

Abstract
A high risk factor for spontaneous and often fatal lobar hemorrhage is cerebral amyloid angiopathy (CAA). We now report that CAA in an amyloid precursor protein transgenic mouse model (APP23 mice) leads to a loss of vascular smooth muscle cells, aneurysmal vasodilatation, and in rare cases, vessel obliteration and severe vasculitis. This weakening of the vessel wall is followed by rupture and bleedings that range from multiple, recurrent microhemorrhages to large hematomas. Our results demonstrate that, in APP transgenic mice, the extracellular deposition of neuron-derived β-amyloid in the vessel wall is the cause of vessel wall disruption, which eventually leads to parenchymal hemorrhage. This first mouse model of CAA-associated hemorrhagic stroke will now allow development of diagnostic and therapeutic strategies.
Keywords: cerebral amyloid angiopathy, hemorrhage, stroke, bleeding, Alzheimer's disease, amyloid, amyloid precursor protein, smooth muscle cells, mouse, brain, CNS
In the rapidly growing segment of elderly people in industrialized countries, hemorrhagic stroke is an increasing threat. Nontraumatic etiologies for cerebral hemorrhage include hypertension and cerebral amyloid angiopathy (CAA). In contrast to hypertensive small-vessel disease, in which bleeding is predominantly found in the basal ganglia, cerebellum, or pons, CAA leads to spontaneous and often fatal lobar hemorrhage (Vinters, 1987;Massaro et al., 1991; Greenberg, 1998; Sacco, 2000). CAA as a major cause of hemorrhagic stroke has not been fully appreciated in the past, with previous estimates in the range of 10% as a cause of all nontraumatic intracerebral hemorrhages (Vinters, 1987; Itoh et al., 1993; Greenberg, 1998).
The most common form of CAA is of the β-amyloid (Aβ) type (Burgermeister et al., 2000; Yamada, 2000). Aβ is a 40 to 42 amino acid peptide derived from the longer amyloid precursor protein (APP) (Price et al., 1998; Selkoe, 1999). CAA occurs sporadically and can be detected to various degrees in approximately half of all individuals beyond 70 years (Yamada et al., 1987; Itoh et al., 1993). In addition, CAA can be detected in up to 90% of Alzheimer's disease (AD) patients (Vinters, 1987; Yamada et al., 1987). In normal aging and AD, CAA occurs in conjunction with parenchymal amyloid plaques. However, CAA can also occur in the absence of compact plaques, as evidenced by patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D) caused by a point mutation within Aβ at codon 693 of APP (E693Q) (Levy et al., 1990). These patients develop a severe form of CAA and suffer recurrent intracerebral hemorrhages, leading to death between the ages of 45 and 55 (Wattendorff et al., 1995).
Progress in CAA and CAA-related spontaneous hemorrhage has been slow because of the lack of useful animal models (Walker, 1997). We have reported recently cerebral deposition of amyloid in plaques and vessels in an APP transgenic mouse model (APP23 mice) (Calhoun et al., 1999). In the present study, we report that CAA in these mice consistently leads to multiple and recurrent spontaneous cerebral hemorrhages. This first mouse model of CAA-associated hemorrhagic stroke provides clues to the mechanism of CAA-related hemorrhage, as well as a needed model for testing diagnostic and therapeutic interventions.
MATERIALS AND METHODS
Animals. Generation of B6,D2-TgN(Thy1-APPSwe)23 transgenic mice (APP23 mice) has been described previously (Sturchler-Pierrat et al., 1997). APP23 mice overexpress APP751 with the Swedish double-mutation under the control of a neuron-specific Thy-1 promoter element (Sturchler-Pierrat et al., 1997; Calhoun et al., 1999). The mice have been backcrossed with C57BL/6J mice. A total of 101 heterozygous male and female APP23 mice and nontransgenic control mice ranging from 8 to 28 months of age from generation F6–F12 have been used in this study. Nontransgenic control mice were either littermate control mice or control mice from another litter of the same generation of backcrossing.
Histology and immunohistochemistry. Mice were overdosed with pentobarbital. Brains were removed, immersion fixed for 2 d in 4% paraformaldehyde, and embedded in paraffin (Calhoun et al., 1998a). Coronal serial sections of 25 μm thickness were cut with a microtome throughout the brain. For three-dimensional (3D) confocal reconstruction (see below), some brains were post-fixed, cryoprotected, frozen, and sectioned at 100 μm with a freezing–sliding microtome (Jucker et al., 1994).
Cresyl violet, hematoxylin and eosin (H&E), and Congo red staining were done according to standard protocols (Carson, 1996). The Berlin Blue method of Perls's was used to visualize ferric iron in hemosiderin (Gomori, 1936; Carson, 1996). Immunohistochemistry on paraffin and fixed-frozen sections was done according to previously published protocols (Jucker et al., 1994; Calhoun et al., 1998a) by using the avidin–biotin–peroxidase complex method (Vector Laboratories, Burlingame, CA) with diaminobenzidine as chromogen. The following antibodies were used: polyclonal antibodies to Aβ (NT-11/12) (Sturchler-Pierrat et al., 1997), polyclonal antibody AS42/14 specifically to Aβ42 (Sturchler-Pierrat et al., 1997); polyclonal antibody FCA3340 and FCA3542 specifically to Aβ40 and to Aβ42, respectively [(Barelli et al., 1997) generous gift from F. Checlair]; mouse monoclonal antibody to α-smooth muscle actin (clone 1A4; Sigma, St. Louis, MO), mouse monoclonal antibody to β-dystroglycan (Novocastra, Newcastle upon Tyne, UK) (Tian et al., 1996); polyclonal antibody to glial fibrillary acidic protein (GFAP) (Dako, Glostrup, Denmark); polyclonal antibodies to cystatin C (Accurate Chemicals, Westbury, NY and Dako); and polyclonal antibody to mouse serum amyloid P component (SAP) (Calbiochem, La Jolla, CA).
Confocal microscopy. Double-labeling for Aβ and smooth muscle cells was achieved by incubating paraffin sections simultaneously with polyclonal antibody to Aβ (NT12) and mouse monoclonal antibody to α-smooth muscle actin. The secondary antibodies were Alexa 568 goat anti-rabbit IgG and Alexa 488 goat anti-mouse IgG (1:500; Molecular Probes, Eugene, OR). Sections were mounted with Vectashield (Vector Laboratories) and analyzed with a Confocal Laser Scanning Microscope LSM 510, inverted Axiovert 100 M (Zeiss, Oberkochen, Germany). For 3D reconstruction of amyloid-laden vessels, thick, fixed frozen sections were incubated with NT12 antibody, followed by Alexa 488 goat anti-rabbit IgG. The 3D reconstruction was done by using the Full3D function of the Imaris 3.0 software (Bitplane AG, Zürich, Switzerland).
Quantitative analysis of CAA and total amyloid burden.Groups of young (8.0 months; n = 10), adult (19.2 ± 0.2 months; n = 15), and aged (27.1 ± 0.2 months; n = 16) APP23 mice were used, with males and females balanced in all groups. Age-matched nontransgenic young (8.0 months; n = 10), adult (19.8 ± 0.4 months;n = 8), and aged (26.9 ± 0.4 months;n = 10) mice were used. Frequency and severity of CAA were quantified on systematically sampled serial Aβ-immunostained sections (NT12 antibody) throughout the region of interest (every 20th section through the neocortex; every 10thsection through the hippocampus; every 10th section through the thalamus; yielding 7–10 sections per region). A rating scale was used that was similar to that described previously (Olichney et al., 1996;Calhoun et al., 1999). “CAA frequency” was calculated by counting the total number of Aβ-positive vessels in the entire set of systematically sampled sections. To calculate “CAA severity,” Aβ-positive vessels were divided in one of three severity grades: 1, Aβ immunoreactivity confined to the vessel wall; 2, granular Aβ immunoreactivity in and around vessel wall with focal infiltration of the amyloid into the neuropil; and 3, extensive infiltration of amyloid into the neuropil with a complete amyloid coat around the vessel (see Fig. 1c–e). The mean for all vessels was taken as CAA severity. Finally, “CAA score” was calculated by multiplying CAA frequency with CAA severity. All of the quantification was done on the right hemisphere only. This grading system was used by two independent raters and yielded similar results. Total amyloid burden (percentage) was quantified on the same set of systematically sampled Aβ-immunostained sections using a point grid as described previously (Calhoun et al., 1998b).
Fig. 1.

Cerebral amyloid angiopathy in APP23 mice. Aβ staining reveals significant CAA (arrowheads) in neocortex (a) and thalamus (b) in aged 27-month-old APP23 mice. Within these regions, CAA showed a great variability (c–e), ranging from vessels with a thin rim of amyloid in the vessel wall (c; severity grade, 1), to vascular amyloid with amyloid infiltrating the surrounding neuropil (d; severity grade, 2), and to dyshoric amyloid with amyloid deposition within the vessel wall and with a thick and complete amyloid coat around the vessel wall (e; severity grade, 3). Scale bars:a, b, 100 μm; c–e, 10 μm.
Quantitation of cerebral hemorrhage. Cerebral hemorrhage is accompanied by a delayed appearance of hemosiderin-positive microglia (Koeppen et al., 1995). Perls's Berlin blue-stained clusters of hemosiderin staining were quantified on sets of systematically sampled sections (every 10th section throughout the neocortex, hippocampus, and thalamus). All numbers are again for the right hemisphere only. An additional set of every 10th section was stained for H&E and screened for acute intraparenchymal bleedings (presence of large accumulation of erythrocytes in brain parenchyma). In addition to the groups of 8-, 19-, and 27-month-old APP23 and control mice, we also assessed hemorrhage number in aged APP23 mice and age-matched controls that were collected after their spontaneous death (APP23, n = 9; mean age, 24.6 ± 0.7 months; controls, n = 4; 24.0 ± 1.5 months). Brains of these mice were immersion-fixed in 4% paraformaldehyde for several weeks, paraffin-embedded, and serially cut.
Assessment of the blood–brain barrier. Three 24-month-old female APP23 mice and three littermate controls were used. Mice received an intravenous injection of horseradish peroxidase (HRP) (type IV-A; Sigma) in the tail vein (0.4 mg/gm body weight). Thirty minutes later, mice were overdosed with pentobarbital and perfused with PBS, followed by 2% paraformaldehyde plus 2% glutaraldehyde. Brains were post-fixed, cryoprotected, frozen, and cut with a freezing–sliding microtome. Blood–brain barrier (BBB) leakage was studied by incubating sections in PBS with 0.05% DAB and 0.03% hydrogen peroxide (Banks and Broadwell, 1994). One transgenic and one aged control mouse were perfused with 10 ml of 0.4% trypan blue (Fluka, Buchs, Switzerland) in PBS, followed by 2% paraformaldehyde plus 2% glutaraldehyde (Reynolds and Morton, 1998). Brains were post-fixed, cut with a vibratome, and examined for BBB leakage of the dye.
Statistical analysis. All statistical analysis was done with STATVIEW 5.01. Significance levels were set atp < 0.05. Indicated is the mean ± SEM.
RESULTS
Age-related increase in CAA frequency and severity in APP23 mice
In 8-month-old APP23 mice, cerebrovascular amyloid was generally absent with the exception of rare focal deposits in leptomeningeal vessels. In contrast, in the 19- and 27-month-old groups, cerebrovascular amyloid was found consistently throughout the neocortex, hippocampus, and thalamus (Fig.1), and to a lesser degree in other regions such as septum, striatum, brainstem, and white matter. Leptomeningeal vessels were always heavily affected (Fig.2). Cerebrovascular amyloid was almost exclusively Congo red-positive, suggesting that amyloid is of a compact β-pleated nature. Robust staining of vascular amyloid was found with both Aβ40- and Aβ42-specific antibodies. Aβ40 exceeded Aβ42 staining intensity, suggesting a predominance of Aβ40 over Aβ42 in vascular amyloid similar to that reported in humans (Alonzo et al., 1998). Antibodies to cystatin C revealed appreciable staining of cerebrovascular amyloid, suggesting that mouse cystatin C is part of the amyloid. However, the cystatin C immunoreactivity was restricted to a subpopulation of amyloid-laden vessels predominantly in the thalamus and was clearly less intense than Aβ staining. Antibodies to SAP did not reveal any appreciable amyloid staining.
Fig. 2.
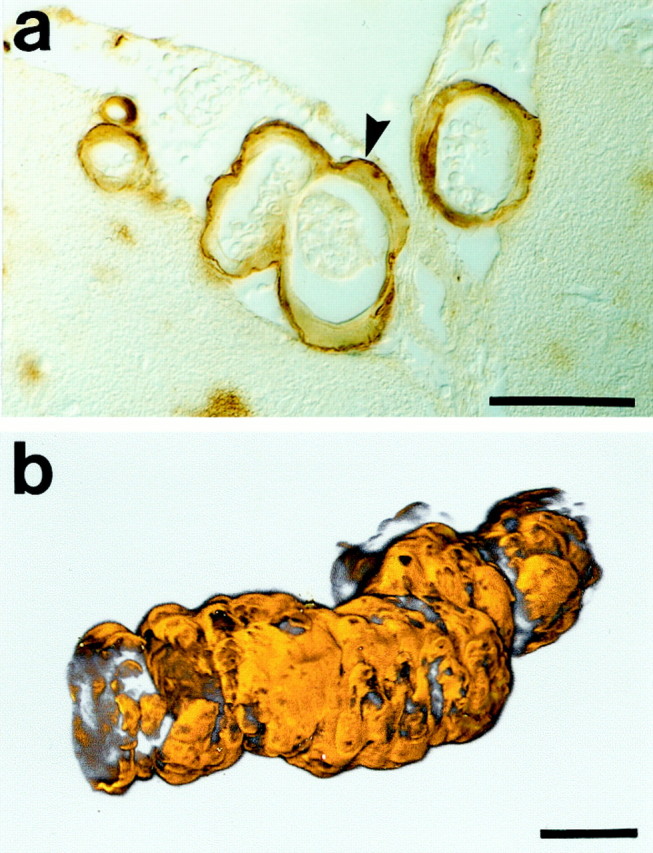
Cerebrovascular amyloid in leptomeningeal vessels. Leptomeningeal vessels are the most consistent and the first to exhibit cerebrovascular amyloid in APP23 mice. Shown in a are leptomeningeal vessels at the surface of the cingulate cortex of a 19-month-old APP23 mouse. Note that the amyloid is mostly confined to the outer vessel wall (arrowhead), consistent with CAA in humans in which initial deposits are found in the outer basement membrane (Yamaguchi et al., 1992). b, 3D reconstruction of an Aβ-stained (orange pseudocolored) heavily affected leptomeningeal vessel in an aged APP23 mouse. Note that nearly the entire surface is covered by a thick amyloid coat. The reconstruction consists of 198 optical slices (<0.7 μm), with a sampling interval of 0.35 μm. Scale bars, 25 μm.
Quantification of CAA frequency in systematically sampled sections revealed a striking age-related increase in neocortex (Fig.3a), hippocampus, and thalamus (data not shown). CAA severity also increased with aging (Fig.3b), indicating that not only are more vessels affected with aging but also that the amyloid burden of individual vessels increased with aging. Interestingly, thalamic vessels revealed a greater CAA severity compared with neocortical vessels in both the 19- and 27-month-old mice (p < 0.001; CAA severity for thalamus, 1.59 ± 0.08 and 1.82 ± 0.05, respectively). This observation was all the more interesting because the thalamus does not express the APP transgene (see Discussion). No difference in CAA frequency and severity was found between males and females (p > 0.05), consistent with no significant sex predilection of CAA in humans (Vinters, 1987; Yamada et al., 1987).
Fig. 3.

Age-related increase in CAA frequency and severity in APP23 mice. a, Number of amyloid affected vessels (CAA frequency) was quantified in systematically sampled sections through the neocortex of young (8 months), adult (19 months), and aged (27 months) APP23 mice. ANOVA revealed a significant affect of age (F(2,38) = 41.6; p< 0.001). b, A grading score was then used to assess severity of affected vessels (for details, see Fig.1c–e and Materials and Methods). The mean CAA severity is indicated for the 19- and 27-month-old groups and revealed a significant age-related increase (t(29)= 2.95; p < 0.01).
Similar to the striking increase in CAA with aging, a robust age-related increase in total amyloid load has been reported in these mice (Sturchler-Pierrat et al., 1997). However, we did not find a significant correlation between CAA frequency or severity and amyloid load within age groups (data not shown), confirming previous age-corrected linear regression analysis (Calhoun et al., 1999).
CAA leads to smooth muscle cell degeneration and aneurysm-like vasodilatation
Confocal microscopy using double-labeling for Aβ and smooth muscle cell actin revealed an extensive loss of smooth muscle cells in the tunica media of amyloid-laden vessels (Fig.4). Whereas in 19-month-old mice a focal discontinuity of the smooth muscle cell layer was typically observed (Fig. 4b), in 27-month-old mice, we often observed a dramatic loss of smooth muscle cells, with only patchy staining for smooth muscle cell actin remaining (Fig. 4c). Such a loss of smooth muscle cells concomitant with an increasing amyloid burden in the vessel wall was evident in leptomeningeal vessels and in vessels throughout neocortex, hippocampus, and thalamus, very similar to CAA in humans (Kawai et al., 1993; Wisniewski and Wegiel, 1994). Interestingly, even in the heavily affected mice, there were often individual smooth muscle cell containing vessels that were not affected by CAA (Fig. 4d,e).
Fig. 4.

Cerebrovascular amyloid leads to smooth muscle cell loss. Confocal microscopy of double-immunolabeled vessels (green, smooth muscle actin; red, amyloid) in APP23 mice. a, Leptomeningeal vessel in an 8-month-old mouse shows no amyloid deposition and a complete layer of smooth muscle cells. b, Leptomeningeal vessel in a 19-month-old mouse shows focal disappearance of smooth muscle cells at the site of cerebrovascular amyloid (arrowheads).c, In 27-month-old mice, smooth muscle cells have greatly disappeared, and a thick sheet of amyloid covers the wall of a leptomeningeal vessel. d, e, Parenchyma in the neocortex of a 19-month-old mouse showing an unaffected (d) and an amyloid-laden vessel (e) in close anatomical proximity. Shown are superpositions of 0.9- to 5-μm-thick optical sections. Scale bars:a, 10 μm; b–e, 20 μm.
We have shown previously a dystroglycan-mediated linkage between perivascular astrocytes and the vascular basement membrane (Tian et al., 1996). Such a tight linkage between the vessel wall and astrocytic end feet is clearly important for vessel stabilization and nutrient trafficking. To study a potential disruption of this glia–vascular interface by cerebrovascular amyloid, we have used double-labeling for GFAP, β-dystroglycan, and Aβ. In cases in which the amyloid was confined to the vessel wall, no apparent changes in perivascular glia staining was apparent. However, when the vascular amyloid infiltrated the parenchyma, GFAP-positive glial processes were no longer tightly associated with the vessel parenchymal basement membrane, and there was a focal loss of β-dystroglycan (data not shown).
Loss of smooth muscle cells and disruption of the glia–vascular interface leads to vessel wall weakening. In the 27-month-old mice, a significant number of vessels with aneurysm-like enlargements were most often found in the thalamus and also neocortex (Fig.5c). In such dilated vessels, the smooth muscle layer was in most cases absent, and vasodilatation often reached dramatic sizes of up to 200 μm (Fig. 5c). No loss of smooth muscle cells or aneurysm type of vasodilatation was found in nontransgenic mice of any age.
Fig. 5.

CAA-related hemorrhage in APP23 mice.a, Hemosiderin staining (blue) in the frontal cortex of a 27-month-old mouse indicative of an old hemorrhage. The section is counterstained with nuclear fast red. B, Perivascular hemosiderin-positive microglia (arrowhead) in close vicinity of a small vessel in a 27-month-old mouse.C, Hemosiderin-positive microglia surrounding an enlarged neocortical vessel of aneurysm-like appearance.d, e, Double-labeling for amyloid (brown) and hemosiderin (blue) localized bleedings to amyloid-laden vessels. f, Evidence for acute hematoma was assessed in H&E-stained sections. A significant hemorrhage (asterisk) in the frontal cortex of a 27-month-old APP23 mouse is shown. g, An adjacent section to f was stained with Berlin blue and revealed an old hemorrhage in the same region. Scale bars: a, 100 μm; b, c, f,g, 50 μm; d, e, 5 μm.
CAA-related cerebral hemorrhage in APP23 mice
The high incidence of cerebrovascular amyloid and the loss of smooth muscle cells led us to examine whether CAA in aged APP23 mice also causes hemorrhage similar to that described in humans (Vinters, 1987; Vonsattel et al., 1991; Itoh et al., 1993; Greenberg, 1998). Old cerebral hemorrhages were studied using Perls's iron staining, which identifies residual hemosiderin. Acute bleeding was assessed in H&E-stained sections. No evidence for both old and acute hemorrhages were found in 8-month-old mice. In contrast, 19-month-old APP23 mice revealed several focal hemosiderin deposits in neocortex and thalamus, most of which were localized to the cytoplasm of perivascular microglial cells. Strikingly, when we looked at 27-month-old mice, we found a dramatic increase in the frequency but also size of such hemosiderin clusters (Figs. 5,6a). Hemosiderin-positive microglia were often in close contact to vessels that had formed aneurysm-like enlargements (Fig. 5c). In several aged mice, we also found evidence for acute bleeding (Fig. 5f). Mice with acute hematomas also revealed numerous hemosiderin deposits throughout the neocortex, suggesting multiple recurrent bleedings over time. Acute and old bleedings were sometimes colocalized, suggesting recurrent bleeding in the same region (Fig.5f,g). No such bleedings were observed in nontransgenic control mice of any age.
Fig. 6.

Age-related increase in hemorrhage in neocortex of APP23 mice. a, Frequency of perivascular hemosiderin-positive staining was assessed in systematically sampled sections through the neocortex. No evidence of old bleedings (hemosiderin) was found in the 8-month-old mice. From 19 to 27 months of age, there appears a striking increase in frequency of intracerebral hemorrhages (ANOVA;F(2,38)= 26.1; p < 0.001). Because the sampling was done in the right hemisphere only and in every 10th section, total incidence of hemorrhages in the neocortex of 27-month-old mice can be estimated to be >100. b, Significant positive relationship between CAA score (frequency × severity) and hemorrhage number in neocortex of the 27-month-old mice (p < 0.01). Similar positive correlations were found between CAA frequency and hemorrhage and between CAA severity and hemorrhage (for bothR2 = 0.44).c, In contrast, no relationship between neocortical amyloid plaque load and hemorrhages was found (p > 0.05).
The anatomical distribution of the hemorrhages (primarily neocortex and thalamus, and to a lesser degree pia, hippocampus, and striatum) appeared very similar to the distribution of CAA. Correlative analysis between hemorrhage number and CAA score (frequency × severity) in neocortex of the 27-month-old group of mice revealed a significant positive correlation (Fig. 6b). A similar significant positive relationship was found in the thalamus (data not shown). These observations are in line with the morphological analysis, in which in most cases hemosiderin could clearly be assigned to amyloid-laden vessels (Fig. 5d,e). Interestingly, and consistent with the independence of amyloid plaque and CAA development, no significant relationship was observed between total amyloid load and hemorrhages (Fig. 6c).
It is difficult to establish whether an acute hemorrhagic stroke was the cause of spontaneous death in some of the aged APP23 mice. Most of the hematomas were small, reaching only a “subclinical” state. However, a large neocortical hematoma may have been the cause of the spontaneous death of at least one mouse.
CAA-associated vasculitis
A granulomatous giant cell vasculitis has been reported in some cases of human CAA. This observation has been attributed to a coexistence of vasculitis and CAA or to an immunological reaction and complication of CAA (Probst and Ulrich, 1985; Mandybur and Balko, 1992;Yamada et al., 1996). In 3 of the 25 aged APP23 mice, all of them with a high CAA score, we have found evidence of CAA-associated vasculitis. In particular, one mouse, examined after its spontaneous death, exhibited a severe lymphocytic vasculitis throughout subcortical, cortical, and leptomeningeal vessels (Fig.7). In this case, lymphocytes were found in the vessel wall, indicative of endovasculitis. Affected vessels appeared thickened, partially necrotic, and sometimes obliterated (Fig.7). There were no multinucleated giant cells or neutrophils. Because vasculitis was not observed in nontransgenic mice and only in transgenic mice with significant CAA, it does not appear to be the cause but an occasional consequence of CAA in our mouse model.
Fig. 7.
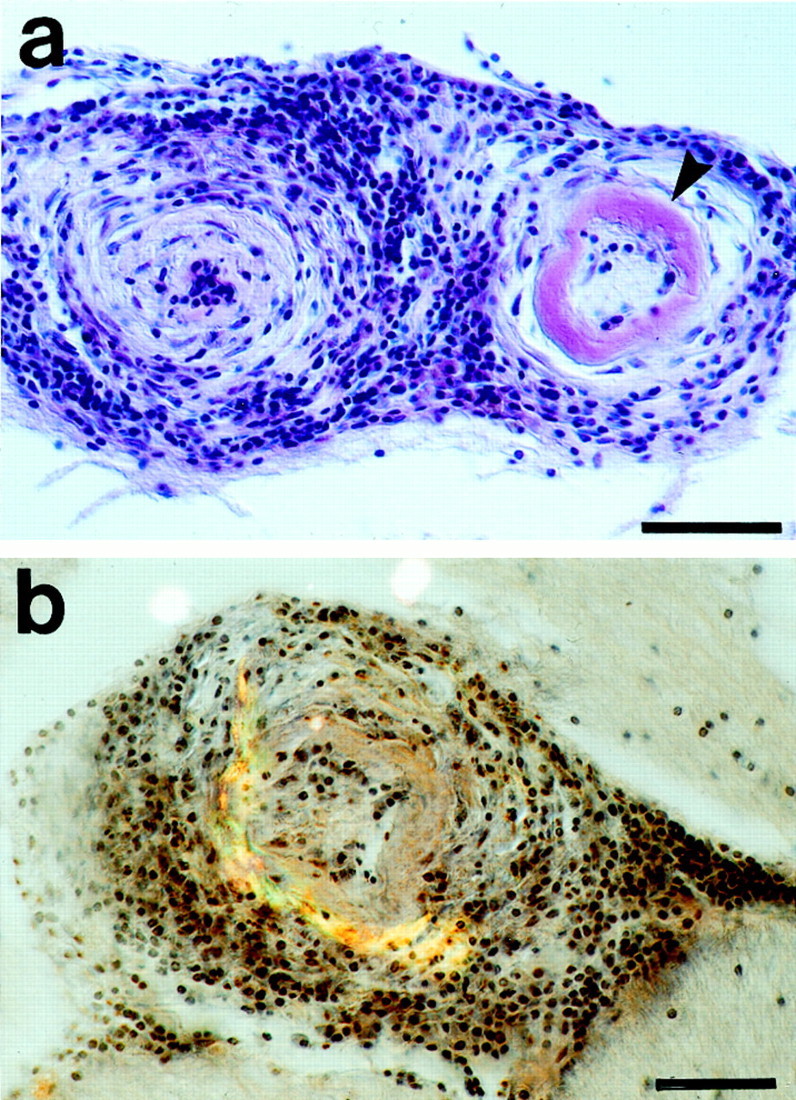
Vasculitis in aged APP23 mice with severe CAA.a, H&E staining of two vessels affected by a chronic lymphocytic vasculitis. Lymphocytic infiltrates are seen throughout the entire vessel walls. The vessel wall on the left appears thickened and the lumen is obliterated. There is severe amyloid deposition in the right vessel wall (arrowhead). b, Double-staining for H&E and for Congo red (green-yellow birefringence) reveals amyloid deposits in a vessel heavily affected by a lymphocytic vasculitis. Scale bars, 50 μm.
BBB leakage
Breakdown of the BBB with transition of blood protein into the vessel wall and brain parenchyma has been implicated as a key step in the pathogenesis leading to cerebral hemorrhage (Maeda et al., 1993). However, the present results using two different methods of BBB testing did not reveal any obvious leakage of the BBB unless an acute bleeding was present. We noticed that trypan blue labeled more vessels in the aged APP23 mice compared with age-matched controls and that the labeling was preferentially associated with amyloid-laden vessels. However, neither the dye nor HRP significantly infiltrated the neuropil, and the punctate staining for HRP in the vessel wall was consistent with the reported normal incorporation of blood-derived HRP by endocytic vesicles of the vascular endothelia (Banks and Broadwell, 1994).
DISCUSSION
Progression of CAA in APP23 mice is similar to CAA in humans
CAA in APP23 mice shows striking similarities to human CAA (Mandybur, 1986; Vinters, 1987; Alonzo et al., 1998). It is mostly congophilic and consists mainly of Aβ40. Initial deposition occurs in the abluminal part of smooth muscle cell-containing vessels, and leptomeningeal vessels are the first to be affected. Later, many smaller vessels and capillaries become affected. CAA in mice and humans occurs in the neocortex and to a lesser degree in hippocampus, striatum, basal forebrain, brainstem, and white matter. There are, however, also differences in the anatomical distribution of CAA between mouse and human. For example, CAA in thalamus is much more prominent in mouse than human. Vice versa, there is almost no CAA in mouse cerebellum, whereas CAA occurs in human cerebellum. These differences might be explained by the anatomically restricted transgene expression (Andra et al., 1996) but also by species-differences in Aβ transport and drainage along perivascular spaces (Weller, 1998; Weller et al., 1998; Calhoun et al., 1999).
In both APP23 mouse and human, there is a striking age-related increase in frequency and severity of CAA (Vinters, 1987; Yamada et al., 1987). Such an increase may reflect a stochastic seeding process in the vessel wall and subsequent Aβ accumulation (Lansbury, 1997). In addition, an age-related decrease in perivascular drainage of Aβ by a thickening of the vessel basement membrane and/or by an impaired vessel motility in the aging brain may significantly contribute to the increase in CAA with aging (Kalaria, 1996; Weller et al., 1998).
Interestingly, in both mouse and human, development of CAA and amyloid plaques appear to be independent processes, both naturally depending on Aβ levels and on age as common risk factors (Greenberg et al., 1995;Calhoun et al., 1999). Consistently, it has been demonstrated that overexpression of TGFβ1 increases CAA but decreases amyloid plaque formation in APP/TGFβ1 double-transgenic mice (Wyss-Coray et al., 1997, 2000). The different pathogeneses of vascular amyloid and parenchymal amyloid have important consequences for therapeutic intervention in CAA-associated hemorrhagic stroke (see below).
CAA is the cause of hemorrhagic stroke in APP23 mice
The strongest causal link between CAA and cerebral hemorrhage in humans comes from the observation that HCHWA-D patients with a point mutation at position 22 of Aβ (position 693 of APP) develop severe CAA and suffer fatal lobar hemorrhagic strokes early in their fifties (Wattendorff et al., 1995; Vinters et al., 1998). Cerebral hemorrhage is also a frequent finding in sporadic CAA and AD. However in these patients, CAA appears to be a prerequisite but not sufficient for vessel rupture, with additional factors such as hypertension, vascular abnormalities, fibrinoid necrosis, infarcts, trauma, vasculitis, and apolipoprotein E (ApoE) genotype playing an important role (Mandybur, 1986; Vonsattel et al., 1991; Itoh et al., 1993; O'Donnell et al., 2000). Whether these factors contribute to vessel rupture independently of CAA or secondary to CAA is not clear.
In APP23 mice, CAA is the only factor to which the hemorrhage could be attributed. Hemorrhage was only found in aged transgenic mice with CAA and was not observed in aged control mice. Both hemorrhage and CAA increase very similarly and almost exponentially with aging. Hemorrhage was predominantly found in brain regions in which CAA is most severe, i.e., in the neocortex and thalamus. In areas with no CAA (such as the cerebellum), no hemorrhages were detected. There was a significant correlation between CAA and hemorrhage in the neocortex and thalamus, and in most cases, bleeding could be clearly allocated to individual amyloid-laden vessels. It may be argued that APP overexpression (albeit restricted to neurons in APP23 mice) or amyloid deposition in the brain parenchyma predispose vessels to rupture. However, the findings of severe CAA and hemorrhages in the thalamus, which lacks transgene expression (Calhoun et al., 1999), and lack of a correlation between amyloid plaques and hemorrhage argues against this possibility. In summary, these observations demonstrate that CAA is the driving force of vessel rupture and hemorrhage in the APP23 mouse.
Pathogenesis of CAA-induced hemorrhage
The present results indicate that the loss of smooth muscle cells is an early and severe consequence of cerebrovascular amyloid deposition, as described in CAA in humans (Kawai et al., 1993;Wisniewski and Wegiel, 1994). In human CAA, it has been suggested that increased Aβ production of smooth muscle cells leads to smooth muscle degeneration (Kawai et al., 1993; Wisniewski and Wegiel, 1994;Davis-Salinas et al., 1995). However, this cannot be the case in APP23 mice because transgenic Aβ in the mice is of neuronal origin and is not produced by smooth muscle cells (Calhoun et al., 1999). Alternatively, it has been suggested that smooth muscle cells internalize neuron-derived Aβ and that release of Aβ may trigger smooth muscle cell degeneration (Urmoneit et al., 1997). Again, this is unlikely to be the mechanism in the mice, because we did not find any evidence for Aβ within smooth muscle cells. In contrast, our results suggest that extracellular Aβ is toxic to smooth muscle cells. This toxicity may either be mediated by soluble Aβ, which drains along perivascular spaces (Davis-Salinas et al., 1995; Weller et al., 1998; Calhoun et al., 1999), or by Aβ fibril assembly at the surface of smooth muscle cells (Van Nostrand et al., 1998). It is also conceivable that smooth muscle cells degenerate by purely mechanical constriction by the surrounding amyloid coat or by focal ischemia. Regardless of the exact mechanism, our results suggest that smooth muscle cell degeneration can be driven by extracellular amyloid of neuronal origin.
The disruption of the tight link between perivascular astrocytic end feet and the vessel wall appears somewhat later in the pathogenesis of CAA-induced hemorrhage and occurs to a significant degree only when the vascular amyloid infiltrates the neuropil. Such dyshoric amyloid also leads to perivascular microglial activation (Calhoun et al., 1999). Disruption of the tight glial–vascular interface, together with the replacement of the media by amyloid, leads to a weakening of the vessel wall, which occasionally leads to aneurysmal dilatations in aged APP23 mice. In addition, the present results show that a severe endovasculitis with vessel obliteration develops in ∼10% of the aged mice with CAA. Both the frequency and morphology of such CAA-associated endovasculitis appears to be very similar to sporadic human CAA (Probst and Ulrich, 1985; Mandybur and Balko, 1992; Yamada et al., 1996) and also greatly contributes to vessel weakening and rupture.
In humans, it has been suggested that cerebrovascular amyloid leads to “cracks” in the vessel wall, with plasma enzymes leaking into and digesting the wall (Mandybur, 1986; Maeda et al., 1993). In APP23 mice, significant BBB leakage and fibrinoid necrosis were absent. Consistent with no gross BBB leakage, we did not find SAP to be a component of cerebrovascular amyloid in the APP23 mice. In contrast, SAP is a component of human CAA and has been implicated in the protection of the amyloid fibrils from degradation (Coria et al., 1988; Tennent et al., 1995; Verbeek et al., 1998).
It has been suggested that fatal hemorrhage in sporadic CAA and HCHWA-D is associated with the presence of cystatin C as a component of the vessel amyloid (Maruyama et al., 1990; Vinters et al., 1990; Itoh et al., 1993; Maat-Schieman et al., 1997). The present results indicate that cystatin C is also a component of CAA in APP23 transgenic mice, but a clear relationship between cystatin C and hemorrhage was not obvious. In future studies, it will be instrumental to develop mouse model of CAA other than of the Aβ type, which will help to illuminate the mechanisms of CAA and CAA-induced hemorrhages (Burgermeister et al., 2000). For example, it is not clear why HCHWA-Iceland type patients, who develop CAA composed of mutated cystatin C, suffer fatal hemorrhages much earlier than HCHWA-D patients (Olafsson et al., 1996). In contrast, patients with dementia of the British and Danish types, who develop severe CAA composed of ABri and ADan, respectively, do not develop significant hemorrhage (Vidal et al., 1999, 2000b). Thus, the risk of hemorrhage may be predicted by the type of amyloid, the amount of amyloid, the participation of cofactors such as pathological chaperones, or the anatomical distribution of the amyloid within the vessel or certain brain regions.
Diagnostic and therapeutic potential of mouse models of CAA
CAA does not naturally occur in rodents but has been reported in aged dogs and nonhuman primates (Walker, 1997). CAA-related spontaneous hemorrhage has only consistently been reported in aged dogs beyond 13 years of age (Dahme and Schroder, 1979; Uchida et al., 1990). The present findings of robust CAA with multiple and recurrent hemorrhages in aged APP23 transgenic mice make this the first useful and genetically defined animal model to study diagnostic and therapeutic strategies of CAA-associated hemorrhage (Greenberg, 1998; Sacco, 2000).
In terms of diagnostic potential, the APP23 mouse model should be well suited for the development of in vivo detection of cerebrovascular amyloid (Skovronsky et al., 2000) and noninvasive markers for the progression of CAA-induced hemorrhages. There is a great need for diagnostic tools because, for example, it has been reported that recurrent bleedings are more severe than initial bleedings and more often fatal (Passero et al., 1995; Greenberg et al., 1999). Recently, progress in noninvasive detection of hemosiderin has been reported using gradient-echo magnetic resonance imaging (Greenberg et al., 1999).
Potential treatments for CAA-related hemorrhage can be divided into strategies of inhibiting the deposition of amyloid in the vessel wall and in blocking subsequent pathogenesis leading to vessel wall rupture (Greenberg, 1998). It has been reported recently that vaccination of PDAPP transgenic mice leads to a significant reduction of amyloid plaques presumably by phagocytotic microglia (Schenk et al., 1999; Bard et al., 2000). Unfortunately, PDAPP transgenic mice do not develop significant CAA, and the outcome of vaccination on CAA is uncertain. If vaccination indeed has the potential to “clear” even vascular amyloid, great caution has to be devoted to potential induction of bleeding attributable to removal of the amyloid coat, which presumably give the amyloid-laden vessel some stability. Regarding therapies aimed at reducing the risk of vessel rupture, the genetically defined APP23 mice offers a great potential to identify molecular factors involved in vessel rupture. For example, it has been suggested recently that the ApoE ε2 genotype predisposes an amyloid-laden vessel to rupture (O'Donnell et al., 2000).
Finally, mouse models of CAA and CAA-related hemorrhagic stroke will now allow to study the functional consequences of CAA and related hemorrhage in more detail. We have shown previously that CAA in adult APP23 mice (in the absence of bleeding) leads to perivascular neurodegeneration, including neuron loss, dystrophic terminals, and microglial activation (Calhoun et al., 1999; Phinney et al., 1999). In the present study, we have demonstrated multiple and recurrent bleeding in APP23 mice as they age, which in turn induces additional neurodegeneration. These observations suggest that a significant portion of the cognitive impairment in APP23 mice (Kelly et al., 1999;Sommer et al., 2000) may be caused by a chronic toxic effect of CAA on the parenchyma and by CAA-induced multiple hemorrhages. It is also striking that several forms of dementia have been described recently, all of which exhibit severe amyloid angiopathy but lack significant neuritic plaque pathology (Vidal et al., 1999, 2000a,b). All of these observations point to the need to reevaluate the role of CAA in AD dementia.
Footnotes
This work was supported by Swiss National Science Foundation Grants 3100-44526.95 and 3130-56753.99, the Fritz Thyssen Foundation (Cologne, Germany), and the Swiss Academy of Medical Sciences. D.T.W. was supported by MD/PhD Grant 3135-54877.98 from the Swiss National Science Foundation. We thank A. Probst (Basel, Switzerland), L. Walker (Ann Arbor, MI), and R. Kalaria (Newcastle, UK) for discussions and comments on this manuscript. We also thank D. Abramowski, C. Stürchler-Pierrat, C. Mistl, W. Kränger (Basel, Switzerland), and M. Pepys (London, UK) for experimental help and advice. The antibody donation of F. Checlair (Valbonne, France) is greatly acknowledged.
D.T.W., L.B., and M.C.H. contributed equally to this work
Correspondence should be addressed to Dr. Mathias Jucker, Department of Neuropathology, Institute of Pathology, University of Basel, Schönbeinstrasse 40, CH-4003 Basel, Switzerland. E-mail:mjucker@uhbs.ch.
REFERENCES
- 1.Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998;57:353–359. doi: 10.1097/00005072-199804000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Andra K, Abramowski D, Duke M, Probst A, Wiederhold KH, Burki K, Goedert M, Sommer B, Staufenbiel M. Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol Aging. 1996;17:183–190. doi: 10.1016/0197-4580(95)02066-7. [DOI] [PubMed] [Google Scholar]
- 3.Banks WA, Broadwell RD. Blood to brain and brain to blood passage of native horseradish peroxidase, wheat germ agglutinin, and albumin: pharmacokinetic and morphological assessments. J Neurochem. 1994;62:2404–2419. doi: 10.1046/j.1471-4159.1994.62062404.x. [DOI] [PubMed] [Google Scholar]
- 4.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 5.Barelli H, Lebeau A, Vizzavona J, Delaere P, Chevallier N, Drouot C, Marambaud P, Ancolio K, Buxbaum JD, Khorkova O, Heroux J, Sahasrabudhe S, Martinez J, Warter JM, Mohr M, Checler F. Characterization of new polyclonal antibodies specific for 40 and 42 amino acid-long amyloid beta peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer's disease and cerebral amyloid angiopathy cases. Mol Med. 1997;3:695–707. [PMC free article] [PubMed] [Google Scholar]
- 6.Burgermeister P, Calhoun ME, Winkler DT, Jucker M. Mechanisms of cerebrovascular amyloid deposition. Lessons from mouse models. Ann NY Acad Sci. 2000;903:307–316. doi: 10.1111/j.1749-6632.2000.tb06381.x. [DOI] [PubMed] [Google Scholar]
- 7.Calhoun ME, Kurth D, Phinney AL, Long JM, Hengemihle J, Mouton PR, Ingram DK, Jucker M. Hippocampal neuron and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol Aging. 1998a;19:599–606. doi: 10.1016/s0197-4580(98)00098-0. [DOI] [PubMed] [Google Scholar]
- 8.Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Neuron loss in APP transgenic mice. Nature. 1998b;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 9.Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carson FL. Histotechnology, Ed 2. ASCP; Chicago: 1996. [Google Scholar]
- 11.Coria F, Castano E, Prelli F, Larrondo-Lillo M, van Duinen S, Shelanski ML, Frangione B. Isolation and characterization of amyloid P component from Alzheimer's disease and other types of cerebral amyloidosis. Lab Invest. 1988;58:454–458. [PubMed] [Google Scholar]
- 12.Dahme E, Schroder B. Kongophile Angiopathie, cerebrovaskuläre Mikroaneurysmen und cerebrale Blutungen beim alten Hund. Zentralbl Veterinarmed A. 1979;26:601–613. [PubMed] [Google Scholar]
- 13.Davis-Salinas J, Saporito-Irwin SM, Cotman CW, Van Nostrand WE. Amyloid beta-protein induces its own production in cultured degenerating cerebrovascular smooth muscle cells. J Neurochem. 1995;65:931–934. doi: 10.1046/j.1471-4159.1995.65020931.x. [DOI] [PubMed] [Google Scholar]
- 14.Gomori G. Microtechnical demonstration of iron. Am J Pathol. 1936;12:655–663. [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg SM. Cerebral amyloid angiopathy: prospects for clinical diagnosis and treatment. Neurology. 1998;51:690–694. doi: 10.1212/wnl.51.3.690. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg SM, Rebeck GW, Vonsattel JP, Gomez-Isla T, Hyman BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–259. doi: 10.1002/ana.410380219. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg SM, O'Donnell HC, Schaefer PW, Kraft E. MRI detection of new hemorrhages: potential marker of progression in cerebral amyloid angiopathy. Neurology. 1999;53:1135–1138. doi: 10.1212/wnl.53.5.1135. [DOI] [PubMed] [Google Scholar]
- 18.Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T. Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci. 1993;116:135–141. doi: 10.1016/0022-510x(93)90317-r. [DOI] [PubMed] [Google Scholar]
- 19.Jucker M, Walker LC, Schwarb P, Hengemihle J, Kuo H, Snow AD, Bamert F, Ingram DK. Age-related deposition of glia-associated fibrillar material in brains of C57BL/6 mice. Neuroscience. 1994;60:875–889. doi: 10.1016/0306-4522(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 20.Kalaria RN. Cerebral vessels in ageing and Alzheimer's disease. Pharmacol Ther. 1996;72:193–214. doi: 10.1016/s0163-7258(96)00116-7. [DOI] [PubMed] [Google Scholar]
- 21.Kawai M, Kalaria RN, Cras P, Siedlak SL, Velasco ME, Shelton ER, Chan HW, Greenberg BD, Perry G. Degeneration of vascular muscle cells in cerebral amyloid angiopathy of Alzheimer disease. Brain Res. 1993;623:142–146. doi: 10.1016/0006-8993(93)90021-e. [DOI] [PubMed] [Google Scholar]
- 22.Kelly PH, Hunziker D, Schlecht HP, Carver K, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B. Progressive impairment in amyloid precursor protein transgenic mouse line APP23. Soc Neurosci Abstr. 1999;25:1291. [Google Scholar]
- 23.Koeppen AH, Dickson AC, McEvoy JA. The cellular reactions to experimental intracerebral hemorrhage. J Neurol Sci [Suppl] 1995;134:102–112. doi: 10.1016/0022-510x(95)00215-n. [DOI] [PubMed] [Google Scholar]
- 24.Lansbury PT., Jr Structural neurology: are seeds at the root of neuronal degeneration? Neuron. 1997;19:1151–1154. doi: 10.1016/s0896-6273(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 25.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 26.Maat-Schieman ML, van Duinen SG, Rozemuller AJ, Haan J, Roos RA. Association of vascular amyloid beta and cells of the mononuclear phagocyte system in hereditary cerebral hemorrhage with amyloidosis (Dutch) and Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:273–284. doi: 10.1097/00005072-199703000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Maeda A, Yamada M, Itoh Y, Otomo E, Hayakawa M, Miyatake T. Computer-assisted three-dimensional image analysis of cerebral amyloid angiopathy. Stroke. 1993;24:1857–1864. doi: 10.1161/01.str.24.12.1857. [DOI] [PubMed] [Google Scholar]
- 28.Mandybur TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol. 1986;45:79–90. [PubMed] [Google Scholar]
- 29.Mandybur TI, Balko G. Cerebral amyloid angiopathy with granulomatous angiitis ameliorated by steroid-cytoxan treatment. Clin Neuropharmacol. 1992;15:241–247. doi: 10.1097/00002826-199206000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Maruyama K, Ikeda S, Ishihara T, Allsop D, Yanagisawa N. Immunohistochemical characterization of cerebrovascular amyloid in 46 autopsied cases using antibodies to beta protein and cystatin C. Stroke. 1990;21:397–403. doi: 10.1161/01.str.21.3.397. [DOI] [PubMed] [Google Scholar]
- 31.Massaro AR, Sacco RL, Mohr JP, Foulkes MA, Tatemichi TK, Price TR, Hier DB, Wolf PA. Clinical discriminators of lobar and deep hemorrhages: the Stroke Data Bank. Neurology. 1991;41:1881–1885. doi: 10.1212/wnl.41.12.1881. [DOI] [PubMed] [Google Scholar]
- 32.O'Donnell HC, Rosand J, Knudsen KA, Furie KL, Segal AZ, Chiu RI, Ikeda D, Greenberg SM. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med. 2000;342:240–245. doi: 10.1056/NEJM200001273420403. [DOI] [PubMed] [Google Scholar]
- 33.Olafsson I, Thorsteinsson L, Jensson O. The molecular pathology of hereditary cystatin C amyloid angiopathy causing brain hemorrhage. Brain Pathol. 1996;6:121–126. doi: 10.1111/j.1750-3639.1996.tb00795.x. [DOI] [PubMed] [Google Scholar]
- 34.Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, Thal LJ. The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer's disease and Lewy body variant. Neurology. 1996;47:190–196. doi: 10.1212/wnl.47.1.190. [DOI] [PubMed] [Google Scholar]
- 35.Passero S, Burgalassi L, D'Andrea P, Battistini N. Recurrence of bleeding in patients with primary intracerebral hemorrhage. Stroke. 1995;26:1189–1192. doi: 10.1161/01.str.26.7.1189. [DOI] [PubMed] [Google Scholar]
- 36.Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer's disease: genetic studies and transgenic models. Annu Rev Genet. 1998;32:461–493. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- 38.Probst A, Ulrich J. Amyloid angiopathy combined with granulomatous angiitis of the central nervous system: report on two patients. Clin Neuropathol. 1985;4:250–259. [PubMed] [Google Scholar]
- 39.Reynolds DS, Morton AJ. Changes in blood–brain barrier permeability following neurotoxic lesions of rat brain can be visualised with trypan blue. J Neurosci Methods. 1998;79:115–121. doi: 10.1016/s0165-0270(97)00168-4. [DOI] [PubMed] [Google Scholar]
- 40.Sacco RL. Lobar intracerebral hemorrhage. N Engl J Med. 2000;342:276–279. doi: 10.1056/NEJM200001273420410. [DOI] [PubMed] [Google Scholar]
- 41.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 42.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 43.Skovronsky DM, Zhang B, Kung MP, Kung HF, Trojanowski JQ, Lee VM. In vivo detection of amyloid plaques in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97:7609–7614. doi: 10.1073/pnas.97.13.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sommer B, Sturchler-Pierrat C, Abramowski D, Wiederhold KH, Calhoun M, Jucker M, Kelly P, Staufenbiel M. Transgenic approaches to model Alzheimer's disease. Rev Neurosci. 2000;11:47–51. doi: 10.1515/revneuro.2000.11.1.47. [DOI] [PubMed] [Google Scholar]
- 45.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tennent GA, Lovat LB, Pepys MB. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci USA. 1995;92:4299–4303. doi: 10.1073/pnas.92.10.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian M, Jacobson C, Gee SH, Campbell KP, Carbonetto S, Jucker M. Dystroglycan in the cerebellum is a laminin alpha 2-chain binding protein at the glial–vascular interface and is expressed in Purkinje cells. Eur J Neurosci. 1996;8:2739–2747. doi: 10.1111/j.1460-9568.1996.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 48.Uchida K, Miyauchi Y, Nakayama H, Goto N. Amyloid angiopathy with cerebral hemorrhage and senile plaque in aged dogs. Jpn J Vet Sci. 1990;52:605–611. doi: 10.1292/jvms1939.52.605. [DOI] [PubMed] [Google Scholar]
- 49.Urmoneit B, Prikulis I, Wihl G, D'Urso D, Frank R, Heeren J, Beisiegel U, Prior R. Cerebrovascular smooth muscle cells internalize Alzheimer amyloid beta protein via a lipoprotein pathway: implications for cerebral amyloid angiopathy. Lab Invest. 1997;77:157–166. [PubMed] [Google Scholar]
- 50.Van Nostrand WE, Melchor JP, Ruffini L. Pathologic amyloid beta-protein cell surface fibril assembly on cultured human cerebrovascular smooth muscle cells. J Neurochem. 1998;70:216–223. doi: 10.1046/j.1471-4159.1998.70010216.x. [DOI] [PubMed] [Google Scholar]
- 51.Verbeek MM, Otte-Holler I, Veerhuis R, Ruiter DJ, De Waal RM. Distribution of A beta-associated proteins in cerebrovascular amyloid of Alzheimer's disease. Acta Neuropathol (Berl) 1998;96:628–636. doi: 10.1007/s004010050944. [DOI] [PubMed] [Google Scholar]
- 52.Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 53.Vidal R, Calero M, Piccardo P, Farlow MR, Unverzagt FW, Mendez E, Jimenez-Huete A, Beavis R, Gallo G, Gomez-Tortosa E, Ghiso J, Hyman BT, Frangione B, Ghetti B. Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele. Acta Neuropathol (Berl) 2000a;100:1–12. doi: 10.1007/s004010051186. [DOI] [PubMed] [Google Scholar]
- 54.Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, Bojsen-Moller M, Braendgaard H, Plant G, Ghiso J, Frangione B. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci USA. 2000b;97:4920–4925. doi: 10.1073/pnas.080076097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 56.Vinters HV, Secor DL, Pardridge WM, Gray F. Immunohistochemical study of cerebral amyloid angiopathy. III. Widespread Alzheimer A4 peptide in cerebral microvessel walls colocalizes with gamma trace in patients with leukoencephalopathy. Ann Neurol. 1990;28:34–42. doi: 10.1002/ana.410280108. [DOI] [PubMed] [Google Scholar]
- 57.Vinters HV, Natte R, Maat-Schieman ML, van Duinen SG, Hegeman-Kleinn I, Welling-Graafland C, Haan J, Roos RA. Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). Acta Neuropathol (Berl) 1998;95:235–244. doi: 10.1007/s004010050793. [DOI] [PubMed] [Google Scholar]
- 58.Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP., Jr Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol. 1991;30:637–649. doi: 10.1002/ana.410300503. [DOI] [PubMed] [Google Scholar]
- 59.Walker LC. Animal models of cerebral beta-amyloid angiopathy. Brain Res Brain Res Rev. 1997;25:70–84. doi: 10.1016/s0165-0173(97)00017-9. [DOI] [PubMed] [Google Scholar]
- 60.Wattendorff AR, Frangione B, Luyendijk W, Bots GT. Hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D): clinicopathological studies. J Neurol Neurosurg Psychiatry. 1995;58:699–705. doi: 10.1136/jnnp.58.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weller RO. Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol. 1998;57:885–894. doi: 10.1097/00005072-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 62.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wisniewski HM, Wegiel J. Beta-amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol (Berl) 1994;87:233–241. doi: 10.1007/BF00296738. [DOI] [PubMed] [Google Scholar]
- 64.Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- 65.Wyss-Coray T, Barcellos L, Oksenberg J, Green A, Lin C, Masliah E, Greenberg S, Mucke L. Transforming growth factor (TGF)-beta1 modifies Alzheimer's-type pathology in transgenic mice and humans. Neurobiol Aging. 2000;21:S125. [Google Scholar]
- 66.Yamada M. Cerebral amyloid angiopathy: an overview. Neuropathology. 2000;20:8–22. doi: 10.1046/j.1440-1789.2000.00268.x. [DOI] [PubMed] [Google Scholar]
- 67.Yamada M, Tsukagoshi H, Otomo E, Hayakawa M. Cerebral amyloid angiopathy in the aged. J Neurol. 1987;234:371–376. doi: 10.1007/BF00314080. [DOI] [PubMed] [Google Scholar]
- 68.Yamada M, Itoh Y, Shintaku M, Kawamura J, Jensson O, Thorsteinsson L, Suematsu N, Matsushita M, Otomo E. Immune reactions associated with cerebral amyloid angiopathy. Stroke. 1996;27:1155–1162. doi: 10.1161/01.str.27.7.1155. [DOI] [PubMed] [Google Scholar]
- 69.Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ. Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer's disease. An immunoelectron microscopic study. Am J Pathol. 1992;141:249–259. [PMC free article] [PubMed] [Google Scholar]