

Abstract
The present study investigated the effect of inhibiting the expression of Nav1.8 (PN3/SNS) sodium channels by an antisense oligodeoxynucleotide (ODN) on bladder nociceptive responses induced by intravesical acetic acid infusion in rats. Animals were injected intrathecally with either Nav1.8 antisense or mismatch ODN. Control cystometrograms under urethane anesthesia during intravesical saline infusion exhibited intercontraction intervals (ICIs) that were significantly longer in antisense-treated rats than in mismatch ODN-treated rats. Intravesical infusion of 0.1% acetic acid induced bladder hyperactivity as reflected by a 68% reduction in ICIs in mismatch ODN-treated rats but did not significantly reduce ICIs in antisense-treated rats. The number of Fos-positive cells after acetic acid administration were significantly reduced in the L6 spinal cord from antisense-treated animals, compared with mismatch ODN-treated animals. In addition, Nav1.8 immunoreactivity was reduced in L6 dorsal root ganglion neurons in the antisense-treated rat. In patch-clamp recordings, the conductance density of TTX-resistant sodium currents in dissociated bladder afferent neurons that were labeled by axonal transport of a fluorescent dye, Fast Blue, injected into the bladder wall was also smaller in antisense-treated rats than in mismatch ODN-treated rats, whereas no changes were observed in TTX-sensitive currents. These results indicate that the Nav1.8 TTX-resistant sodium channels are involved in the activation of afferent nerves after chemical irritation of the bladder. These channels represent a new target for the treatment of inflammatory pain from visceral organs such as the urinary bladder.
Keywords: dorsal root ganglion, tetrodotoxin, Nav1.8 sodium channels, urinary bladder, acetic acid, inflammation, antisense oligodeoxynucleotide
It has been shown in humans as well as in animals that tissue inflammation in visceral organs such as the urinary bladder can sensitize afferent nerves and thereby elicit painful sensations as well as hyperactivity of the inflamed organs (Stillwell and Benson, 1988; Häbler et al., 1990; Sengupta and Gebhart, 1994; Dmitrieva and McMahon, 1996; Dmitrieva et al., 1997). For example, interstitial cystitis, a chronic inflammatory disorder of the urinary bladder, is characterized by urinary frequency, urgency, and severe suprapubic pain (Ho et al., 1997). Intravesical application of capsaicin or resiniferatoxin, substances that desensitize C-fiber afferents, reduced the painful bladder symptoms in these patients (Lazzeri et al., 1996, 2000). It is also known that bladder hyperreflexia induced by chemical irritation of the bladder in rats is suppressed by pretreatment with capsaicin (Maggi et al., 1992; Yu and de Groat, 1999). Thus it seems reasonable to assume that suppression of activity of C-fiber afferent pathways can be effective in treating pain arising from visceral organs such as the urinary bladder.
Our previous studies showed that capsaicin-sensitive C-fiber afferent neurons innervating the bladder and colon predominantly express high-threshold sodium currents and action potentials that are resistant to tetrodotoxin (TTX) (Yoshimura et al., 1996; Yoshimura and de Groat, 1997; Yoshimura, 1999) and that C-fiber bladder afferent neurons with TTX-resistant action potentials become hyperexcitable in rats with chronic cystitis induced by cyclophosphamide (Yoshimura and de Groat, 1999). Therefore, suppression of TTX-resistant action potentials might be a useful approach for treating visceral pain.
The TTX-resistant sodium channel Nav1.8 (PN3/SNS) is preferentially expressed in small-sized nociceptive dorsal root ganglion (DRG) neurons (Arbuckle and Docherty, 1995; Akopian et al., 1996; Sangameswaran et al., 1996; Goldin et al., 2000; Waxman et al., 2000). It has been reported that suppression of Nav1.8 channels was effective in reducing pain induced by injury of somatic afferent axons or tissue inflammation (Khasar et al., 1998; Porreca et al., 1999). However, it is not known whether Nav1.8 sodium channels are involved in activation of TTX-resistant sodium currents in visceral afferent neurons and in sensitization of visceral nociceptors. Thus we examined the effect of suppressing Nav1.8 TTX-resistant sodium channels with an antisense oligodeoxynucleotide (ODN) on a rodent model of visceral pain induced by intravesical instillation of acetic acid (Birder and de Groat, 1992). We observed that intrathecal antisense ODN treatment decreased expression of Nav1.8 sodium channels in lumbosacral DRG neurons, reduced TTX-resistant sodium conductances in bladder afferent neurons, and suppressed bladder nociceptive responses induced by bladder irritation.
Preliminary results of this study have been reported previously in abstract form (Seki et al., 2000; Yoshimura et al., 2000).
MATERIALS AND METHODS
Animal preparation. Experiments were performed on adult female Sprague Dawley rats (170–200 gm). Care and handling of animals were in accordance with institutional guidelines and approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
An intrathecal catheter (PE-10) was implanted at the level of the L6–S1 spinal cord after a laminectomy at the Th11 vertebra under halothane anesthesia. The distal end of the catheter was fire sealed and placed subcutaneously. The rats were injected intrathecally with either Nav1.8 antisense or mismatch phosphodiester ODN (Life Technologies, Gaithersburg, MD) 3–4 d after intrathecal catheter implantation. Antisense ODN (5′-TCC-TCT-GTG-CTT-GGT-TCT-GGC-CT-3′) was directed against a unique sequence of the Nav1.8 sodium channel (Porreca et al., 1999). The mismatch ODN (5′-TCC-TTC-GTG-CTG-TGT-TCG-TGC-CT-3′) consisted of the antisense sequence, except that five pairs of bases were switched. A dose of 45 μg of ODN dissolved in 8 μl of artificial CSF was injected through the intrathecal catheter under halothane anesthesia twice a day for 3 d. The position of the intrathecal catheter was checked after the experiment to confirm the location at the level of L6–S1 spinal cord.
Cystometry. Within 3–6 hr after the last injection of ODN solution, cystometry was performed under urethane anesthesia (1.2 mg/kg, s.c.). A catheter (PE-50) was inserted via a midline abdominal incision into the bladder through the bladder dome, and then intravesical pressure was measured to monitor bladder activity during continuous infusion of saline solution (0.04 ml/min). After a control period of 1–2 hr, the solution was switched to saline containing 0.1% acetic acid. Intercontraction intervals (ICIs), maximal contraction pressure, and pressure thresholds inducing reflex bladder contraction were measured before and after intravesical infusion of acetic acid in both Nav1.8 mismatch and antisense ODN-treated rats.
Animal perfusion and tissue preparation. Two hours after infusion of acetic acid into the bladder, the animals were killed via intracardiac perfusion with Krebs buffer followed by 4% paraformaldehyde fixative. The L6 spinal cord and L6 DRGs were then removed and post-fixed for 10–12 hr in the same fixative at 4°C. The spinal cords were placed in phosphate-buffered 30% sucrose solution overnight for cryoprotection and embedded in OCT Tissue Tek. Alternate 42-μm-thick cryosections were cut and mounted on gelatin-coated slides for Fos staining. DRGs were processed in xylene and ethanol solutions and embedded in paraffin blocks for Nav1.8 staining. Sections (4 μm thick) were cut, mounted, and air dried, first at room temperature and then in a forced-air dryer (20 min, 60°C).
Fos staining. Spinal cord sections were processed by an avidin–biotin complex (ABC) method for the Fos protein with antibodies purchased from Oncogene Science (Cambridge, MA), as described previously (Birder and de Groat, 1992; Kakizaki et al., 1996). Briefly, sections were incubated in Fos antiserum (1:15,000) for 30 min at room temperature and then for 72 hr at 4°C. Sections were then exposed to biotinylated secondary antibody (Vector, Burlingame, CA; 1:600) and ABC reagent (Vector), each for 1 hr at room temperature. Tissue sections were then mounted on gelatin-coated slides, dehydrated in graded ethanol rinses, cleared in xylene, and coverslipped with Permount. Control sections in which primary antibody was replaced with 0.4% Triton X-100 were negative. Analysis was performed on the L6 spinal cord segment, because in the previous studies the largest number of Fos-positive cells after bladder irritation was located in this segment (Birder and de Groat, 1992; Kakizaki et al., 1996). Cells exhibiting Fos immunoreactivity were counted in four spinal cord regions: medial dorsal horn (MDH), lateral dorsal horn (LDH), dorsal commissure (DCM), and lateral laminas V–VII containing the sacral parasympathetic nucleus (SPN). Counts of Fos-positive cells were performed on both sides of the spinal cord and presented as average numbers per section.
Nav1.8 staining. After deparaffinization in xylene and ethanol solutions, DRG sections were preincubated in potassium PBS (KPBS) containing 20% normal goat serum (NGS) and 0.2% Triton X-100 (1 hr, room temperature), and then incubated in KPBS containing 5% NGS, 0.2% Triton X-100, and Nav1.8 antibodies at 1:50 dilution (overnight, 4°C). The specificity of this antibody for Nav1.8 has been demonstrated previously (Novakovic et al., 1998). The next day, sections were washed in KPBS containing 0.1% bovine serum albumin and 0.1% Triton X-100 (2 hr, room temperature), incubated in the secondary antibody solution (biotinylated anti-rabbit IgG 1:200, Vecstatin Elite Kit; 1 hr, room temperature), washed again, and then incubated with peroxidase ABC (1:50, 90 min, room temperature). After a few washes with 0.1m KPB, the staining pattern was visualized with a DAB substrate reaction (Zymed Laboratories, San Francisco, CA). Tissue sections were then washed, dehydrated in a series of ethanols and xylenes, and mounted with Krystalon. Images were obtained with a Nikon Microphot SA microscope and the IPLab Spectram (Sigma Analytics). Thereafter, in randomly selected DRG sections, cell size and labeling intensity measurements were made on all cell profiles that exhibited a nucleus (∼120–150 cells per animal; n = 4 animals) using Scion Image software (Scion Corporation). The perimeter of each neuron profile was traced, and the cross-sectional area including the nuclear region was calculated. Neurons were then analyzed for mean labeling intensity of Nav1.8 staining. For the measurement of labeling intensity, the nucleus region was excluded. Nav1.8 staining intensity of each neuron was estimated by subtracting nonspecific staining. The latter was determined by sampling the staining intensities of large-diameter neurons (average of six neurons per section) that were similar to those of cells labeled with preabsorbed antibodies (i.e., negative staining for Nav1.8 protein).
Electrical recording. In four animals in each group treated with either Nav1.8 mismatch or antisense ODN, L6–S1 DRGs were dissociated enzymatically into single neurons, and TTX-resistant sodium currents were measured using patch-clamp recording techniques. The population of DRG neurons that innervate the urinary bladder were labeled by retrograde axonal transport of a fluorescent dye, Fast Blue (4% w/v) (Polyloy, Gross Umstadt, Germany) injected into the wall of the bladder in halothane-anesthetized animals (Yoshimura et al., 1994, 1998). The dye was injected with a 29 gauge needle at three to six sites on the dorsal surface of the bladder (5–6 μl per site; total volume of 20–30 μl). Each injection site was washed with saline to minimize contamination of adjacent organs with the dye. Particular care was taken to avoid injections into the lumen, major blood vessels, or overlying fascial layers. Our previous studies (Keast and de Groat, 1992; Yoshimura et al., 1998) using these techniques showed that nonspecific labeling of neurons caused by the leakage of dye is negligible.
One week after dye injection into the bladder, freshly dissociated neurons from DRGs were removed from halothane-anesthetized animals (Yoshimura et al., 1996; Yoshimura and de Groat, 1997). Briefly, L6 and S1 DRGs were dissected and incubated in a bath for 25 min at 35°C with 5 ml of DMEM (Sigma, St. Louis, MO) containing 0.3 mg/ml trypsin (Type 3, Sigma), 1 mg/ml collagenase (Type 1, Sigma), and 0.1 mg/ml deoxyribonuclease (Type 4, Sigma). Trypsin inhibitor (Type 2a, Sigma) was then added to neutralize the activity of trypsin. Individual DRG cell bodies were isolated by trituration and then plated on poly-l-lysine-coated 35 mm Petri dishes. Dye-labeled primary afferent neurons that innervate the urinary bladder were identified using an inverted phase-contrast microscope (Nikon, Tokyo, Japan) with fluorescent attachments (UV-1A filter; excitation wavelength, 365 nm). GΩ-seal whole-cell recordings were performed at room temperature (20–22°C) on each freshly dissociated labeled neuron in a culture dish that usually contained two to five labeled cells among a few hundred unlabeled neurons. The internal solution used during current-clamp recordings of sodium currents contained (in mm) NaCl 10, CsF 125, CaCl2 1, MgCl2 2, EGTA 10, HEPES 10, Mg-ATP 4, and GTP (Tris Salt) 0.3, adjusted to pH 7.4 with CsOH (310 mOsm). Patch electrodes had resistances of 1–4 MΩ when filled with the internal solution. Neurons were superfused at a flow rate of 1.5 ml/min with an external solution containing (in mm): NaCl 40, tetraethylammonium (TEA)-Cl 95, MgCl2 10, CaCl2 0.03, 4-aminopyridine 5, HEPES 10, andd-glucose 10, adjusted to pH 7.4 with TEA-OH (340 mOsm). All recordings were made with an Axopatch-1D patch-clamp amplifier (Axon Instruments, Foster City, CA), and data were acquired and analyzed by PCLAMP software (Axon Instruments). The peak amplitudes of the currents were measured and converted to sodium conductances by means of the following equation: GNa =INa/(Vtest− Vrev), whereGNa is the sodium conductance,INa is the sodium current,Vtest is the test potential, andVrev is the reversal potential for the sodium current. To correct the sodium conductance for cell size, the TTX-resistant and TTX-sensitive sodium conductances for each cell were normalized with respect to cell membrane capacitances that were obtained by reading the value for whole-cell input capacitance neutralization directly from the amplifier.
Analysis. All data are expressed as means ± SEM. Statistical differences between data were determined by Mann–WhitneyU test. A level of p < 0.05 was considered to be statistically significant.
RESULTS
Cystometry
Control cystometrograms performed by infusing saline into the bladder revealed that ICIs were significantly (p< 0.05) longer in antisense-treated rats than in mismatch ODN-treated rats (9.7 ± 1.2 vs 6.7 ± 0.8 min) (Figs.1, 2). Subsequent intravesical infusion of 0.1% acetic acid induced bladder hyperactivity, reducing ICIs to 2.2 ± 0.3 min (68% reduction;p < 0.01) in mismatch ODN-treated rats (n = 8). However, in Nav1.8 antisense ODN-treated rats (n = 8), the ICI after intravesical acetic acid infusion was reduced by 38% to 6.0 ± 1.0 min, which was not significantly different (p = 0.65) from the ICI before acetic acid treatment (9.7 ± 1.2 min) (Figs. 1, 2). Mean pressure thresholds (8.7 ± 0.7 vs 9.3 ± 0.8 cmH2O) and bladder contraction pressure (36.7 ± 3.7 vs 34.2 ± 2.9 cmH2O) were not significantly different in mismatch and antisense ODN-treated animals.
Fig. 1.

Effects of intravesical infusion of 0.1% acetic acid on cystometrograms in rats treated with mismatch or antisense oligodeoxynucleotide (ODN) for Nav1.8 sodium channels. Note that in an antisense-treated rat, acetic acid-induced bladder hyperactivity was suppressed.
Fig. 2.

Effects of intravesical infusion of acetic acid on intercontraction intervals (ICI) in rats treated with mismatch (n = 8) or antisense ODN (n = 8). Note that in antisense ODN-treated rats, the ICI in the control period is significantly longer and a reduction in the ICI after acetic acid infusion is significantly smaller, when compared with mismatch ODN-treated rats. *p < 0.05, **p < 0.01.
Fos staining
After bladder irritation by intravesical infusion of acetic acid, Fos-positive cells were identified in the MDH, LDH, DCM, and SPN areas of the L6 spinal cord (Fig. 3). The largest number of Fos-positive cells was located in the DCM area in both groups of animals. The total number of Fos-positive cells in the L6 spinal cord after bladder irritation in antisense ODN-treated animals was significantly smaller (p < 0.01) than in mismatch ODN-treated animals (20.6 ± 1.1 vs 61.2 ± 2.6 cells per section). The numbers of Fos-positive cells were significantly (p < 0.01) smaller in the DCM (6.3 ± 1.1 vs 25.8 ± 2.4), MDH (2.5 ± 0.6 vs 13.0 ± 2.6), LDH (1.5 ± 0.6 vs 6.1 ± 1.2), and SPN (8.3 ± 1.0 vs 14.9 ± 2.1 cells per section) regions of the L6 spinal cord from antisense-treated animals, compared with mismatch ODN-treated animals (Fig. 3).
Fig. 3.
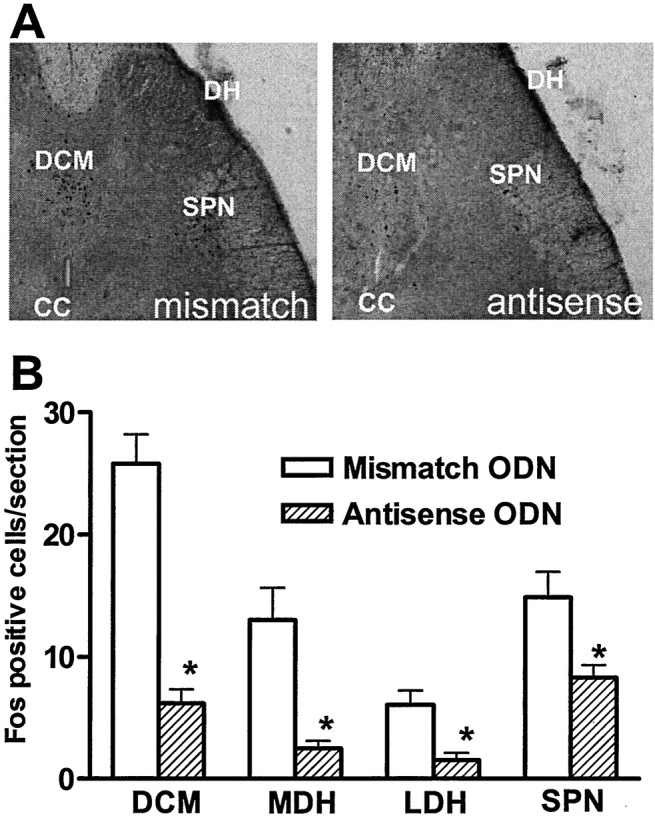
Effects of Nav1.8 antisense ODN on c-fos expression in the L6 spinal cord after intravesical infusion of acetic acid. A, Photomicrographs of Fos staining in transverse sections of the L6 spinal cord from rats treated with mismatch (left panel) and antisense (right panel) ODN. B, Histograms of the number of Fos-positive cells after intravesical acetic acid infusion.DCM, Dorsal commissure; DH, dorsal horn;SPN, sacral parasympathetic nucleus; CC, central canal. *p < 0.01.
Nav1.8 staining
In four mismatch and four antisense ODN-treated rats, 445 and 421 DRG neuron profiles were measured, respectively. The cross-sectional areas of cell profiles were divided into three groups according to their measured area: small (<700 μm2; 174 and 169 cells), medium (700–1200 μm2; 133 and 121 cells), and large (>1200 μm2; 138 and 131 cells) in mismatch and antisense ODN-treated rats. In mismatch ODN-treated rats, the highest intensity of labeling for Nav1.8 was found in small-sized DRG neurons, whereas medium-sized neurons had lower intensity of staining, and the staining intensity of large-sized neurons was minimal (Fig. 4), as reported previously in untreated rats (Novakovic et al., 1998). In Nav1.8 antisense-treated rats, the mean intensity of Nav1.8 labeling in small- and medium-sized neurons was significantly decreased by 44 and 46%, respectively, compared with the labeling intensity in mismatch ODN-treated rats, indicating the reduction of Nav1.8 channel protein expression after antisense ODN treatment (Fig.4B).
Fig. 4.
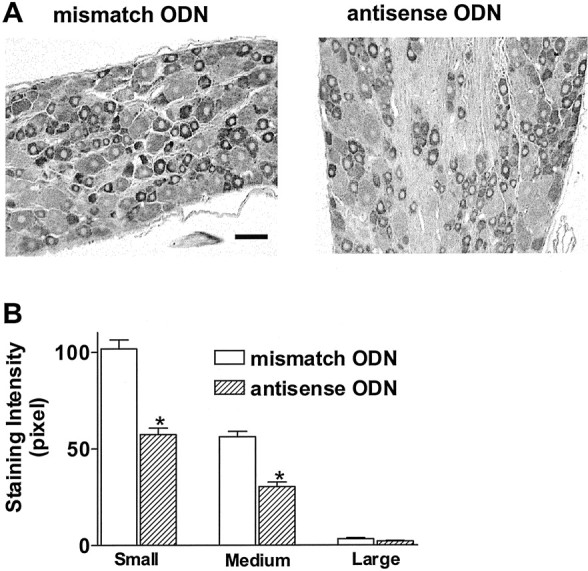
Immunolabeling of Nav1.8 TTX-resistant sodium channels in L6 DRG. A, Photomicrographs of Nav1.8 immunostaining in rat treated with mismatch (left panel) or antisense (right panel) ODN. Scale bar, 50 μm. B, Averaged Nav1.8 labeling intensity in L6 DRG neurons with small, medium, and large cross-sectional somal areas. Note that labeling intensity is greater in small-sized neurons and significantly reduced after antisense treatment in small- to medium-sized neurons. *p < 0.01.
Sodium currents in bladder afferent neurons
As reported previously, in the majority of small-sized (<30 μm diameter or 765 μm2 cross-sectional area) bladder afferent neurons, inward sodium currents elicited from the holding potential of −60 mV were mainly insensitive to TTX (1 μm) (Yoshimura et al., 1996; Yoshimura and de Groat, 1997). In this population of bladder afferent neurons, the peak amplitude of sodium currents was measured in presence of 1 μm TTX. The calculated sodium conductances were then normalized with respect to cell membrane capacitances. The maximum conductance density of TTX-resistant sodium currents at the holding potential of −60 mV in bladder afferent neurons was significantly (p < 0.05) smaller in antisense ODN-treated rats than in mismatch ODN-treated rats (1.9 ± 0.5 vs 4.1 ± 0.9 nS/pF; n = 15) (Fig.5). The values for membrane voltages at half-maximal activation of TTX-resistant currents calculated by the Boltzmann equation in antisense and mismatch ODN-treated rats (−11.2 and −10.7 mV, respectively) were not different from those reported in our previous studies (Yoshimura et al., 1996; Yoshimura and de Groat, 1997). In contrast, large-sized (>30 μm or 765 μm2) bladder afferent neurons expressed TTX-sensitive sodium currents that comprised >60% of the total sodium currents elicited by depolarizing pulses to 0 mV from the holding potential of −60 mV (Yoshimura and de Groat, 1997). In these neurons (n = 12), TTX-sensitive sodium currents were isolated by the subtraction of the currents after TTX application from those before TTX application. The maximum conductance density of TTX-sensitive sodium currents at the holding potential of −60 mV in antisense ODN-treated rats (3.9 ± 0.8 nS/pF) was not different from the conductance density (4.0 ± 0.7 nS/pF) in mismatch ODN-treated rats.
Fig. 5.
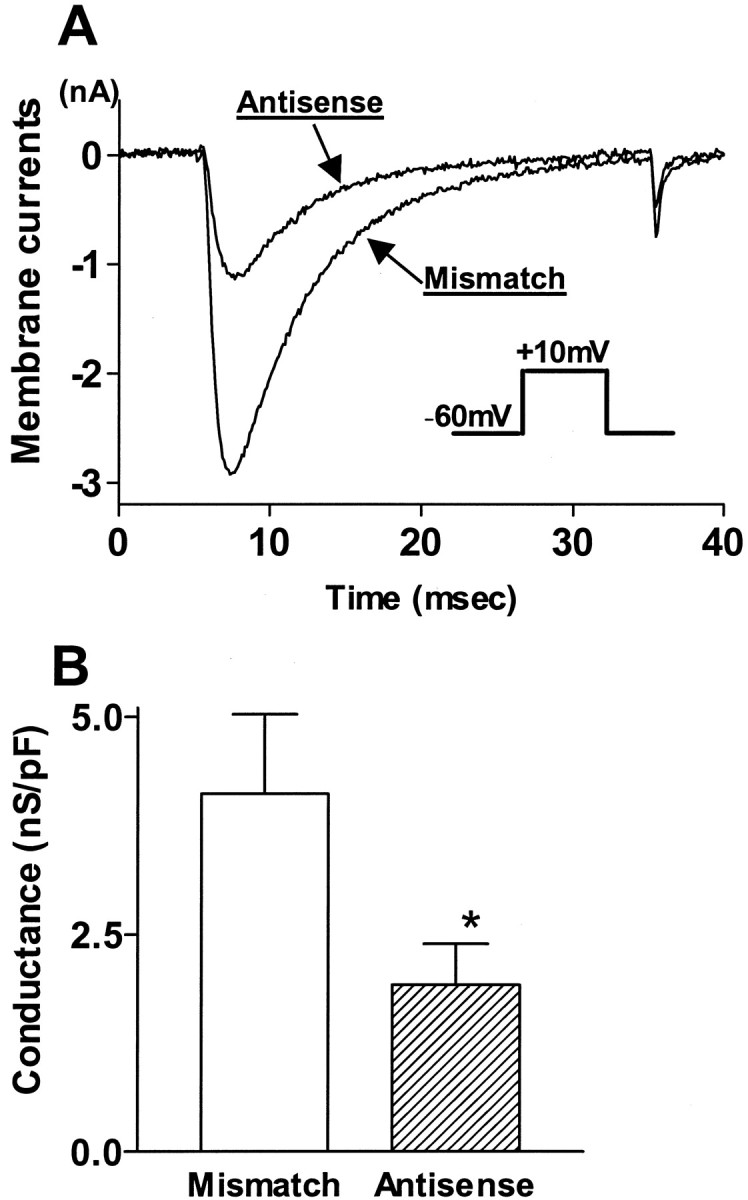
Effects of Nav1.8 antisense ODN on TTX-resistant sodium currents in bladder afferent neurons.A, Superimposed traces of TTX-resistant sodium currents elicited by depolarizing pulses to 10 mV from the holding potential of −60 mV in neurons from rats treated with mismatch and antisense ODN.B, Maximum conductance densities of TTX-resistant sodium currents. In rats treated with antisense ODN, the conductance density of TTX-resistant sodium currents (n = 15 neurons) was significantly smaller compared with rats with mismatch ODN treatment (n = 15 neurons). *p< 0.05.
DISCUSSION
The results obtained in this study indicate that intrathecal Nav1.8 antisense ODN treatment reduced TTX-resistant sodium channel Nav1.8 expression in lumbosacral DRG neurons as well as the TTX-resistant sodium conductance in bladder afferent neurons. The treatment also suppressed bladder hyperactivity and c-fos expression in the spinal cord induced by chemical irritation of the urinary bladder. Because the ICIs during cystometry under urethane anesthesia in mismatch ODN-treated rats were not different from those in untreated rats reported in our previous studies (Ozawa et al., 1999), inhibitory effects of antisense ODN on bladder pain responses are not likely to be caused by toxic or nonspecific effects of repeated injections of ODN. This is also supported by previous studies (Porreca et al., 1999), which revealed that intrathecal injections of the same sequence of Nav1.8 antisense ODN produced only a transient depression of the allodynia and hyperalgesia induced in rats by sciatic nerve injury. Thus it is assumed that Nav1.8 channels are involved in sensitization of bladder afferent nerves induced by noxious stimuli and that suppression of Nav1.8 channels could be a new modality for the treatment of painful bladder symptoms and/or bladder hyperactivity.
Previous studies demonstrated that TTX-resistant sodium channels are responsible for action potentials in a subpopulation of afferent neurons (Elliott and Elliott, 1993; Ogata and Tatebayashi, 1993,Arbuckle and Docherty, 1995; Yoshimura et al., 1996). The TTX-resistant sodium channel (Nav1.8) has been cloned from rat DRG neurons, and the expression of these channels is confined to the capsaicin-sensitive, small-sized DRG neurons (Arbuckle and Docherty, 1995; Akopian et al., 1996; Sangameswaran et al., 1996; Dib-Hajj et al., 1998; Waxman et al., 2000). It has also been documented that TTX-resistant sodium channels are present in central axon terminals of afferent nerves (Novakovic et al., 1998) and participate in action potential initiation in slow-conducting C-fiber afferents (Jeftinija, 1994; Gu and MacDermott, 1997).
Our previous studies revealed that most bladder afferent neurons exhibit TTX-resistant sodium currents and action potentials and are capsaicin-sensitive (Yoshimura et al., 1996; Yoshimura, 1999; Yoshimura and de Groat, 1999). In addition, most (>95%) of the capsaicin-sensitive bladder afferent neurons from normal rats were the C-fiber type (unmyelinated) that did not stain with antibodies for the 200 kDa subunit of neurofilament (Yoshimura et al., 1998). The latter marker is specifically expressed in DRG neurons with myelinated axons (Lawson et al., 1993). Only a small proportion (5%) of neurofilament-positive, myelinated Aδ-fiber bladder afferent neurons are sensitive to capsaicin (Yoshimura et al., 1998). Thus, it is likely that bladder afferent neurons that predominantly express TTX-resistant sodium currents are capsaicin-sensitive C-fiber neurons.
The present study showed that treatment with Nav1.8 antisense ODN, which decreased expression of Nav1.8 channel protein in DRG neurons and reduced the TTX resistant sodium conductance in bladder afferent neurons, suppressed the nociceptive responses induced by chemical irritation of the bladder. This anti-nociceptive effect in Nav1.8 antisense-treated rats is similar to that observed in previous studies in rats in which C-fiber bladder afferents were desensitized by pretreatment with systemic capsaicin (Maggi et al., 1992; Yu and de Groat, 1999). Therefore, it is reasonable to assume that activation of bladder nociceptors and/or nociceptive input to the spinal cord is dependent on activation of Nav1.8 channels in C-fiber bladder afferent nerves.
The present study also demonstrated that, in addition to nociceptive mechanisms, Nav1.8 channels are likely to be involved in activation of mechanosensitive afferents induced by non-noxious bladder distension. This conclusion is based on the finding that antisense treatment increased the ICIs during control cystometry when saline was infused into the bladder. Previous studies revealed that systemic capsaicin pretreatment produced a similar increase in ICIs or bladder capacity, or both, in urethane-anesthetized rats (Santicioli et al., 1985; Yu and de Groat, 1999), indicating that activity in C-fiber bladder afferents modulates reflex voiding. This is consistent with the reports that some C-fiber bladder afferent fibers in rats are not only nociceptive, but also mechanosensitive (Sengupta and Gebhart, 1994; Dmitrieva and McMahon, 1996).
Various evidence has indicated that alterations in sodium channel expression in afferent neurons contribute at least in part to neuronal hyperexcitability after nerve injury. For example, in an animal model of neuropathic pain induced by sciatic nerve ligation, the redistribution and accumulation of Nav1.8 sodium channel protein from DRG cell bodies to nerve fibers proximal to the injury site have been implicated as an important mechanism for ectopic discharges of injured nerves. Under the same conditions, the level of Nav1.8 mRNA in DRG neurons was not altered (Novakovic et al., 1998). A recent study (Porreca et al., 1999) provided further support for this idea by demonstrating that Nav1.8 antisense treatment that reduced the level of Nav1.8 channel protein in DRG neurons suppressed hyperalgesia in the same neuropathic pain model. However, in another model of neuropathic pain induced by sciatic nerve transection (axotomy), TTX-resistant sodium currents were significantly reduced in small-sized DRG neurons along with the reduction in the level of Nav1.8 mRNA and protein expression. In concert with the reduction in TTX-resistant sodium channel expression, TTX-sensitive sodium channel expression was upregulated (Black et al., 1999; Waxman et al., 2000). A similar increase in TTX-sensitive sodium channels and decrease in TTX-resistant sodium channels occurred in bladder afferent neurons in spinal cord injured rats that exhibited bladder hyperreflexia (Yoshimura and de Groat, 1997). Because TTX-sensitive sodium channels are activated at lower thresholds (Elliott and Elliott, 1993; Ogata and Tatebayashi, 1993; Yoshimura et al., 1996; Yoshimura and de Groat, 1997), a switch in sodium channel expression from TTX-resistant to TTX-sensitive types would be expected to contribute to the phenotypic changes in bladder afferent neurons after spinal cord injury (Yoshimura and de Groat, 1997; Yoshimura, 1999) or the increased excitability in injured DRG neurons in an axotomy-induced neuropathic pain model (Black et al., 1999; Waxman et al., 2000). Therefore, plasticity in TTX-resistant and -sensitive sodium channels might contribute to sensory and reflex changes in various pathophysiological conditions.
An involvement of Nav1.8 sodium channels has also been implicated in the hyperalgesia induced by tissue inflammation. In rat models of inflammatory pain induced by carrageenan injection into the hindpaw, Nav1.8 expression in DRG neurons increased along with an increase in TTX-resistant sodium current amplitude (Tanaka et al., 1998). It has also been reported that pretreatment with Nav1.8 antisense ODN suppressed prostaglandin E2 (PGE2)- and complete Freund's adjuvant (CFA)-induced hyperalgesia in the hindpaw of rats (Khasar et al., 1998; Porreca et al., 1999). PGE2 is known to be a chemical mediator that can induce changes in the voltage dependence of TTX-resistant sodium channels, resulting in hyperexcitability of DRG neurons (England et al., 1996; Gold et al., 1996). In addition, Nav1.8 knock-out mice that lack functional Nav1.8 channels exhibited analgesia to noxious mechanical stimuli and reduced inflammatory hyperalgesia (Akopian et al., 1999). Thus it seems likely that Nav1.8 channels are involved at least in part in the development of inflammatory pain in somatic afferent pathways.
We have reported previously that chronic bladder inflammation induced hyperexcitability of capsaicin-sensitive C-fiber bladder afferent neurons (Yoshimura and de Groat, 1999). Although neuronal hyperexcitability in this visceral pain model seemed to be related to the reduction in potassium channel expression in the afferent neurons, the firing of hyperexcitable C-fiber bladder afferent neurons was still dependent on TTX-resistant sodium channels. Thus, the TTX-resistant sodium channel Nav1.8 could be a target for treatment of inflammatory pain in visceral organs such as the urinary bladder.
In the present study, Nav1.8 antisense treatment did not completely suppress bladder hyperactivity induced by bladder irritation. This is probably attributable to incomplete suppression of Nav1.8 channel protein in DRG neurons after antisense treatment, as revealed by ∼50% reduction in Nav1.8 staining intensity in small- to medium-sized DRG neurons from antisense-treated rats. However, an alternative explanation could be the presence of another type of TTX-resistant sodium channel (Nav1.9 or NaN/SNS2), which is expressed in capsaicin-sensitive, small-sized DRG neurons (Dib-Hajj et al., 1998; Tate et al., 1998). Nav1.9 sodium channels are reportedly upregulated in a chronic inflammatory condition induced by CFA injection into the rat hindpaw (Tate et al., 1998; Waxman et al., 2000). On the other hand, Nav1.9 channels are not likely to contribute to the development of neuropathic pain because antisense treatment that reduced the expression of Nav1.9 channel protein in DRG neurons did not change the behavioral responses in nerve-injured rats (Porreca et al., 1999).
The relative contribution of the two types of TTX-resistant sodium channels (Nav1.8 and Nav1.9) to bladder sensory mechanisms can only be inferred from indirect evidence. Studies by other investigators (Fjell et al., 1999, 2000) revealed that the two types of TTX-resistant channels were expressed in different types of C-fiber afferent neurons: (1) Nav1.8 in peptidergic, isolectin B4 (IB4)-negative neurons and (2) Nav1.9 in nonpeptidergic, IB4-positive neurons. We and other investigators (Bennett et al., 1996; Yoshimura, 2001) found that IB4 staining was present in a smaller number of bladder afferent neurons (10–20%) than in somatic afferent neurons (40%) innervating skin or striated muscles. Thus, it seems likely that Nav1.8 channels are more important than Nav1.9 channels in bladder nociceptive mechanisms.
In conclusion, the present study provides the first evidence that Nav1.8 sodium channels are involved in sensitization of C-fiber bladder afferents and in the triggering of nociceptive responses. It has been well documented that tissue inflammation in visceral organs can induce C-fiber afferent hyperexcitability, which contributes to painful sensations as well as hyperactivity of the inflamed visceral organs (Häbler et al., 1990; Sengupta and Gebhart, 1994; Dmitrieva and McMahon, 1996;Lazzeri et al., 1996, 2000; Yoshimura and de Groat, 1999). Therefore, the Nav1.8 TTX-resistant sodium channel could be a new target for the treatment of chronic visceral painful disorders such as interstitial cystitis.
Footnotes
This work was supported by grants from the National Institutes of Health (DK 49430 and DK 57267) and the Spinal Cord Research Foundation, Paralyzed Veterans of America (1861-01/02).
Correspondence should be addressed to Dr. Naoki Yoshimura, Departments of Urology and Pharmacology, University of Pittsburgh School of Medicine, Kaufmann Medical Building, Suite 700, 3471 Fifth Avenue, Pittsburgh, PA 15213. E-mail: nyos@pitt.edu.
REFERENCES
- 1.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 2.Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- 3.Arbuckle JB, Docherty RJ. Expression of tetrodotoxin-resistant sodium channels in capsaicin-sensitive dorsal root ganglion neurons of adult rats. Neurosci Lett. 1995;185:70–73. doi: 10.1016/0304-3940(94)11227-a. [DOI] [PubMed] [Google Scholar]
- 4.Bennett DL, Dmietrieva N, Priestley JV, Clary D, McMahon SB. trkA, CGRP and IB4 expression in retrogradely labelled cutaneous and visceral primary sensory neurones in the rat. Neurosci Lett. 1996;206:33–36. doi: 10.1016/0304-3940(96)12418-6. [DOI] [PubMed] [Google Scholar]
- 5.Birder LA, de Groat WC. Increased c-fos expression in spinal neurons after irritation of the lower urinary tract in the rat. J Neurosci. 1992;12:4878–4889. doi: 10.1523/JNEUROSCI.12-12-04878.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black JA, Cummins TR, Plumpton C, Chen YH, Hormuzdiar W, Clare JJ, Waxman SG. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol. 1999;82:2776–2785. doi: 10.1152/jn.1999.82.5.2776. [DOI] [PubMed] [Google Scholar]
- 7.Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci USA. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dmitrieva N, McMahon SB. Sensitisation of visceral afferents by nerve growth factor in the adult rat. Pain. 1996;66:87–97. doi: 10.1016/0304-3959(96)02993-4. [DOI] [PubMed] [Google Scholar]
- 9.Dmitrieva N, Shelton D, Rice ASC, McMahon SB. The role of nerve growth factor in a model of visceral inflammation. Neuroscience. 1997;78:449–459. doi: 10.1016/s0306-4522(96)00575-1. [DOI] [PubMed] [Google Scholar]
- 10.Elliott AA, Elliott JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol (Lond) 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fjell J, Cummins TR, Dib-Hajj SD, Fried K, Black JA, Waxman SG. Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Mol Brain Res. 1999;67:267–282. doi: 10.1016/s0169-328x(99)00070-4. [DOI] [PubMed] [Google Scholar]
- 13.Fjell J, Hjelmstrom P, Hormuzdiar W, Milenkovic M, Aglieco F, Tyrrell L, Dib-Hajj S, Waxman SG, Black JA. Localization of the tetrodotoxin-resistant sodium channel NaN in nociceptors. NeuroReport. 2000;11:199–202. doi: 10.1097/00001756-200001170-00039. [DOI] [PubMed] [Google Scholar]
- 14.Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 16.Gu JG, MacDermott AB. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- 17.Häbler HJ, Jänig W, Koltzenburg M. Activation of unmyelinated afferent fibres by mechanical stimuli and inflammation of the urinary bladder in the cat. J Physiol (Lond) 1990;425:545–562. doi: 10.1113/jphysiol.1990.sp018117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho N, Koziol JA, Parsons CL. Epidemiology of interstitial cystitis. In: Sant GR, editor. Interstitial cystitis. Lippincott-Raven; Philadelphia: 1997. pp. 9–16. [Google Scholar]
- 19.Jeftinija S. The role of tetrodotoxin-resistant sodium channels of small primary afferent fibers. Brain Res. 1994;639:125–134. doi: 10.1016/0006-8993(94)91772-8. [DOI] [PubMed] [Google Scholar]
- 20.Kakizaki H, Yoshiyama M, de Groat WC. Role of NMDA and AMPA glutamatergic transmission in spinal c-fos expression after urinary tract irritation. Am J Physiol. 1996;270:R990–R996. doi: 10.1152/ajpregu.1996.270.5.R990. [DOI] [PubMed] [Google Scholar]
- 21.Keast JR, de Groat WC. Segmental distribution and peptide content of primary afferent neurons innervating the urogenital organs and colon of male rats. J Comp Neurol. 1992;319:615–623. doi: 10.1002/cne.903190411. [DOI] [PubMed] [Google Scholar]
- 22.Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- 23.Lawson SN, Perry MJ, Prabhakar E, McCarthy PW. Primary sensory neurones: neurofilament, neuropeptides, and conduction velocity. Brain Res Bull. 1993;30:239–243. doi: 10.1016/0361-9230(93)90250-f. [DOI] [PubMed] [Google Scholar]
- 24.Lazzeri M, Beneforti P, Benaim G, Maggi CA, Lecci A, Turini D. Intravesical capsaicin for treatment of severe bladder pain: a randomized placebo controlled study. J Urol. 1996;156:947–952. [PubMed] [Google Scholar]
- 25.Lazzeri M, Beneforti P, Spinelli M, Zanollo A, Barbagli G, Turini D. Intravesical resiniferatoxin for the treatment of hypersensitive disorder: a randomized placebo controlled study. J Urol. 2000;164:676–679. doi: 10.1097/00005392-200009010-00014. [DOI] [PubMed] [Google Scholar]
- 26.Maggi CA, Lecci A, Santicioli P, Bianco ED, Giuliani S. Cyclophosphamide cystitis in rats: involvement of capsaicin-sensitive primary afferents. J Auton Nerv Syst. 1992;38:201–208. doi: 10.1016/0165-1838(92)90031-b. [DOI] [PubMed] [Google Scholar]
- 27.Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR, Eglen RM, Hunter JC. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18:2174–2187. doi: 10.1523/JNEUROSCI.18-06-02174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogata N, Tatebayashi H. Kinetic analysis of two types of Na+ channels in rat dorsal root ganglia. J Physiol (Lond) 1993;466:9–37. [PMC free article] [PubMed] [Google Scholar]
- 29.Ozawa H, Chancellor MB, Jung SY, Yokoyama T, Fraser MO, Yu Y, de Groat WC, Yoshimura N. Effect of intravesical nitric oxide therapy on cyclophosphamide-induced cystitis. J Urol. 1999;162:2211–2216. doi: 10.1016/S0022-5347(05)68161-X. [DOI] [PubMed] [Google Scholar]
- 30.Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci USA. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sangameswaran L, Delgado SG, Fish LM, Koch BD, Jakeman LB, Stewart GR, Sze P, Hunter JC, Eglen RM, Herman RC. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J Biol Chem. 1996;271:5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- 32.Santicioli P, Maggi CA, Meli A. The effect of capsaicin pretreatment on the cystometrograms of urethane anesthetized rats. J Urol. 1985;133:700–703. doi: 10.1016/s0022-5347(17)49164-6. [DOI] [PubMed] [Google Scholar]
- 33.Seki S, Erickson V, Kassotakis L, Novakovic S, Rohrer D, de Groat WC, Yoshimura N. Suppression of the TTX-resistant sodium channel (PN3/SNS) in rat DRG neurons by an antisense oligodeoxynucleotide reduces acetic acid-induced bladder hyperactivity. Soc Neurosci Abstr. 2000;26:1905. [Google Scholar]
- 34.Sengupta JN, Gebhart GF. Mechanosensitive properties of pelvic nerve afferent fibres innervating the urinary bladder of the rat. J Neurophysiol. 1994;72:2420–2430. doi: 10.1152/jn.1994.72.5.2420. [DOI] [PubMed] [Google Scholar]
- 35.Stillwell TJ, Benson RC. Cyclophosphamide-induced cystitis. A review of 100 patients. Cancer. 1988;61:451–457. doi: 10.1002/1097-0142(19880201)61:3<451::aid-cncr2820610308>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M, Cummins TR, Ishikawa K, Dib-Hajj SD, Black JA, Waxman SG. SNS Na channel expression increases in dorsal root ganglion neurons in the carrageenan inflammatory pain model. NeuroReport. 1998;9:967–972. doi: 10.1097/00001756-199804200-00003. [DOI] [PubMed] [Google Scholar]
- 37.Tate S, Benn S, Hick C, Trezise D, John V, Mannion RJ, Costigan M, Plumpton C, Grose D, Gladwell Z, Kendall G, Dale K, Bountra C, Woolf CJ. Two sodium channels contribute to the TTX-R sodium current in primary sensory neurons. Nat Neurosci. 1998;1:653–655. doi: 10.1038/3652. [DOI] [PubMed] [Google Scholar]
- 38.Waxman SG, Cummins TR, Dib-Hajj SD, Black JA. Voltage-gated sodium channels and the molecular pathogenesis of pain: a review. J Rehabil Res Dev. 2000;37:517–528. [PubMed] [Google Scholar]
- 39.Yoshimura N. Bladder afferent pathway and spinal cord injury: possible mechanisms inducing hyperreflexia of the urinary bladder. Prog Neurobiol. 1999;57:583–606. doi: 10.1016/s0301-0082(98)00070-7. [DOI] [PubMed] [Google Scholar]
- 40.Yoshimura N. Afferent pathways to the urethra. Int Bladder Symp Abstr. 2001;1:6. [Google Scholar]
- 41.Yoshimura N, de Groat WC. Plasticity of Na+ channels in afferent neurones innervating rat urinary bladder following spinal cord injury. J Physiol (Lond) 1997;503:269–276. doi: 10.1111/j.1469-7793.1997.269bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshimura N, de Groat WC. Increased excitability of afferent neurons innervating rat urinary bladder after chronic bladder inflammation. J Neurosci. 1999;19:4644–4653. doi: 10.1523/JNEUROSCI.19-11-04644.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshimura N, White G, Weight FF, de Groat WC. Patch-clamp recordings from subpopulations of autonomic and afferent neurons identified by axonal tracing techniques. J Auton Nerv Syst. 1994;49:85–92. doi: 10.1016/0165-1838(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimura N, White G, Weight FF, de Groat WC. Different types of Na+ and K+ currents in rat dorsal root ganglion neurones innervating the urinary bladder. J Physiol (Lond) 1996;494:1–16. doi: 10.1113/jphysiol.1996.sp021471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshimura N, Erdman SL, Snider MW, de Groat WC. Effects of spinal cord injury on neurofilament immunoreactivity and capsaicin sensitivity in rat dorsal root ganglion neurons innervating the urinary bladder. Neuroscience. 1998;83:633–643. doi: 10.1016/s0306-4522(97)00376-x. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimura N, Erickson V, Novakovic S, de Groat WC. Reduction of TTX-resistant sodium channel PN3 expression by antisense oligodeoxynucleotide suppresses acetic acid-induced bladder hyperactivity in the rat. Auton Neurosci. 2000;82:50. [Google Scholar]
- 47.Yu Y, de Groat WC. Effects of ZD6169, a KATP channel opener, on the micturition reflex in the rat. J Pharmacol Exp Ther. 1999;290:825–831. [PubMed] [Google Scholar]