

Abstract
Epilepsy is a disease of neuronal hyperexcitability, and pharmacological and genetic studies have identified norepinephrine (NE) and neuropeptide Y (NPY) as important endogenous regulators of neuronal excitability. Both transmitters signal through G-protein-coupled receptors, are expressed either together or separately, and are abundant in brain regions implicated in seizure generation. NPY knock-out (NPY KO) and dopamine β-hydroxylase knock-out (DBH KO) mice that lack NE are susceptible to seizures, and agonists of NE and NPY receptors protect against seizures. To examine the relative contributions of NE and NPY to neuronal excitability, we testedDbh;Npy double knock-out (DKO) mice for seizure sensitivity. In general, DBH KO mice were much more seizure-sensitive than NPY KO mice and had normal NPY expression, demonstrating that an NPY deficiency did not contribute to the DBH KO seizure phenotype. DKO mice were only slightly more sensitive than DBH KO mice to seizures induced by kainic acid, pentylenetetrazole, or flurothyl, although DKO mice were uniquely prone to handling-induced seizures. NPY contributed to the seizure phenotype of DKO mice at high doses of convulsant agents and advanced stages of seizures. These data suggest that NE is a more potent endogenous anticonvulsant than NPY, and that NPY has the greatest contribution under conditions of extreme neuronal excitability.
Keywords: norepinephrine, NPY, dopamine β-hydroxylase, mice, epilepsy, seizure, pentylenetetrazole, flurothyl, kainic acid, mice, knock-out, in situ hybridization, neurotransmitter
Pharmacological and genetic studies have provided insight into how single neurotransmitters affect the activity of target cells. However, most neurons in the brain receive concurrent signals from more than one neurotransmitter, and little is known about how multiple signals are integrated and drive behavioral output. Transmitters could have synergistic, opposing, or completely separate effects on target cells.
Norepinephrine (NE) and neuropeptide Y (NPY) are abundant in the nervous system and are expressed both together and separately (Colmers and Wahlestedt, 1993; Cooper et al., 1996). In the periphery, they are co-released from neurons of the sympathetic nervous system and regulate multiple physiological processes (Lundberg et al., 1990). NPY potentiates NE-induced contractions in arteries via postsynaptic receptors (Itoi et al., 1986; Cortés et al., 1999) but can oppose NE-inducted contraction of the vas deferens by inhibiting NE release presynaptically (Lundberg and Stjarne, 1984; Bitran et al., 1996). A majority of central noradrenergic neurons originate in the locus coeruleus (LC) and send projections widely throughout the brain (Cooper et al., 1996). NPY is co-expressed in 20–40% of LC neurons (Everitt et al., 1984; Holets et al., 1988; Xu et al., 1998) and is also expressed in other regions that receive noradrenergic innervation, such as the hippocampus, cortex, and hypothalamus (Morris, 1989; Colmers and Wahlstedt, 1993). The interactions between NE and NPY that act on common targets in the brain, whether released together or separately, have not been extensively studied.
Pharmacological and genetic experiments have demonstrated that NE and NPY are potent endogenous anticonvulsants (Löscher and Czuczwar, 1987; Erickson et al., 1996; Baraban et al., 1997; Woldbye et al., 1997; Szot et al., 1999). Because epilepsy is a disease of neuronal hyperexcitability, both transmitters likely dampen excessive excitation of neurons in regions of the brain implicated in epileptic seizures. Seizure susceptibility is very sensitive to species, strain, and seizure-inducing paradigm used. For example, slightly altering the genetic background of NPY Y5 receptor knock-out mice can drastically change sensitivity to kainic acid-induced seizures (Marsh et al., 1999). Although many papers have described the anticonvulsant effects of NE and NPY, each study focused on only one of the two transmitters. Therefore, it is not possible to predict the relative contribution of each transmitter to seizure susceptibility or the possible synergistic or antagonistic interactions between them.
We took a genetic approach to identifying NE/NPY interactions and their relative contributions to cell excitability by examining seizure susceptibility of mice lacking NE, NPY, or both. A targeted disruption of the Dbh gene, which is required for NE synthesis, was used to remove NE (Thomas et al., 1995), and NPY was removed by a targeted disruption of the Npy gene (Erickson et al., 1996). Mice that lack both NE and NPY were generated by crossing theDbh- and Npy-deficient strains. The advantage of this system is that wild-type, single, and double mutant mice on the same genetic background were simultaneously tested in multiple seizure paradigms, and direct comparisons between transmitters were possible.
MATERIALS AND METHODS
Animals. Dbh;Npy double knock-out mice were bred as follows: Npy −/− males maintained on a pure 129/SvEv background were bred to Dbh +/− females maintained on a 129/SvEv and C57BL/6J mixed background. Then, F1 Dbh +/−;Npy +/− males and females were bred to each other. F2 Dbh −/−;Npy +/− males were bred to Dbh +/−;Npy +/− females to produce the four genotypes used in this study:Dbh −/−;Npy −/− [double knock-out (DKO)], Dbh −/−;Npy +/+ [dopamine β-hydroxylase knock-out (DBH KO)], Dbh +/−;Npy −/− (NPY KO), and Dbh +/−;Npy +/+ [wild type (WT)]. This breeding strategy was chosen to maximize production of DKO mice and because the seizure phenotype of Dbh +/− mice is indistinguishable fromDbh +/+ littermates (Szot et al., 1999), whereas Npy +/− mice are slightly more seizure-sensitive than Npy +/+ littermates (D. Marsh, personal communication). All mice were genotyped by PCR analysis from tail genomic DNA. Treatment withl-threo-3,4-dihydroxyphenylserine (DOPS; Sumitomo Pharmaceuticals, Osaka, Japan) was used to enhance fertility ofDbh −/− males and to rescue the embryonic lethal phenotype during development as described (Thomas et al., 1995,1998).
Mice were reared in a specific pathogen-free facility with a 12 hr light/dark cycle at the University of Washington, although mice were moved to a conventional facility for some experiments. Food and water were available ad libitum, and mice between 3 and 6 months of age were used for all experiments. Experimental protocols were approved by the Animal Care Committee at the University of Washington and met the guidelines of the American Association for Accreditation of Laboratory Animal Care.
Tissue preparation. Naive male mice were killed, and brains were removed, quick-frozen on dry ice, and stored at −80°C. Coronal sections (18 μm) of the hindbrain were cut on a cryostat and mounted onto three sets of Superfrost/Plus microscope slides (Fisher Scientific, Pittsburgh, PA), thereby placing every third section into a given set. One set was used for the NPY in situhybridization. Slides were post-fixed in 4% paraformaldehyde, washed in PBS, treated with acetic anhydride (0.25% in 0.1m triethanolamine), dehydrated, delipidated, and air-dried as described (Szot and Dorsa, 1994).
In Situ Hybridization. The plasmid used to make the NPY riboprobe was generously provided by Dr. Steven Sabol (National Heart, Lung, and Blood Institute, Bethesda, MD). A full-length 511 bp pre-pro-NPY rat cDNA cloned into the EcoRI site of the Bluescript (−) vector (Stratagene, La Jolla, CA) was linearized withPvuII, and antisense RNA was synthesized using T3 RNA polymerase (Promega, Madison, WI) [35S]-(α-thio)-UTP (NEN, Boston, MA). The reaction was performed for 90 min at 37°C, then extracted with phenol-chloroform and chloroform, and precipitated with ammonium acetate (1.5 m final concentration) and isopropyl alcohol. The pellet was resuspended in Tris-EDTA-dithiothreitol buffer. Then, the riboprobe was diluted as described (Szot et al., 1994), but with 200 mminstead of 10 mm dithiothreitol. The riboprobe (specific activity, 6.9 × 106 cpm/50 μl) was applied to the tissue and incubated overnight at 62°C. The next day, coverslips were removed by soaking in 2× SSC. Slides were treated with RNase A for 30 min at 37°C, followed by a series of washes in 0.1× SSC at 65°C. Then, slides were dehydrated, air-dried, and dipped in Kodak NTB2 emulsion (VWR, Seattle, WA). Slides were developed 14 d later in Kodak D-19 developer, rinsed in water, and fixed in Kodak general fixer. Then, the slides were stained with cresyl violet acetate, dehydrated, air-dried, and mounted with coverslips. The locus coeruleus region was identified using a mouse brain atlas (Franklin and Paxinos, 1997). Slides were coded and analyzed in random order with an automated image-processing system by an operator blind to genotype. NPY-positive cells were counted manually, and the number of silver grains per cell was determined with a grain-counting program, as described (Marks et al., 1992). Pictures were taken with a digital camera and cropped and sized using Adobe Photoshop (Adobe Systems, San Jose, CA).
Reverse transcription-PCR. Total RNA was extracted from whole brains without hindbrain, which was used for in situ analysis, with Trizol reagent (Life Technologies, Rockville, MD) and reverse transcribed using oligo-dT. An aliquot of each reverse transcription (RT) reaction was used for PCR reactions, which were primed with oligonucleotides complementary to sequences in the 3′ UTR of the mouse Npygene. After 27 cycles of PCR, which preliminary experiments indicated was within the linear range, products were diluted 1:5 and 1:10 and electrophoresed on a 2% NuSieve GTG agarose (ISC Bioexpress, Denver, CO) plus 0.8% agarose (Life Technologies) gel and stained with ethidium bromide. A picture of the gel was scanned into Adobe Photoshop, cropped, and sized.
Pentylenetetrazole seizures. Pentylenetetrazole (PTZ) (25–50 mg/kg) was dissolved in water and administered (4 ml/kg, i.p.) to seizure-naive mice. After injection, animals were placed into a clear container and closely monitored for 10 min. Latencies to first myoclonic jerk (MJ) (focal seizure) and clonic–tonic (C/T) (generalized) seizure served as measures of seizure susceptibility. Animals not having seizures were assigned latencies of 10 min. Data were analyzed by Student's t test or Wilcoxon–Mann–Whitney U test when comparing two groups, and ANOVA followed by Student–Newman–Keuls or Bonferroni post hoc tests when comparing more than two groups.
Flurothyl seizures. Seizure-naive mice were placed in an air-tight Plexiglas chamber, and flurothyl (Aldrich, Milwaukee, WI) was infused (20 μl/min) onto filter paper from which it vaporized. Each mouse was tested individually, was removed immediately from the chamber after onset of generalized seizure, and received only one exposure to flurothyl. Latencies to first myoclonic jerk and to clonic–tonic seizure were measured, and data were analyzed as described above.
Kainic acid seizures. Kainic acid (Sigma, St. Louis, MO) was dissolved in PBS at 4 mg/ml, brought to neutral pH with 1m NaOH, and administered to seizure-naive mice (20 mg/kg, i.p.). After injection, animals were placed into a clear container and closely monitored for 40 min. The latency to the first clonic–tonic seizure was measured, and maximal seizure severity was scored on a Racine scale (Racine, 1972) as follows: 0, no response; 1, staring; 2, myoclonic jerk; 3, forelimb clonus; 4, rearing; 5, loss of posture–generalized seizure; 6, death. Data were analyzed as described above. Animals not having clonic–tonic seizures were assigned latencies of 40 min.
Handling-induced seizures. Handling-induced seizures were quantitated in the course of the PTZ experiments. Before being injected with PTZ, mice were weighed, placed on a cage top, and observed for 2 min. Animals that displayed seizure-like behavior were not included in the PTZ study.
Cerebral blood flow. Cerebral blood flow (CBF) was measured using the [14C]iodoantipyrine method described by Maeda et al. (2000) with the following modifications. DBH KO mice were placed in the flurothyl chamber, and flurothyl was administered as described above. When the first myoclonic jerk was observed, the mice were quickly removed and injected intraperitoneally with 19 μCi of [14C]iodoantipyrine (Amersham Pharmacia Biotech, Little Chalfont, UK) in a volume of 152 μl of 0.9% NaCl. Mice were then placed back in the flurothyl chamber, and flurothyl was administered for 60 sec, during which time the mice continued to have seizure-like activity, after which mice were removed from the chamber and killed. WT mice were time-matched for flurothyl exposure to DBH KO mice and displayed no MJs or other seizure-like activity. Blood was collected in heparinized tubes, centrifuged, frozen on dry ice, and stored at −80°C. A scintillation counter was used to quantify the [14C]iodoantipyrine present in 10 μl of serum. Brains were removed, frozen on dry ice, and stored at −80°C. Coronal brain sections (20 μm) were mounted on slides and exposed to autoradiographic film next to a [14C] standard; hippocampal [14C]iodoantipyrine was quantitated by an observer blind to genotype using MicroComputer Imaging Device Systems (MCID; Imaging Research, St. Catharines, Ontario, Canada). Relative CBF was calculated by dividing the amount of [14C]iodoantipyrine in the hippocampus (mCi/gm) by the amount of [14C]iodoantipyrine in the serum (cpm/μl) and expressed as a ratio. Data were analyzed by Student'st test.
RESULTS
Handling-induced seizures
During normal handling of the mice, we noticed that DKO mice occasionally had seizures. We quantitated this phenotype before our PTZ assay (see Materials and Methods). Handling-induced seizures were apparent only in DKO mice and ranged in severity from cessation of locomotion and staring in a hunched position to clonic–tonic seizures. We found that 25% (4 of 16) of the DKO mice had handling-induced seizures. In contrast, none of the WT, DBH KO, or NPY KO mice showed any seizure-like activity, suggesting that NE and NPY act together to prevent handling-induced seizures.
PTZ seizures
Mice were treated with 30 mg/kg PTZ, and latency to focal seizure (MJ) and generalized seizure (C/T) was measured. We found that although NPY KO mice were slightly more sensitive to PTZ seizures than WT mice, DBH KO mice were drastically more so (Fig.1A). DKO mice tended to be slightly more sensitive than DBH KO mice in terms of latency to generalized seizure, although the differences were not significant (Fig. 1A). Because WT and NPY KO mice had very little seizure activity at 30 mg/kg PTZ, we administered a higher dose, and the small difference between NPY KO and WT mice became more pronounced at 40 mg/kg PTZ (Fig. 1B). To rule out the possibility that seizure sensitivity was similar between DBH KO and DKO mice because of a ceiling effect at this dose of PTZ, a lower dose (25 mg/kg) was administered. However, 25 mg/kg PTZ failed to reveal a significant difference between DKO and DBH KO mice (Fig.1C), suggesting that DKO mice are only slightly more PTZ-sensitive than DBH KO mice.
Fig. 1.

PTZ-induced seizures. A, PTZ administered at 30 mg/kg (WT, n= 6; NPY KO, n = 7; DBH KO, n = 6; DKO,n = 6); B, 40 mg/kg (WT, n = 7; NPY KO,n = 8); C, 25 mg/kg (DBH KO, n = 13; DKO,n = 8). Latencies to first myoclonic jerk (MJ) and clonic/tonic (C/T) seizures shown. *p < 0.01, compared with WT.
Flurothyl seizures
The results from flurothyl-induced seizures mirrored those of PTZ-induced seizures. NPY KO mice were slightly more sensitive than WT mice, DBH KO mice were much more sensitive than WT and NPY KO mice, and DKO mice were only slightly more sensitive than DBH KO mice (Fig.2). Latency to C/T seizure was significantly shorter in the DKO mice compared with DBH KO mice.
Fig. 2.

Flurothyl-induced seizures. Latencies to first myoclonic jerk (MJ) and clonic/tonic (C/T) seizures shown (WT,n = 7; NPY KO, n= 7; DBH KO, n = 6;DKO, n = 7). *p< 0.05, **p < 0.01, compared with WT. †p < 0.05, compared with DBH KO.
Kainic acid seizures
We found no differences in seizure severity or latency to generalized seizure between NPY KO and WT mice in a 40 min assay (Fig.3A,B). However, we observed that more NPY KO mice were dead 24 hr after kainic acid administration (4 of 8 NPY KO, 0 of 8 WT), in agreement with previous results (Baraban et al., 1997; DePrato Primeaux et al., 2000). DBH KO and DKO mice were comparatively more sensitive than WT mice in terms of both seizure severity and latency (Fig.3A,B).
Fig. 3.

Kainic acid-induced seizures. A, Maximal seizure severity; B, latency to generalized seizure in response to 20 mg/kg kainic acid (WT,n = 8; NPY KO, n= 8; DBH KO, n = 8;DKO, n = 7). *p< 0.05.
Cerebral blood flow
Because NE and NPY are co-expressed in sympathetic neurons and have profound effects on heart rate and blood pressure, we measured CBF to determine whether the seizure susceptibility phenotype of DBH KO mice was caused by changes in cardiovascular function. CBF during flurothyl-induced seizures, determined by the [14C]iodoantipyrine method and expressed as a ratio of hippocampal [14C]iodoantipyrine to serum [14C]iodoantipyrine, did not significantly differ between WT (0.37 ± 0.03; n = 3) and DBH KO (0.42 ± 0.1; n = 3) mice, despite greater seizure activity in the DBH KO mice (see Materials and Methods). Similarly, no differences in cortical CBF between genotypes were found (data not shown). These results suggest that the increased sensitivity to flurothyl-induced seizures in Dbh−/− mice is likely attributable to direct effects on neuronal excitability rather than differences in CBF. It follows that the interactions observed between NE and NPY in response to seizures occur in the brain.
NPY levels are normal in DBH KO mice
The lack of a profound difference in seizure susceptibility between DBH KO and DKO mice could be explained if DBH KO mice were already deficient in NPY. To determine whether NPY expression is regulated by NE, we examined NPY mRNA levels in the LC by in situ hybridization and the rest of the brain by RT-PCR. Similar to rats (Holets et al., 1988; Xu et al., 1998), NPY is expressed in a subset of mouse LC neurons (Fig.4A). The number of cells expressing NPY and the intensity of expression per cell was similar between WT and DBH KO mice (Fig. 4A–D). There was a trend for fewer cells to show NPY expression in theDbh −/− mice, but it was not significant. Although there was some variability from animal to animal in NPY expression in the rest of the brain as measured by RT-PCR, there was no difference between genotypes (Fig. 4E). In addition, no gross differences in NPY immunostaining were observed (data not shown). These results suggest that normal NPY expression does not require NE.
Fig. 4.
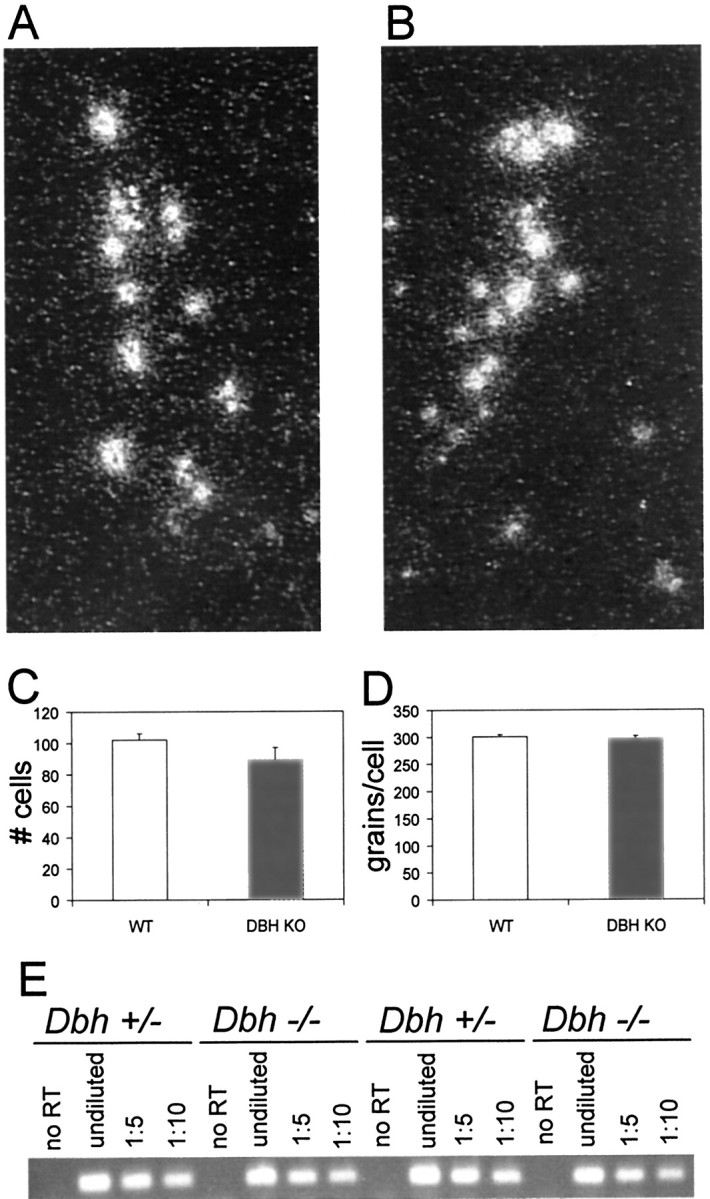
NPY mRNA expression in DBH KO mice. Representative atlas-matched locus coeruleus in situ hybridization darkfield images (10×) of NPY in WT (A) and DBH KO (B) mice. Quantitation of number of cells (C) and grains per cell (D) for NPY in situhybridization in the locus coeruleus (WT,n = 10; DBH KO,n = 10). E, Representative gel of NPY RT-PCR in brains of WT (n = 6) and DBH KO (n = 6) mice. Shown for each animal: no RT (no reverse transcriptase in reaction), undiluted(0.5 μl of RT reaction in PCR reaction), 1:5 (0.5 μl of 1:5 diluted RT reaction in PCR reaction), and 1:10 (0.5 μl of 1:10 diluted RT reaction in PCR reaction).
DISCUSSION
Although NE and NPY are released in many of the same brain regions, including from some of the same neurons, the way that their signals converge and are integrated has not been well characterized in the CNS. The data presented here demonstrate that measuring behavioral outputs in single and double neurotransmitter mutants provides a straightforward means of assessing their single and combined contributions to neuronal excitability.
Seizure inhibition by NE and NPY
Previously, the only information regarding interactions between NE and NPY in the brain suggested that NPY inhibited noradrenergic neuronal firing and NE release. This was demonstrated in the LC (Illes and Regenold, 1990) and the hypothalamus (Tsuda et al., 1989). In both cases, NPY appeared to enhance the action of NE acting on presynaptic α2 autoreceptors. However, because NE and NPY are both endogenous anticonvulsants, this type of interaction cannot be representative in brain regions that mediate seizure susceptibility. For example, if the inhibition of NE release in the hippocampus was the primary anticonvulsant mechanism of NPY action, then either NE would be expected to promote seizures or a lack of NPY would protect against seizures by allowing more NE release.
Because DBH KO mice are significantly more seizure-sensitive than NPY KO mice in most seizure paradigms tested, we conclude that NE is a more potent endogenous inhibitor of neuronal excitability than NPY. The lack of NPY contributed most to seizure susceptibility at high doses of convulsants and at late stages of seizures, when neuronal firing is highest. For example, differences in seizure latency between NPY KO and WT mice reached significance at 40 mg/kg, but not 30 mg/kg PTZ (Fig.2), and for generalized seizure, but not myoclonic jerk, for flurothyl (Fig. 3). These results are consistent with literature demonstrating that NPY, and neuropeptides in general, require high neuronal activity for their release (for review, see Bartfai et al., 1988). Because NPY gene expression is upregulated in the hippocampus after seizures (Gruber et al., 1994; Kragh et al., 1994; Tønder et al., 1994), the primary anticonvulsant action of NPY may be to inhibit recurring seizures rather than a single epileptic episode. In contrast, DBH KO mice have reduced latencies to the first behavioral signs of seizure (Szot et al., 1999) (Figs. 2, 3). Furthermore, tyrosine hydroxylase, the rate-limiting enzyme for NE biosynthesis (Cooper et al., 1996), is upregulated in the LC by kainic acid- and PTZ-induced seizures (Szot et al., 1997; Bengzon et al., 1999), and NE is critical for inhibiting seizures in kindling paradigms (Corcoran et al., 1974;Ehlers et al., 1980; Barry et al., 1987; Gellman et al., 1987; Weiss et al., 1990). These results suggest that NE is critical both for inhibiting seizure onset and protecting against recurring seizures.
In the periphery, several cases of synergy between NE and NPY have been described (Cortés et al., 1999; Hoyo et al., 2000; Pellieux et al., 2000). For example, NPY alone has no effect, but potently contracts arteries in the presence of NE. NPY enhances noradrenergic α1 signaling in this system and appears to converge with NE at the level of phospholipase C (PLC), because PLC-coupled NE (α1) and NPY (Y1) receptors are involved in this response (Selbie et al., 1995). However, we did not observe any synergy between the two transmitters with regard to PTZ-, flurothyl-, and kainic acid-induced seizures; DKO mice were only slightly more seizure-sensitive than DBH KO mice. This is more consistent with an additive model of interaction. The one exception was handling-induced seizures, for which there was clear evidence for synergy.
What is the source of the anticonvulsant NPY in the brain? Although a majority of the NPY in the hippocampus is synthesized and released from GABAergic interneurons (Morris, 1989), a subset of LC neurons projecting to the hippocampus and entorhinal cortex, two regions central to seizure generation and propagation, co-express NPY (Wilcox and Unnerstall, 1990). Further experiments, such as depleting NPY specifically from noradrenergic neurons and examining seizure susceptibility, will be required to determine where the seizure-inhibiting NPY is produced.
Regulation of neurotransmitter levels
Most neurons produce multiple neurotransmitters, and many different combinations of transmitters exist. For example, multiple neuropeptides, one or more neuropeptides plus a classical neurotransmitter, and multiple classical neurotransmitters have been found to coexist in different classes of neurons (for review, seeBartfai et al., 1988). However, only a few functional interactions have been described. We have focused on central neurons co-expressing a neuropeptide (NPY) and a small molecule neurotransmitter (NE). Interestingly, a majority of LC neurons also co-express the neuropeptide galanin, and a subset of LC neurons expresses all three neurotransmitters (Skofitsch and Jacobowitz, 1985; Moore and Gustafson, 1989; Xu et al., 1998). We expected that NPY might be upregulated in LC neurons of DBH KO mice to compensate for the lack of NE, because drugs that deplete NE induce NPY in the LC (Gundlach et al., 1990). However, the NE-depleting agents used (e.g., reserpine) can also deplete cotransmitters from the noradrenergic neurons (Xu et al., 1998); therefore, the increased NPY expression may not be attributable only to a decrease in NE. The RT-PCR assay may not have been sensitive enough to detect small changes in discrete brain regions, but our in situ hybridization experiments clearly showed that NPY was not induced in LC neurons by specific NE depletion. We conclude that NPY is not upregulated in the LC to compensate for the loss of a co-expressed neurotransmitter. The analysis of co-expressed neurotransmitters in other knock-out mice will be required to determine whether this result can be extended to other sets of neurotransmitters.
Implications for epilepsy therapeutics
Because animal models of epilepsy are potentially useful for developing new anticonvulsant drugs, it is critical to determine which neurotransmitter systems will make the most potent targets. Some anticonvulsant drugs have been compared in wild-type rodents (Dalby and Nielsen, 1997), but this is the first genetic comparison of endogenous anticonvulsant neurotransmitters. Our results demonstrate that, in terms of seizure susceptibility, a lack of NE is more deleterious than a lack of NPY. Because most people suffering from epilepsy probably have normal NE and NPY levels, it will be interesting to determine whether drugs that increase NE signaling are more potent anticonvulsants than NPY agonists in wild-type animals. If so, the efficacy in treating epilepsy will likely be greater for noradrenergic drugs than those that target the NPY system.
Although it is clear that noradrenergic agonists are anticonvulsant in many acute seizure paradigms, it is unclear how well these drugs can protect animals with recurring seizures. Some groups have shown that α2 adrenergic agonists delay the development of amygdaloid kindling in rats but cannot decrease seizure susceptibility in rats that have already been kindled (Gellman et al., 1987), although other groups report an anticonvulsant effect of adrenergic agonists on fully kindled rats (Löscher and Czuczwar, 1987) and kittens (Shouse et al., 1996). In addition, noradrenergic agonists have an anticonvulsant effect in animals that have a genetic susceptibility to seizures (Chermat et al., 1981; Micheletti et al., 1987; Tsuda et al., 1990; Yan et al., 1998). Some anticonvulsants that are used in the clinic have been shown to increase central NE or require an intact noradrenergic system for their efficacy in animal seizure models (Waller and Buterbaugh, 1985; Baf et al., 1994; Krahl et al., 1998; our unpublished data). In addition, drugs that decrease NE release exacerbate seizures in humans with intractable focal epilepsy (Kirchberger et al., 1998), and epileptic foci from patients with intractable partial epilepsy contain decreased levels of adrenergic α1 receptors (Brière et al., 1986). We conclude that the possibility of using noradrenergic drugs to treat epilepsy warrants further consideration.
Footnotes
This work was supported by the Howard Hughes Medical Institute (D.W., N.S.M., and N.C.R.), the National Alliance for Research on Schizophrenia and Depression, the Department of Veterans Affairs (P.S. and S.S.W.), and National Science Foundation Grant IBN97201 (J.G.H.). We thank Sumitomo Pharmaceuticals for the generous donation of DOPS, R. Steiner for use of his facilities and reagents, C. Bjornson and D. Kimelman for the use of their digital camera and technical advice, G. Mies for technical advice, and D. Kim and M. Szczypka for critical reading of this manuscript.
Correspondence should be addressed to Dr. Richard D. Palmiter, Howard Hughes Medical Institute, Biochemistry, Box 357370, University of Washington, Seattle, WA 98195. E-mail:palmiter@u.washington.edu.
REFERENCES
- 1.Baf MH, Subhash MN, Lakshmana KM, Rao BS. Sodium valproate induced alterations in monoamine levels in different regions of the rat brain. Neurochem Int. 1994;24:67–72. doi: 10.1016/0197-0186(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 2.Baraban SC, Hollopeter G, Erickson JC, Schwartzkroin PA, Palmiter RD. Knock-out mice reveal a critical antiepileptic role for neuropeptide Y. J Neurosci. 1997;17:8927–8936. doi: 10.1523/JNEUROSCI.17-23-08927.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barry DI, Kikvadze I, Brundin P, Bolwig TG, Bjorklund A, Lindvall O. Grafted noradrenergic neurons suppress seizure development in kindling-induced epilepsy. Proc Natl Acad Sci USA. 1987;84:8712–8715. doi: 10.1073/pnas.84.23.8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartfai T, Iverfeldt K, Fisone G, Serfozo P. Regulation of the release of coexisting neurotransmitters. Annu Rev Pharmacol Toxicol. 1988;28:285–310. doi: 10.1146/annurev.pa.28.040188.001441. [DOI] [PubMed] [Google Scholar]
- 5.Bengzon J, Hansson SR, Hoffman BJ, Lindvall O. Regulation of norepinephrine transporter and tyrosine hydroxylase mRNAs after kainic acid-induced seizures. Brain Res. 1999;842:239–242. doi: 10.1016/s0006-8993(99)01874-0. [DOI] [PubMed] [Google Scholar]
- 6.Bitran M, Torres G, Tapia W, Huidobro-Toro JP. Neuropeptide Y inhibits 3[H]noradrenaline release in the rat vas deferens independently of cAMP levels. Neurochem Int. 1996;28:309–317. doi: 10.1016/0197-0186(95)00084-4. [DOI] [PubMed] [Google Scholar]
- 7.Brière R, Sherwin AL, Robitaille Y, Olivier A, Quesney LF, Reader TA. Alpha-1 adrenoceptors are decreased in human epileptic foci. Ann Neurol. 1986;19:26–30. doi: 10.1002/ana.410190106. [DOI] [PubMed] [Google Scholar]
- 8.Chermat R, Doare L, Lachapelle F, Simon P. Effects of drugs affecting the noradrenergic system on convulsions in the quaking mouse. Naunyn Schmiedebergs Arch Pharmacol. 1981;318:94–99. doi: 10.1007/BF00508832. [DOI] [PubMed] [Google Scholar]
- 9.Cooper JR, Bloom FE, Roth RH. Norepinephrine and epinephrine. In: House J, editor. The biochemical basis of neuropharmacology. Oxford; New York: 1996. pp. 226–292. [Google Scholar]
- 10.Corcoran ME, Fibiger HC, McCaughran JA, Jr, Wada J. Potentiation of amygdaloid kindling and metrazol-induced seizures by 6-hydroxydopamine in rats. Exp Neurol. 1974;45:118–133. doi: 10.1016/0014-4886(74)90105-8. [DOI] [PubMed] [Google Scholar]
- 11.Cortés V, Donoso MV, Brown N, Fanjul R, Lopez C, Fournier A, Huidobro-Toro JP. Synergism between neuropeptide Y and norepinephrine highlights sympathetic cotransmission: studies in rat arterial mesenteric bed with neuropeptide Y, analogs, and BIBP 3226. J Pharmacol Exp Ther. 1999;289:1313–1322. [PubMed] [Google Scholar]
- 12.Dalby NO, Nielsen EB. Comparison of the preclinical anticonvulsant profiles of tiagabine, lamotrigine, gabapentin and vigabatrin. Epilepsy Res. 1997;28:63–72. doi: 10.1016/s0920-1211(97)00031-4. [DOI] [PubMed] [Google Scholar]
- 13.DePrato Primeaux S, Holmes PV, Martin RJ, Dean RG, Edwards GL. Experimentally induced attenuation of neuropeptide-Y gene expression in transgenic mice increases mortality rate following seizures. Neurosci Lett. 2000;287:61–64. doi: 10.1016/s0304-3940(00)01137-x. [DOI] [PubMed] [Google Scholar]
- 14.Ehlers CL, Clifton DK, Sawyer CH. Facilitation of amygdala kindling in the rat by transecting ascending noradrenergic pathways. Brain Res. 1980;189:274–278. doi: 10.1016/0006-8993(80)90028-1. [DOI] [PubMed] [Google Scholar]
- 15.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 16.Everitt BJ, Hökfelt T, Terenius L, Tatemoto K, Mutt V, Goldstein M. Differential co-existence of neuropeptide Y (NPY)-like immunoreactivity with catecholamines in the central nervous system of the rat. Neuroscience. 1984;11:443–462. doi: 10.1016/0306-4522(84)90036-8. [DOI] [PubMed] [Google Scholar]
- 17.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1997. [Google Scholar]
- 18.Gellman RL, Kallianos JA, McNamara JO. Alpha-2 receptors mediate an endogenous noradrenergic suppression of kindling development. J Pharmacol Exp Ther. 1987;241:891–898. [PubMed] [Google Scholar]
- 19.Gruber B, Greber S, Rupp E, Sperk G. Differential NPY mRNA expression in granule cells and interneurons of the rat dentate gyrus after kainic acid injection. Hippocampus. 1994;4:474–482. doi: 10.1002/hipo.450040409. [DOI] [PubMed] [Google Scholar]
- 20.Gundlach AL, Rutherfurd SD, Louis WJ. Increase in galanin and neuropeptide Y mRNA in locus coeruleus following acute reserpine treatment. Eur J Pharmacol. 1990;184:163–167. doi: 10.1016/0014-2999(90)90677-x. [DOI] [PubMed] [Google Scholar]
- 21.Hendry SHC. Organization of neuropeptide Y neurons in the mammalian central nervous system. In: Colmers WF, Wahlestedt C, editors. The biology of neuropeptide Y. Humana; Totowa, NJ: 1993. pp. 65–135. [Google Scholar]
- 22.Holets VR, Hökfelt T, Rokaeus A, Terenius L, Goldstein M. Locus coeruleus neurons in the rat containing neuropeptide Y, tyrosine hydroxylase or galanin and their efferent projections to the spinal cord, cerebral cortex and hypothalamus. Neuroscience. 1988;24:893–906. doi: 10.1016/0306-4522(88)90076-0. [DOI] [PubMed] [Google Scholar]
- 23.Hoyo Y, McGrath JC, Vila E. Evidence for Y1-receptor-mediated facilitatory, modulatory cotransmission by NPY in the rat anococcygeus muscle. J Pharmacol Exp Ther. 2000;294:38–44. [PubMed] [Google Scholar]
- 24.Illes P, Regenold JT. Interaction between neuropeptide Y and noradrenaline on central catecholamine neurons. Nature. 1990;344:62–63. doi: 10.1038/344062a0. [DOI] [PubMed] [Google Scholar]
- 25.Itoi K, Mouri T, Takahashi K, Sasaki S, Imai Y, Yoshinaga K. Synergistic pressor action of neuropeptide Y and norepinephrine in conscious rats. J Hypertens Suppl. 1986;4:S247–S250. [PubMed] [Google Scholar]
- 26.Kirchberger K, Schmitt H, Hummel C, Peinemann A, Pauli E, Kettenmann B, Stefan H. Clonidine- and methohexital-induced epileptiform discharges detected by magnetoencephalography (MEG) in patients with localization-related epilepsies. Epilepsia. 1998;39:1104–1112. doi: 10.1111/j.1528-1157.1998.tb01297.x. [DOI] [PubMed] [Google Scholar]
- 27.Kragh J, Tønder N, Finsen BR, Zimmer J, Bolwig TG. Repeated electroconvulsive shocks cause transient changes in rat hippocampal somatostatin and neuropeptide Y immunoreactivity and mRNA in situ hybridization signals. Exp Brain Res. 1994;98:305–313. doi: 10.1007/BF00228418. [DOI] [PubMed] [Google Scholar]
- 28.Krahl SE, Clark KB, Smith DC, Browning RA. Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia. 1998;39:709–714. doi: 10.1111/j.1528-1157.1998.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 29.Löscher W, Czuczwar SJ. Comparison of drugs with different selectivity for central alpha 1- and alpha 2-adrenoceptors in animal models of epilepsy. Epilepsy Res. 1987;1:165–172. doi: 10.1016/0920-1211(87)90037-4. [DOI] [PubMed] [Google Scholar]
- 30.Lundberg JM, Stjarne L. Neuropeptide Y (NPY) depresses the secretion of 3H-noradrenaline and the contractile response evoked by field stimulation, in rat vas deferens. Acta Physiol Scand. 1984;120:477–479. doi: 10.1111/j.1748-1716.1984.tb07410.x. [DOI] [PubMed] [Google Scholar]
- 31.Lundberg JM, Franco-Cereceda A, Lacroix JS, Perrow J. Neuropeptide Y and sympathetic neurotransmission. Ann NY Acad Sci. 1990;611:166–174. doi: 10.1111/j.1749-6632.1990.tb48930.x. [DOI] [PubMed] [Google Scholar]
- 32.Maeda K, Mies G, Oláh L, Hossmann K-A. Quantitative measurement of local cerebral blood flow in the anesthetized mouse using intraperitoneal [14C]iodoantipyrine injection and final arterial heart blood sampling. J Cereb Blood Flow Metab. 2000;20:10–14. doi: 10.1097/00004647-200001000-00003. [DOI] [PubMed] [Google Scholar]
- 33.Marks DL, Weimann JN, Burton KA, Lent KL, Clifton DK, Steiner RA. Simultaneous visualization of two cellular mRNA species in individual neurons by use of a new double in situ hybridization method. Mol Cell Neurosci. 1992;3:395–405. doi: 10.1016/1044-7431(92)90051-3. [DOI] [PubMed] [Google Scholar]
- 34.Marsh DJ, Baraban SC, Hollopeter G, Palmiter RD. Role of the Y5 neuropeptide Y receptor in limbic seizures. Proc Natl Acad Sci USA. 1999;96:13518–13523. doi: 10.1073/pnas.96.23.13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Micheletti G, Warter JM, Marescaux C, Depaulis A, Tranchant C, Rumbach L, Vergnes M. Effects of drugs affecting noradrenergic neurotransmission in rats with spontaneous petit mal-like seizures. Eur J Pharmacol. 1987;135:397–402. doi: 10.1016/0014-2999(87)90690-x. [DOI] [PubMed] [Google Scholar]
- 36.Moore RY, Gustafson EL. The distribution of dopamine-beta-hydroxylase, neuropeptide Y and galanin in locus coeruleus neurons. J Chem Neuroanat. 1989;2:95–106. [PubMed] [Google Scholar]
- 37.Morris BJ. Neuronal localisation of neuropeptide Y gene expression in rat brain. J Comp Neurol. 1989;290:358–368. doi: 10.1002/cne.902900305. [DOI] [PubMed] [Google Scholar]
- 38.Pellieux C, Sauthier T, Domenighetti A, Marsh DJ, Palmiter RD, Brunner HR, Pedrazzini T. Neuropeptide Y (NPY) potentiates phenylephrine-induced mitogen-activated protein kinase activation in primary cardiomyocytes via NPY Y5 receptors. Proc Natl Acad Sci USA. 2000;97:1595–1600. doi: 10.1073/pnas.030533197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 40.Selbie LA, Darby K, Schmitz-Peiffer C, Browne CL, Herzog H, Shine J, Biden TJ. Synergistic interaction of Y1-neuropeptide Y and alpha 1b-adrenergic receptors in the regulation of phospholipase C, protein kinase C, and arachidonic acid production. J Biol Chem. 1995;270:11789–11796. doi: 10.1074/jbc.270.20.11789. [DOI] [PubMed] [Google Scholar]
- 41.Shouse MN, Langer J, Bier M, Farber PR, Alcalde O, Moghimi R, Richkind M, Szymusiak R. The alpha 2 adrenoreceptor agonist clonidine suppresses seizures, whereas the alpha 2 adrenoreceptor antagonist idazoxan promotes seizures: pontine microinfusion studies of amygdala-kindled kittens. Brain Res. 1996;731:203–207. doi: 10.1016/0006-8993(96)00594-x. [DOI] [PubMed] [Google Scholar]
- 42.Skofitsch G, Jacobowitz DM. Immunohistochemical mapping of galanin-like neurons in the rat central nervous system. Peptides. 1985;6:509–546. doi: 10.1016/0196-9781(85)90118-4. [DOI] [PubMed] [Google Scholar]
- 43.Szot P, Dorsa DM. Expression of cytoplasmic and nuclear vasopressin RNA following castration and testosterone replacement: evidence for transcriptional regulation. Mol Cell Neurosci. 1994;5:1–10. doi: 10.1006/mcne.1994.1001. [DOI] [PubMed] [Google Scholar]
- 44.Szot P, White SS, Veith RC. Effect of pentylenetetrazol on the expression of tyrosine hydroxylase mRNA and norepinephrine and dopamine transporter mRNA. Brain Res Mol Brain Res. 1997;44:46–54. doi: 10.1016/s0169-328x(96)00217-3. [DOI] [PubMed] [Google Scholar]
- 45.Szot P, Weinshenker D, White SS, Robbins CA, Rust NC, Schwartzkroin PA, Palmiter RD. Norepinephrine-deficient mice have increased susceptibility to seizure-inducing stimuli. J Neurosci. 1999;19:10985–10992. doi: 10.1523/JNEUROSCI.19-24-10985.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374:643–646. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- 47.Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem. 1998;70:2468–2476. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- 48.Tønder N, Kragh J, Finsen BR, Bolwig TG, Zimmer J. Kindling induces transient changes in neuronal expression of somatostatin, neuropeptide Y, and calbindin in adult rat hippocampus and fascia dentata. Epilepsia. 1994;35:1299–1308. doi: 10.1111/j.1528-1157.1994.tb01802.x. [DOI] [PubMed] [Google Scholar]
- 49.Tsuda K, Yokoo H, Goldstein M. Neuropeptide Y and galanin in norepinephrine release in hypothalamic slices. Hypertension. 1989;14:81–86. doi: 10.1161/01.hyp.14.1.81. [DOI] [PubMed] [Google Scholar]
- 50.Tsuda H, Ito M, Oguro K, Mutoh K, Shiraishi H, Shirasaka Y, Mikawa H. Involvement of the noradrenergic system in the seizures of epileptic El mice. Eur J Pharmacol. 1990;176:321–330. doi: 10.1016/0014-2999(90)90026-3. [DOI] [PubMed] [Google Scholar]
- 51.Waller SB, Buterbaugh GG. Convulsive thresholds and severity and the anticonvulsant effect of phenobarbital and phenytoin in adult rats administered 6-hydroxydopamine or 5,7-dihydroxytryptamine during postnatal development. Pharmacol Biochem Behav. 1985;23:473–478. doi: 10.1016/0091-3057(85)90024-3. [DOI] [PubMed] [Google Scholar]
- 52.Weiss GK, Lewis J, Jimenez-Rivera C, Vigil A, Corcoran ME. Antikindling effects of locus coeruleus stimulation: mediation by ascending noradrenergic projections. Exp Neurol. 1990;108:136–140. doi: 10.1016/0014-4886(90)90020-s. [DOI] [PubMed] [Google Scholar]
- 53.Wilcox BJ, Unnerstall JR. Identification of a subpopulation of neuropeptide Y-containing locus coeruleus neurons that project to the entorhinal cortex. Synapse. 1990;6:284–291. doi: 10.1002/syn.890060308. [DOI] [PubMed] [Google Scholar]
- 54.Woldbye DP, Larsen PJ, Mikkelsen JD, Klemp K, Madsen TM, Bolwig TG. Powerful inhibition of kainic acid seizures by neuropeptide Y via Y5-like receptors. Nat Med. 1997;3:761–764. doi: 10.1038/nm0797-761. [DOI] [PubMed] [Google Scholar]
- 55.Xu ZQ, Shi TJ, Hökfelt T. Galanin/GMAP- and NPY-like immunoreactivities in locus coeruleus and noradrenergic nerve terminals in the hippocampal formation and cortex with notes on the galanin-R1 and -R2 receptors. J Comp Neurol. 1998;392:227–251. doi: 10.1002/(sici)1096-9861(19980309)392:2<227::aid-cne6>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 56.Yan QS, Dailey JW, Steenbergen JL, Jobe PC. Anticonvulsant effect of enhancement of noradrenergic transmission in the superior colliculus in genetically epilepsy-prone rats (GEPRs): a microinjection study. Brain Res. 1998;780:199–209. doi: 10.1016/s0006-8993(97)01139-6. [DOI] [PubMed] [Google Scholar]