

Abstract
We developed an in situ model to investigate the hypothesis that AMPA/kainate (AMPA/KA) receptor activation contributes to hypoxic–ischemic white matter injury in the adult brain. Acute coronal brain slices, including corpus callosum, were prepared from adult mice. After exposure to transient oxygen and glucose deprivation (OGD), white matter injury was assessed by electrophysiology and immunofluorescence for oligodendrocytes and axonal neurofilaments. White matter cellular components and the stimulus-evoked compound action potential (CAP) remained stable for 12 hr after preparation. OGD for 30 min resulted in an irreversible loss of the CAP as well as structural disruption of axons and subsequent loss of neurofilament immunofluorescence. OGD also caused widespread oligodendrocyte death, demonstrated by the loss of APC labeling and the gain of pyknotic nuclear morphology and propidium iodide labeling. Blockade of AMPA/KA receptors with 30 μm NBQX or the AMPA-selective antagonist 30 μm GYKI 52466 prevented OGD-induced oligodendrocyte death. Oligodendrocytes also were preserved by the removal of Ca2+, but not by a blockade of voltage-gated Na+ channels. The protective action of NBQX was still present in isolated corpus callosum slices. CAP areas and axonal structure were preserved by Ca2+ removal and partially protected by a blockade of voltage-gated Na+ channels. NBQX prevented OGD-induced CAP loss and preserved axonal structure. These observations highlight convergent pathways leading to hypoxic–ischemic damage of cerebral white matter. In accordance with previous suggestions, the activation of voltage-gated Na+ channels contributes to axonal damage. Overactivation of glial AMPA/KA receptors leads to oligodendrocyte death and also plays an important role in structural and functional disruption of axons.
Keywords: oligodendrocyte, white matter injury, AMPA/kainate receptors, axonal injury, brain slices, compound action potential, ischemia, glutamate
Approaches to pharmacological intervention in acute ischemic stroke most often have focused on the protection of neuronal cell bodies in gray matter. Because focal cerebral ischemia in humans damages both gray and white matter, an understanding of white matter injury is important in devising potential therapeutic approaches. In animal models early ischemic damage in white matter involves axons and oligodendrocytes (Pantoni et al., 1996;Dietrich et al., 1998; Valeriani et al., 2000). Axonal conduction is crucial to the functional integrity of the CNS. Studies of anoxia in optic nerve and spinal cord slice preparations suggest that axonal injury is mediated by the activation of voltage-gated Na+ channels (VGSC) and the subsequent reversal of Na+/Ca2+exchange, leading to lethal axoplasmic Ca2+ overload (Stys, 1998).
These mechanisms are less likely to contribute directly to hypoxic–ischemic death of glial cells, which do not express the voltage-sensitive channels that are found on excitable cells (Barres et al., 1988). Alternative pathways for oligodendrocytes include generation of toxic free radicals, loss of trophic support, and glutamate-mediated cell death. Glial cells do not have NMDA receptors (Gallo et al., 1994; Patneau et al., 1994) but do express functional AMPA/kainate (AMPA/KA) receptors (Steinhauser and Gallo, 1996;Garcia-Barcina and Matute, 1998) and can be killed by glutamate agonists. AMPA/KA antagonists reduce neuronal injury in cerebral ischemia (Sheardown et al., 1990), decrease damage to both gray and white matter in spinal cord contusion models (Wrathall et al., 1994), and reduce calpain activation in spinal cord myelin and oligodendrocytes (Li and Stys, 2000). Effects of AMPA/KA receptor blockade in white matter have not been reported for cerebral ischemic injury. Although synaptic neurotransmitter is not present in white matter, there are ample sources of glutamate release during energy deprivation. In spinal cord slices the anoxia-induced loss of CAPs is mediated by a reversal of Na+-dependent glutamate transport, with a reduction of intra-axonal glutamate (Li et al., 1999). Astrocytes (Anderson and Swanson, 2000) and oligodendrocytes (Fern and Möller, 2000) are also potential sources of glutamate release.
Mechanisms of glutamate toxicity in cultured oligodendrocytes include nonreceptor-mediated inhibition of cysteine uptake and oxidative stress (Oka et al., 1993) and direct AMPA/KA receptor activation (Yoshioka et al., 1996; Garcia-Barcina and Matute, 1998; McDonald et al., 1998). In cell culture models AMPA/KA receptor blockade prevents the death of oligodendrocytes during oxygen–glucose deprivation (McDonald et al., 1998; Fern and Möller, 2000; Yoshioka et al., 2000). Thesein vitro studies raise the possibility that AMPA/KA receptor activation may contribute to hypoxic–ischemic death of oligodendrocytes in vivo. However, cultured oligodendrocytes differ from their in vivo counterparts in several important respects, including maturational state, myelin production, receptor expression, and axonal–glial cellular interactions.
Our study investigated whether the death of mature oligodendrocytesin situ is mediated by the overactivation of AMPA/KA receptors. We developed an adult brain slice model to assess white matter conduction and cellular vulnerability after oxygen and glucose deprivation.
MATERIALS AND METHODS
Preparation of slices and oxygen–glucose deprivation
After we induced deep halothane anesthesia, adult female Swiss Webster mice were perfused transcardially with artificial CSF (aCSF) with the addition of 2 mm kynurenic acid (Sigma, St. Louis, MO). aCSF was composed of (in mm) 126 NaCl, 3.5 KCl, 1.3 MgCl2, 2 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, and 10 glucose, pH 7.4. The osmolality (∼300 mOsm) was checked with a micro-osmometer (Precision Systems, Natick, MA). The brains were dissected out immediately into ice-cold aCSF oxygenated with a mixture of 95% O2/5% CO2. The whole brain was placed on the platform of the vibroslicer (Vibratome 1000, Technical Products, St. Louis, MO), and 400-μm-thick coronal slices were cut. Only the slices (8–10/brain) in which the anatomical structure of the corpus callosum was visualized clearly were included in the experiments. Slices were allowed at least 2 hr at room temperature to stabilize (Kirov et al., 1999) before they were transferred to a Haas-type slice chamber (Harvard Apparatus, South Natick, MA).
For oxygen–glucose deprivation (OGD), aCSF was replaced by glucose-free aCSF (containing 10 mm sucrose to keep the osmolality constant) saturated with a 95% N2/5% CO2 mixture. After OGD the slices were superfused in glucose containing oxygenated aCSF for up to 9 hr after the end of OGD.
In some experiments (perfusion-fixed slices), after transcardial aCSF, the perfusion was switched to a fixative composed of 4% paraformaldehyde and 0.025% glutaraldehyde in PBS. The brains were dissected out and kept in fixative for 2 more hr at 4°C before being sliced. The 400-μm-thick coronal slices obtained from these brains were incubated further in fixative individually for another 2 hr at 4°C before they were placed in 10, 20, and 30% sucrose solution for 4, 6–8, and 16–18 hr, respectively. In another group (immediately fixed slices), after an animal was perfused with aCSF and kynurenic acid the brains were sliced, and the slices were fixed immediately in 4% paraformaldehyde and 0.025% glutaraldehyde in PBS. The immunohistochemical staining properties of the slices obtained from each group were analyzed and quantified comparatively to confirm that the acute brain slice model is a useful and representative tool for the assessment of cellular and cytoskeletal structure of white matter injury induced by OGD.
Electrophysiology
A single slice was placed on a piece of lens paper, transferred to the recording chamber, and kept at the interface between the warm humidified gas (95% O2/5% CO2, 1 l/min), and oxygenated aCSF at 33 ± 1°C, with a flow rate of 3–3.5 ml/min. Each slice was kept in the chamber for at least 30 min before baseline responses were recorded. Extracellular compound action potentials (CAPs) across the corpus callosum were evoked by using a bipolar stimulation electrode. Typically 50 μsec long, supramaximal pulses were delivered every 30 sec, and the responses were recorded with microelectrodes filled with 2m NaCl. The evoked responses were digitized and stored, and the changes in the CAP integral were analyzed (pClamp, Axon Instruments, Foster City, CA).
In initial experiments, OGD was applied in regular aCSF for 15 or 30 min to determine the duration of OGD that caused an irreversible loss of electrical activity. Later, the control slices were exposed to OGD always in aCSF containing 10 μm MK-801 (Research Biochemicals, Natick, MA; 10 mm stock dissolved in distilled water) to limit the activation of NMDA receptors in gray matter. The AMPA/KA receptor antagonist 30 μm NBQX (Research Biochemicals; 30 mm stock dissolved in distilled water, care being taken to prevent from direct light) was applied throughout the experiments, starting at least 30 min before OGD. For Ca2+-free experiments the slices were superfused with Ca2+-free aCSF (containing 200 μm EGTA) for a total of 90 min, starting 30 min before and during and 30 min after the end of OGD. Control experiments for this group consisted of recordings of CAPs in slices kept Ca2+-free for the same duration without OGD exposure.
In experiments investigating the role of voltage-gated Na+ channels, the slices were superfused with 10 μm tetrodotoxin (TTX; 100 mm stock dissolved in distilled water) starting 30 min before and during and 30 min after OGD. Control experiments were done in slices kept normoxic in TTX for similar durations.
Immunohistochemistry
The Haas-type slice chamber was divided into two compartments. Four to five slices were placed in each half of the chamber on lens paper (8–10 slices all from the same brain). All slices were superfused with aCSF at 33 ± 1°C for 60 min with a flow rate of 3–3.5 ml/min. At the end of 1 hr the slices in one of the compartments (control group) and the slices in the other compartment (experimental group) were superfused with aCSF containing 30 μm NBQX for at least another 30 min before the induction of OGD. Similar experiments also were performed at 37°C to determine the effects of temperature on the protective role of NBQX. Pretreatment of slices in Ca2+-free media and TTX before OGD was done as described above.
A sample slice from each compartment before and 1, 3, 5, and 9 hr after OGD was fixed and cryoprotected as described above. The 9 hr observation period, which represents a total slice preparation duration of 12 hr (2 hr preincubation at room temperature, 30 min equilibration at 34°C, 30 min OGD or normoxia, and 9 hr reperfusion), was chosen as the latest time point at which we could be certain that compound action potentials and cellular elements were preserved optimally. Then 16-μm-thick sections (10–12/slice) from each brain slice were cut by cryotome and collected onto Superfrost slides (Fisher Scientific, Pittsburgh, PA) for immunohistochemical analysis. Sections from the outer 50–80 μm surfaces were excluded to avoid damaged cells on the surface.
The brain sections were blocked and permeabilized in 40% normal goat serum (Sigma) and 0.4% Triton X-100 (Sigma) for 30 min at room temperature. All primary antibodies were prepared in the same mixture. Sections were labeled for oligodendrocytes with monoclonal antibodies CC1 (APC-7; Oncogene Research Products, Cambridge, MA) (Bhat et al., 1996) or anti-2,3-cyclic-nucleotide 3-phosphodiesterase (CNPase; Promega, Madison, WI) (Trapp et al., 1988), for oligodendrocyte progenitors with a polyclonal chondroitin sulfate proteoglycan antibody NG2 (Chemicon, Temecula, CA) (Levine and Nishiyama, 1996), astrocytes with a polyclonal antibody to glial fibrillary acidic protein (GFAP; DiaSorin, Stillwater, MN), and microglia with monoclonal CD11b (CR3) (711, Serotec, Raleigh, NC) (Rosen and Gordon, 1990). Axonal neurofilaments (NFs) were labeled with monoclonal antibodies SMI-31 and SMI-32 (Sternberger Monoclonals, Lutherville, MD) (Sternberger et al., 1982) against epitopes of phosphorylated and nonphosphorylated NF 200, respectively. Monoclonal antibody NR4 (Sigma) (Debus et al., 1983) was used to label against NF 68. For each slice, immunolabeling was examined and quantified in two or three sections for each experimental condition.
Primary antibodies were used at a dilution of 1:100 for APC, 1:500 for CNPase, 1:100 for NG2, 1:2 for GFAP (prediluted), 1:10,000 for SMI-31 and SMI-32, and 1:100 for NR4. Sections were incubated in primary antibodies together for 2–3 hr at room temperature or overnight at 4°C. After a thorough wash in PBS, the tissue was exposed to a secondary antibody, one by one for each primary, prepared in 2% normal goat serum for 1 hr at room temperature. Goat anti-mouse Alexa 488 (Molecular Probes, Eugene, OR) and noncross-reactive donkey anti-rabbit Cy3 (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:100 were used. Sections were examined via fluorescence microscopy (Nikon Diaphot) with digital image acquisition (Spot camera, Diagnostic Instruments, Sterling Heights, MI).
Assessment of cell death
Hoechst 33258 staining. Nuclear staining was achieved by dipping brain sections into 5 μg/ml Hoechst 33258 (Sigma) for several minutes at room temperature. After a final wash in PBS the sections were treated with Prolong Antifade (Molecular Probes) before they were coverslipped (Sigma). Before being imaged, the slides were kept at 4°C overnight to assure antifade treatment. Sections were examined by conventional fluorescence (Nikon Diaphot microscope, Spot digital camera) under ultraviolet light. Live cells were identified by their oval nuclei and characteristic eccentric three to five inclusion bodies (Skoff et al., 1976) and low fluorescent intensity with Hoechst stain. Pyknotic nuclei were identified by round, smaller, clumped nuclei and condensed chromatin with bright blue fluorescence (Deckwerth and Johnson, 1993) (see Figs. 4, 6, 7).
Fig. 4.
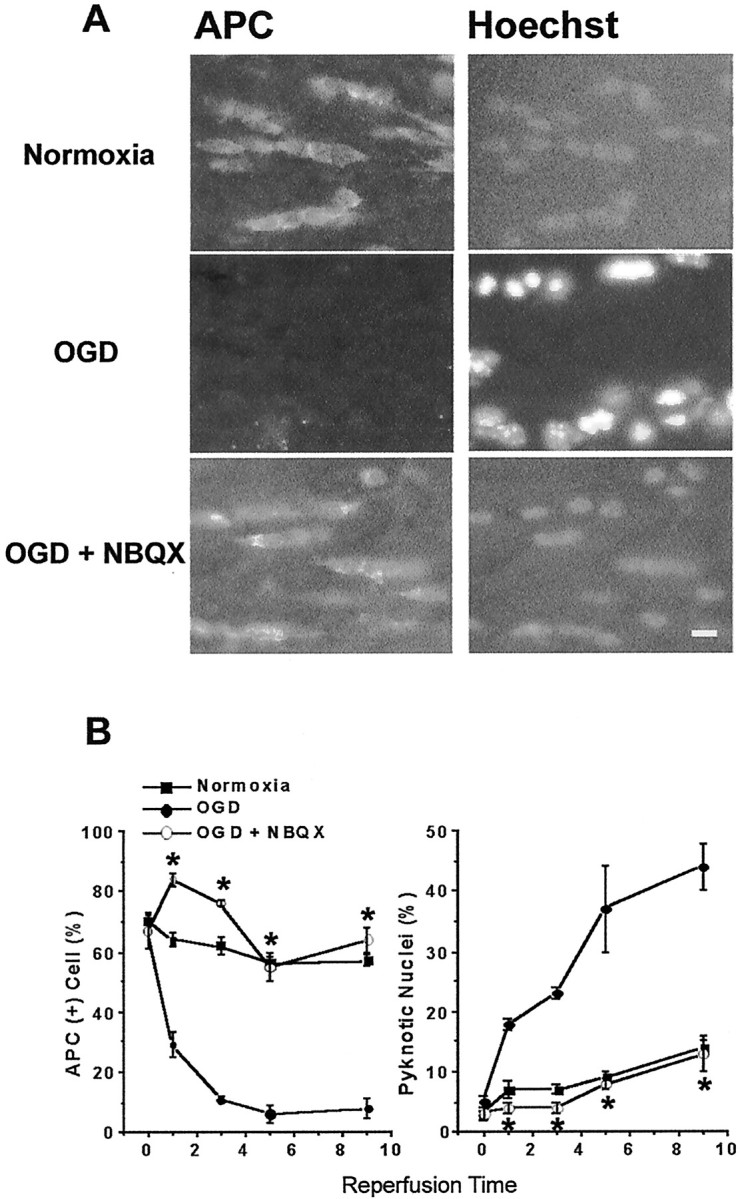
AMPA/KA receptor blockade prevents OGD-induced oligodendrocyte death. A, Numerous APC+ oligodendrocytes (left) and their nuclei stained with Hoechst (right) are seen in slices kept normoxic for 12 hr (corresponding to 9 hr of reperfusion time). OGD 30 min (in the presence of MK-801) results in the loss of APC immunoreactivity (middle left panel). Pyknotic nuclei are detected with Hoechst staining by increased fluorescence intensity and smaller nuclei (middle right panel). When AMPA/KA receptors are blocked with 30 μm NBQX, many APC+ cells are observed (bottom left panel), and nuclei are not pyknotic. Scale bar, 10 μm. B, Quantitative effects of NBQX. Plot on the left summarizes APC+oligodendrocyte counts over time in normoxia (filled squares), 30 min OGD (filled circles), and 30 min OGD with NBQX (open circles). On theright, pyknotic cell counts in the same conditions are shown. Note the increase in pyknotic nuclei after OGD; *p < 0.05 compared with OGD at matched time points (two-way ANOVA and Dunnett's post test).
Fig. 6.
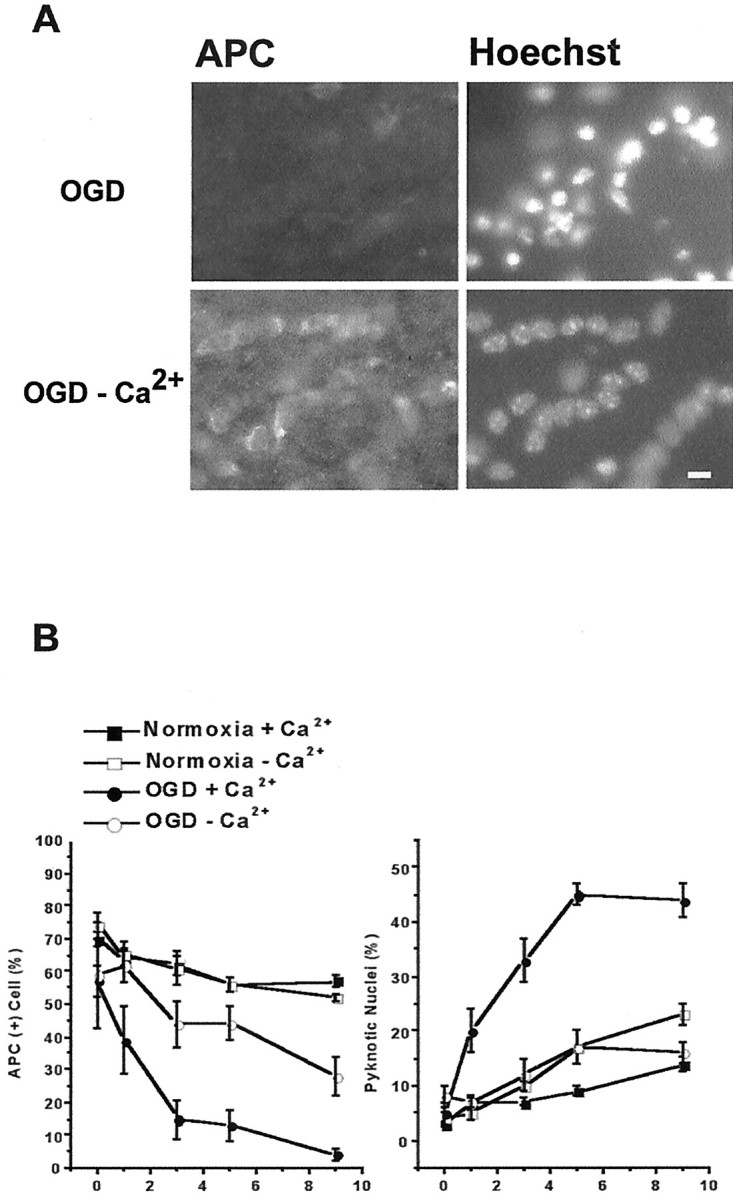
OGD-induced oligodendrocyte death depends on extracellular Ca2+. Slices are exposed to 30 min OGD in normal aCSF (containing 2.0 mm Ca2+) or aCSF with no Ca2+ including 200 μmEGTA. Perfusion conditions are maintained for 30 min before and during and 30 min after OGD. A, APC immunofluorescence of slices fixed 9 hr after OGD. Exposing slices to OGD in Ca2+-free media reduces oligodendrocyte death (left) and pyknotic nuclei formation (right) compared with OGD with normal Ca2+. Scale bar, 10 μm. B, Oligodendrocyte cell counts in slices from normoxia (filled squares), Ca2+-free normoxia (open squares), OGD (filledcircles), and Ca2+-free OGD (open circles) are summarized in the plot on the left. Ca2+-free aCSF in normoxic conditions does not alter oligodendrocyte numbers. OGD in Ca2+-free aCSF remarkably reduces OGD-induced oligodendrocyte death. B,Right, Pyknotic nuclei number in slices exposed to Ca2+-free OGD is significantly less than in slices exposed to OGD. Values represent mean ± SEM.
Fig. 7.

Blockade of voltage-gated sodium channels does not prevent OGD-induced oligodendrocyte death. Slices are exposed to 10 μm TTX or control aCSF for 30 min before and during and 30 min after OGD. A, APC immunofluorescence of slices fixed 9 hr after OGD. TTX does not reduce oligodendrocyte loss or formation of pyknotic nuclei. Scale bar, 10 μm. B, Plots summarize the number of APC+ cells (left) and pyknotic nuclei (right). TTX does not change counts significantly at any time point, as in Figure6.
Propidium iodide. To verify that pyknotic nuclei labeled with Hoechst 33258 represented dead cells, we performed additional experiments with propidium iodide (PI) labeling. Slices were grouped in individual chambers as wash control, OGD for 360 min (to determine maximal cellular death), OGD 30 min, and OGD 30 min with 30 μm NBQX or 30 μm GYKI 52466 (Research Biochemicals; 10 mm stock dissolved in 0.1N HCl) and were placed in a water bath at 33 ± 1°C bubbled with 95% O2/5% CO2. Slices in all groups were allowed 1 hr to stabilize in their chambers. Slices in OGD and OGD with NBQX or GYKI 52466 groups were transferred to respective chambers containing glucose-free aCSF saturated with 95% argon/5% CO2 for 30 min and then returned to chambers for another 6 hr with regular aCSF and 95% O2/5% CO2. All groups contained 10 μmMK-801 to minimize the possible effects of NMDA receptor activation acting indirectly on brain slice gray matter. At 30 min before the end of the experiment, 0.25 μg/ml PI was added into each condition. The corpus callosum of each slice was imaged by using a fluorescence microscope and digital camera at constant exposure times (adjusted to OGD 360 min conditions) at 40×. The sequence of sampling of slices was kept constant. Images were transferred to an image analysis program (MetaMorph, Universal Imaging, West Chester, PA), and the average pixel intensity was measured within the outlined corpus callosum area. Cell deaths in OGD versus OGD with NBQX or GYKI 52466 conditions were expressed and compared as a percentage of maximum fluorescence (OGD 360 min) after wash control levels were subtracted from each condition (see Fig. 5). Although care was taken to compare slices with similar anatomy, we also monitored other parameters such as total corpus callosum area that was outlined to assure that the results were not affected by differences among slices.
Fig. 5.
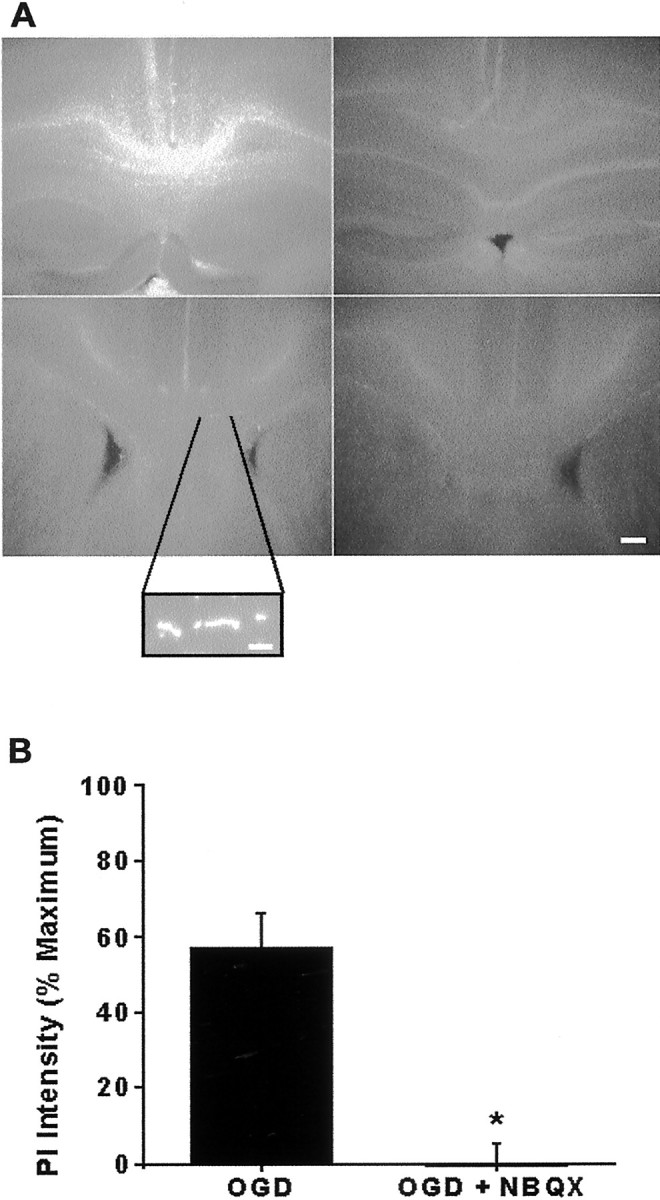
Propidium iodide (PI) detection of OGD-induced cell death. A, Low-magnification (4× objective) images show PI labeling in the corpus callosum of slices exposed to 360 min OGD (maximal injury) or to wash control for 360 min, 30 min OGD followed by 6 hr normoxia, or 30 min OGD with 30 μm NBQX followed by 6 hr normoxia. PI (0.25 μg/ml) was added to the bath 1 hr before imaging. PI intensity was highest in slices from 360 min OGD and lowest in control and in 30 min OGD with NBQX. Micrographs also include neighboring cortex, hippocampus, and ventricles. Scale bar, 50 μm.Inset, High magnification showing PI-positive oligodendrocyte nuclei. Scale bar, 10 μm. B, Quantification of PI intensity. The 30 min OGD results in 56% increase in PI intensity, which is fully preventable by 30 μmNBQX. Values represent mean fluorescence intensity ± SEM of the white matter region of interest, normalized by subtracting wash control and dividing by 360 min OGD values (=100%). *p < 0.05 (Student's t test).
Data analysis
Corpus callosum function was monitored quantitatively as the area under the supramaximal CAP. The area under the CAP represents the best measure of the number of active axons because activities of individual axons within a fiber tract are considered to sum linearly (Cummins et al., 1979). The areas under the CAPs from each slice were normalized to baseline control levels. Slices in the same group were pooled together, and the percentage of recovery was calculated in the normalized group time course. In slices treated with Ca2+-free media, the percentage of recovery was calculated by taking the Ca2+-free responses as the baseline. Recovery of the CAP was defined as a return to >70% of the initial value.
For morphological evaluation, representative microscopic fields in the corpus callosum of each brain section (see Fig. 2A,top panel) were imaged by 60× objective lens with constant exposure times. Exposure times were calculated in sections obtained from perfusion-fixed brain, which was labeled simultaneously with the experimental sections. Scoring for cell counts and nuclear morphology was performed with MetaMorph, using a predetermined grid (5 × 5). Of 25 fields, immunoreactive cells in every other field were counted only if their nuclei were visible with Hoechst 33258 staining in the specific section. Cell numbers were expressed as the percentage of immunoreactive cells to the total number of nuclei present. In each experimental condition the total number of nuclei also was compared to ensure that differences observed between the groups were not attributable to a change in the number of cells. Hoechst 33258-stained pyknotic nuclei were counted in the same fields as immunoreactive cells.
Fig. 2.
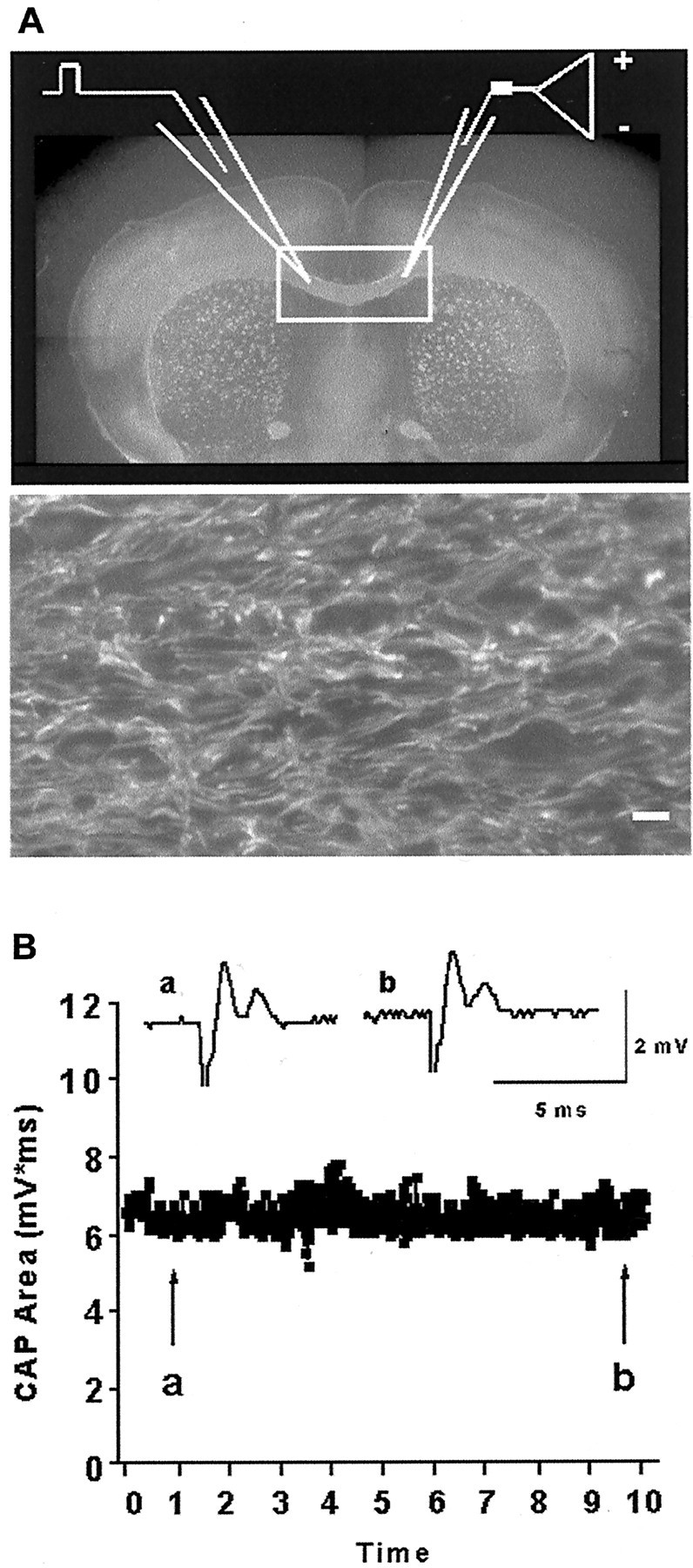
Axonal structural and functional assessment in brain slices. A, Placement of recording (right) and stimulation (left) electrodes across the corpus callosum (rectangle) in a perfusion-fixed slice labeled with SMI-31 for neurofilaments.Bottom panel shows higher magnification (60×) of SMI-31-labeled axons from same region. Note parallel linear structure of axons. Scale bar: Top, 125 μm;bottom, 10 μm. B, Compound action potential (CAP) area measurements remain stable over time. Insets are sample traces chosen at the beginning (a) and at the end (b) of a 10 hr recording period.
Axonal injury evaluation was based on a composite axon score for SMI-31 staining (Table 1). Tissue structure was described by a number between 0 and 12 after one to three positive sign(s) were assigned for each of the following parameters: brightness of labeling intensity, preservation of anatomically parallel and linear organization of axonal fibers, continuity in individual fibers, and lack of axonal head and bulb formation (Adams et al., 1983). Each section was scored independently by two observers. SMI-31 labeling intensity was quantified further in 600× images by measurement of the average pixel intensity within a defined region of the corpus callosum. Values were normalized to the average intensity of wash control slices from the same animals.
Table 1.
Comparison of axonal function and structure at 9 hr after OGD
| Condition | SMI-31 labeling | CAP recovery | |||||
|---|---|---|---|---|---|---|---|
| Pixel intensity (%) | Brightness | Individual fibers | Linearity | Lack of axonal head/bulbs | Score (n) | Percentage (n) | |
| Perfusion-fixed | 100 ± 0 | +++ | +++ | +++ | +++ | 12 ± 0 (11)1-a | NA |
| Normoxia | 88 ± 1 | +++ | ++ | ++ | +++ | 10 ± 1 (9)1-a | 97 ± 1 (6)1-a |
| OGD 15 min | 85 ± 4 | ++ | +++ | +++ | +++ | 11 ± 0 (2)1-a | 97 ± 8 (4)1-a |
| OGD 30 min | 38 ± 3 | − | − | − | − | 1 ± 1 (6)1-b | 0 (10)1-b |
| OGD + MK-801 | 40 ± 2 | − | − | − | − | 1 ± 1 (6)1-b | 0 (8)1-b |
| OGD + NBQX | 81 ± 4 | ++ | ++ | ++ | +++ | 9 ± 1 (6)1-a | 81 ± 7 (6)1-a,1-b |
| OGD − Ca2+ | 76 ± 3 | +++ | ++ | ++ | +++ | 9 ± 1 (5)1-a | 112 ± 23 (6)1-a |
| OGD + TTX | 56 ± 4 | + | + | + | + | 5 ± 1 (7)1-a,1-b | 49 ± 2 (6)1-a,1-b |
SMI-31 neurofilament immunofluorescence in corpus callosum was measured for average fluorescence intensity and axon score calculated as described in Materials and Methods. CAPs were normalized to baseline values. CAP recovery was measured at 9 hrs, except OGD 15 min and OGD + TTX, which were followed for 5 hrs. *
Significantly different from OGD 30 min with MK-801.
Significantly different from normoxia control (one-way ANOVA and Dunnett's post test). NA, Not available.
Statistical analysis
All data are expressed as means ± SEM. Statistical differences were assessed by Student's t test, one-way or two-way ANOVA, followed by the Dunnett's post test where appropriate to detect the significantly different groups. Statistical significance level was set at p < 0.05 and was denoted by an asterisk. Reported n values represent the number of slices in electrophysiological experiments (only the first slice of the day). In immunohistochemical analysis two to three brain sections were quantified for each experiment. The reported n values refer to the experiments that were analyzed.
RESULTS
Brain slices as a model for OGD-induced white matter injury
We tested whether brain slices are a reliable and reproducible model to study white matter injury on the basis of immunohistochemical properties. First we identified various glial cell types by immunofluorescence. Oligodendrocytes were identified as APC+ or CNPase+ cells with oval cell bodies. Numerous APC+ cells were aligned linearly along the fiber axis of the corpus callosum, meeting the description of intrafascicular oligodendrocytes (Szuchet, 1995) (Fig.1A, green). All APC+ cells also labeled with antibody to CNPase (data not shown). GFAP+astrocytes had round cell bodies with many processes extending along the myelinated axons (Fig. 1A, red). NG2+ cells (adult oligodendrocyte progenitor cells) were located between tightly packed myelinated axons and had elongated cell bodies with processes emanating predominantly from the poles of the cells. NG2+ cells were more abundant in cortical gray matter than in white matter; the proportion of NG2+ cells was ∼6% in the corpus callosum (data not shown). Spindle-shaped 711+ microglia cell counts did not exceed 5% of the cells in the corpus callosum. Neither NG2 nor 711 immunofluorescence overlapped with APC+ or CNPase+ cells (data not shown).
Fig. 1.

Immunohistochemical localization of white matter components in brain slices. A, Fluorescence photomicrograph shows oligodendrocytes (APC;green) astrocytes (GFAP;red), and cell nuclei (Hoechst;blue) in the corpus callosum. Intrafascicular oval oligodendrocyte cell bodies and stellate astrocytes are recognized easily and do not overlap. Scale bar, 10 μm. B, Glial cell counts in the corpus callosum are stable over 12 hr in the acute brain slice preparation. Histograms summarize the APC+ (dark gray) and GFAP+ (gray) cell counts in slices from perfusion-fixed, immediately fixed, and normoxic (12 hr) groups. Values represent the mean proportion ± SEM of Hoechst-positive nuclei that express the cell-specific marker. *Significantly different (p < 0.05) from perfusion-fixed conditions. **Significantly different from perfusion-fixed but not immediately fixed conditions.
The consistency of APC+ oligodendrocyte and GFAP+ astrocyte counts in perfusion-fixed and immediately fixed slices was compared with slices treated under normoxic wash conditions. Normoxic controls were obtained by superfusing the slices with oxygenated aCSF at 33 ± 1°C, with a flow rate of 3–3.5 ml/min for up to 12 hr after slicing (corresponding to 9 hr reperfusion time in experimental conditions). Cell nuclei were detected as blue by Hoechst fluorescence (Fig.1A). Histograms in Figure 1Bsummarize the quantification of specific cell types when slices were prepared from perfusion-fixed brains (perfusion) or fixed immediately after slicing (immediate) or perfused under normoxic conditions for 12 hr at 33 ± 1°C (12 hr). There was no difference in the proportion of APC+ cells (dark gray bars) among the slices from the perfusion-fixed (62 ± 2,n = 11), immediately fixed (62 ± 3,n = 6), and 12 hr groups (57 ± 2,n = 9). Slicing caused a small decrease in GFAP+ astrocyte counts (gray bars) (28 ± 2, n = 11 vs 17 ± 2,n = 6; p < 0.05) as seen in immediately fixed and 12 hr groups.
To monitor the functional and structural integrity of axons, we recorded evoked CAPs across the corpus callosum (for the recording arrangement, see Fig.2A), and the axons were labeled with SMI-31 (Fig. 2A, top andbottom). The brightness of SMI-31 labeling and the number of individual linear axons that could be seen were evaluated and quantified in every experimental condition (Table 1). Recording CAPs showed that axonal electrical responses remained stable in slices up to 10 hr in normoxic conditions (Fig. 2B).
The 30 min OGD causes irreversible axonal injury
To determine the effects of OGD on axonal conduction, we monitored CAPs in individual brain slices kept at the interface of the slice chamber. After a stable baseline of CAPs was obtained, OGD was induced by switching to glucose-free aCSF saturated with 95% N2/5% CO2. In slices exposed to 15 min OGD (Fig.3A, filled squares), CAP area during OGD disappeared completely in three slices and was reduced by 68% in one slice. Returning to glucose-containing oxygenated aCSF resulted in complete recovery of CAP area within 10 min, and this remained stable for up to 9 hr after the end of OGD (97 ± 8% of control; n = 4). In slices exposed to 30 min OGD (Fig. 3A, open circles), the time to complete block of CAPs was 19 ± 1 min (n = 6). Switching to oxygen–glucose-containing media caused an initial maximum recovery by 45 ± 4% of control within 10 min after the end of OGD. By 1 hr after OGD the CAP area recovery was 22 ± 8% of control and decreased to 17 ± 3% by 2 hr after OGD. Axonal conduction irreversibly vanished in all slices well before 3 hr in this group.
Fig. 3.
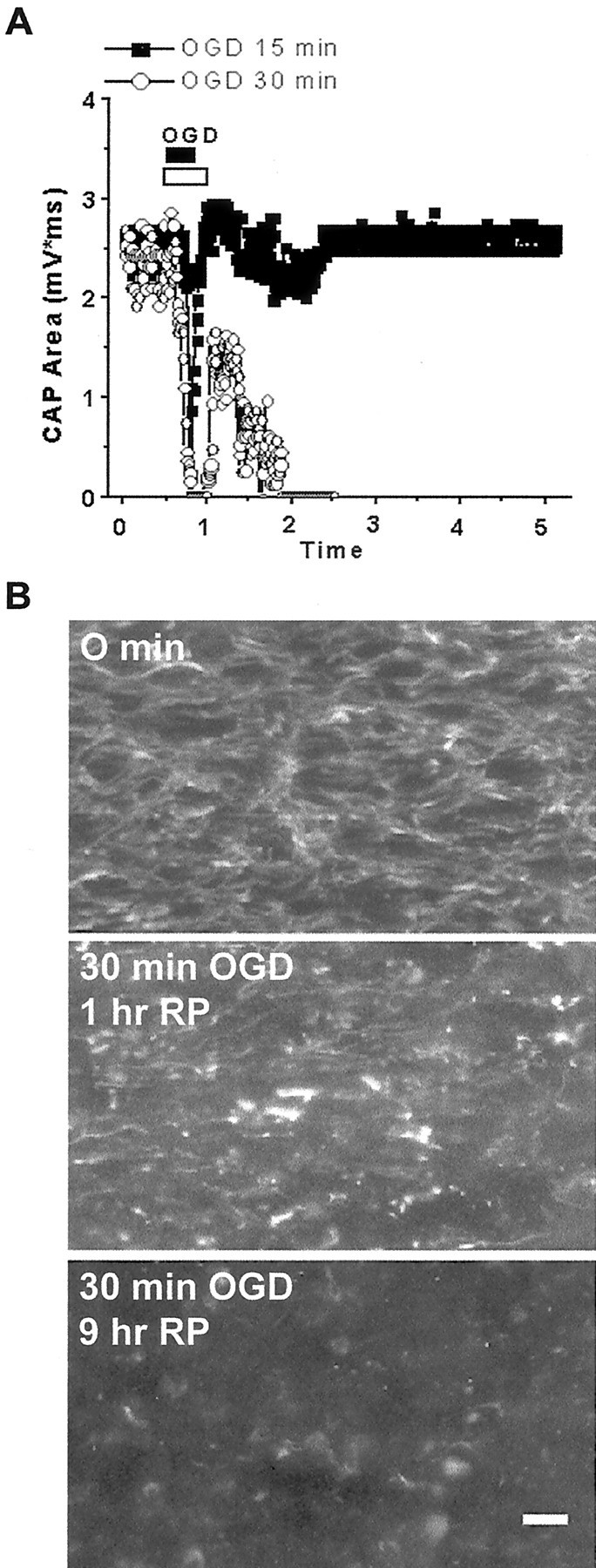
The 30 min OGD causes loss of axonal function and structural injury. A, CAP recovers completely after 15 min OGD (filledsquares), but not after 30 min OGD (open circles). B, Immunofluorescence micrograph shows SMI-31 labeling of axons in control (top), 1 hr after 30 min OGD (1 hr RP;middle), and 9 hr after OGD (9 hr RP;bottom). Note linear fibers in control, formation of axonal heads and retraction bulbs 1 hr after OGD, and almost complete loss of SMI-31 labeling 9 hr after OGD. Scale bar, 10 μm.
On determining that 30 min OGD caused irreversible conduction failure along the corpus callosum, we investigated whether functional axonal impairment also was associated with cytoskeletal changes in the axonal structure. The bright intensity of neurofilament labeling seen in 0 min slice (Fig. 3B, top panel) was replaced by very bright spots of axonal head and retraction ball formation in slices imaged 1 hr after OGD. Few individual linear axons with dim staining were observed in this group of slices (Fig. 3B,middle panel). SMI-31 labeling was completely lost (Fig. 3B, bottom panel) and was scored 0 in slices kept 9 hr after 30 min OGD (Table 1). Loss of SMI-31 labeling was associated with axonal structural disruption in semi-thin histological sections (S. Baltan Tekkök, B. T. Faddis, M. P. Goldberg, unpublished data).
We evaluated the nuclear morphology and number of oligodendrocytes under normoxic conditions. There was no significant change in oligodendrocyte nuclear morphology or counts over 12 hr in vitro (Fig. 4A). Uninjured oligodendrocyte cell nuclei labeled with Hoechst were observed as dimly fluorescent oval cell bodies with clear cytoplasm containing three to five nuclear inclusions (Fig. 4A,top panel). Exposing slices to 30 min OGD resulted in a loss of APC+ cells and a gain in the appearance of pyknotic nuclei with brighter Hoechst fluorescence (Fig.4A, middle panel). Some of the nuclei had a fragmented appearance (e.g., Figs. 4, 7). We did not perform experiments to establish apoptotic or necrotic mechanisms of injury.
The effects of glutamate receptor antagonists on OGD-induced oligodendrocyte death
Next we investigated whether OGD-induced oligodendrocyte injury in the corpus callosum of brain slices involved AMPA/KA receptor activation. Blockade of AMPA/KA receptor with 30 μm NBQX fully prevented the loss of APC+ cells and the appearance of pyknotic nuclei (Fig. 4A,bottom panel). The counts of APC+ cells in slices kept normoxic (filled squares), in slices exposed to OGD (filled circles), and in slices treated with NBQX (open circles) before exposure to 30 min OGD were compared (Fig. 4B, left). The number of APC+ oligodendrocytes remained stable over time in slices kept in normoxic conditions (n = 7). The number of pyknotic nuclei (Fig. 4B, right) in this group did not exceed 14 ± 1% by 9 hr after OGD (n = 7). OGD resulted in rapid reduction in the APC+ cell counts starting from 1 hr after OGD (67 ± 6% at 0 hr vs 29 ± 4%; p < 0.05), reaching a minimum of 8 ± 3% by 9 hr after OGD (n = 6). In parallel, pyknotic nuclei number increased from 5 ± 1 to 44 ± 4% (n = 6;p < 0.05). Treatment with NBQX fully preserved APC+ cell counts (64 ± 4% by 9 hr RP; n = 6; p < 0.05) and kept pyknotic nuclei counts similar to those in control conditions (13 ± 3% by 9 hr RP).
To determine whether OGD-induced oligodendrocyte death was a direct effect of the overactivation of white matter AMPA/KA receptors rather than an indirect effect of gray matter, we performed experiments by using isolated corpus callosum slices (Berger et al., 1991). Slices were prepared as described in Materials and Methods, except that the angle of the section was ∼20° more rostrocaudal than coronal (to increase the cross-sectional area of the corpus callosum). Cortical tissue was removed immediately with a series of four razor cuts, and the remaining tissue was handled as described above. The proportion of APC+ cells (56 ± 2%) and pyknotic nuclei (3 ± 1) in isolated corpus callosum slices under normoxic conditions was similar to that previously observed for routinely prepared coronal brain slices. OGD (30 min) resulted in a loss of APC+ cells (18 ± 4%) and a gain in pyknotic nuclei number (38 ± 2). Blockade of AMPA/KA receptors preserved oligodendrocyte numbers (52 ± 1%; p < 0.05) and prevented the formation of pyknotic nuclei (11 ± 1;p < 0.05). These results in isolated corpus callosum slices suggest that OGD-induced oligodendrocyte death is mediated primarily by the overactivation of AMPA/KA receptors in the white matter.
Assessment of OGD-induced cell death with propidium iodide
The loss of APC+ immunoreactivity suggests oligodendrocyte damage, but not necessarily cell death. We confirmed oligodendrocyte death, as defined by a loss of cell membrane integrity, by PI uptake of white matter cells. PI labeling in the corpus callosum of representative slices was assessed after maximally toxic OGD exposure (360 min), wash control, 30 min OGD, and 30 min OGD plus NBQX (Fig. 5A). Slices from the OGD 360 min group demonstrated the highest PI intensity. Slices in the OGD 30 min group had significantly higher fluorescence intensity than wash control slices, with PI staining intensity 57 ± 9% of OGD 360 min. Slices treated with NBQX were similar to control. Similar results also were obtained when the slices were pretreated with 30 μm GYKI 52466 before OGD, suggesting that AMPA rather than kainate receptors contributed to OGD-induced oligodendrocyte death.
Ionic dependence of OGD-induced oligodendrocyte death
Because increased intracellular Ca2+is a critical step of the excitotoxic injury cascade, we investigated the role of Ca2+ in OGD-induced AMPA/KA receptor-mediated oligodendrocyte death in slices. Exposing slices in Ca2+-free media (containing 200 μm EGTA) substantially reduced OGD-induced loss of APC+ oligodendrocytes at every time point (Fig. 6). In parallel, fewer pyknotic nuclei were observed in slices exposed to OGD in Ca2+-free aCSF (Fig. 6B,right, 44 ± 3 with Ca2+vs 16 ± 2% without Ca2+;p < 0.05). Keeping slices in Ca2+-free media under normoxic conditions (Fig. 6B, open squares) did not alter oligodendrocyte counts or nuclear morphology.
We next examined the role of VGSCs on oligodendrocyte viability. The addition of 10 μm TTX (30 min before and during and 30 min after OGD) did not prevent a loss of APC+ cells or an increase in pyknotic nuclei (Fig. 7A,B).
Ionic dependence of OGD-induced axonal injury
We examined the dependence of OGD-induced axon injury on extracellular Ca2+ by exposing slices to OGD in Ca2+-free media (including 200 μm EGTA). The application of Ca2+-free aCSF caused an initial increase in the CAP area by 40 ± 22% (n = 6). This may be attributable to a reduction in extracellular Ca+2-dependent surface potential screening (Hille, 1992) (Fig.8A). Switching to glucose-free and Ca2+-free aCSF saturated with 95% N2/5% O2 reduced the CAP area to 1 ± 1% in ∼29 min. Once regular aCSF and oxygen were resumed, the responses recovered to baseline and showed slight potentiation such that, 1 hr after OGD, CAP area was 127 ± 24% of Ca2+-free control and 112 ± 23% by 9 hr after OGD (p > 0.05). Superfusion with Ca2+-free aCSF during OGD resulted in good preservation of axonal structure (Fig. 8B,bottom). The SMI-31 labeling score of axons in Ca2+-free media was 9 ± 1 (see Table1), significantly different from the score in Ca2+-containing media (Fig.8B, top).
Fig. 8.
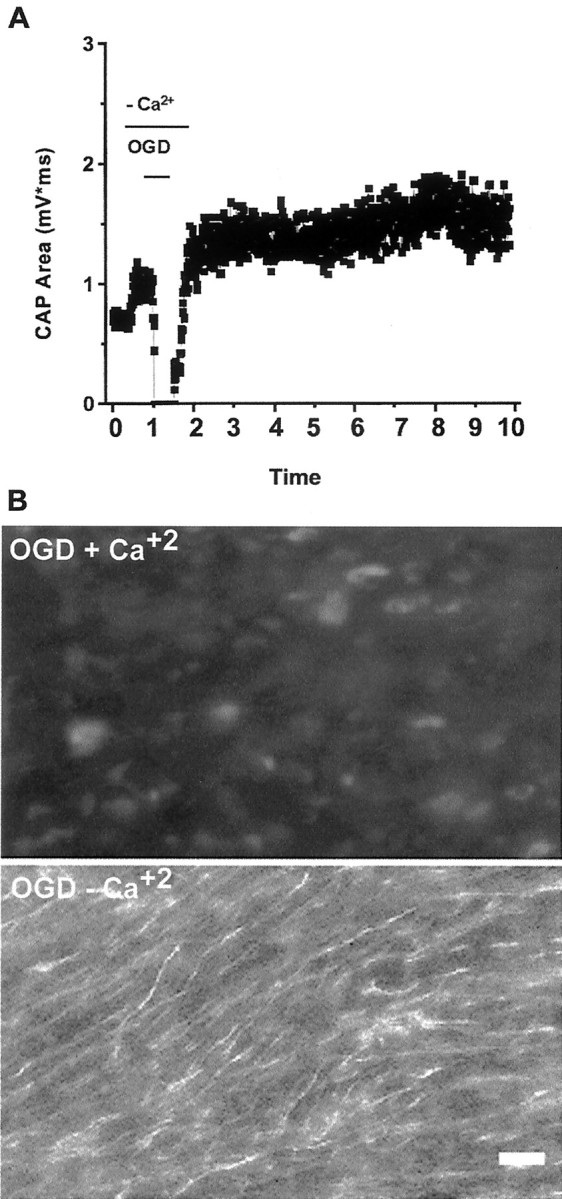
Ca2+ dependence of OGD-induced axonal injury. Slices are exposed to Ca2+-free or control aCSF as described in Figure 6. A, Pretreatment of slices with Ca2+-free aCSF preserves stimulus-evoked CAPs. B, SMI-31 immunofluorescence of slices 9 hr after 30 min OGD with (top) or without (bottom) Ca2+. Removal of Ca2+ results in preservation of axonal function (compare with Fig. 3) and neurofilament labeling. Scale bar, 10 μm.
The role of voltage-gated Na+ channels in axonal injury
Previous studies have shown that Na+flux through VGSCs plays a major role in impaired axonal conduction during anoxia (Stys et al., 1992a; Agrawal and Fehlings, 1997). We examined the effects of TTX (10 μm) in our model. CAPs were depressed completely with 3–5 min of TTX application (Fig.9, left, open circles). Slices were exposed to OGD (30 min) and were kept in TTX for another 30 min after the end of OGD. After TTX was washed out, the CAP area recovered slowly to 48 ± 1% (n = 6) of control levels and remained stable. When CAPs were recorded in slices (n = 3) treated with 10 μm TTX for identical durations without OGD, the percentage of recovery of CAP area was 61 ± 1%, which was significantly higher than in slices exposed to OGD (Fig. 9, filled squares). In accordance with partial functional preservation, TTX also reduced OGD-induced axonal injury. Although SMI-31 labeling demonstrated the presence of linearly projecting axons (Fig. 9B), overall labeling brightness was dim and a majority of axons lost their continuity, with numerous axonal bulbs and retraction heads. The semi-quantitative injury score was 5 ± 1 (see Table 1).
Fig. 9.
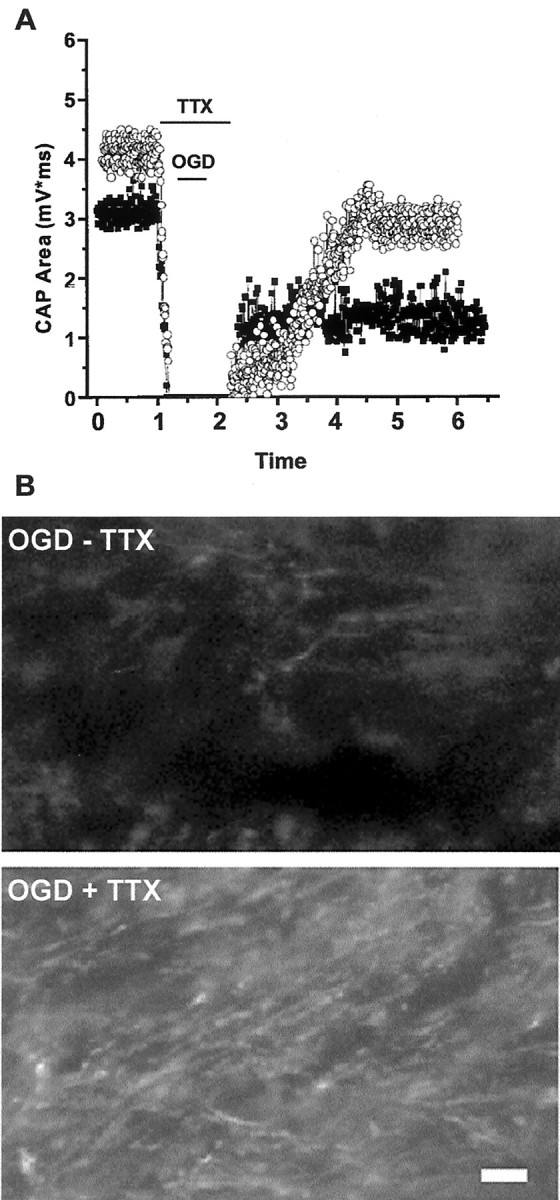
Blockade of voltage-gated sodium channels offers partial protection of axonal function and structure. A, CAP recovery in slices exposed to TTX for 90 min, with normoxic perfusion (open circles) or 30 min OGD (filled squares). TTX exposure alone results in complete loss of CAP, which recovers to ∼61% of initial values 5 hr after removal. CAP recovery in slices exposed to OGD + TTX is better than OGD with no drug (see Fig. 3) but does not reach levels of TTX in normoxia. B, Compared with OGD without TTX (top), SMI-31 labeling is preserved partially in slices exposed to OGD + TTX, but some axonal head and retraction bulb formation occurs (bottom).
The role of glutamate receptor antagonist in OGD-induced axonal injury
To evaluate the contribution of glutamate receptor activation in axonal injury, we recorded CAPs in slices pretreated with MK-801 (10 μm) or MK-801 and NBQX (30 μm) for 30 min before OGD. Blockade of NMDA receptors with MK-801 (Fig.10, top) partially preserved residual CAP during OGD and delayed the time to irreversible CAP loss after OGD. MK-801 did not prevent irreversible loss of CAPs by the end of the recording period (eight of eight slices). In parallel experiments the axonal structure assessed by SMI-31 immunoreactivity was poor, with a score of 0. Figure 10, top right, shows the loss of SMI-31 labeling in a representative slice from this group. In contrast, a blockade of AMPA/KA receptors with 30 μm NBQX provided full recovery of axonal conduction after OGD. Figure 10, bottom, shows the time course of CAP recording in a typical slice pretreated with NBQX. In the presence of NBQX, OGD suppressed the CAP area to 52 ± 6% (n = 6) of baseline levels, followed by rapid recovery to control values within ∼9 min (96 ± 7%). There were one to two further depression–recovery periods in this group of slices before a stable recording of CAPs was achieved that remained stable up to 9 hr after OGD (81 ± 7%; Fig.11). SMI-31 labeling in similarly treated slices was bright, with many individual linear axons (see Fig.10, bottom) and with a score of 9 ± 1 (see Table1).
Fig. 10.

AMPA/KA receptor blockade prevents OGD-induced loss of axonal function. Slices are exposed to 30 min OGD in control aCSF (containing 10 μm MK-801) or aCSF with the further addition of 30 μm NBQX. Images show SMI-31 labeling 9 hr after OGD. Top, Blockade of NMDA receptors with MK-801 delays the immediate OGD-induced reduction of CAPs but has no effect on CAP recovery beyond 3 hr (left) and does not preserve SMI-31 labeling (right). Bottom, NBQX prevents acute CAP loss and preserves CAP for 6 hr after OGD. Axonal structure is protected substantially. Scale bar, 10 μm.
Fig. 11.

Proposed model for hypoxic–ischemic injury pathways in cerebral white matter. Schematic shows myelinated axon (blue), oligodendrocytes (green), and astrocytes (red).A, Hypoxia, ischemia, or glucose deprivation results in energy depletion and loss of ATP. B, Failure of Na+/K+-ATPase and depolarization leads to opening of noninactivating axonal voltage-gated Na+ channels. Ca2+ enters axons by reversal of Na+/Ca2+ exchange and activation of voltage-gated Ca2+ channels. Action potentials are halted reversibly by loss of ionic gradients.C, Excessive axoplasmic Ca2+ levels trigger destructive pathways, leading to degradation of axonal cytoskeleton and organelles, focal axonal swelling, and eventual interruption of axonal integrity. D, Another effect of energy deprivation is release of glutamate into extracellular space (likely by reversal of Na+-dependent glutamate transport from axons and possibly astrocytes, oligodendrocytes). Glutamate activates ionotropic AMPA/KA receptors on oligodendrocytes (and possibly astrocytes). E, Sustained glutamate receptor activation triggers excitotoxic damage of oligodendrocyte processes (myelin) and subsequent death of oligodendrocytes. Myelin damage might result in conduction delay or block.F, Activation of glial AMPA/KA receptors or oligodendrocyte death triggers further damage to axons under energy-depleted conditions. Mechanisms linking glial glutamate receptors to axonal damage might include release of toxic substances from injured cells, increase in tissue energy use, loss of substrate or trophic support, exposure of protected membrane or ion channels, or failure of glial homeostatic functions. Blockade of stepD by NBQX protects oligodendrocytes and axons, whereas blockade of step B by TTX provides partial protection for axons, but not for oligodendrocytes. Other interactions are not shown. Ion homeostatic failure in axons may be responsible for glutamate release; Na+ removal and a blockade of Na+/Ca2+ exchange likely act on both axons and oligodendrocytes.
There was good agreement between axonal functional preservation and axonal structural integrity (see Table 1). In conditions in which the CAP did not recover, there was derangement of neurofilaments detected as a loss of SMI-31 labeling (McCracken et al., 1999).
DISCUSSION
This study reports delayed effects of oxygen–glucose deprivation in a myelinated central fiber tract. In acutely prepared brain slices from adult mice, OGD caused an irreversible loss of CAPs, followed by a loss of APC-immunoreactive oligodendrocytes and disruption of axonal neurofilaments in the corpus callosum. AMPA/KA receptor blockade and Ca2+ removal preserved oligodendrocytes and axonal structure and function. In contrast, a blockade of VGSCs provided only partial protection for axons and did not reduce oligodendrocyte death. To our knowledge this is the first demonstration that the activation of AMPA/KA receptors in white matter causes the death of mature oligodendrocytes in hypoxic–ischemic brain. Our results also show that AMPA/KA receptors as well as VGSCs can mediate axonal disruption during hypoxic white matter injury.
Brain slices as a model of white matter ischemia
Brain slice preparations are used widely to study neuronal injury (Schurr and Rigor, 1995). They also offer important advantages for the investigation of white matter injury. Slices preserve the anatomical and structural integrity of the mature nervous system. In contrast, dissociated cell cultures exclude many cell–cell interactions, and isolated white matter preparations may lack continuity of the axon with the cell body. Control of the tissue environment enables ionic and pharmacological manipulations. Finally, unlike most cultures, slices can be prepared from adult animals, allowing for the assessment of myelinated axons and fully differentiated oligodendrocytes. In our experiments the coronal brain slices remained structurally and functionally intact as long as 12 hr at ∼34°C, sufficient to assess slowly developing processes such as oligodendrocyte death and axonal structural disintegration. The observed damage to oligodendrocytes and axons in brain slices matches patterns of white matter injury in experimental models (Pantoni et al., 1996; Valeriani et al., 2000).
Excitotoxic oligodendrocyte death
OGD for 30 min resulted in the selective death of ∼90% of oligodendrocytes after 9 hr, accompanied by a parallel increase in pyknotic nuclei counts and propidium iodide cellular uptake (see Figs.4, 5). There was minimal change in GFAP+astrocytes. Oligodendrocyte death was blocked by NBQX, but not by the NMDA antagonist MK-801 (see Fig. 4). The protective effect of NBQX was sustained when experiments were repeated at 37°C. An AMPA receptor-selective blocker, GYKI 52466, was also protective.
AMPA receptor agonists can lead to the injury of oligodendrocytes in culture (Yoshioka et al., 1995, 1996; Garcia-Barcina and Matute, 1998;McDonald et al., 1998), in situ (Yoshioka et al., 1996; Li and Stys, 2000), and in vivo (Dusart et al., 1992; McDonald et al., 1998). AMPA/KA receptor-mediated toxicity contributes to the death of hypoxic oligodendrocytes in vitro (McDonald et al., 1998; Fern and Möller, 2000; Yoshioka et al., 2000). The significance of the cell culture observations might be challenged because of the relatively immature phenotype of cultured oligodendrocytes and the absence of cell–cell interactions. The current experiments provide novel support for an important role of AMPA/KA receptors in hypoxic–ischemic oligodendrocyte death in the mature brain.
TTX did not reduce oligodendrocyte loss, suggesting that Na+ flux through VGSC does not contribute to excitotoxic oligodendrocyte death. This agrees with observations in traumatic spinal cord injury (Rosenberg et al., 1999). Oligodendrocyte death was also dependent on extracellular Ca2+. The routes of toxic Ca2+ entry in oligodendrocytes (Yoshioka et al., 1995) remain to be explored and might include direct permeation through AMPA/KA receptors (Meucci et al., 1996; Underhill et al., 2000), voltage-gated calcium channels (Barres et al., 1988), and reversal of Na+/Ca2+exchange (Baltan Tekkök et al., 2000).
Excitotoxic axonal injury
Anoxia or glucose deprivation in optic nerve and spinal cord white matter interrupts the electrical activity of myelinated axons (Ransom et al., 1990; Stys et al., 1992b; Fern et al., 1998; Li et al., 1999;Li and Stys, 2000) and causes ultrastructural changes in axonal mitochondria and cytoskeleton (Waxman et al., 1992). In our experiments OGD for 30 min (but not for 15 min) caused an irreversible loss of CAPs. Axonal head and bulb formation were visualized by SMI-31 labeling 1 hr after OGD, followed by a loss of SMI-31, SMI-32, and NR4 immunoreactivity. Thus, axonal injury after OGD is similar to the pathology of diffuse axonal injury in humans and animals (Pantoni et al., 1996; Dietrich et al., 1998; Yam et al., 1998; Graham et al., 2000).
In agreement with previous studies (Stys et al., 1990; Waxman et al., 1993), Ca2+ removal preserved CAPs and axonal structure (see Fig. 8). However, our results diverge from previous work (Stys et al., 1992a; Waxman et al., 1994; Agrawal and Fehlings, 1996; Garthwaite et al., 1999) in that we found only partial protection of axonal structure and function when the slices were pretreated with 10 μm TTX. This concentration of TTX was sufficient to cause complete CAP blockade under normoxic conditions (see Fig. 7).
OGD-induced axonal injury was blocked by the addition of NBQX. Previous studies demonstrated that an AMPA/KA blockade preserves electrical conduction in spinal cord tissue exposed to anoxia or compression injury (Agrawal and Fehlings, 1997; Li et al., 1999; Li and Stys, 2000). However, earlier studies did not report axon structural damage or preservation by AMPA/KA antagonists. Possible differences in the current model that might account for these differences include differential susceptibility of brain, as opposed to spinal cord, more severe insult accorded by combined oxygen and glucose deprivation (Garthwaite et al., 1999), or a longer observation period after OGD.
Interactions between oligodendrocytes and axons in hypoxic white matter
Previous studies of optic nerve and spinal cord preparations support a model of axon injury (Stys, 1998), as illustrated in Figure11A–C. Loss of ATP-dependent Na+/K+exchange leads to excessive Na+ influx through noninactivating VGSCs; this is followed by axoplasmic Ca2+ accumulation that is mediated by reverse Na+/Ca2+exchange and other Ca2+ channels. Disruption of axonal ion homeostasis may lead to acute conduction failure, whereas intra-axonal Ca2+accumulation may trigger subsequent cascades leading to the destruction of axonal structural elements. Although the present data support a role for VGSCs and extracellular Ca2+, our results are not explained fully by this model. TTX provided only a partial reduction of axonal injury and did not block oligodendrocyte death. Furthermore, a blockade of AMPA/KA receptors preserved both oligodendrocytes and axons, suggesting an important alternative pathway for white matter injury (see Fig. 11D–F).
Excitotoxic death of oligodendrocytes might be predicted from their vulnerability in vitro. However, the structural preservation of axons by NBQX is surprising. How do AMPA/KA antagonists act in this setting? NBQX preserves white matter histology and ameliorates behavioral deficits after spinal contusion (Wrathall et al., 1994) and spinal ischemia (Kanellopoulos et al., 2000). These studies demonstrate a role of AMPA/KA receptor activation in spinal cord injury but cannot establish whether receptor blockade occurs in the gray or white matter. Glutamate receptor activation in neuronal somata of gray matter might enhance axon and oligodendrocyte injury by acting indirectly (Schabitz et al., 2000). Mechanisms could include diffusion of toxic substances such as glutamate or free radicals, modulation of axonal activity and energy requirements, retrograde axon degeneration, or activation of inflammatory responses (Matute, 1998; Pitt et al., 2000; Smith et al., 2000). It does not appear that the primary effect of NBQX in our experiments was on neuronal cell bodies. NBQX did not reduce neuronal damage after OGD (data not shown) and was protective even in isolated corpus callosum slices. Therefore, NBQX protected white matter oligodendrocytes and axons in these experiments even in the absence of neighboring gray matter or circulating inflammatory cells.
The cellular targets of excitotoxic damage within the white matter remain to be established. Astrocytes express functional non-NMDA receptors but are not directly susceptible to excitotoxic death [in part because of rapid AMPA receptor desensitization (David et al., 1996)]. Activation of astrocyte AMPA receptors could contribute to white matter injury by disrupting ionic regulation. In contrast to astrocytes, the activation of oligodendrocyte AMPA/KA receptors leads directly to excitotoxicity. An initial consequence of AMPA/KA receptor activation may be localized myelin damage (Li and Stys, 2000), leading to action potential slowing or even conduction block. Further damage may cause the death of oligodendrocyte cell bodies and myelin loss (see Fig. 11). Although oligodendrocytes have the potential to regenerate (at least in some settings), axon loss is likely to be irreversible.
Our data suggest that the overactivation of glutamate receptors in white matter contributes to axonal injury. Is glutamate directly toxic to axons? Immunocytochemical studies found glutamate receptor subunits within axon cylinders (Li and Stys, 2000), but there is no evidence that functional receptors are expressed at the axolemma or can participate in axonal injury. Intracerebral administration of excitatory amino acid analogs is characteristically “axon-sparing” (Simson et al., 1977). Likewise, AMPA or kainate application does not cause disruption of axons in cortical culture (Hasbani et al., 1998) or spinal cord slice (Li and Stys, 2000). Therefore, AMPA/KA receptor-dependent axon injury more likely occurs via glial receptors and may require simultaneous energy depletion (see Fig. 11).
Glutamate receptor-mediated injury may contribute to damage in a spectrum of clinical conditions in which white matter is the target, including stroke, cerebral and spinal trauma, periventricular leukoencephalopathy, and multiple sclerosis. Our results suggest that excitotoxic compromise of white matter glia adversely affects structure and function of axon fibers as well.
Footnotes
This work was supported by National Institutes of Health Grants NS36265 and NS32636, by the Juvenile Diabetes Foundation, and by a postdoctoral fellowship from the American Heart Association (S.B.T.). We thank Olga Strots for technical assistance, Kelvin A. Yamada and Steven Rothman for the use of electrophysiology equipment, and John Choi for early development of this project. Laura L. Dugan, Gabriel De Erausquin, and Steven Rothman provided helpful critiques.
Correspondence should be addressed to Dr. Mark P. Goldberg, Department of Neurology, Washington University, Campus Box 8111, 660 South Euclid, St. Louis, MO 63110-1193. E-mail: goldberg@neuro.wustl.edu.
REFERENCES
- 1.Adams JH, Graham DI, Gennarelli TA. Head injury in man and experimental animals: neuropathology. Acta Neurochir Suppl (Wien) 1983;32:15–30. doi: 10.1007/978-3-7091-4147-2_2. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na+-K+-ATPase, the Na+–H+ exchanger, and the Na+–Ca2+ exchanger. J Neurosci. 1996;16:545–552. doi: 10.1523/JNEUROSCI.16-02-00545.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agrawal SK, Fehlings MG. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci. 1997;17:1055–1063. doi: 10.1523/JNEUROSCI.17-03-01055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- 5.Baltan Tekkök S, Hyrc KL, Underhill SM, Goldberg MP. Na+/Ca2+ exchange blocker KB-R7943 protects axons and oligodendrocytes during oxygen glucose deprivation. Soc Neurosci Abstr. 2000;26:2065. [Google Scholar]
- 6.Barres BA, Chun LL, Corey DP. Ion channel expression by white matter glia. I. Type 2 astrocytes and oligodendrocytes. Glia. 1988;1:10–30. doi: 10.1002/glia.440010104. [DOI] [PubMed] [Google Scholar]
- 7.Berger T, Schnitzer J, Kettenmann H. Developmental changes in the membrane current pattern, K+ buffer capacity, and morphology of glial cells in the corpus callosum slice. J Neurosci. 1991;11:3008–3024. doi: 10.1523/JNEUROSCI.11-10-03008.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 9.Cummins KL, Perkel DH, Dorfman LJ. Nerve fiber conduction velocity distributions. I. Estimation based on the single-fiber and compound action potentials. Electroencephalogr Clin Neurophysiol. 1979;46:634–646. doi: 10.1016/0013-4694(79)90101-9. [DOI] [PubMed] [Google Scholar]
- 10.David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debus E, Weber K, Osborn M. Monoclonal antibodies specific for glial fibrillary acidic (GFA) protein and for each of the neurofilament triplet polypeptides. Differentiation. 1983;25:193–203. doi: 10.1111/j.1432-0436.1984.tb01355.x. [DOI] [PubMed] [Google Scholar]
- 12.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietrich WD, Kraydieh S, Prado R, Stagliano NE. White matter alterations following thromboembolic stroke: a beta-amyloid precursor protein immunocytochemical study in rats. Acta Neuropathol (Berl) 1998;95:524–531. doi: 10.1007/s004010050833. [DOI] [PubMed] [Google Scholar]
- 14.Dusart I, Marty S, Peschanski M. Demyelination, and remyelination by Schwann cells and oligodendrocytes after kainate-induced neuronal depletion in the central nervous system. Neuroscience. 1992;51:137–148. doi: 10.1016/0306-4522(92)90478-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fern R, Möller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fern R, Davis P, Waxman SG, Ransom BR. Axon conduction and survival in CNS white matter during energy deprivation: a developmental study. J Neurophysiol. 1998;79:95–105. doi: 10.1152/jn.1998.79.1.95. [DOI] [PubMed] [Google Scholar]
- 17.Gallo V, Patneau DK, Mayer ML, Vaccarino FM. Excitatory amino acid receptors in glial progenitor cells: molecular and functional properties. Glia. 1994;11:94–101. doi: 10.1002/glia.440110204. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Barcina JM, Matute C. AMPA-selective glutamate receptor subunits in glial cells of the adult bovine white matter. Brain Res Mol Brain Res. 1998;53:270–276. doi: 10.1016/s0169-328x(97)00318-5. [DOI] [PubMed] [Google Scholar]
- 19.Garthwaite G, Brown G, Batchelor AM, Goodwin DA, Garthwaite J. Mechanisms of ischaemic damage to central white matter axons: a quantitative histological analysis using rat optic nerve. Neuroscience. 1999;94:1219–1230. doi: 10.1016/s0306-4522(99)00389-9. [DOI] [PubMed] [Google Scholar]
- 20.Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma. J Neuropathol Exp Neurol. 2000;59:641–651. doi: 10.1093/jnen/59.8.641. [DOI] [PubMed] [Google Scholar]
- 21.Hasbani MJ, Hyrc KL, Faddis BT, Romano C, Goldberg MP. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Exp Neurol. 1998;154:241–258. doi: 10.1006/exnr.1998.6929. [DOI] [PubMed] [Google Scholar]
- 22.Hille B. Ionic channels of excitable membranes. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 23.Kanellopoulos GK, Xu XM, Hsu CY, Lu X, Sundt TM, Kouchoukos NT. White matter injury in spinal cord ischemia: protection by AMPA/kainate glutamate receptor antagonism. Stroke. 2000;31:1945–1952. doi: 10.1161/01.str.31.8.1945. [DOI] [PubMed] [Google Scholar]
- 24.Kirov SA, Sorra KE, Harris KM. Slices have more synapses than perfusion-fixed hippocampus from both young and mature rats. J Neurosci. 1999;19:2876–2886. doi: 10.1523/JNEUROSCI.19-08-02876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine JM, Nishiyama A. The NG2 chondroitin sulfate proteoglycan: a multifunctional proteoglycan associated with immature cells. Perspect Dev Neurobiol. 1996;3:245–259. [PubMed] [Google Scholar]
- 26.Li S, Stys PK. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neurosci. 2000;20:1190–1198. doi: 10.1523/JNEUROSCI.20-03-01190.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci. 1999;19:RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matute C. Characteristics of acute and chronic kainate excitotoxic damage to the optic nerve. Proc Natl Acad Sci USA. 1998;95:10229–10234. doi: 10.1073/pnas.95.17.10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCracken E, Hunter AJ, Patel S, Graham DI, Dewar D. Calpain activation and cytoskeletal protein breakdown in the corpus callosum of head-injured patients. J Neurotrauma. 1999;16:749–761. doi: 10.1089/neu.1999.16.749. [DOI] [PubMed] [Google Scholar]
- 30.McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- 31.Meucci O, Fatatis A, Holzwarth JA, Miller RJ. Developmental regulation of the toxin sensitivity of Ca2+-permeable AMPA receptors in cortical glia. J Neurosci. 1996;16:519–530. doi: 10.1523/JNEUROSCI.16-02-00519.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27:1641–1646. doi: 10.1161/01.str.27.9.1641. [DOI] [PubMed] [Google Scholar]
- 34.Patneau DK, Wright PW, Winters C, Mayer ML, Gallo V. Glial cells of the oligodendrocyte lineage express both kainate- and AMPA-preferring subtypes of glutamate receptor. Neuron. 1994;12:357–371. doi: 10.1016/0896-6273(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 35.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 36.Ransom BR, Stys PK, Waxman SG. The pathophysiology of anoxic injury in central nervous system white matter. Stroke. 1990;21[Suppl 11]:11152–11157. [PubMed] [Google Scholar]
- 37.Rosen H, Gordon S. The role of the type 3 complement receptor in the induced recruitment of myelomonocytic cells to inflammatory sites in mouse. Am J Respir Cell Mol Biol. 1990;3:3–10. doi: 10.1165/ajrcmb/3.1.3. [DOI] [PubMed] [Google Scholar]
- 38.Rosenberg LJ, Teng YD, Wrathall JR. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline reduces glial loss and acute white matter pathology after experimental spinal cord contusion. J Neurosci. 1999;19:464–475. doi: 10.1523/JNEUROSCI.19-01-00464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schabitz WR, Li F, Fisher M. The N-methyl-d-aspartate antagonist CNS 1102 protects cerebral gray and white matter from ischemic injury following temporary focal ischemia in rats. Stroke. 2000;31:1709–1714. doi: 10.1161/01.str.31.7.1709. [DOI] [PubMed] [Google Scholar]
- 40.Schurr A, Rigor BM. Brain slices in basic and clinical research. CRC; Boca Raton, FL: 1995. [Google Scholar]
- 41.Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- 42.Simson EL, Gold RM, Standish LJ, Pellett PL. Axon-sparing brain lesioning technique: the use of monosodium-l-glutamate and other amino acids. Science. 1977;198:515–517. doi: 10.1126/science.910144. [DOI] [PubMed] [Google Scholar]
- 43.Skoff RP, Price DL, Stocks A. Electron microscopic autoradiographic studies of gliogenesis in rat optic nerve. II. Time of origin. J Comp Neurol. 1976;169:313–334. doi: 10.1002/cne.901690304. [DOI] [PubMed] [Google Scholar]
- 44.Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- 45.Steinhauser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–345. doi: 10.1016/0166-2236(96)10043-6. [DOI] [PubMed] [Google Scholar]
- 46.Sternberger LA, Harwell LW, Sternberger NH. Neurotypy: regional individuality in rat brain detected by immunocytochemistry with monoclonal antibodies. Proc Natl Acad Sci USA. 1982;79:1326–1330. doi: 10.1073/pnas.79.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab. 1998;18:2–25. doi: 10.1097/00004647-199801000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Stys PK, Ransom BR, Waxman SG, Davis PK. Role of extracellular calcium in anoxic injury of mammalian central white matter. Proc Natl Acad Sci USA. 1990;87:4212–4216. doi: 10.1073/pnas.87.11.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stys PK, Ransom BR, Waxman SG. Tertiary and quaternary local anesthetics protect CNS white matter from anoxic injury at concentrations that do not block excitability. J Neurophysiol. 1992a;67:236–240. doi: 10.1152/jn.1992.67.1.236. [DOI] [PubMed] [Google Scholar]
- 50.Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+–Ca2+ exchanger. J Neurosci. 1992b;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szuchet S. The morphology and ultrastructure of oligodendrocytes and their functional implications. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. [Google Scholar]
- 52.Trapp BD, Bernier L, Andrews SB, Colman DR. Cellular and subcellular distribution of 2′,3′-cyclic nucleotide 3′-phosphodiesterase and its mRNA in the rat central nervous system. J Neurochem. 1988;51:859–868. doi: 10.1111/j.1471-4159.1988.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 53.Underhill SM, Baltan Tekkök S, Hyrc KL, Goldberg MP. Role of calcium-permeable AMPA receptors in oligodendrocyte excitotoxicity. Soc Neurosci Abstr. 2000;26:2064. [Google Scholar]
- 54.Valeriani V, Dewar D, McCulloch J. Quantitative assessment of ischemic pathology in axons, oligodendrocytes, and neurons: attenuation of damage after transient ischemia. J Cereb Blood Flow Metab. 2000;20:765–771. doi: 10.1097/00004647-200005000-00002. [DOI] [PubMed] [Google Scholar]
- 55.Waxman SG, Black JA, Stys PK, Ransom BR. Ultrastructural concomitants of anoxic injury and early post-anoxic recovery in rat optic nerve. Brain Res. 1992;574:105–119. doi: 10.1016/0006-8993(92)90806-k. [DOI] [PubMed] [Google Scholar]
- 56.Waxman SG, Black JA, Ransom BR, Stys PK. Protection of the axonal cytoskeleton in anoxic optic nerve by decreased extracellular calcium. Brain Res. 1993;614:137–145. doi: 10.1016/0006-8993(93)91027-p. [DOI] [PubMed] [Google Scholar]
- 57.Waxman SG, Black JA, Ransom BR, Stys PK. Anoxic injury of rat optic nerve: ultrastructural evidence for coupling between Na+ influx and Ca2+-mediated injury in myelinated CNS axons. Brain Res. 1994;644:197–204. doi: 10.1016/0006-8993(94)91680-2. [DOI] [PubMed] [Google Scholar]
- 58.Wrathall JR, Choiniere D, Teng YD. Dose-dependent reduction of tissue loss and functional impairment after spinal cord trauma with the AMPA/kainate antagonist NBQX. J Neurosci. 1994;14:6598–6607. doi: 10.1523/JNEUROSCI.14-11-06598.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yam PS, Dewar D, McCulloch J. Axonal injury caused by focal cerebral ischemia in the rat. J Neurotrauma. 1998;15:441–450. doi: 10.1089/neu.1998.15.441. [DOI] [PubMed] [Google Scholar]
- 60.Yoshioka A, Hardy M, Younkin DP, Grinspan JB, Stern JL, Pleasure D. Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors mediate excitotoxicity in the oligodendroglial lineage. J Neurochem. 1995;64:2442–2448. doi: 10.1046/j.1471-4159.1995.64062442.x. [DOI] [PubMed] [Google Scholar]
- 61.Yoshioka A, Bacskai B, Pleasure D. Pathophysiology of oligodendroglial excitotoxicity. J Neurosci Res. 1996;46:427–437. doi: 10.1002/(SICI)1097-4547(19961115)46:4<427::AID-JNR4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 62.Yoshioka A, Yamaya Y, Saiki S, Kanemoto M, Hirose G, Beesley J, Pleasure D. Non-N-methyl-d-aspartate glutamate receptors mediate oxygen–glucose deprivation-induced oligodendroglial injury. Brain Res. 2000;854:207–215. doi: 10.1016/s0006-8993(99)02359-8. [DOI] [PubMed] [Google Scholar]