

Abstract
Interleukin-10 (IL-10) has been shown to reduce neuronal degeneration after CNS injury. However, the molecular mechanisms underlying the neuroprotective properties of this cytokine are still under investigation. Glutamate exacerbates secondary injury caused by trauma. Thus, we examined whether IL-10 prevents glutamate-mediated cell death. We used rat cerebellar granule cells in culture because these neurons undergo apoptosis upon exposure to toxic concentrations of glutamate (100–500 μm) or NMDA (300 μm). Pretreatment of cerebellar granule cells with IL-10 (1–50 ng/ml) elicited a dose- and time-dependent reduction of glutamate-induced excitotoxicity. Most importantly, IL-10 reduced the number of apoptotic cells when added to the cultures together or 1 hr after glutamate. Using patch-clamping and fluorescence Ca2+ imaging techniques, we examined whether IL-10 prevents glutamate toxicity by blocking the function of NMDA channel. IL-10 failed to affect NMDA channel properties and to reduce NMDA-mediated rise in intracellular Ca2+. Thus, this cytokine appears to prevent glutamate toxicity by a mechanism unrelated to a blockade of NMDA receptor function. Various proteases, such as caspase-3, and transcription factors, such as nuclear factor κB (NF-κB), have been proposed to participate in glutamate-mediated apoptosis. Thus, we examined whether IL-10 modulates the activity of these apoptotic markers. IL-10 blocked both the glutamate-mediated induction of caspase-3 as well as NF-κB DNA binding activity, suggesting that the neuroprotective properties of IL-10 may rely on its ability to block the activity of proapoptotic proteins.
Keywords: apoptosis, Ca2+, caspase-3, EAA, IL-10, NF-κB, NMDA receptors
Injury to the CNS triggers an abnormal release of glutamate and other excitatory amino acids (EAAs) that contribute significantly to the neurological outcome (Wielock, 1985; Rothman and Olney, 1986). The released glutamate causes an excessive activation of glutamate receptors of the NMDA subtype, leading to an abnormal influx of Ca2+ in viable neurons (Garthwaite et al., 1986; MacDermott et al., 1986) and a subsequent neuronal death (Choi, 1988; Hahn et al., 1988). The type of cell death caused seems to depend on the nature of the injury. Necrosis occurs after an acute insult, whereas apoptotic cell death is involved in propagation of the secondary injury (Bonfoco et al., 1995; Liu et al., 1997; Yakovlev et al., 1997). Because apoptotic neurons can be rescued, because they remain viable for a period of time, compounds that prevent apoptosis may have a therapeutic significance.
The cytokine interleukin-10 (IL-10) has been shown to improve neurological outcome after CNS injury (Knoblach and Faden, 1998; Bethea et al., 1999) and to render neurons in culture less vulnerable to ischemic and EAA-mediated damage (Grilli et al., 2000). IL-10 is notoriously known as an inhibitor of the synthesis of inflammatory cytokine, including tumor necrosis factor-α (TNF-α) and IL-1β (Bogdan et al., 1992; Wang et al., 1994; Kline et al., 1995; Di Santo et al., 1997; Bethea et al., 1999; Sawada et al., 1999). Some of these cytokines can exacerbate neuronal damage after CNS trauma (Mocchetti and Wrathall, 1995; Feuerstein et al., 1998); therefore, it has been suggested that the IL-10 ability to improve neurological outcome after CNS injury relies on its anti-inflammatory effects. However, these properties cannot fully explain why IL-10 can also reduce glutamate-mediated cell death (Grilli et al., 2000). Thus, the mechanisms underlying the neuroprotective properties of IL-10 are not fully understood.
Evidence has accumulated suggesting that neurotrophic factors, such as brain-derived neurotrophic factor (BDNF) and basic fibroblast growth factor (FGF2), prevent glutamate-mediated neuronal cell death in culture by blocking the sustained increase in cytosolic free Ca2+ concentration evoked by toxic concentrations of glutamate (Mattson et al., 1989; Cheng et al., 1995). This effect has been shown to depend on the ability of these neurotrophic factors to reduce the synthesis of specific subunits of NMDA receptors (Brandoli et al., 1998). In contrast, the proinflammatory cytokine IL-6 increases excitotoxicity by enhancing NMDA receptor function (Qiu et al., 1998). IL-10 may exert a neuroprotective effect by a mechanism similar to that of growth factors and opposite to that of IL-6. However, this hypothesis remains primarily speculative. In addition, IL-10 has been shown to slow down progression of apoptosis in immuno-derived cells (Schottelius et al., 1999). Hence, IL-10 may improve neurological outcome after CNS trauma by reducing apoptosis and, thus, secondary injury processes.
The current study was undertaken to examine the ability of IL-10 to prevent EAA-mediated neuronal cell death in cerebellar granule cells and gain insights into the mechanisms underlying this effect. We report that IL-10 prevents glutamate-mediated apoptotic cell death by blocking the activity of proapoptotic markers.
MATERIALS AND METHODS
Cell culture
Cerebellar granule cells were prepared from 8-d-old Sprague Dawley rat pups (Taconic Farms, Germantown, NY) as described previously (Brandoli et al., 1998; Marini et al., 1998). Briefly, neurons were plated onto poly-l-lysine (1%) precoated 100 mm plastic dishes at a density of 2.5 × 106 cells/ml and grown in Basal Medium Eagle (Life Technologies, Gaithersburg, MD) containing glutamine (2 mm), fetal calf serum (10%), KCl (25 mm), gentamicin (100 μg/ml), and penicillin–streptomycin (10,000U/ml). Cells were maintained at 37°C in 5% CO2–95% air. Cytosine arabinoside (10 μm) was added 24 hr after cell plating to inhibit glial proliferation. At the time of the experiments, these cultures were composed of ∼95% neurons and ∼5% of non-neuronal cells, such as astrocytes, oligodendrocytes, and endothelial cells. Human recombinant IL-10 (a gift from Dr. S. Narula, Schering-Plough, Kenilworth, NJ), glutamate, or NMDA (Sigma, St. Louis, MO) were added to the cultures at 8 d in vitro. After the addition of glutamate or other compounds, cultures were kept in the same medium until analysis of cell viability. Sister cultures that received medium alone were used as a control.
Cell survival
The percent of surviving neurons in the presence of IL-10 and/or glutamate was estimated by determining the activity of mitochondrial dehydrogenases [3(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide (MTT) assay] and the number of apoptotic cells [in situ terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)].
MTT assay. The conversion of the yellow tetrazolium salt (MTT) to the purple formazan dye is dependent on the activity of mitochondrial dehydrogenases and is, therefore, reflective of the viability of the cell and the cytotoxicity of glutamate. The assay was performed according to the specifications of the manufacturer (MTT Kit I; Boehringer Mannheim, Indianapolis, IN). Briefly, neurons were cultured on 96-well plates, 10 μg of the 5 mg/ml MTT labeling reagent was added to each well containing neurons in 100 μl of medium, and the plate was incubated for 4 hr in a humidified atmosphere. After the incubation, 100 μl of the solubilization solution were added to each well for 18 hr. The absorbance of the samples was measured at a wavelength of 570 and 700 nm (reference wavelength). Unless otherwise indicated, the extent of MTT conversion in cells exposed to glutamate is expressed as a percentage of control.
In situ TUNEL. Cerebellar granule cells were plated onto 25-mm-round, 1-mm-thick glass coverslips (Fisher Scientific, Houston, TX) precoated with poly-l-lysine (1%). Cells were washed with PBS, fixed with 4% paraformaldehyde for 30 min, and rinsed three times with PBS. Cells were then permeabilized with 0.1% Triton X-100 in PBS and then treated with 0.3% H2O2 for 30 min to eliminate endogenous peroxidases. The DNA nick labeling reaction was performed using 50 U/ml Klenow (Boehringer Mannheim) and 2 mm dNTP with 0.5 nmbiotin-16-dUTP in buffer A (0.05 m Tris HCl, pH 5.5, 5 mm MgCl2, 14.5 mm 2-mercaptoethanolsulfonic acid, and 50 mg/ml BSA) for 60 min at 37°C. Cells were then rinsed in PBS and incubated with streptavidin-peroxidase-HRP (50 μg/ml) for 30 min at 37°C. After rinsing, the labeling was visualized using diaminobenzidine. The viable neurons were quantified by counting TUNEL-positive cell bodies, and results are expressed as percent of cell survival.
Double labeling
For caspase-3 and TUNEL double labeling, neurons were plated onto 12-mm-round, 1-mm-thick precoated glass coverslips. Cells were fixed with 4% paraformaldehyde, post-fixed in ethanol/acetic acid 2:1, washed, and incubated with caspase-3-p20 antibody (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). After rinsing with PBS, cells were equilibrated according to the instructions of the manufacturer (ApoTag; Intergen, Purchase, NY), incubated with TdT enzyme in the presence of digoxigenin-labeled dNTP, followed by anti-digoxigenin (fluorescein conjugate) antibody. Cells were then incubated with secondary antibody for caspase-3, Texas Red anti-goat (1:500; Vector Laboratories, Burlingame, CA) and mounted using Vectashield Mounting Medium with 4′,6′-diamidino-2-phenylindole (Vector Laboratories) to detect viable cells. Reaction was visualized with the Nikon (Tokyo, Japan) inverted fluorescent microscope ECLIPSE TE300. Optronics Magnafire software (Optronics, Goleta, CA) was used to analyze positive cells.
Caspase-3-like activity
Neurons were plated onto 100 mm dishes. Caspase-3-like activity was measured in lysates of cerebellar granule cells using the caspase-3 colorimetric assay protease kit (Chemicon, Temecula, CA) following the instructions of manufacturer. In brief, neurons were lysed in ice-cold lysis buffer (150 mm NaCl, 20 mm Tris HCl, pH 7.2, 1% Triton X-100, and 1 mm DTT) for 10 min. After removal of cellular debris by centrifugation, protein levels in the lysates (cytosolic extract) were measured by the Bradford Coomassie blue colorimetric assay (Bio-Rad, Hercules, CA) and equalized accordingly to obtain 150 μg of cytosolic extract per sample. Samples were incubated with 200 μm caspase-3 substrateN-acethyl-Asp-Glu-Val-Asp (DEVD)-p-nitroanilide at 37°C for 2 hr. Samples were analyzed at 400 nm in a microtiter plate reader. Data are expressed as fold increase–decrease in caspase-3 activity compared with control cells.
Fluorescence Ca2+ imaging
Cytosolic free Ca2+ concentration ([Ca2+]i) was measured by single-cell fura-2 fluorescence ratio imaging as described previously (De Bernardi et al., 1996). For this purpose, neurons were plated onto 25-mm-round, 1-mm-thick glass coverslips (Fisher Scientific) precoated with poly-l-lysine (1%). For the acute (10 min) treatment, cells were labeled with fura-2 (fura-2 AM; Molecular Probes, Eugene, OR) in growth medium for 30 min at 37°C in an atmosphere of 5% CO2, washed in Mg2+-free Locke's solution (in mm: 154 NaCl, 5.6 KCl, 3.6 NaHCO3, 2.3 CaCl2, 5.6 glucose, and 15 HEPES, pH 7.4) and imaged. Resting [Ca2+]i was recorded for ∼60 sec, vehicle (medium alone) or IL-10 (50 ng/ml) was added, and [Ca2+]iwas followed for 10 min. Cells were then exposed to NMDA (50 μm), and [Ca2+]i was monitored over a 20–30 min period. For the 24 hr treatment, neurons were incubated with growth medium or IL-10 (50 ng/ml) for 24 hr. Cells were then labeled with fura-2, imaged and challenged with NMDA as described above. Ca2+ imaging was performed at room temperature using an Attofluor RatioVision digital fluorescence microscopy system (Atto Instruments, Rockville, MD) equipped with a Zeiss Axiovert 135 microscope and a F-Fluar 40×, 1.3 numerical aperture oil-immersion objective, as described previously (De Bernardi et al., 1996). Briefly, fura-2 was excited at 334 and 380 nm with its emission monitored at 510–530 nm; the 334/380 nm excitation ratio increases as a function of the [Ca2+]i. Before the experiments, the instrument was calibrated (calibration was donein vitro with fura-2 pentapotassium salt in the presence of high concentration of Ca2+ or EGTA), and the 334/380 nm excitation ratio was converted to [Ca2+]inm values (Grynkiewicz et al., 1985). For each coverslip, 50–99 neurons were simultaneously imaged in a given microscopic field, and single-cell Ca2+responses were collected and averaged to yield [Ca2+]i population means ± SEM) that were plotted versus time.
Electrophysiology
Electrodes were pulled from thin-walled borosilicate glass (Drummond Scientific, Broomall, PA) using a Narashige (Tokyo, Japan) PP-83 vertical puller. Electrodes had open-tip resistances of 5–8 MΩ. Recordings were made on the stage of a CK2 inverted phase-contrast microscope (Olympus Optical, Lake Success, NY) at room temperature (22–25°C). Cultured granule cells were voltage clamped at −60 mV in the whole-cell configuration using the patch-clamp technique after series resistance compensation (typically 15–20 MΩ). Series resistance was monitored for constancy, and cell capacitance was estimated from the average of 10 transient relaxation currents produced by 5 mV hyperpolarizing voltage pulses. The recording pipette contained (in mm): 145 K-gluconate, 5 EGTA, 5 MgATP, 0.2 NaGTP, and 10 mm HEPES at pH 7.2 with KOH. Cells were bathed in 145 mm NaCl, 5 mm KCl, 1 mmCaCl2, 5 mm glucose, 5 mmHEPES, and 20 μm glycine at pH 7.4. Osmolarity was adjusted to 325 mOsm with sucrose. The culture dish in the recording chamber (<500 μl total volume) was continuously perfused (5 ml/min) to prevent accumulation of drugs.
Drug application. All of the drugs were diluted in bath solution. NMDA (200 μm) was applied directly by a gravity-fed Y-tubing delivery system (Murase et al., 1989) placed within 100 μm of the recorded cell. Drug application had fast onset (<10 msec) and achieved a completely local perfusion of the recorded cell. Ifenprodil (Reseach Biochemicals, Natick, MA) or IL-10 were coapplied with NMDA after at least 1 min of preperfusion. The response recovery was achieved after 5–7 min of wash from the last application. Recordings were performed in the presence of GABAA and AMPA receptor antagonists bicuculline methiodide (10 μm; Sigma) and 5 μm6-nitro-7-sulfamoilbenzo[f]quinoxaline-2,3-dione (Tocris Coockson, St. Louis, MO), diluted in bath solution from stock solutions prepared in water and DMSO, respectively.
Data acquisition and analysis. Currents were monitored with a patch amplifier (EPC-7; List Electronics, Darmstadt, Germany), filtered at 1.5 kHz (eight-pole low-pass Bessel; Frequency Devices, Haverhill, MA), and digitized using a IBM-compatible microcomputer equipped with the Digidata 1200 data acquisition board and pClamp 8 software (Axon Instruments, Foster City, CA). Off-line data analysis, dose–response fitting, and figure preparation were performed with Origin (MicroCal Software, Northampton, MA) and pClamp 8 software (Axon Instruments). Data values are expressed as mean ± SEM. Significance was assessed with ANOVA followed by independentt tests, unless otherwise indicated.
Electrophoretic mobility shift assay
Nuclear factor κB (NF-kB) binding activity was analyzed in nuclear extracts from cerebellar granule cells prepared as described previously (Colangelo et al., 1998). Nuclei were incubated with a double-stranded oligonucleotide containing a consensus NF-kB binding site (5′-GGCAGAGGGGACTTTCCGAGAGGC-3′) labeled with32P-dCTP by Klenow polymerase (Boehringer Mannheim). Binding reactions were performed for 20 min at room temperature in a 25 μl of reaction containing 10 mmHEPES, pH 7.6, 134 mm NaCl, 4% (w/v) Ficoll, 5% (v/v) glycerol, 1 mm EDTA, 10 mm DTT, 0.25 μg of BSA, 0.06% bromophenol blue, 1 μg poly(dI-dC), and 0.5 ng of probe. Protein:DNA complexes were separated on 6% PAGE. For supershift assays, nuclear extracts were preincubated with 1 μl of antisera at 4°C for 20 min before addition of the probe. Quantitation of binding activity was done by densitometry as described previously (Colangelo et al., 1998).
RESULTS
IL-10 prevents glutamate-mediated cell death
Before examining whether IL-10 limits glutamate excitotoxicity, we first established time- and dose-dependent excitotoxicity in cerebellar granule cells exposed continuously to glutamate. Thus, cultures were exposed to medium alone or containing increasing concentrations of glutamate for various times without replacing the medium. Cell death–survival was measured by MTT assay. Glutamate (300 μm) induced a time-dependent decrease in cell viability starting at 6 hr and culminating at 24 hr (Fig.1A). Glutamate-mediated excitotoxicity was blocked by the NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (Fig. 1A), supporting previous findings that glutamate toxicity depends on stimulation of NMDA receptors (Schramm et al., 1990; Marini et al., 1997). Glutamate also evoked a dose-dependent excitotoxic effect starting from a concentration of 100 μm and peaking at 500 μm (Fig. 1B). Neurons were then exposed to IL-10 (50 ng/ml) for 14 hr before the addition of glutamate, and cell survival was measured 14 hr later. IL-10 prevented glutamate-mediated cell death even when glutamate was used at the highest concentration (Fig. 1B).
Fig. 1.
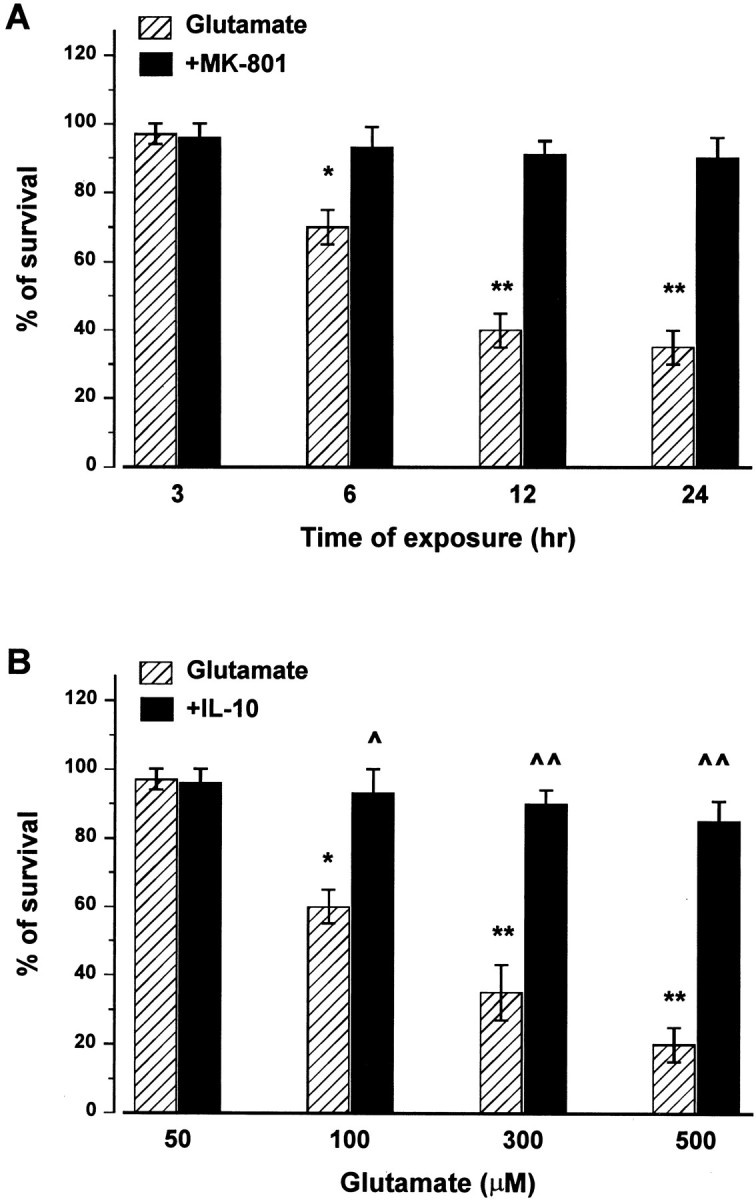
Glutamate induces a time- and dose-dependent excitotoxicity blocked by IL-10. A, Cerebellar granule cells were exposed to glutamate (300 μm) in the presence or absence of MK-801 (10 μm) for the indicated times, and then cell viability was measured at the indicated times by MTT assay.B, Neurons were exposed to IL-10 (50 ng/ml) for 14 hr before the addition of different concentrations of glutamate. Cell viability was measured by MTT assay 14 hr after glutamate addition. Data, expressed as percentage of control, are the mean ± SEM of four separate experiments. *p < 0.01, **p < 0.005 versus control; ^p < 0.01, ^^p < 0.005 versus glutamate (ANOVA and Dunnett's test).
IL-10 inhibits glutamate-mediated apoptosis
In cerebellar granule cells, glutamate evokes necrosis and/or apoptosis depending on the experimental conditions used (Resink et al., 1994; Ankarcrona et al., 1995; Du et al., 1997). Thus, we determine whether IL-10 prevented glutamate-mediated apoptosis by in situ TUNEL (Fig. 2). In control neurons, few cells (at the most 5%) were TUNEL-positive (Fig.2A). Exposure of cells to glutamate for 24 hr increased the number of TUNEL-positive cells (Fig.2B). Pretreatment of neurons with IL-10 for 24 hr, a treatment that per se did not alter cell viability (Fig.2C), blocked glutamate-mediated increase in TUNEL-positive cells (Fig. 2D). To confirm that, in our experimental condition glutamate evokes cell death by apoptosis, caspase-3 staining and in situ TUNEL were performed in the same cells. Caspase-3 is a protease that plays a role in the EAA-mediated apoptosis in cerebellar granule cells (Du et al., 1997; Tenneti and Lipton, 2000). Figure 3 shows that all TUNEL-positive cells were also caspase-3-positive, suggesting that, in our experimental conditions, apoptosis could be the main cause of cell death by glutamate.
Fig. 2.

IL-10 reduces the increase of TUNEL-positive cells glutamate mediated. Cerebellar granule cells were exposed to medium alone (A), glutamate (B; 300 μm), or IL-10 (C; 50 ng/ml) for 14 hr, or IL-10 for 14 hr followed by glutamate for 14 hr (D). Cells were then fixed and stained for TUNEL for the determination of apoptosis. In both control and IL-10-treated cultures, 95% of cells were TUNEL-negative, whereas ∼65% of neurons after glutamate treatment were TUNEL-positive (dark brown). Scale bar, 15 μm.
Fig. 3.

Caspase-3 immunoreactivity in TUNEL-positive cells. Neurons were exposed to medium alone or glutamate (300 μm) for 3 hr. Determination of apoptotic neurons was performed by TUNEL (A, D) and caspase-3-p20 (B, E) staining.A–C, Control cells; D–F, glutamate-treated cells. Analysis by Magnafire revealed that all TUNEL-positive cells were also positive for caspase-3 (overlay). Scale bar, 15 μm.
IL-10 prevents NMDA-mediated cell death
Cell death of cerebellar granule neurons is attributable to the overactivation of the NMDA subtype of receptors (Schramm et al., 1990;Marini et al., 1997). We then examined whether IL-10 prevented glutamate and NMDA-mediated cell death by TUNEL (Fig.4A) and MTT (Fig.4B) assays. Exposure of cerebellar granule cells to IL-10 for 24 hr elicited a dose-dependent neuronal protection against both EAAs (Fig. 4). Indeed, a significant effect of IL-10 was seen already at a concentration of 10 ng/ml (∼70% survival), whereas the maximal neuroprotection (95% of survival) was obtained with a concentration of 50 ng/ml (Fig. 4). Similar neuroprotection was observed when neurons were exposed to MK-801 (1 μm) 30 min before glutamate (data not shown).
Fig. 4.

The neuroprotective effect of IL-10 is dose-dependent. Cerebellar granule cells were exposed to different concentrations of IL-10 for 14 hr, and then glutamate or NMDA (300 μm each) was added for additional 14 hr. Cell survival was determined by in situ TUNEL (A) and MTT (B) assay. Data, expressed as percentage of control, are the mean ± SEM of four separate experiments (n = 12 each group). *p < 0.01, **p < 0.005 versus glutamate or NMDA alone (ANOVA and Dunnett's test).
To establish the temporal profile of IL-10 neuroprotective effect, neurons were exposed to IL-10 (50 ng/ml) for various times (6, 12, and 24 hr) before glutamate. In addition, to examine the effect of an acute exposure to IL-10, neurons were incubated with glutamate either concomitantly with IL-10 or 1 hr before IL-10. Cell death was then measured by both MTT and TUNEL assays 14 hr later. IL-10 evoked a time-dependent neuroprotection that was maximal when IL-10 was added several hours before glutamate (Fig. 5). When IL-10 was added concomitantly or after glutamate, a modest but significant neuroprotective effect was still observed (Fig. 5). These data suggest that IL-10 is an effective neuroprotective agent against glutamate-mediated cell death.
Fig. 5.

IL-10 elicits a time-dependent neuroprotection against glutamate. Neurons were exposed to IL-10 alone (50 ng/ml) for 6, 12, and 24 hr before glutamate, to IL-10 and glutamate simultaneously (0), or to glutamate 1 hr before IL-10 (−1). Cell survival was measured 14 hr after glutamate by TUNEL assay. Data are the mean ± SEM of three independent experiments (n = 15 each group). ^p < 0.005 versus control; *p < 0.05, **p < 0.005 versus glutamate (ANOVA and Dunnett's test).
Effect of IL-10 on NMDA receptor channel
The effect of IL-10 in preventing glutamate and NMDA toxicity is rapid because it occurs even if the addition of IL-10 is delayed after glutamate. These data suggest that IL-10 may interact directly with the NMDA receptor. To test this hypothesis, we examined whether IL-10 alters NMDA currents. Cerebellar granule cells in culture were voltage clamped at −60 mV using whole-cell recordings with a potassium gluconate-based solution. On average, the current recorded from granule cells normalized by the cell capacitance was 25 ± 7.3 pA/pF (n = 65). NMDA applications produced desensitizing whole-cell currents (Fig.6A). Coapplication of IL-10 (50 ng/ml) with NMDA did not change the NMDA response (Fig.6A). The ratio of the maximal current densities recorded in each cell in the presence or the absence of IL-10 (50 ng/ml) revealed that the current density did not change in cells exposed concomitantly to IL-10 and NMDA (Fig. 6B,first bar) or preexposed to IL-10 for 30 min, 180 min, or 14 hr before NMDA (Fig. 6B).
Fig. 6.

IL-10 fails to inhibit NMDA-activated currents.A, Current traces from cerebellar granule cells elicited by 200 μm NMDA in the presence and absence of IL-10 (50 ng/ml). NMDA was applied by a Y-tubing device for the duration indicated by the bars. Holding potential, −60 mV.B, Summary of the action of IL-10 treatments in cerebellar granule cells. The left histogram bar labeledIL-10 represents the percentage control of peak currents normalized to the cell capacitance recorded from 31 individual cells during application of NMDA in the presence of IL-10 (50 ng/ml). The other histogram bars represent the percentage control peak current density from at least 10 distinct granule neurons in three distinct sets of experiments in which cells were pretreated with IL-10 (50 ng/ml) for 30 min, 180 min, or 14 hr. Each pointrepresents the mean ± SEM of the ratios of normalized current. No significant differences were found between control and treated cells.
Because some neuroprotective neurotrophic factors have been shown to alter the synthesis of NMDA receptor subunits (Brandoli et al., 1998), NMDA currents were also studied in the presence of Ifenprodil, a selective antagonist of NMDA receptor that include the NR1 and NR2B subunits (Williams, 1993). Similar to a previous report (Corsi et al., 1998), 10 μm Ifenprodil reduced the peak currents elicited by 200 μm NMDA by 50 ± 16%. In 12 granule cells from three distinct experiments, IL-10 (50 ng/ml) incubation did not alter the Ifenprodil effect, which was 49 ± 11% after 30 min of IL-10 treatment, 52 ± 19% after 180 min of treatment, and 48 ± 18% for the overnight treatments. These results indicate that IL-10 treatment failed to alter the functional expression of distinct subunits of NMDA receptor in cerebellar granule cells.
IL-10 does not prevent EAA-evoked [Ca2+]i increase
The relatively rapid effect of IL-10 in preventing glutamate and NMDA toxicity suggests that IL-10 might interact directly with the NMDA receptor. Because activation of NMDA receptor promotes influx of extracellular Ca2+ through its own channel, the functional state of the NMDA receptor can be assessed by measuring its ability to evoke an [Ca2+]i increase after stimulation with a proper ligand. Thus, we investigated whether IL-10 blocks glutamate- or NMDA-mediated surge in [Ca2+]i. Cerebellar granule cells were exposed to either vehicle or IL-10 (50 ng/ml) for 10 min or 24 hr, NMDA (50 μm) was then added, and [Ca2+]i was measured over time. Exposure of neurons to IL-10 did not affect [Ca2+]i, and the resting [Ca2+]imonitored before the addition of NMDA showed no statistically significant differences between neurons treated with vehicle or IL-10 for either 10 min or 24 hr (in nm: vehicle-treated, 40 ± 8.6, n = 10; IL-10-treated, 52 ± 7.8,n = 10). [Ca2+]i increase induced by NMDA peaked within 10 min from EAA addition and remained elevated for at least 20 min. The [Ca2+]i rise evoked by NMDA in neurons pretreated with IL-10 for 10 min or 24 hr (the latter treatment maximally preventing excitotoxicity) was comparable, in both magnitude and kinetics, with that elicited in control, vehicle-treated cells (Fig.7A,B). The magnitude of the EAA-evoked Ca2+response varied among the four cerebellar granule neuron preparations used in this study, regardless of the pretreatment (vehicle or IL-10, 10 min or 24 hr). Peak [Ca2+]i increase (expressed as fold above basal) was as follows: NMDA-treated cells, 10.7 ± 3.6, n = 6; IL-10 plus NMDA-treated cells, 12.5 ± 5.6, n = 6. These results show that IL-10 does not prevent the EAA-mediated increase in [Ca2+]i through NMDA receptor channels.
Fig. 7.
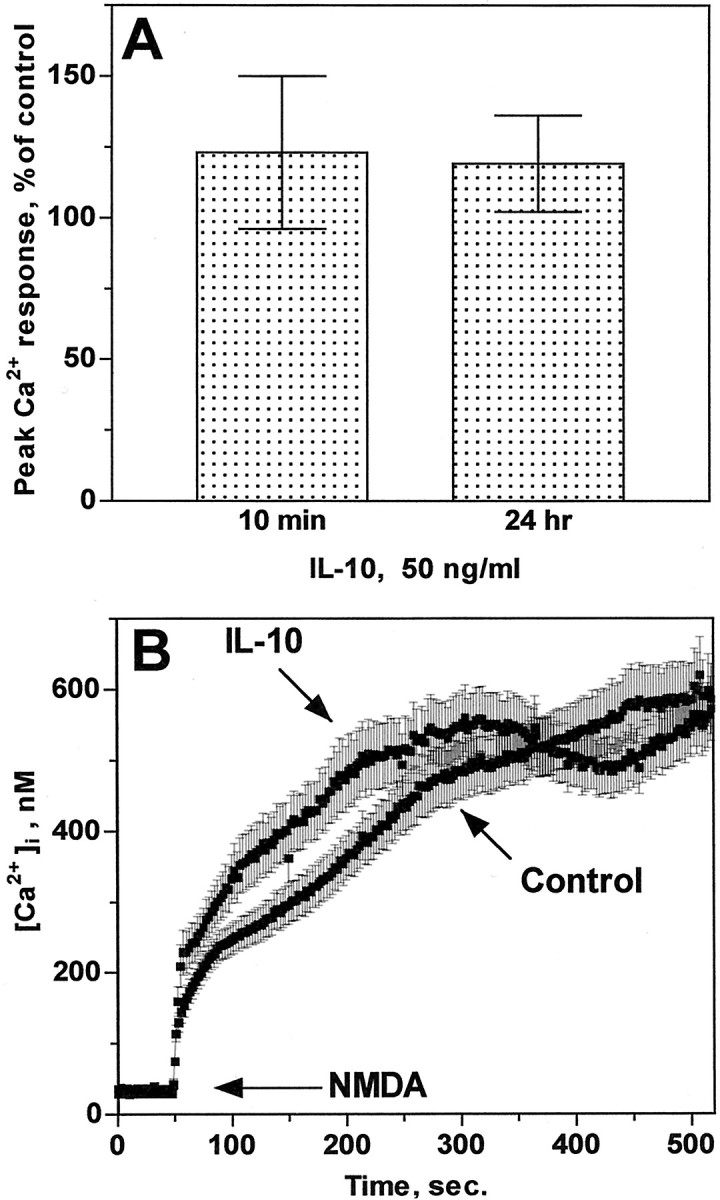
IL-10 fails to block EAA-mediated [Ca2+]i increase. Ca2+ imaging in cerebellar granule neurons from four independent preparations was performed as described in Materials and Methods. A, Peak [Ca2+]i increase evoked by NMDA in IL-10-treated cells and expressed as percentage of the Ca2+ response in vehicle-treated (control) cells that were imaged in parallel experiments. Data represent mean ± SEM (n = 5) for both 10 min and 24 hr (n = 1 coverslip with 50–99 neurons being imaged simultaneously). B, Ca2+ traces representative of NMDA-evoked [Ca2+]iincrease in neurons pretreated for 24 hr with either vehicle (control) or IL-10. Data (mean ± SEM) represent the [Ca2+]i population mean from 96 (control) and 99 (IL-10) neurons imaged simultaneously.
IL-10 prevents the increase in NF-κB binding activity evoked by glutamate
One way to prevent EAA-mediated apoptosis is to block the activity of proapoptotic proteins. The transcription factor NF-κB is associated with differentiation and apoptosis (O'Neill and Kaltschmidt, 1997). In neurons, induction of NF-κB DNA binding by glutamate is believed to play a role in programmed neuronal cell death (Kaltschmidt et al., 1995; Grilli et al., 1996). Thus, we examined whether IL-10 affected NF-κB nuclear activity in cerebellar granule cells. Electrophoretic mobility shift assay (EMSA) of nuclear extracts of cells exposed for 1 hr to glutamate (300 μm) showed higher NF-κB binding activity than control cells (Fig.8A). Supershift experiments with specific antibodies (Fig. 8A) indicated that the active complex consisted of the typical heterodimer of NF-κB p52 and p65 found in mammalian cells (Baeuerle and Baltimore, 1996). The effect of glutamate began at 30 min and lasted for at least up to 3 hr (Fig. 8B). We then examined whether IL-10, added immediately before (10 min) glutamate, could affect the glutamate-mediated increase in NF-κB DNA binding activity. IL-10, which per se decreased NF-κB DNA binding activity slightly below control levels (Fig. 8A), blocked the glutamate-mediated increase in NF-κB binding activity tested at 30 min, 1 hr, and 3 hr after EAA application (Fig.8B).
Fig. 8.
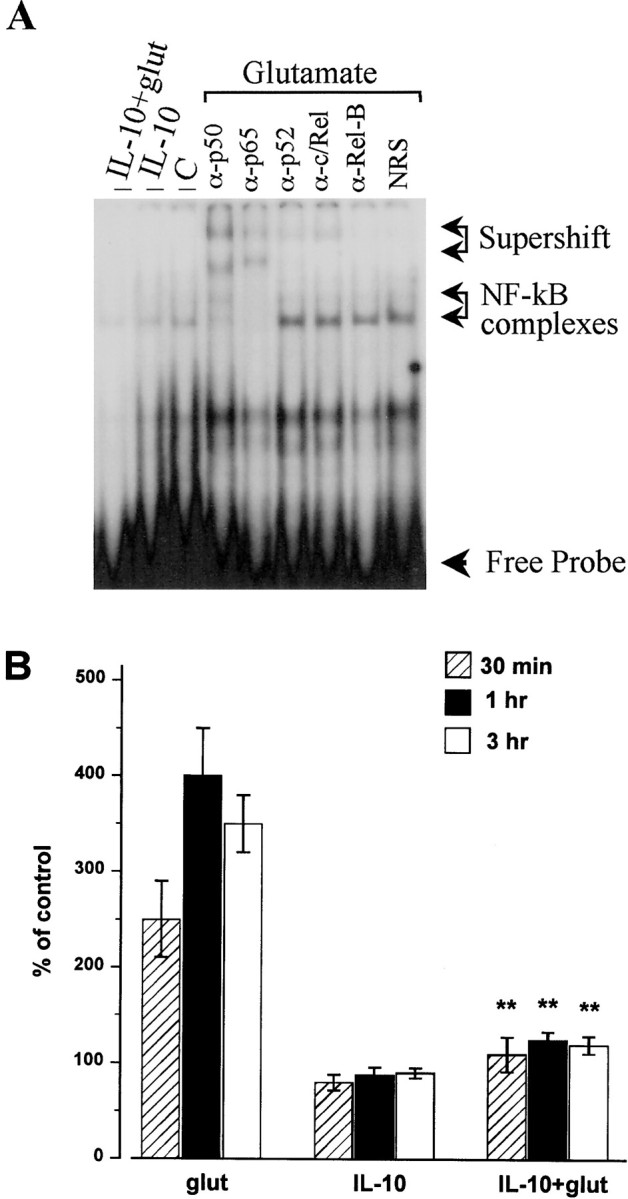
IL-10 blocks the glutamate-mediated increase in NF-kB binding activity. Cells were exposed to glutamate (300 μm), IL-10 (50 ng/ml), or a combination of glutamate and IL-10 for various times, nuclear extracts were prepared, and then NF-kB binding activity was measured by EMSA. A, Typical EMSA showing NF-κB complexes (arrows). These complexes were supershifted by α-p50 and p65 but not p52 and c/Rel antibodies. C, Control; glut, glutamate; NRS, normal rabbit serum. B, Relative levels of NF-κB binding activity were quantified by phosphorimager analysis of the band corresponding to NF-κB complexes. Data are the mean ± SEM of three separate experiments, with two to three independent samples each experiment. **p< 0.01 versus glutamate (ANOVA and Dunnett's test).
IL-10 and caspase-3-like activity
The inhibition of NF-κB activity is very often associated with inactivation of caspases (O'Neill and Kaltschmidt, 1997), proteases that participate in the pathogenesis of CNS disorders associated with apoptosis. Several lines of independent investigations have demonstrated that, in cerebellar granule cells, caspase-3 but not caspase-1 plays a role in the NMDA-mediated apoptosis (Du et al., 1997;Tenneti and Lipton, 2000). In addition, we have shown that TUNEL-positive cells are also caspase-3-positive (Fig. 3). Thus, we evaluated whether the neuroprotective properties of IL-10 might involve an inhibition of caspase-3-like activity. To provide quantitative data, caspase-3 was measured by a colorimetric assay. Exposure of cerebellar granule cells to glutamate (300 μm) evoked an increase in caspase-3-like activity within 1 hr (Fig.9), an effect that lasted at least up to 3 hr (data not shown). In lysates of cells exposed to IL-10 for 1 hr, the basal levels of caspase-3-like activity were reduced (Fig. 9). This effect was similar to that obtained with the relatively specific, irreversible caspase-3-like protease inhibitor acetyl-DEVD-chloromethylketone (DEVDK) (Fig. 9). Most importantly, IL-10, similar to DEVDK, blocked the glutamate-mediated rise in caspase-3-like activity (Fig. 9), suggesting that IL-10 may be an effective caspase-3 inhibitor.
Fig. 9.
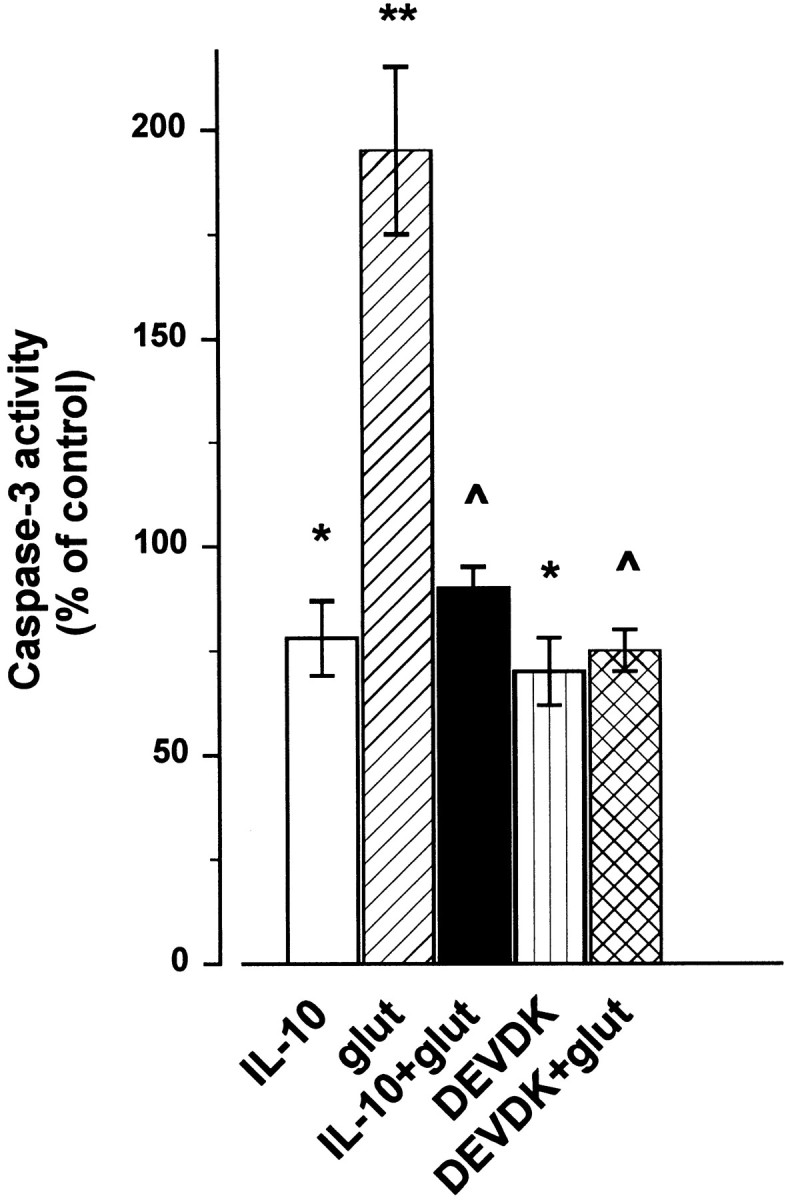
IL-10 prevents the increase in caspase-3-like activity evoked by glutamate (glut). Cerebellar granule cells exposed to glutamate (300 μm), IL-10 (50 ng/ml), or DEVDK (100 μm) alone or in combination. IL-10 and DEVDK were added 10 min before glutamate. Caspase-3-like activity was measured 1 hr later. Data are the mean ± SEM of three independent preparations (n = 6 each group). *p < 0.01, **p < 0.005 versus control; ^p < 0.005 versus glutamate (ANOVA and Dunnett's test).
To further examine the role of caspase-3 in the glutamate-mediated cell death, cells were incubated with IL-10 or DEVDK (100 μm) 10 min before glutamate (300 μm), and cell death was measured 14 hr later. Both compounds blocked the glutamate-mediated neuronal cell death (Fig. 10), further suggesting that in these neurons excitotoxicity involves a caspase-mediated pathway.
Fig. 10.

Inhibition of caspases prevents glutamate-mediated excitotoxicity. Cerebellar granule cells were exposed to glutamate (glut; 300 μm), IL-10 (50 ng/ml), or DEVDK (100 μm) alone or in combination. IL-10 and DEVDK were added 10 min before glutamate. Cell survival was measured 14 hr after by TUNEL assay. Data are the mean ± SEM of three independent preparations (n = 6 each group). *p < 0.005 versus control; **p < 0.005 versus glutamate (ANOVA and Dunnett's test).
DISCUSSION
The anti-inflammatory cytokine IL-10 has been shown to reduce vulnerability of neurons to CNS ischemia and trauma (Knoblach and Faden, 1998; Bethea et al., 1999; Grilli et al., 2000). However, the mechanisms underlying its neuroprotective activity remain to be fully elucidated. In these studies, we demonstrated that IL-10 prevents glutamate and NMDA-mediated cell death in primary cultures of cerebellar granule cells by blocking apoptosis. Remarkably, IL-10 prevents EAA-mediated apoptotic cell death, even when added after glutamate. Apoptosis is an event that plays an important pathophysiological role in CNS after trauma, stroke, or ischemia. In fact, apoptosis has been demonstrated to contribute to neuronal or glial cell death occurring several hours after brain or spinal cord injury (Hara et al., 1997; Liu et al., 1997; Yakovlev et al., 1997). Moreover, several lines of independent investigations have shown that apoptosis may be the major form of cell death following EAA accumulating after an insult to the CNS (for review, see Bettmann and Henderson, 1998). Thus, the ability of IL-10 to reduce EAA-mediated apoptosis could explain the therapeutic efficacy of this cytokine and shed some insights into the mechanisms whereby IL-10 reduces infarct volume after middle cerebral artery occlusion (Spera et al., 1998;Grilli et al., 2000), enhances neurological recovery after traumatic brain injury (Knoblach and Faden, 1998), or spinal cord lesion (Bethea et al., 1999) in rats. We suggest that IL-10 is an effective compound against EAA-mediated excitotoxicity.
Several growth factors–cytokines, in addition to IL-10, have been shown to prevent glutamate toxicity in cerebellar granule cells, among others BDNF and FGF2 (Fernandez-Sanchez and Novelli, 1993; Lindholm et al., 1993; Courtney et al., 1997; Marini et al., 1998). Indeed, we have reported recently that the neuroprotective activity of BDNF and FGF2 correlates with their ability to evoke a downregulation of the synthesis of NMDA receptor subunit NR2A and NR2C (Brandoli et al., 1998). Consequently, these growth factors decreased the abnormally sustained [Ca2+]itypically seen after excessive stimulation of NMDA receptors (Brandoli et al., 1998) and implicated in neuronal cell death (Garthwaite et al., 1986; Rothman and Olney, 1986; Choi, 1988; Hahn et al., 1988; Anegawa et al., 1995). In contrast, neurotoxic cytokines, such as IL-6, has been shown to significantly potentiate NMDA-mediated increase intracellular calcium in cerebellar granule cells and, consequently, NMDA-mediated excitotoxicity (Qiu et al., 1998). Thus, it was important to ascertain whether IL-10, a physiological anti-inflammatory cytokine, could prevent NMDA-mediated neurotoxicity by reducing NMDA current responses or Ca2+ influx. IL-10, used at a concentration and time effective against EAA-evoked cell death, failed to block glutamate or NMDA-mediated Ca2+influx or NMDA-mediated Ifenprodil-sensitive membrane depolarization. Thus, our data exclude that the neuroprotective effect of IL-10 is attributable to a direct effect on the NMDA channel.
Apoptosis, in addition to toxic concentrations of EAA, can be caused experimentally by a wide variety of stimuli. However, each given cell type may use different molecular mechanisms to activate the cell death pathway. Cell death may be caspase-dependent or -independent and may involve a particular type of caspases. For instance, in cerebellar granule cells, caspase-3 does not appear to be involved in cell death evoked by serum deprivation (Miller et al., 1997). Instead, caspase-3, but not caspase-1, plays an important role in glutamate-mediated apoptosis (Du et al., 1997; Tenneti and Lipton, 2000). Evidence has also suggested that caspase-3 participates in apoptotic cell death after brain injury (Yakovlev et al., 1997) or ischemia (Cheng et al., 1998; Namura et al., 1998), indicating that caspase-3 plays a critical role in the terminal stage of the apoptotic pathway after CNS injury. In this report, we have shown that IL-10 prevents the increase in caspase-3-like activity mediated by glutamate with a temporal profile and magnitude similar to that of DEVDK, a typical caspase inhibitor. In addition, IL-10 induced a rapid (within hours) decrease in caspase-3-like activity, regardless of its lack of effect on [Ca2+]i, whose levels modulate caspase-3 induction (Moran et al., 1999). Thus, it appears that IL-10 has an intrinsic ability to inhibit directly or indirectly caspase-3-like activity; this mechanism could explain the neuroprotective properties of IL-10 in vivo (Knoblach and Faden, 1998; Bethea et al., 1999; Grilli et al., 2000). It remains to be established whether IL-10 affects other caspases as well and the mechanisms whereby IL-10 reduces caspase-3 activity in neurons.
Recent findings have implicated the transcription factor NF-κB as a mediator of neuronal apoptosis. Also, NF-κB appears to have a deleterious role on neuronal survival. In fact, cell death occurs in neurons when NF-κB is permanently activated, such as after trauma (Bethea et al., 1998), global ischemia (Clemens et al., 1998), or toxic concentrations of glutamate (Kaltschmidt et al., 1995; Grilli et al., 1996). On the other hand, inhibition of NF-κB activity results in inactivation of caspases (Gill and Windebank, 2000). IL-10 blocked the glutamate-mediated NF-κB DNA binding activity, shown previously to be causally involved in glutamate-mediated cell death (Kaltschmidt et al., 1995; Grilli et al., 1996). Thus, we provided additional support that the neuroprotective mechanism of this cytokine relies on its ability to interfere with the cellular mechanism involved in apoptosis. Interestingly, the suppression of NF-κB DNA binding activity plays a role in the anti-inflammatory effect of IL-10 also in non-neuronal cells (Schottelius et al., 1999). Therefore, we speculate that IL-10, by reducing or preventing the activity of caspase-3 and NF-κB evoked by EAA, reestablishes the physiological basal ratio of proapoptotic/antiapoptotic proteins that, in turn, may render neurons less vulnerable to apoptosis.
Our results might be of crucial neurological significance because effective therapies against post-trauma secondary injury in humans are still scarce. The findings reported here provide strong support to the belief that IL-10 may have a therapeutic significance for neurodegenerative diseases by blocking EAA-mediated excitotoxicity. However, we cannot definitively rule out the existence of other mechanisms that could account for the neuroprotective effect of IL-10. For example, the anti-inflammatory properties of IL-10 and its ability to decrease the synthesis of TNF-α, IL-1β (Bogdan et al., 1992;Wang et al., 1994; Kline et al., 1995; Di Santo et al., 1997; Bethea et al., 1999, Sawada et al., 1999), or other cytokines such as FGF2 (Zocchi et al., 1997) or macrophage inflammatory protein-1 α (Berkman et al., 1995), may also help to reduce apoptosis. However, when we examined anti-inflammatory neuroprotective compounds, such as methylprednisolone, we did not observed neuronal protection against glutamate (our unpublished observations). Moreover, astrocytes and microglia are mostly the primary sources of proinflammatory cytokines. Our neuronal cultures contain only few non-neuronal cells (at the most 5%), thus, most likely do not contain toxic concentration of inflammatory cytokines. In conclusion, we propose that the neuroprotective effects of IL-10 against EAA-mediated excitotoxicity is, at least in cerebellar granule cells, primary because of the ability of IL-10 to inhibit the activity of proapoptotic proteins and in particular caspase-3.
Footnotes
This work was supported by grants from American Heart Association Nation's Affiliate (I.M.), Health and Human Services Grants HL 28940 (G.B.) and MH58946 and MH01680 (S.V.), and a fellowship from Schering Plough Research Institute (A.B.). We thank Randi Goodnight for her help in computer programs and image analysis and Dr. S. Narula for the gift of IL-10.
Correspondence should be addressed to Dr. Italo Mocchetti, Department of Neuroscience, Research Building, Georgetown University, 3900 Reservoir Road NW, Washington, DC 20007. E-mail:moccheti@gunet.georgetown.edu.
REFERENCES
- 1.Anegawa NJ, Lynch DR, Verdoorn TA, Pritchett DB. Transfection of N-methyl-d-aspartate receptors in a nonneuronal cell line leads to cell death. J Neurochem. 1995;64:2004–2012. doi: 10.1046/j.1471-4159.1995.64052004.x. [DOI] [PubMed] [Google Scholar]
- 2.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 3.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 4.Berkman N, John M, Roesems G, Jose PJ, Barnes PJ, Chung KF. Inhibition of macrophage inflammatory protein-1 α expression by IL-10. J Immunol. 1995;155:4412–4418. [PubMed] [Google Scholar]
- 5.Bethea JR, Castro M, Keane RW, Dietrich WD, Yezeirski RP. Traumatic spinal cord injury induces nuclear factor-kB activation. J Neurosci. 1998;18:3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, Loor K, Green J, Dietrich D. Systemically administered interleukin-10 reduces tumor necrosis factor alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma. 1999;16:851–863. doi: 10.1089/neu.1999.16.851. [DOI] [PubMed] [Google Scholar]
- 7.Bettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 8.Bogdan C, Paik J, Vodovotz Y, Nathan C. Contrasting mechanisms for suppression of macrophages cytokine release by transforming growth factor–β and interleukin-10. J Biol Chem. 1992;267:23301–23308. [PubMed] [Google Scholar]
- 9.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced respectively by mild and intense insults with NMDA or nitric oxide/superoxide in cortical cell culture. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandoli C, Sanna A, De Bernardi MA, Follesa P, Brooker G, Mocchetti I. Brain-derived neurotrophic factor and basic fibroblast growth factor downregulate NMDA receptor function in cerebellar granule cells. J Neurosci. 1998;18:7953–7961. doi: 10.1523/JNEUROSCI.18-19-07953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng B, Furukawa K, O'Keefe JA, Goodman Y, Kihiko M, Fabian T, Mattson MP. Basic fibroblast growth factor selectively increases AMPA-receptor subunit GluR1 protein level and differentially modulates Ca2+ responses to AMPA and NMDA in hippocampal neurons. J Neurochem. 1995;65:2525–2536. doi: 10.1046/j.1471-4159.1995.65062525.x. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday J, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic ischemic brain injury. J Clin Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 14.Clemens JA, Stephenson DT, Yin T, Smaltstig B, Panetta JA, Little SP. Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-kB in rats. Stroke. 1998;29:677–682. doi: 10.1161/01.str.29.3.677. [DOI] [PubMed] [Google Scholar]
- 15.Colangelo AM, Johnson P, Mocchetti I. β-Adrenergic receptor-induced activation of Nerve Growth Factor gene transcription in rat cerebral cortex involves CCAAT/Enhancer binding protein δ. Proc Natl Acad Sci USA. 1998;95:10920–10925. doi: 10.1073/pnas.95.18.10920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corsi L, JinHong L, Krueger KE, Wang YH, Wolfe BB, Vicini S. Up-regulation of NR2B subunit of NMDA receptors in cerebellar granule neurons by Ca2+ calmodulin kinase inhibitor KN93. J Neurochem. 1998;70:1–9. doi: 10.1046/j.1471-4159.1998.70051898.x. [DOI] [PubMed] [Google Scholar]
- 17.Courtney MJ, Akeman KEO, Coffey ET. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci. 1997;17:4201–4211. doi: 10.1523/JNEUROSCI.17-11-04201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Bernardi MA, Rabin SJ, Colangelo AM, Brooker G, Mocchetti I. TrkA mediates the nerve growth factor-induced intracellular calcium accumulation. J Biol Chem. 1996;271:6092–6098. doi: 10.1074/jbc.271.11.6092. [DOI] [PubMed] [Google Scholar]
- 19.Di Santo E, Adami M, Bertorelli R, Ghezzi P. Systemic interleukin 10 administration inhibits brain tumor necrosis factor production in mice. Eur J Pharmacol. 1997;336:197–202. doi: 10.1016/s0014-2999(97)01225-9. [DOI] [PubMed] [Google Scholar]
- 20.Du Y, Bales KR, Dodel RC, Hamilton-Byrf E, Horn JW, Czilli DL, Simmons LK, Binhui N, Paul SM. Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc Natl Acad Sci USA. 1997;94:11657–11662. doi: 10.1073/pnas.94.21.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez-Sanchez MT, Novelli A. Basic fibroblast growth factor protects cerebellar neurons in primary cultures from NMDA and non-NMDA receptor-mediated neurotoxicity. FEBS Lett. 1993;335:124–131. doi: 10.1016/0014-5793(93)80453-2. [DOI] [PubMed] [Google Scholar]
- 22.Feuerstein GZ, Wang XK, Barone FC. Inflammatory mediators and brain injury. In: Ginsberg M, Bogouslasky J, editors. The role of cytokine and chemokines in stroke and CNS diseases. Blackwell Science; Oxford: 1998. pp. 507–531. [Google Scholar]
- 23.Garthwaite G, Hajos F, Garthwaite J. Ionic requirements for neurotoxic effects of excitatory amino acid analogues in rat cerebellar slices. Neuroscience. 1986;18:437–447. doi: 10.1016/0306-4522(86)90164-8. [DOI] [PubMed] [Google Scholar]
- 24.Gill JS, Windebank AJ. Ceramide initiates NF-kB mediated caspase activation in neuronal apoptosis. Neurobiol Dis. 2000;7:448–461. doi: 10.1006/nbdi.2000.0312. [DOI] [PubMed] [Google Scholar]
- 25.Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 26.Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, Ongini E. Interleukin-10 modulates neuronal threshold of vulnerability to ischemic damage. Eur J Neurosci. 2000;12:1–8. doi: 10.1046/j.1460-9568.2000.00090.x. [DOI] [PubMed] [Google Scholar]
- 27.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 28.Hahn JS, Aizenman E, Lipton S. Central mammalian neurons normally resistant to glutamate toxicity are made sensitive by elevated Ca2+: toxicity is blocked by N-methyl-d-aspartate antagonist, MK801. Proc Natl Acad Sci USA. 1988;85:6556–6560. doi: 10.1073/pnas.85.17.6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin-1 beta converting enzyme family of proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Stimulation of ionotropic glutamate receptors activates transcription factor NF-kB in primary neurons. Proc Natl Acad Sci USA. 1995;92:9618–9622. doi: 10.1073/pnas.92.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kline JN, Fisher PA, Monick MM, Hunninghake GW. Regulation of interleukin-1 receptor antagonist by Th1 and Th2 cytokines. Am J Physiol. 1995;269:92–98. doi: 10.1152/ajplung.1995.269.1.L92. [DOI] [PubMed] [Google Scholar]
- 32.Knoblach SM, Faden AI. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Exp Neurol. 1998;153:143–151. doi: 10.1006/exnr.1998.6877. [DOI] [PubMed] [Google Scholar]
- 33.Lindholm D, Dechant G, Heisenberg CP, Thoenen H. Brain derived neurotrophic factor is a survival factor for cultured rat cerebellar granule neurons and protects against glutamate-induced neurotoxicity. Eur J Neurosci. 1993;5:1455–1464. doi: 10.1111/j.1460-9568.1993.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 34.Liu XZ, Xu XM, Hu R, Du C, Zhang S, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentrations in cultured spinal cord neurons. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 36.Marini AM, Spiga G, Mocchetti I. Toward the development of strategies to prevent ischemic neuronal injury. Ann NY Acad Sci. 1997;825:209–219. doi: 10.1111/j.1749-6632.1997.tb48431.x. [DOI] [PubMed] [Google Scholar]
- 37.Marini AM, Rabin SJ, Lipski R, Mocchetti I. Activity-dependent release of BDNF underlies the neuroprotective effect of NMDA. J Biol Chem. 1998;273:29394–29399. doi: 10.1074/jbc.273.45.29394. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3732. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller TM, Moulder KL, Knudson M, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mocchetti I, Wrathall JR. Neurotrophic factors in central nervous system trauma. J Neurotrauma. 1995;12:853–870. doi: 10.1089/neu.1995.12.853. [DOI] [PubMed] [Google Scholar]
- 41.Moran J, Itoh T, Reddy UR, Chen M, Almeri ES, Pleasure D. Caspase-3 expression by cerebellar granule neurons is regulated by calcium and cyclic AMP. J Neurochem. 1999;73:568–577. doi: 10.1046/j.1471-4159.1999.0730568.x. [DOI] [PubMed] [Google Scholar]
- 42.Murase K, Ryu PD, Randic M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely isolated rat spinal dorsal horn neurons. Neurosci Lett. 1989;103:56–63. doi: 10.1016/0304-3940(89)90485-0. [DOI] [PubMed] [Google Scholar]
- 43.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Neill LAJ, Kaltschmidt C. NFkB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–257. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 45.Qiu Z, Sweeney DD, Netzeband JG, Gruol DL. Chronic interleukin-6 alters NMDA receptor-mediated membrane responses and enhances neurotoxicity in developing CNS neurons. J Neurosci. 1998;18:10445–10456. doi: 10.1523/JNEUROSCI.18-24-10445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Resink A, Hack N, Boer GJ, Balázs R. Growth conditions differentially modulate the vulnerability of developing cerebellar granule cells to excitatory amino acids. Brain Res. 1994;655:222–232. doi: 10.1016/0006-8993(94)91617-9. [DOI] [PubMed] [Google Scholar]
- 47.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 48.Sawada M, Suzumura A, Hosoya H, Marunouchi T, Nagatsu T. Interleukin-10 inhibits both production of cytokines and expression of cytokine receptors in microglia. J Neurochem. 1999;72:1466–1471. doi: 10.1046/j.1471-4159.1999.721466.x. [DOI] [PubMed] [Google Scholar]
- 49.Schottelius AJG, Mayo M, Sartor BR, Baldwin AS. Interleukin-10 signaling blocks inhibitor of kB kinase activity and nuclear factor kB DNA binding. J Biol Chem. 1999;45:31868–31874. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 50.Schramm M, Eimerl S, Costa E. Serum and depolarizing agents cause acute neurotoxicity in cultured cerebellar granule cells: role of glutamate receptor responsive to N-methyl-d-aspartate. Proc Natl Acad Sci USA. 1990;87:1193–1197. doi: 10.1073/pnas.87.3.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett. 1998;251:189–192. doi: 10.1016/s0304-3940(98)00537-0. [DOI] [PubMed] [Google Scholar]
- 52.Tenneti L, Lipton SA. Involvement of activated caspase-3like proteases in N-methyl-d-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem. 2000;74:134–142. doi: 10.1046/j.1471-4159.2000.0740134.x. [DOI] [PubMed] [Google Scholar]
- 53.Wang P, Wu P, Siegel MI, Egan RW, Billah MM. IL-10 inhibits transcription of cytokine genes in human peripheral blood mononuclear cells. J Immunol. 1994;153:811–816. [PubMed] [Google Scholar]
- 54.Wielock T. Hypoglycemia-induced neuronal damage prevented by a N-methyl-d-aspartate antagonists. Science. 1985;230:681–683. doi: 10.1126/science.2996146. [DOI] [PubMed] [Google Scholar]
- 55.Williams K. Ifenprodil discriminates subtypes of N-methyl-d-aspartate receptors: selectivity and mechanism at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- 56.Yakovlev AG, Knoblach SM, Fan Lei, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zocchi C, Spiga G, Rabin SJ, Grekova M, Richert J, Chernyshev O, Colton C, Mocchetti I. Biological activity of interleukin-10 in the central nervous system. Neurochem Int. 1997;30:433–439. doi: 10.1016/s0197-0186(96)00079-4. [DOI] [PubMed] [Google Scholar]