

Abstract
Superior cervical ganglion (SCG) sympathetic neurons die by apoptosis when deprived of nerve growth factor (NGF). It has been shown previously that the induction of apoptosis in these neurons at NGF withdrawal requires both the activity of the small GTP-binding protein Cdc42 and the activation of the c-Jun N-terminal kinase (JNK) pathway. The mixed lineage kinase 3 (MLK3) belongs to a family of mitogen-activated protein (MAP) kinase kinase kinases. MLK3 contains a Cdc42/Rac interactive-binding (CRIB) domain and activates both the JNK and the p38 MAP kinase pathways. In this study the role of MLK3 in the induction of apoptosis in sympathetic neurons has been investigated. Overexpression of an active MLK3 induces activation of the JNK pathway and apoptosis in SCG neurons. In addition, overexpression of kinase dead mutants of MLK3 blocks apoptosis as well as c-Jun phosphorylation induced by NGF deprivation. More importantly, MLK3 activity seems to increase by 5 hr after NGF withdrawal in both differentiated PC12 cells and SCG neurons. We also show that MLK3 lies downstream of Cdc42 in the neuronal death pathway. Regulation of MLK3 in neurons seems to be dependent on MLK3 activity and possibly on an additional cellular component, but not on its binding to Cdc42. These results suggest that MLK3, or a closely related kinase, is a physiological element of NGF withdrawal-induced activation of the Cdc42–c-Jun pathway and neuronal death. MLK3 therefore could be an interesting therapeutic target in a number of neurodegenerative diseases involving neuronal apoptosis.
Keywords: apoptosis, Cdc42, MLK3, signal transduction, sympathetic neurons, Jun kinase
Activation of the c-Jun transcriptional pathway has been shown to be an important regulator of apoptosis of sympathetic neurons induced by nerve growth factor (NGF) withdrawal by both microinjection of antibodies against c-Jun or expression of a c-Jun dominant negative mutant (Estus et al., 1994; Ham et al., 1995). Consistent with these observations, removal of survival factors leads to the activation of c-Jun N-terminal kinase (JNK), either alone or together with p38 mitogen-activated kinase, in a number of neuronal death paradigms, including PC12 cells (Xia et al., 1995), superior cervical ganglion (SCG) neurons (Ham et al., 1995; Eilers et al., 1998), and embryonic motoneurons (Maroney et al., 1998). In addition, phosphorylation of c-Jun and activation of JNK have been observed after neuronal injury in the adult rat brain (Herdegen et al., 1998) (for review, see Mielke and Herdegen, 2000). Taken together, these studies demonstrate that the pathways regulating both the level of c-Jun and its phosphorylation are crucial for the induction of neuronal cell death. Therefore, we were interested in identifying the upstream signaling pathways regulating the activation of the JNK–c-Jun pathway in neurons.
Recently, we have shown that, in rat SCG neurons, the Rho-like GTPases, Cdc42 and Rac1, are required for NGF withdrawal-induced death and that they induce apoptosis via activation of the c-Jun transcriptional pathway (Bazenet et al., 1998). Despite a plethora of complex results (for review, see Johnson, 1999), it is still unclear how Cdc42 directly activates the JNK pathway, especially in neuronal systems. The small GTP-binding proteins act via various, multiple downstream mitogen-activated protein (MAP) kinase kinase kinases, depending on the cell type, which bind the active GTP-bound form of the small G-proteins and induce JNK activity and transcriptional activation (Lim et al., 1996; Tapon and Hall, 1997; Van Aelst and D'Souza-Schorey, 1997) (for review, see Johnson, 1999). There are several likely intermediates in the signaling cascades linking Cdc42 to the JNK pathway, including the p21-activated kinases (rat p65PAK/PAK1/PAK α, PAK-2/PAK γ, PAK-3/PAK β, and PAK4; Bagrodia et al., 1995; Knaus et al., 1995;Manser et al., 1995; Martin et al., 1995; Brown et al., 1996; Abo et al., 1998), the germinal center kinase (GCK; Pombo et al., 1995), the nick-interacting protein (NIK; Su et al., 1997), MEKK1 and MEKK4 (Fanger et al., 1997; Gerwins et al., 1997), plenty of SH3 (POSH) (Tapon et al., 1998), and the mixed lineage kinase family (MLK1, MLK2, MLK3, DLK, LZK) (Dorow et al., 1993; Holzman et al., 1994; Reddy and Pleasure, 1994; Fan et al., 1996; Hirai et al., 1996; Rana et al., 1996; Teramoto et al., 1996; Tibbles et al., 1996; Hirai et al., 1997;Sakuma et al., 1997; Nagata et al., 1998).
In this study we concentrated our efforts on mediators containing a Cdc42/Rac interactive-binding (CRIB) region (Burbelo et al., 1995) and more specifically on members of the MLK family. The MLKs have a distinctive protein kinase domain that shows structural features of both tyrosine and serine/threonine protein kinases, and they all have two leucine zipper-like motifs, located at the C terminus of the kinase domain, which are necessary for dimerization that then precedes the activation of MLKs (Dorow et al., 1993; Ezoe et al., 1994; Gallo et al., 1994; Ing et al., 1994; Leung and Lassam, 1998). The MLK family has been divided into two subgroups, based on their structural differences: the MLK subgroup includes MLK1, MLK2/MST, and MLK3/SPRK/PTK1; the DLK subgroup includes DLK/MUK/ZPK and LZK (which do not contain an SH3 domain or a CRIB domain). Of the three known MLKs, MLK2 and MLK3 interact with GTP-Cdc42 and activate the JNK pathway (Nagata et al., 1998). When overexpressed in mammalian cells, MLK3 activates JNK via the phosphorylation and activation of SEK/MKK4 (Rana et al., 1996) or MKK7 (Whitmarsh et al., 1998). Moreover, putative scaffold proteins, JIP-1 (JNK interacting protein-1) and JIP-2 have recently been identified and shown to interact in a specific manner with members of the MLK family and with MKK7 and JNK, but not with Cdc42, thereby linking these kinase-signaling components (Dickens et al., 1997; Whitmarsh et al., 1998; Yasuda et al., 1999). MLK3 has also been shown to activate the MAPK p38 kinase pathway via MKK3/6 (Tibbles et al., 1996) and the extracellular signal-related protein kinase (ERK) pathway (Hartkamp et al., 1999). In addition, expression of a dominant negative mutant of MLK3 led to a reduction in the Cdc42-dependent activation of JNK (Teramoto et al., 1996), and MLK3 CRIB domain has been shown to be required for its association with and activation by Cdc42 in human embryonic kidney 293 cells (Bock et al., 2000). Altogether, these studies strongly suggest that MLK3 may be an important element within the mechanism of stress-induced activation of the JNK pathway and of neuronal death.
Herein, using wild type (WT) and various MLK3 mutants [kinase dead (KD), CRIB(−), and KD CRIB(−)], we showed that overexpression of a kinase-active MLK3 is sufficient to induce apoptosis in rat sympathetic neurons and that this induction of cell death is accompanied by an increase in the level of phosphorylated c-Jun. In addition, both kinase dead, dominant negative mutants of MLK3 could significantly block NGF withdrawal-induced cell death and its concomitant increase in the phosphorylation of c-Jun. Overexpression of both kinase dead mutants of MLK3 could also block death induced by Cdc42. Finally, an increase in MLK3 activity was observed 5 hr after the induction of apoptosis in both differentiated PC12 cells and SCG neurons. The present study strongly supports the notion that MLK3, or a closely related kinase, plays a crucial and physiological role in neuronal apoptotic cell death.
MATERIALS AND METHODS
Cell culture. Sympathetic neurons were isolated from SCG of 1-d-old Sprague Dawley rats (supplied by the Biological Services Unit, University College London) as described previously (Ham et al., 1995). Briefly, neurons were plated on 13 mm glass coverslips coated with poly-l-lysine and laminin (both from Sigma, Poole, UK) at a density of 8000–10,000 cells per coverslip and cultured in SCG growth medium [DMEM (Sigma), 10% heat inactivated fetal calf serum (FCS; Globepharm, Surrey, UK), 2 mm each of glutamine and penicillin–streptomycin (both from Life Technologies, Paisley, UK), 100 ng/ml NGF, and 20 μm each of the anti-mitotic agents fluoro-deoxyuridine and uridine (Sigma)]. Neurons were kept in culture in the presence of NGF for 5–7 d before being used for experiments. NGF withdrawal was performed by refeeding the cells with the SCG growth medium that lacked NGF but was supplemented with 100 ng/ml of neutralizing anti-NGF antibody (Boehringer Mannheim, Mannheim, Germany). In some experiments, SCG neurons were pretreated for 2 hr with 100 μm of the broad caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp (O-methyl)-fluoromethylketone (zVAD-fmk, Enzyme Systems Products, Dublin, CA) before being withdrawn from NGF. zVAD-fmk was present in the culture medium throughout the experiment.
PC12 cells were seeded at a density of 2 × 106 cells per 9-cm-diameter dish and coated with poly-l-lysine (Sigma) and rat tail collagen (Upstate Biotechnology, Buckingham, UK). The cells were cultured in defined medium supplemented with 10 μg/ml insulin and 2% FCS as described previously (Doherty et al., 1988). PC12 cells were differentiated by culturing them with defined medium supplemented with 100 ng/ml 2.5S NGF (Promega, Southampton, UK). The cells were used within 7–8 d of NGF differentiation. NGF withdrawal was performed by refeeding the cells with the defined medium containing 100 ng/ml of anti-NGF antibody (Boehringer Mannheim).
Jurkat cells were diluted to a density of 2.5 × 104 cells/ml and cultured in DMEM supplemented with 10% FCS, 2 mm glutamine, and penicillin–streptomycin for 24 hr.
Cos-7 cells were grown in DMEM supplemented with 10% FCS, 2 mm glutamine, and penicillin–streptomycin (both from Life Technologies).
Immunoblotting. The culture medium of differentiated PC12 cells and SCG neurons was removed and centrifuged at 2000 rpm for 10 min at 4°C to collect the detached cells. The cells remaining on the dish were rinsed twice with PBS and lysed with 250 μl of cold lysis buffer [ containing (in mm) 50 Tris-HCl, pH 7.4, 250 NaCl, 5 EDTA, 50 NaF, 1 Na3VO4, and 1 PMSF plus 0.1% Nonidet P-40, 10 μg/ml aprotinin, 1 μg/ml leupeptin, and 10 μg/ml TPCK]. The cells were scraped, and the lysate was added to the pellet of detached apoptotic cells and further incubated on ice for 10 min. The Jurkat cells were centrifuged for 5 min at 2000 rpm and resuspended in 100 μl of lysis buffer. The nuclei and cell membranes were removed by centrifugation at 5000 rpm for 15 min at 4°C. Protein concentration in the supernatants was determined by the Bradford method.
Thirty micrograms of protein were resolved by electrophoresis, using 12.5% SDS-polyacrylamide gels. Then the proteins were transferred to nitrocellulose. The membranes were incubated for 1 hr at room temperature in blocking buffer (5% nonfat dry milk, 0.1% Tween 20 in PBS) and then overnight at 4°C with 200 ng/ml of an anti-MLK3 primary antibody (C-20, Santa Cruz, Santa Cruz, CA) in blocking buffer. In peptide competition experiments the diluted antibody was incubated with a 10-fold excess (in micrograms) of the antigen peptide (Santa Cruz) for 2 hr at room temperature before being used for immunoblotting. After rinsing, the membranes were washed and incubated with a horseradish peroxidase-conjugated goat anti-rabbit (Amersham, Buckinghamshire, UK) diluted 1:2000 in blocking buffer. The blots were developed with the ECL system (Amersham).
In vitro MLK3 kinase assay. MLK3 was immunoprecipitated from differentiated PC12 cells or SCG neurons maintained in the presence or absence of NGF; the lysates were precleared by the addition of 15 μl of 1:1 slurry of protein A-agarose beads previously washed in lysis buffer [containing (in mm) 50 HEPES, pH 7.5, 150 NaCl, 1 EGTA, 150 NaF, 1.5 MgCl2, 1 Na3VO4, 10 β-glycerophosphate, and 1 PMSF plus 1% Triton X-100, 10% glycerol, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin]. After 15 min of incubation at 4°C, 7.5 μg of MLK3 antibody was added to the lysate of 4 × 106 cells (PC12 cells) or 1.5 × 105 cells (SCG neurons) together with 15 μl of 1:1 slurry of prewashed protein A-agarose beads. Then the mixture was incubated end over end for 1 hr. The beads were spun down for 2 min at 4°C and washed twice with lysis buffer and once with MLK3 buffer (50 mm Tris-HCl, pH 7.4, 10 mm MgCl2, and 1 mm EGTA). Finally, the beads were resuspended in 10 μl of kinase buffer and incubated with 20 μg/ml of myelin basic protein (MBP) as an exogenous substrate for 10 min at 4°C, after which 10 μCi of [γ-32P]ATP was added to the immunoprecipitates. The reaction was allowed to proceed for 10 min at 30°C and was stopped by the addition of 20 μl of 2× sample buffer (4% SDS, 5.6 m 2-mercaptoethanol, 20% glycerol, 200 mm Tris-HCl, pH 6.8, and 1% bromophenol blue) containing 20 mm EDTA. The samples were heated up at 98°C and then separated on a 12.5% SDS-PAGE polyacrylamide gel. The gel was stained with colloidal blue (Invitrogen, Groningen, The Netherlands) for 3 hr at room temperature, destained with water, and dried. The labeled proteins were revealed by autoradiography or by using the STORM PhosphorImager analysis (Molecular Dynamics, Amersham Pharmacia Biotech, Buckinghamshire, UK); the intensity of each band was determined by using ImageQuant software (Molecular Dynamics, Amersham Pharmacia Biotech). The results were plotted as a graph.
Microinjection and survival assay. Sympathetic neurons were microinjected as described previously (Bazenet et al., 1998). All plasmids were double-purified on a cesium chloride gradient and resuspended in H2O. The injection mix comprised plasmid DNA in 0.5× PBS (−Ca2+ and −Mg2+) and 5 mg/ml 70,000 kDa Texas Red-dextran (survival assay; Molecular Probes, Eugene, OR) or 2.5 mg/ml purified guinea pig IgG (immunostaining analysis; Sigma) to identify the injected cells. Four hours after injection the neurons were re-fed as described above. The percentage of cell survival was assessed as described previously (Bazenet et al., 1998). Briefly, only the injected cells displaying a normal nucleus and intact neurites were counted as alive.
Immunofluorescence analysis. At 24 hr after microinjection, SCG neurons were fixed in 3% para-formaldehyde, permeabilized with 0.5% Triton X-100 in PBS, and blocked with 50% goat serum and 0.5% BSA in PBS. Then they were stained with a specific monoclonal phospho-c-Jun antibody (raised against a phosphopeptide, encompassing amino acids 57–68) and with phosphoserine 63 (Watson et al., 1998) or a monoclonal antibody against the FLAG epitope (Sigma) and then with a FITC-conjugated secondary antibody and a rhodamine-conjugated anti-guinea pig IgG antibody (Stratech Scientific, Luton, UK) to detect the injected cells. Only those cells showing a clear increase over the background staining were scored as positive. To examine nuclear morphology, were stained with Hoechst dye (Hoechst 33342, Sigma) at 10 μg/ml. Shrunken, condensed, or fragmented nuclei were considered pyknotic.
Preparation of total RNA from SCG neurons. The growth medium of SCG neurons was transferred to a centrifuge tube and kept to collect the detached apoptotic cells. The cells remaining on the dish were harvested by rinsing them in a small volume of ice-cold PBS. The adherent and floating cells were pooled and spun at 2000 rpm for 10 min at 4°C and rinsed twice with PBS; total RNA was prepared with the RNeasy mini kit (Qiagen, West Sussex, UK).
Reverse transcription-PCR. First-strand cDNA was prepared from the total RNA extracts from SCG neurons with Superscript II RNase H Reverse Transcriptase (Life Technologies, Paisley, UK). Briefly, 2 μg of total SCG RNA was incubated with 0.5 μg of Oligo-dT12–18 primer (Life Technologies) and 3 μg of random primers (Life Technologies) in a total volume of 20 μl for 10 min at 70°C. The reaction mix was placed on ice for 1–2 min before the addition of 4 μl of First-Strand Buffer, 2 μl 0.1m DTT (all from Life Technologies), and 1 μl of 10 mm dNTP mix. The contents of the tube were mixed gently and incubated at 42°C for 2 min. Two hundred units of SuperScript II were added, and the reaction mix was incubated for 50 min at 42°C. The reaction was inactivated by heating to 70°C for 15 min. The PCR reactions were performed with the Elongase Enzyme Mix kit (Life Technologies) by following the manufacturer's recommendations. The following primers were used: forward primer, 5′-GTCATGGAATGGCAGTGG-3′; reverse primer, 5′-GGCTGTAGTCGAACAGG-3′. The PCR products were analyzed on a 1% agarose gel.
Plasmids. pcDNA-FLAG MLK3 and MLK3 KD were obtained from Jim Woodgett (Ontario Cancer Institute, Toronto, Canada) (Tibbles et al., 1996). The CRIB(−) mutants MLK3 S493P, P495A, and H500L were made by using the Gene Editor in vitro site-directed mutagenesis system (Promega). V12Cdc42 was a gift from Alan Hall (University College London, London, UK).
In vivo binding of MLK3 proteins to Cdc42. Cos-7 cells were transiently transfected with 0.5 mg of myc-tagged Cdc42 (pRK5) or vector control along with 2 mg of the various FLAG-tagged MLK3 constructs by the Lipofectamine method (Life Technologies). Cells were harvested 48 hr after transfection in a Nonidet P-40 lysis buffer [1% Nonidet P-40, 10% glycerol, and (in mm) 20 Tris, pH 8.0, 137 NaCl, 50 NaF, 10 β-glycerophosphate, 1 PMSF, and 10 sodium orthovanadate plus 10 μg/ml aprotinin]. The lysates were adjusted for equal protein, and each sample was divided so that two-thirds of the lysate was incubated with anti-myc antibodies (A-14, Santa Cruz, Santa Cruz, CA) to immunoprecipitate Cdc42 along with bound MLK3 proteins; one-third of the lysate was incubated with anti-FLAG antibodies (M2, Scientific Imaging Systems, Kodak, Rochester, NY) to immunoprecipitate MLK3 protein. The immune complexes were collected with protein A-agarose beads (Pierce, Rockford, IL), washed with the above lysis buffer without protease inhibitors, and analyzed by Western blotting to detect MLK3 protein with anti-FLAG antibodies.
RESULTS
MLK3 is expressed in neuronal cells
MLK3 expression in rat neurons was determined at both mRNA and protein levels. Primers, based on the human MLK3 sequence for a 120 base pair glycine-rich region that is highly specific for MLK3 (amino acids 15–54; Sakuma et al., 1997), were designed and RNA-extracted from either PC12 cells or purified cultures of SCG neurons and then amplified by RT-PCR. A single PCR product of 120 bases from both PC12 cells and SCG neurons, as predicted for MLK3, was obtained (Fig.1A). Sequencing of these products confirmed that they were derived from MLK3. The identified rat sequence shares >83% homology with the human sequence at the amino acid level. Our second approach was to analyze cell extracts for the presence of MLK3 protein. Extracts from Jurkat cells (positive control for the antibody), PC12 cells, and SCG neurons were prepared. Western blotting that used the MLK3 antibody detected a 95 kDa band in the Jurkat cells, PC12 cells, and SCG extracts (Fig.1B), confirming that MLK3 indeed is expressed in neurons. The immunodetection of MLK3 could be competed by a 10-fold excess by weight of the peptide antigen, confirming the specificity of this antibody.
Fig. 1.

MLK3 is expressed in sympathetic neurons.A, MLK3 RNA is expressed in sympathetic neurons. Sympathetic neurons cultured for 6 d in the presence of NGF were lysed in RNA lysis buffer, and total RNA was extracted. RT-PCR was performed on the total RNA, and the products were analyzed on a 1.0% agarose gel. The amplified 120 bp fragment was sequenced and is consistent with MLK3. B, MLK3 protein is expressed in sympathetic neurons and PC12 cells. Cell extracts were prepared from SCG neurons, cultured for 6 d in the presence of NGF, 7-d-differentiated PC12 cells, and Jurkat cells. Then 30 μg of protein was resolved on a 12.5% SDS-PAGE polyacrylamide gel, transferred onto nitrocellulose, and probed with a polyclonal antibody to MLK3. The 95 kDa MLK3 band is competed away by the addition of the peptide antigen.
Characterization of MLK3 mutants and expression in SCG neurons
To investigate the role of MLK3 in neuronal death induced either by NGF withdrawal or the expression of Cdc42, we examined the effect of different MLK3 mutants [K144E, CRIB(−), and CRIB(−)K144E]. The K144E mutation in the ATP-binding site inactivates MLK3, thereby acting as a kinase dead (KD), dominant negative mutant (Tibbles et al., 1996). To abrogate the binding of MLK3 to the small GTP-binding proteins, we introduced additional mutations, S493P, P495A, and H500L (three of five crucial amino acids in the consensus CRIB sequence; Burbelo et al., 1995). These amino acids were replaced in both wild type (WT) and KD MLK3 to generate the corresponding CRIB(−) mutants. To verify the binding properties of these mutants, we transiently transfected Cos-7 cells with myc-tagged Cdc42, along with the various FLAG-tagged MLK3 constructs. Then 2 d later the immunoprecipitates of the myc-tagged Cdc42 were analyzed by Western blotting for the presence of bound MLK3. In addition, the FLAG-tagged MLK3 proteins were immunoprecipitated to check their level of expression. WT and KD MLK3, but not WT CRIB(−) or KD CRIB(−), coimmunoprecipitated with Cdc42, demonstrating that the CRIB(−) mutants fail to bind the activated Cdc42 and function as expected (Fig.2A).
Fig. 2.

Characterization of the MLK3 mutants and expression in SCG neurons. A, In vivobinding of MLK3 proteins with Cdc42. Cos-7 cells were transiently transfected with 2 μg of various FLAG-tagged MLK3 constructs along with 0.5 μg of myc-tagged Cdc42. Cells were harvested and lysed 48 hr after transfection. Cdc42 and MLK3 were immunoprecipitated with anti-myc and anti-FLAG antibodies, respectively. Then the immune complexes were analyzed by Western blotting, using the FLAG antibody to detect MLK3 protein. B, Subcellular localization of the different MLK3 mutants in SCG neurons. Sympathetic neurons, cultured for 5–7 d in the presence of NGF, were microinjected with 0.3 mg/ml plasmid DNA and 5 mg/ml guinea pig IgG to mark the injected cells. At 24 hr after injection the cells were stained with an anti-FLAG antibody to detect MLK3 expression, an anti-guinea pig IgG antibody (anti-GPIgG) to follow the injected cells, and with Hoechst to visualize the nuclei. Scale bar, 30 μm. Thearrows point to cells displaying pyknotic nuclei.
Next, we microinjected rat sympathetic neurons, cultured for 5–7 d in the presence of NGF, with 0.3 mg/ml of an empty expression vector or the various MLK3 mutants. At 24 hr after injection the cells were stained with an anti-FLAG antibody to check the level of expression. We consistently found that in all cases 80–100% of the injected cells expressed the MLK3 construct. Interestingly, we noticed that the kinase-active forms [WT and CRIB(−)] of MLK3 were expressed mainly at the plasma membrane, whereas the kinase dead mutants [KD and KD CRIB(−)] were seen throughout the cytoplasm of the neurons (Fig.2B). The leucine zipper motifs of MLK3 are necessary and sufficient for the dimerization of the protein, which in turn is a prerequisite for the transphosphorylation and autoactivation of MLK3 (Leung and Lassam, 1998). It appears that overexpression of MLK3 mutants is sufficient to drive dimerization as long as they contain the leucine zipper motifs. However, translocation of MLK3 from the cytoplasm to the plasma membrane occurred only with a kinase-active mutant. Surprisingly, the mutations in the CRIB domain had no effect on the subcellular localization of MLK3. This suggests that transphosphorylation recruits MLK3 to a membrane component other than or in addition to Cdc42.
MLK3 induces neuronal cell death
We then examined the effect of these mutants on the survival of SCG neurons cultured in the presence of NGF. SCG neurons were microinjected with 0.3 mg/ml of an empty expression vector or of the different MLK3 mutants. The percentage of survival was assessed 48 hr after injection. Expression of the kinase-active forms [WT and CRIB(−)] of MLK3 significantly decreased the survival of SCG neurons in a dose-dependent manner (Fig.3A; data not shown), whereas the empty vector and the kinase dead mutants [KD and KD CRIB(−)] had no effect (Fig. 3A). The nuclear morphology of the injected neurons was characterized by Hoechst staining. At 24 hr after injection the cells expressing WT and WT CRIB(−) clearly displayed pyknotic nuclei, a hallmark of apoptotic cells (Figs. 2B,3B), whereas neither of the KD mutants had an effect on the nuclear morphology of the injected cells. These results demonstrate that MLK3 can induce neuronal apoptosis in the presence of NGF and that its kinase activity is required for its proapoptotic effect.
Fig. 3.

MLK3 induces neuronal apoptosis. A, Induction of neuronal cell death by MLK3 in SCG neurons. Rat sympathetic neurons, cultured for 5–7 d in the presence of NGF, were microinjected with 0.3 mg/ml empty expression vector (E) or the different MLK3 constructs [WT, KD, CRIB(−), andKDCRIB(−)] along with 70 kDa Texas Red-dextran to mark the injected cells. Then 48 hr later the percentage of surviving cells was assessed. In each experiment 200 cells were injected. The results are the mean of three independent experiments ± SEM. B, MLK3 increases the number of pyknotic nuclei in SCG neurons. SCG neurons were injected with 0.3 mg/ml control empty expression vector or the various constructs of MLK3. After 24 hr the nuclear morphology was visualized by Hoechst staining. The results are the mean of three independent experiments ± SEM. C, zVAD-fmk protects SCG neurons from MLK3-induced death. The 5- to 7-d-old sympathetic neurons were pretreated with 100 μm zVAD-fmk for 2 hr or were left untreated before microinjection with 0.3 mg/ml WT MLK3. The percentage of surviving cells was assessed 48 hr later. The results are the mean of three independent experiments ± SEM.
To confirm further the apoptotic nature of this death, we examined whether an inhibitor of caspases could protect SCG neurons from MLK3-induced death. Sympathetic neurons were pretreated with 100 μm of zVAD-fmk for 2 hr or maintained in their culture medium before microinjection with WT MLK3. The percentage of surviving cells was assessed 48 hr later. As a control, we treated SCG neurons with zVAD-fmk and maintained them in the presence of NGF or deprived them of NGF. zVAD-fmk had no toxic effect and rescued SCG neurons from NGF withdrawal (data not shown). We also found that zVAD-fmk could rescue neurons from MLK3-induced death to levels almost similar to +NGF control, suggesting that MLK3 can activate a caspase-dependent apoptotic pathway in SCG neurons (Fig. 3C).
MLK3 activity is increased after NGF deprivation in differentiated PC12 cells and in SCG neurons
More importantly, we wanted to examine whether the induction of neuronal apoptosis by NGF deprivation had any effect on the endogenous MLK3. To address this issue, we determined MLK3 activity in both differentiated PC12 cells and SCG neurons. PC12 cells were differentiated for 7 d and either maintained in the presence of NGF or withdrawn from NGF, as described in Materials and Methods. After 3, 5, and 7 hr the cells were lysed, and MLK3 was immunoprecipitated with a specific antibody raised against the C-terminal region of MLK3. The kinase activity of the resulting immunoprecipitates was assayed against the myelin basic protein. A 2.5-fold increase in MLK3 activity above basal level was observed and peaked at ∼5 hr after NGF withdrawal, whereas no significant variations were seen when the cells were maintained in the presence of NGF (Fig.4A,B).
Fig. 4.
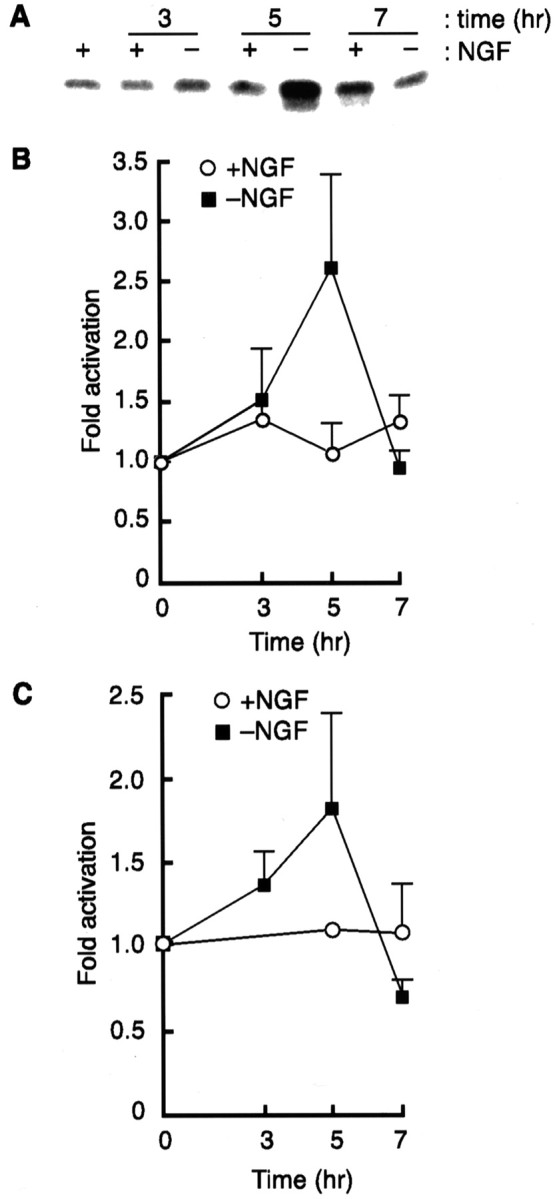
MLK3 activity is increased after NGF withdrawal in PC12 cells and in SCG neurons. MLK3 kinase assays were performed in differentiated PC12 cells (A, B) and in SCG neurons (5 DIV; C) that had been maintained in the presence of NGF or withdrawn from NGF for 3, 5, and 7 hr, as described in Materials and Methods. Fold activation of MLK3 activity was obtained by scanning the gels on a STORM PhosphorImager. A representative autoradiograph of a MLK3 kinase assay in PC12 cells is shown inA. The level of MLK3 activity at time 0was set at 1. The results are the mean of three independent experiments ± SEM.
We next measured MLK3 activity in SCG neurons [5 d in vitro(5 DIV)] either maintained in the presence of NGF or withdrawn from NGF for 3, 5, and 7 hr. A comparable increase in the basal level of MLK3 activity was observed in apoptotic SCG neurons (Fig.4C). Our results suggest that NGF withdrawal leads to the activation of MLK3 in both differentiated PC12 cells and SCG neurons, implicating a role for MLK3 as a physiological mediator of neuronal apoptosis.
MLK3 activity is required for NGF withdrawal- and Cdc42-induced neuronal death
To confirm that MLK3 plays a role in NGF withdrawal-induced death, we microinjected each of the MLK3 constructs as well as an empty expression vector control (negative control) and Bcl-2 (positive control) into SCG neurons. The cells were withdrawn from NGF, and the percentage of surviving cells was assessed 48 hr later. Both dominant negative mutants [KD and KD CRIB(−)] protected the neurons from NGF withdrawal-induced death to levels similar to those obtained with Bcl-2 (Fig. 5A). Neither WT forms of MLK3 [WT and WT CRIB(−)] rescued the sympathetic neurons (Fig.5A). In addition, we looked at the effect of these mutants on the nuclear morphology of the injected neurons by Hoechst staining at 24 hr after injection. The cells injected with the empty vector control had clearly started to display pyknotic nuclei, whereas the cells injected with the kinase dead mutants of MLK3 had a much lower percentage of condensed or fragmented nuclei (Fig. 5B). These results confirm that MLK3 is involved in the mediation of apoptosis in SCG neurons and that its kinase activity seems to be an important requirement for the execution of neuronal cell death.
Fig. 5.

MLK3 is required for NGF withdrawal- and Cdc42-induced deaths. A, MLK3-KD mutants prevent NGF withdrawal-induced cell death in SCG neurons. SCG neurons, cultured for 5–7 d in the presence of NGF, were microinjected with 70 kDa Texas Red-dextran and 0.3 mg/ml of the various MLK3 mutants, an empty vector, or 0.05 mg/ml Bcl-2. Then 4 hr later the cells were withdrawn from NGF, and the number of injected cells was scored (100% value). The percentage of surviving cells was assessed after 48 hr, as described in Materials and Methods. The results are the mean of three independent experiments ± SEM. B, MLK3-KD mutants decrease the number of pyknotic nuclei after NGF withdrawal from SCG neurons. Neurons were injected with 0.3 mg/ml control empty expression vector or the different MLK3 mutants and withdrawn from NGF. After 24 hr the nuclear morphology was visualized by Hoechst staining. The results are the mean of three independent experiments ± SEM.C, Cdc42-induced apoptosis requires MLK3 activity. We coinjected 0.3 and 0.5 mg/ml KD MLK3 along with 0.1 mg/ml V12Cdc42 and 70 kDa Texas Red-dextran into SCG neurons. The cells were maintained in the presence of NGF, and the percentage of surviving cells was assessed 48 hr after injection as described previously. The results are the mean of three independent experiments ± SEM. D, Cdc42-induced apoptosis does not require binding to MLK3. KD CRIB(−)MLK3, at the indicated concentrations (in mg/ml), and 0.1 mg/ml V12Cdc42 were microinjected into 5- to 7-d-old SCG neurons maintained in the presence of NGF. The percentage of surviving cells was assessed 48 hr later. The results are the mean of three independent experiments ± SEM.
We have recently shown that constitutively activated forms of the Rho family of GTPases, Cdc42 and Rac1, could induce apoptosis of SCG neurons via the activation of the c-Jun transcriptional pathway (Bazenet et al., 1998). To examine whether MLK3 and Cdc42 were on the same death-signaling pathway, we coinjected an activated mutant of Cdc42 (V12Cdc42) with both the CRIB(+) or CRIB(−) kinase dead mutants of MLK3 into SCG neurons. The percentage of surviving cells was assessed 48 hr after injection. Regardless of its ability to bind Cdc42, a kinase-inactive mutant of MLK3 efficiently blocks V12Cdc42-induced death (Fig. 5C,D), suggesting that the kinase activity of MLK3, but not its binding to Cdc42, is crucial for the induction of apoptosis by Cdc42 and that MLK3 is a downstream mediator of Cdc42 signaling in sympathetic neurons. These results demonstrate that blocking MLK3 activity is sufficient to inhibit both NGF withdrawal- and Cdc42-induced death.
Overexpression of MLK3 induces an increase in the level of phosphorylated c-Jun
MLK3 has been shown previously to activate the JNK pathway in non-neuronal cells (Teramoto et al., 1996; Tibbles et al., 1996). To investigate whether this is the case in sympathetic neurons, we examined the effect of MLK3 on the phosphorylation of c-Jun in both the presence and the absence of NGF. The 5- to 7-d-old SCG neurons were microinjected with an empty vector or with each of the different MLK3 mutants. The cells were fixed, permeabilized, and stained with a specific phospho-c-Jun antibody. Cells injected with either the empty vector control or the kinase dead mutants of MLK3 did not show any increase in the level of phosphorylated c-Jun compared with the noninjected cells (Fig.6A,B). However, overexpression of the kinase-active forms of MLK3 induced a clear increase in the levels of nuclear phosphorylated c-Jun (Fig.6A,B). Moreover, we examined whether dominant negative mutants of MLK3 could prevent the increase in phosphorylated c-Jun levels that occurs after NGF withdrawal. Overexpression of both kinase dead mutants in sympathetic neurons blocked the increase in the level of phosphorylated c-Jun induced by NGF withdrawal, whereas the empty vector and the kinase-active forms of MLK3 did not (Fig.6B).
Fig. 6.
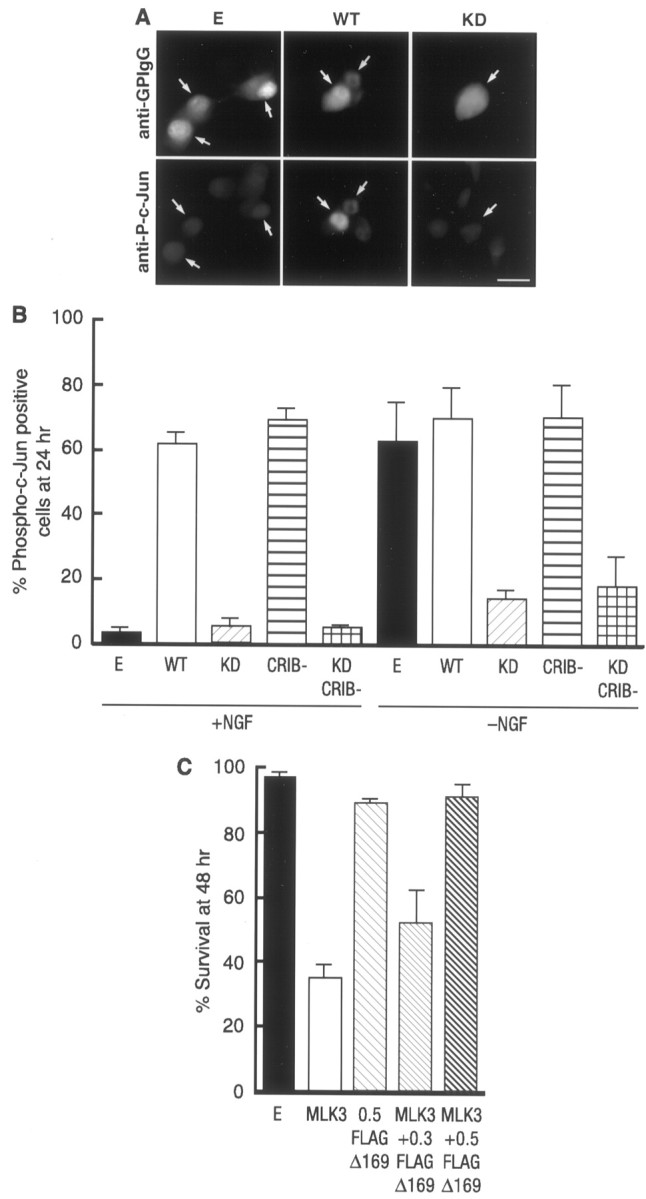
MLK3 activates the JNK pathway in neurons. Shown is the MLK3-dependent phosphorylation of c-Jun in SCG neurons. SCG neurons were microinjected either with 0.3 mg/ml of an empty expression vector or the different MLK3 mutants, together with 5 mg/ml guinea pig IgG to detect the injected cells, and maintained in the presence of NGF or withdrawn from NGF as indicated. After 24 hr the cells were fixed, permeabilized, and stained with an anti-guinea pig IgG antibody (A, top) and with a specific anti-phospho-c-Jun antibody (A, bottom). Only the cells in which phospho-c-Jun staining was clearly above background were scored as positive. Arrows indicate injected cells. Scale bar, 30 μm. The results were quantified and represented as a bar graph (B). They are the mean of three independent experiments ± SEM. C, FLAG-Δ169 blocks MLK3-induced apoptosis. FLAG-Δ169 at the indicated concentrations (in mg/ml) and 0.1 mg/ml of WT MLK3 were microinjected into 5- to 7-d-old SCG neurons and maintained in the presence of NGF. The percentage of surviving cells was assessed 48 hr later. The results are the mean of three independent experiments ± SEM.
An accumulation of phosphorylated c-Jun in the nucleus should lead to the activation of the c-Jun transcriptional pathway. To investigate whether MLK3-induced apoptosis requires the activation of the c-Jun transcriptional pathway, we coinjected sympathetic neurons with WT MLK3 and FLAG-Δ169, a dominant negative mutant of c-Jun that lacks the N-terminal transactivation domain and acts as a dominant inhibitor of AP-1 activity (Ham et al., 1995). Coexpression of FLAG-Δ169 completely blocked MLK3-induced death (Fig. 6C). Taken together, these results demonstrate that the MLK3 activity is important for the induction of c-Jun phosphorylation in sympathetic neurons deprived of NGF and that the death signal induced by MLK3 is mediated by the JNK–c-Jun transcriptional pathway.
DISCUSSION
The Cdc42–JNK–c-Jun pathway is a crucial component of the induction of apoptosis of sympathetic neurons by growth factor deprivation (Estus et al., 1994; Ham et al., 1995; Bazenet et al., 1998). Here, we investigated the relationship between MLK3 and the activation of the Cdc42–JNK–c-Jun pathway in this paradigm. In contrast to Merritt et al. (1999), who could not detect MLK3 in neurons of rat brain slices by immunofluorescence with the same MLK3 antibody, we found that MLK3 is expressed endogenously in rat pheochromocytoma cells and SCG neurons at both mRNA (Fig.1A) and protein levels (Fig. 1B) as well as by immunocytochemistry (data not shown).
Recently, Leung and Lassam (1998) have shown that the leucine zipper-like motifs of MLK3 are sufficient for its dimerization. In addition, they demonstrated that dimerization of MLK3 is a prerequisite for its autophosphorylation and, thereby, activation. Furthermore,Leung and Lassam (1998) found that Cdc42 led to an increase in MLK3 dimerization, suggesting that recruitment of MLK3 to the plasma membrane by Cdc42 might increase the local concentration of MLK3 and therefore the chances of dimerization. To study the regulation by Cdc42 and the role of MLK3 in neurons, we constructed a series of mutants. The KD mutants have an inactive kinase domain, thereby acting as dominant negatives (Tibbles et al., 1996), and the CRIB(−) mutants can no longer bind to activated Cdc42, as shown by our immunoprecipitation studies (Fig. 2A). In the light of the above findings, dimerization should still occur in the kinase dead mutants, and the translocation of the CRIB-containing mutants to the membrane would be increased compared with the CRIB(−) ones. However, we found that neither of the KD mutants locates to the plasma membrane, whereas the mutants with an intact kinase domain [CRIB(+) and CRIB(−)] do (Fig. 2B), suggesting that autophosphorylation, but not interaction with Cdc42, is required for the translocation of MLK3. We cannot exclude, however, that overexpression of MLK3 may override the necessity for upstream activators of dimerization.
When we looked at the functional effect of these mutants on the survival of SCG neurons in the presence of NGF, we found that overexpression of both WT and WT CRIB(−) MLK3 dramatically increased the death of the neurons, whereas both the kinase dead mutants had no effect (Fig. 3A). The MLK3-injected [WT and WT CRIB(−)] neurons clearly displayed pyknotic nuclei (Figs. 2B,3B), a hallmark of apoptosis. In addition, zVAD-fmk, a broad caspase inhibitor that has been shown to protect SCG neurons from NGF withdrawal (McCarthy et al., 1997), protected sympathetic neurons from MLK3-induced death (Fig. 3C). This supports the notion that MLK3 has a role in the induction of apoptosis in SCG neurons. More importantly, a rapid increase in MLK3 activity was observed in both differentiated PC12 cells and SCG neurons after NGF deprivation (Fig.4A–C), suggesting that MLK3 may act as a physiological activator of the death pathway in neuronal cells.
Blocking MLK3 activity by overexpression of both MLK3 kinase dead mutants is sufficient to prevent neuronal apoptosis (Fig.5A,B), confirming the apoptotic effect of MLK3. Although it is possible that the KD MLK3 interferes with a related kinase, because MLK3 can form complexes with coexpressed MLK2 or DLK (Leung and Lassam, 1998; Tanaka and Hanafusa, 1998), our observations strongly suggest a role of MLK3 in the induction of neuronal apoptosis.
Because overexpression of MLK3 has been shown to specifically activate JNK in non-neuronal cells (Teramoto et al., 1996; Tibbles et al., 1996), we investigated the effect of the MLK3 mutants on the phosphorylation of c-Jun in both the presence and absence of NGF. We showed that overexpression of MLK3 [WT and CRIB(−)] induced an increase in the phosphorylation of c-Jun and that kinase dead mutants of MLK3 [KD and KD CRIB(−)] significantly blocked the increase in the level of phosphorylated c-Jun induced by NGF withdrawal (Fig.6A,B). In addition, we showed that AP-1 activity is necessary for MLK3-induced apoptosis, because expression of FLAG-Δ169, a c-Jun dominant negative mutant that inhibits AP-1 activity (Ham et al., 1995), efficiently blocked MLK3-induced death (Fig. 6C). These results demonstrate that not only does MLK3 mediate activation of the JNK–c-Jun transcriptional pathway but also, in dying SCG neurons, MLK3 activity is required for the activation of that pathway.
A hypothetical model, consistent with the above observations, is presented in Figure 7. We propose that NGF withdrawal leads to the activation of Cdc42, which then activates MLK3, thereby turning on the JNK–c-Jun pathway. To test our model, we coinjected KD and KD CRIB(−) MLK3 mutants and V12Cdc42 into SCG neurons and assessed the percentage of survival 48 hr later. Figure 5,C and D, shows that kinase dead mutants of MLK3 block apoptosis induced by overexpression of Cdc42, suggesting that MLK3 activity is important for the induction of cell death by Cdc42 and acts as a downstream mediator of Cdc42 in SCG neurons. The inhibitory activity of the kinase dead MLK3 mutants is not related to sequestration of Cdc42 in neurons. Consistent with this, Bock and coworkers (2000) showed that, although Cdc42 activates MLK3, it is not necessary to maintain MLK3 in an activated state and that activation of MLK3 by Cdc42 requires an additional cellular component.
Fig. 7.

A model for the apoptotic signaling pathway mediated by NGF withdrawal. Removal of the survival agent, NGF, activates the small GTPase Cdc42. Activated Cdc42 leads to the activation of ASK1 and MLK3. Activation of MLK3 requires two steps: dimerization (1) and autophosphorylation (2); then it translocates to the plasma membrane. Activation of ASK1 and/or MLK3 leads to an increase in JNK activity via the formation of a multiprotein complex. Phosphorylation of c-Jun activates its transcriptional activity, which then turns on the transcription of genes that are necessary for neuronal apoptosis to proceed.
Recently, we have demonstrated that the apoptosis signal-regulating kinase (ASK1) is a crucial element of NGF withdrawal-induced activation of the Cdc42–c-Jun pathway and neuronal apoptosis (Kanamoto et al., 2000). ASK1 has also been found in complexes including the JIPs (JNK-interacting proteins) but to a much lower extent than MLK3 (Dickens et al., 1997; Yasuda et al., 1999). The formation of such multiprotein complexes might be a convergence point of various pathways mediated by ASK1 and by MLK3. This hypothesis could explain the fact that expression of dominant negative mutants of either kinase blocks the death pathway in neurons as they may compete for common downstream partners. The relationship between ASK1 and MLK3 and a mechanism of how these kinases are activated remain to be elucidated. So we completed our model (Fig. 7) by suggesting that both ASK1 and MLK3, or a related MLK protein, are activated after NGF withdrawal downstream of Cdc42, thereby inducing apoptosis of SCG neurons via the common activation of the JNKK1/2–JNK–c-Jun pathway. Blocking the activity of these kinases could then be of therapeutic benefit in a number of neurodegenerative disorders thought to involve neuronal apoptosis.
Footnotes
We thank J. Woodgett for providing the MLK3 constructs and A. Hall for the Cdc42 plasmid. We also thank Cesare Spadoni and Jonathan Whitfield for technical advice and Joanne Taylor, Jim Staddon, and Stephen Neame for a critical reading of this manuscript and helpful discussions.
Correspondence should be addressed to Dr. Chantal Bazenet, Eisai London Research Laboratories, Bernard Katz Building, University College London, Gower Street, London WC1E 6BT, UK. E-mail:Chantal_Bazenet@eisai.net.
REFERENCES
- 1.Abo A, Qu J, Cammarano MS, Dan C, Fritsch A, Baud V, Belisle B, Minden A. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 1998;17:6527–6540. doi: 10.1093/emboj/17.22.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bagrodia S, Derijard B, Davis RJ, Cerione RA. Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J Biol Chem. 1995;270:27995–27998. doi: 10.1074/jbc.270.47.27995. [DOI] [PubMed] [Google Scholar]
- 3.Bazenet CE, Mota MA, Rubin LL. The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc Natl Acad Sci USA. 1998;95:3984–3989. doi: 10.1073/pnas.95.7.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bock BC, Vacratsis PO, Qamirani E, Gallo KA. Cdc42-induced activation of the mixed-lineage kinase SPRK in vivo. J Biol Chem. 2000;275:14231–14241. doi: 10.1074/jbc.275.19.14231. [DOI] [PubMed] [Google Scholar]
- 5.Brown JL, Stowers L, Baer M, Trejo J, Coughlin S, Chant J. Human Ste20 homologue hPAK1 links GTPases to the JNK MAP kinase pathway. Curr Biol. 1996;6:598–605. doi: 10.1016/s0960-9822(02)00546-8. [DOI] [PubMed] [Google Scholar]
- 6.Burbelo PD, Drechsel D, Hall A. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J Biol Chem. 1995;270:29071–29074. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- 7.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 8.Doherty P, Seaton P, Flanigan TP, Walsh FS. Factors controlling the expression of the NGF receptor in PC12 cells. Neurosci Lett. 1988;92:222–227. doi: 10.1016/0304-3940(88)90065-1. [DOI] [PubMed] [Google Scholar]
- 9.Dorow DS, Devereux L, Dietzsch E, De Kretser T. Identification of a new family of human epithelial protein kinases containing two leucine/isoleucine-zipper domains. Eur J Biochem. 1993;213:701–710. doi: 10.1111/j.1432-1033.1993.tb17810.x. [DOI] [PubMed] [Google Scholar]
- 10.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-Jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ezoe K, Lee ST, Strunk KM, Spritz RA. PTK1, a novel protein kinase required for proliferation of human melanocytes. Oncogene. 1994;9:935–938. [PubMed] [Google Scholar]
- 13.Fan G, Merritt SE, Kortenjann M, Shaw PE, Holzman LB. Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk, but not ERK2. J Biol Chem. 1996;271:24788–24793. doi: 10.1074/jbc.271.40.24788. [DOI] [PubMed] [Google Scholar]
- 14.Fanger GR, Johnson NL, Johnson GL. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J. 1997;16:4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallo KA, Mark MR, Scadden DT, Wang Z, Gu Q, Godowski PJ. Identification and characterization of SPRK, a novel src-homology 3 domain-containing proline-rich kinase with serine/threonine kinase activity. J Biol Chem. 1994;269:15092–15100. [PubMed] [Google Scholar]
- 16.Gerwins P, Blank JL, Johnson GL. Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J Biol Chem. 1997;272:8288–8295. doi: 10.1074/jbc.272.13.8288. [DOI] [PubMed] [Google Scholar]
- 17.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 18.Hartkamp J, Troppmair J, Rapp UR. The JNK/SAPK activator mixed lineage kinase 3 (MLK3) transforms NIH 3T3 cells in a MEK-dependent fashion. Cancer Res. 1999;59:2195–2202. [PubMed] [Google Scholar]
- 19.Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirai S, Izawa M, Osada S, Spyrou G, Ohno S. Activation of the JNK pathway by distantly related protein kinases, MEKK and MUK. Oncogene. 1996;12:641–650. [PubMed] [Google Scholar]
- 21.Hirai S, Katoh M, Terada M, Kyriakis JM, Zon LI, Rana A, Avruch J, Ohno S. MST/MLK2, a member of the mixed lineage kinase family, directly phosphorylates and activates SEK1, an activator of c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 1997;272:15167–15173. doi: 10.1074/jbc.272.24.15167. [DOI] [PubMed] [Google Scholar]
- 22.Holzman LB, Merritt SE, Fan G. Identification, molecular cloning, and characterization of dual leucine zipper-bearing kinase. A novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. J Biol Chem. 1994;269:30808–30817. [PubMed] [Google Scholar]
- 23.Ing YL, Leung IW, Heng HH, Tsui LC, Lassam NJ. MLK-3: identification of a widely-expressed protein kinase bearing an SH3 domain and a leucine zipper-basic region domain. Oncogene. 1994;9:1745–1750. [PubMed] [Google Scholar]
- 24.Johnson DI. Cdc42: an essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol Mol Biol Rev. 1999;63:54–105. doi: 10.1128/mmbr.63.1.54-105.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanamoto T, Mota M, Takeda K, Rubin LL, Miyazono K, Ichijo H, Bazenet CE. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol. 2000;20:196–204. doi: 10.1128/mcb.20.1.196-204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knaus UG, Morris S, Dong HJ, Chernoff J, Bokoch GM. Regulation of human leukocyte p21-activated kinases through G-protein-coupled receptors. Science. 1995;269:221–223. doi: 10.1126/science.7618083. [DOI] [PubMed] [Google Scholar]
- 27.Leung IW, Lassam N. Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J Biol Chem. 1998;273:32408–32415. doi: 10.1074/jbc.273.49.32408. [DOI] [PubMed] [Google Scholar]
- 28.Lim L, Manser E, Leung T, Hall C. Regulation of phosphorylation pathways by p21 GTPases. The p21 Ras-related Rho subfamily and its role in phosphorylation signaling pathways. Eur J Biochem. 1996;242:171–185. doi: 10.1111/j.1432-1033.1996.0171r.x. [DOI] [PubMed] [Google Scholar]
- 29.Manser E, Chong C, Zhao ZS, Leung T, Michael G, Hall C, Lim L. Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J Biol Chem. 1995;270:25070–25078. doi: 10.1074/jbc.270.42.25070. [DOI] [PubMed] [Google Scholar]
- 30.Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Jr, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin GA, Bollag G, McCormick F, Abo A. A novel serine kinase activated by Rac1/CDC42Hs-dependent autophosphorylation is related to PAK65 and STE20. EMBO J. 1995;14:1970–1978. doi: 10.1002/j.1460-2075.1995.tb07189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy MJ, Rubin LL, Philpott KL. Involvement of caspases in sympathetic neuron apoptosis. J Cell Sci. 1997;110:2165–2173. doi: 10.1242/jcs.110.18.2165. [DOI] [PubMed] [Google Scholar]
- 33.Merritt SE, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. The mixed lineage kinase DLK utilizes MKK7 and not MKK4 as substrate. J Biol Chem. 1999;274:10195–10202. doi: 10.1074/jbc.274.15.10195. [DOI] [PubMed] [Google Scholar]
- 34.Mielke K, Herdegen T. JNK and p38 stress kinases: degenerative effectors of signal transduction cascades in the nervous system. Prog Neurobiol. 2000;61:45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- 35.Nagata K, Puls A, Futter C, Aspenstrom P, Schaefer E, Nakata T, Hirokawa N, Hall A. The MAP kinase kinase kinase MLK2 colocalizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO J. 1998;17:149–158. doi: 10.1093/emboj/17.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pombo CM, Kehrl JH, Sanchez I, Katz P, Avruch J, Zon LI, Woodgett JR, Force T, Kyriakis JM. Activation of the SAPK pathway by the human STE20 homologue germinal centre kinase. Nature. 1995;377:750–754. doi: 10.1038/377750a0. [DOI] [PubMed] [Google Scholar]
- 37.Rana A, Gallo K, Godowski P, Hirai S, Ohno S, Zon L, Kyriakis JM, Avruch J. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J Biol Chem. 1996;271:19025–19028. doi: 10.1074/jbc.271.32.19025. [DOI] [PubMed] [Google Scholar]
- 38.Reddy UR, Pleasure D. Cloning of a novel putative protein kinase having a leucine zipper domain from human brain. Biochem Biophys Res Commun. 1994;205:1494–1495. doi: 10.1006/bbrc.1994.2834. [DOI] [PubMed] [Google Scholar]
- 39.Sakuma H, Ikeda A, Oka S, Kozutsumi Y, Zanetta JP, Kawasaki T. Molecular cloning and functional expression of a cDNA encoding a new member of mixed lineage protein kinase from human brain. J Biol Chem. 1997;272:28622–28629. doi: 10.1074/jbc.272.45.28622. [DOI] [PubMed] [Google Scholar]
- 40.Su YC, Han J, Xu S, Cobb M, Skolnik EY. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 1997;16:1279–1290. doi: 10.1093/emboj/16.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka S, Hanafusa H. Guanine-nucleotide exchange protein C3G activates JNK1 by a ras-independent mechanism. J Biol Chem. 1998;273:1281–1284. doi: 10.1074/jbc.273.3.1281. [DOI] [PubMed] [Google Scholar]
- 42.Tapon N, Hall A. Rho, Rac, and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- 43.Tapon N, Nagata K, Lamarche N, Hall A. A new Rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-κB signaling pathways. EMBO J. 1998;17:1395–1404. doi: 10.1093/emboj/17.5.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J Biol Chem. 1996;271:27225–27228. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- 45.Tibbles LA, Ing YL, Kiefer F, Chan J, Iscove N, Woodgett JR, Lassam NJ. MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO J. 1996;15:7026–7035. [PMC free article] [PubMed] [Google Scholar]
- 46.Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 47.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 49.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 50.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]