB. Regulation of amyloid precursor protein cleavage. J Neurochem. 1999;72:443–460. doi: 10.1046/j.1471-4159.1999.0720443.x. [DOI] [PubMed] [Google Scholar]
- 29.Mochizuki A, Tamaoka A, Shimoata A, Komatsuzaki Y, Shoji S. Aβ42-positive non-pyramidal neurons around amyloid plaques in Alzheimer's disease. Lancet. 2000;355:42–43. doi: 10.1016/S0140-6736(99)04937-5. [DOI] [PubMed] [Google Scholar]
- 30.Mortel KF, Wood S, Pavol MA, Meyer JS, Rexer JL. Analysis of familial and individual risk factors among patients with ischemic vascular dementia and Alzheimer's disease. Angiology. 1993;44:599–605. doi: 10.1177/000331979304400802. [DOI] [PubMed] [Google Scholar]
- 31.Nielson KA, Nolan JH, Berchtold NC, Sandman CA, Mulnard RA, Cotman CW. Apolipoprotein-E genotyping of diabetic dementia patients: is diabetes rare in Alzheimer's disease? J Am Geriatr Soc. 1996;44:897–904. doi: 10.1111/j.1532-5415.1996.tb01857.x. [DOI] [PubMed] [Google Scholar]
- 32.Ott A, Stolk RP, Hofman A, van Harskemp F, Grobbee DE, Breteler MMB. Association of diabetes mellitus and the risk of dementia. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- 33.Ott A, Stolk RP, van Harskamp F, Pols HAP, Hofman A, Breteler MMB. Diabetes mellitus and the risk of dementia. The Rotterdam study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 34.Perry HM. The endocrinology of aging. Clin Chem. 1999;45:1369–1376. [PubMed] [Google Scholar]
- 35.Pessin JE, Thurmond DC, Elmendorf JS, Coker KJ, Okada S. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J Biol Chem. 1999;274:2593–2596. doi: 10.1074/jbc.274.5.2593. [DOI] [PubMed] [Google Scholar]
- 36.Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner RM, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 37.Refolo LM, Salton SRJ, Anderson JP, Mehta P, Robakis NK. Nerve and epidermal growth factors induce the release of the Alzheimer amyloid precursor from PC12 cell cultures. Biochem Biophys Res Commun. 1989;164:664–670. doi: 10.1016/0006-291x(89)91511-8. [DOI] [PubMed] [Google Scholar]
- 38.Schubert D, Jin L-W, Saitoh T, Cole G. The regulation of amyloid β-protein precursor secretion and its modulatory role in cell adhesion. Neuron. 1989;3:689–694. doi: 10.1016/0896-6273(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 39.Seeger M, Norstedt C, Petacenska S, Kovacs DM, Gouras GK, Hahne S, Fraser P, Levesque L, Czernik AJ, St. George-Hyslop P, Sisodia SS, Thinakaran G, Tanzi RE, Greengard P, Gandy S. Evidence for phosphorylation and oligomeric assembly of presenilin1. Proc Natl Acad Sci USA. 1997;94:5090–5094. doi: 10.1073/pnas.94.10.5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Selkoe DJ. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 41.Shii K, Roth RA. Inhibition of insulin degradation by hepatoma cells after microinjection of monoclonal antibodies to a specific cytosolic protease. Proc Natl Acad Sci USA. 1986;83:4147–4151. doi: 10.1073/pnas.83.12.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skovronsky DM, Doms RW, Lee VM-Y. Detection of intraneuronal pool of insoluble amyloid β-protein that accumulates with time in culture. J Biol Chem. 1998;141:1031–1039. doi: 10.1083/jcb.141.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solano DC, Sironi M, Bonfini C, Solerte B, Govoni S, Racchi M. Insulin regulates amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000;14:1015–1022. doi: 10.1096/fasebj.14.7.1015. [DOI] [PubMed] [Google Scholar]
- 44.Vekrellis K, Ye Z, Qiu WQ, Walsh DM, Hartley D, Chesneau V, Rosner RM, Selkoe DJ. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000;20:1657–1665. doi: 10.1523/JNEUROSCI.20-05-01657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- 46.Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, Mendelsohn FA. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology. 1987;121:1562–1570. doi: 10.1210/endo-121-4-1562. [DOI] [PubMed] [Google Scholar]
- 47.Wickelgren I. Tracking insulin to the mind. Science. 1998;280:517–519. doi: 10.1126/science.280.5363.517. [DOI] [PubMed] [Google Scholar]
- 48.Wild-Bode C, Yamazaki T, Capell A, Leimer U, Steiner H, Ihara Y, Haass C. Intracellular generation and accumulation of amyloid β-peptide terminating at amino acid 42. J Biol Chem. 1997;272:16085–16088. doi: 10.1074/jbc.272.26.16085. [DOI] [PubMed] [Google Scholar]
- 49.Wilson CA, Doms RW, Lee VM-Y. Intracellular APP processing and Aβ production in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:787–794. doi: 10.1097/00005072-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 50.Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, Greengard P, Gandy S. Generation of Alzheimer β-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc Natl Acad Sci USA. 1997;94:3748–3752. doi: 10.1073/pnas.94.8.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic JN, Seeger M, Relkin NR, Liao F, Checler F, Buxbaum JD, Charr BT, Thinakaran G, Sisodia SS, Wang R, Greengard P, Gandy S. Estrogen reduces neuronal generation of Alz-heimer β-amyloid peptides. Nat Med. 1998;4:447–451. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]
- 52.Yoshitake T, Kiyoara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, Ohmori S, Nomiyama K, Kawano K, Ueda K. Incidence and risk factors of vascular dementia and Alzheimer's disease in a defined elderly Japanese population: the Hisayama study. Neurology. 1995;45:1161–1168. doi: 10.1212/wnl.45.6.1161. [DOI] [PubMed] [Google Scholar]
- 53.Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, Alkon DJ. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem. 1999;274:34893–34902. doi: 10.1074/jbc.274.49.34893. [DOI] [PubMed] [Google Scholar]