

Abstract
Caspase-9, an initiator caspase, and caspase-3, an effector caspase, have been suggested to mediate the terminal stages of neuronal apoptosis, but little is known about their activation in vivo. We examined temporal and spatial aspects of caspase-9 and -3 activation in olfactory receptor neurons (ORNs) undergoing apoptosis after target removal in vivo. After removal of the olfactory bulb, enhanced expression of procaspase-9 and -3 is observed in ORNs, followed by activation initially at the level of the lesion, then in axons, and only later in the ORN soma. We established the amyloid precursor-like protein-2 (APLP2) as a caspase substrate that is cleaved in an identical spatiotemporal pattern, suggesting its cleavage is the result of retrograde propagation of a pro-apoptotic signal in a caudorostral wave from the synapse through the axon to the ORN cell body. A null mutation in caspase-3 causes a change in axonal patterning indicative of an overall developmental expansion of the ORN population, and mature ORNs of caspase-3 knock-outs do not undergo caspase-dependent terminal dUTP nick end labeling-positive apoptosis after olfactory bulb removal. These results demonstrate that ORNs require caspase-3 activation to undergo normal developmental and mature target-deprived apoptosis. In addition, we demonstrate an axonal site of action for caspase-3 and -9 and show that regulation and activation of caspase-3 and -9 leading to apoptosis is a highly ordered process that occurs initially at the presynaptic level and only later at the cell body after deafferentation.
Keywords: olfactory receptor neuron, caspases, amyloid precursor-like protein-2, neuronal apoptosis, retrograde signaling, degeneration
Olfactory receptor neurons (ORNs) in the olfactory epithelium (OE) of the nasal cavity are the only described neurons in the mature nervous system that functionally turn over (Graziadei and Graziadei, 1979; Farbman, 1990). Individual ORNs die (by an as yet unexplained mechanism) every 4–6 weeks and are replaced by new neurons derived from a precursor population at the base of the OE (Schwartz Levey et al., 1991; Carr and Farbman, 1992; Calof et al., 1996; Roskams et al., 1996; Huard et al., 1998). Surgical removal of the ORN target, the olfactory bulb, causes a massive and synchronous wave of retrograde apoptosis in the distant population of 8–10 million ORNs within 72 hr of lesion (Michel et al., 1994; Roskams et al., 1996). This lesion-induced apoptosis of ORNs can be modulated by Bcl-2 (Jourdan et al., 1998), a regulator of the cytochrome c–Apaf-1–caspase-9 pathway (Earnshaw and Kaufmann, 1999; Gross et al., 1999; Krajewski et al., 1999). Thus, the OE has become a valuable model for studying neuronal degeneration and regeneration in vivo because of (1) its accessibility, (2) its exclusive location, distancing its cell body from all other neuronal cell types, and (3) its unique ability to constantly regenerate neurons throughout mammalian life (Moulton, 1974; Graziadei and Graziadei, 1979; Farbman, 1997).
Neuronal apoptosis is important in the developmental sculpting of a normal nervous system and also in the loss of neurons caused by neurodegenerative disease, ischemia, or trauma (Rink et al., 1995;Choi, 1996; Stefanis et al., 1997) (for review, see Oppenheim, 1991;Stefanis et al., 1997). Despite considerable progress in elucidating the cellular pathways of apoptosis, it is not yet known whether immature and mature neurons from the same lineage use identical cellular pathways to mediate apoptosis. Developmental neuronal cell death is thought to occur when an excess of immature neurons compete for limiting quantities of neurotrophic factors in their immediate and target environment (Burek and Oppenheim, 1998). Trophic support is also required from various sources throughout the neuraxis to ensure neuronal survival both before and after the critical period of naturally occurring cell death (Davies, 1997; Wang and Tessier-Lavigne, 1999). Regardless of the initiating stimulus, the terminal stages of apoptosis involve activation and action of caspases, a family of aspartate-directed intracellular cysteine proteases. This highly conserved protease family contains 14 mammalian family members, at least six of which have a clearly defined role in apoptosis (Earnshaw and Kaufmann, 1999). Knock-out mice have been used to highlight the critical importance of caspase-9, an initiator caspase, and caspase-3, an effector caspase, in neuronal apoptosis during development (Kuida et al., 1996, 1998; Hakem et al., 1998). These same caspases have been implicated in distant neuronal and glial apoptosis after ischemia and spinal cord injury, although the manner in which they transmit apoptotic signals in these in vivo models has yet to be revealed (Endres et al., 1998; Clark et al., 1999; Springer et al., 1999).
Despite significant progress in understanding the enzymological process of neuronal apoptosis, essentially nothing is known about how a neuron, which exists in a complex three-dimensional environment in vivo, integrates the pro- and anti-apoptotic signals that are received from different parts of the neuraxis. In the present study, we have taken advantage of the unique structural and organizational features of the primary olfactory neuraxis to examine where caspases are activated in mature ORNs after deafferentation and how this activity is propagated in space and time.
MATERIALS AND METHODS
Bulbectomies and tissue preparation. Olfactory bulbectomies were performed on adult CD-1 mice as described previously (Roskams et al., 1996). Bulbs were removed unilaterally for subsequent analysis of OE by immunohistochemistry, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), orin situ hybridization (ISH) (n = 3–4 per time point); or bilaterally for OE protein preparation (n = 6 per time point). Animals were killed 0, 4, 8, 12, 24, 36, 48, or 72 hr after surgery. For partial bulbectomies, the center of the olfactory bulb (including the mitral cell bodies) was removed stereotaxically, leaving the nerve fiber layer and glomerular layer relatively intact (confirmed both visually on dissection and histologically).
To prepare tissue for immunohistochemistry, TUNEL, or in situ hybridization, mice (n = 3–4 per time point, genotype, or treatment group) were anesthetized with Xylaket [25% ketamine HCl (MTC Pharmaceuticals), 2.5% xylazine (Bayer, Wuppertal, Germany), 15% ethanol, 0.55% NaCl) and rapidly perfused with PBS. Brains, olfactory bulbs, and olfactory epithelia were dissected, equilibrated in Tissue-Tek embedding medium (Sakura Finetek, Torrance, CA) for 10 min, and frozen in liquid nitrogen. Coronal and sagittal sections (10 μm) of OE and brain were prepared by cryostat sectioning and stored at −20°C for subsequent analysis.
For protein preparation, mice were killed by decapitation after anesthesia as above. Olfactory tissue and control brain areas were dissected out without perfusion, snap frozen in liquid nitrogen, and homogenized in buffer A (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% Triton X-100, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 100 μg/ml PMSF). Each set (time point, genotype, or treatment group) consisted of pooled tissues of three animals. Western blot analysis was performed on at least two different sets of animals (total n = 6 per time point). After estimation of protein concentration by the BCA method (Pierce), tissue homogenates were frozen at −80°C for subsequent analysis.
Caspase-3-deficient mice. Caspase-3-deficient mice were generated as previously described (Keramaris et al., 2000). Briefly, exons 2–7 of the caspase-3 gene were targeted for deletion by homologous recombination in C 57BL/6J mice. Homozygous disruption of the Casp 3 gene was confirmed by PCR genotyping (30 sec at 94°C, 1 min at 60°C, and 1 min at 72°C) with primers F108 and WCPR21. Heterozygous males and females that were generated from this line were backcrossed through two generations to produce the long-livedCasp 3 −/− variant reported here, which has now been propagated by sibling mating through an additional 12 generations. Absence of detectable caspase-3 expression was also confirmed by reverse transcription (RT)-PCR and Western blotting as described below. Developmental studies within this line have confirmed that a low percentage of the null mice (<10%) do not survive embryonic development.
Immunoblotting and immunoprecipitation. Aliquots of tissue homogenates (25 μg of protein) were subjected to SDS-PAGE and transferred to Immobilon membrane (Millipore, Bedford, MA). Equal loading of lanes was confirmed by Ponceau S staining. Membranes were blocked for 1 hr at room temperature with 5% nonfat milk in Tris-buffered saline (TBS), incubated 12–20 hr at 4°C in primary antibody in 2% milk/TBS, washed three times for 5 min each in 0.1% Tween 20 in TBS, and incubated for 1 hr at room temperature in peroxidase-coupled goat anti-rabbit IgG (Bio-Rad, Hercules, CA) diluted in 2% milk/TBS. Signals were detected with chemiluminescence reagents (Pierce, Rockford, IL). The following primary antisera were used: rabbit-anti-caspase-3 (MF393) (Samali et al., 1998) or anti-human p17 (Mesner et al., 1999), rabbit-anti-caspase-9 (MF 443) and C9/Bur 49 (raised against cleaved recombinant human caspase-9), rabbit antiserum raised against the caspase-9 peptide neoepitope PEPD (Mesner et al., 1999) and the Δ-C neoepitope antibody (Gervais et al., 1999) that recognizes the C-terminal VEVD peptide product generated from amyloid precursor protein (APP) and APLP2 by caspase-3 cleavage.
Immunoprecipitation of Δ-C APP cleavage products was performed by incubating 200 μg of tissue lysate (prepared as described above) with affinity-purified D2-II APLP2 specific antibody (gift from Gopal Thinakaran, University of Chicago, Chicago, IL) at a dilution of 1:250 in a final volume of 500 μl of modified RIPA buffer (150 mm NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 100 μg/ml PMSF). After incubation at 4°C, APLP2-containing immunocomplexes were isolated by further incubation at 4°C with protein A-conjugated Sepharose CL-2B beads (Sigma, St. Louis, MO), washed, and separated on gel containing a 4–20% polyacrylamide gradient.
Immunohistochemistry and TUNEL. Fresh frozen sections were fixed in 4% paraformaldehyde, permeablized in 0.1% Triton X-100, blocked with 4% normal serum, and incubated at 4°C for 12–20 hr in primary antibody (Roskams et al., 1994). Immunohistochemistry was performed using the Vectastain Elite anti-rabbit and anti-mouse kits (Vector Laboratories, Burlingame, CA) and Vector VIP peroxidase substrate chromagen to give a pink–purple color. Immunofluorescence was performed by incubating sections with primary antibody as described above, followed by a 1 hr incubation at room temperature with the following secondary antibodies: Cy2-conjugated goat or donkey anti-rabbit IgG, Texas Red-labeled rabbit anti-goat or anti-mouse IgG (all from Jackson ImmunoResearch, West Grove, PA), or cascade blue-conjugated goat anti-mouse-IgG (Molecular Probes, Eugene, OR). Where multiple secondary antibodies were used, they were used sequentially in the order given above. Then, sections were coverslipped using citiflour mounting medium (Marivac, Halifax, NS, Canada). Primary antisera used for immunocytochemistry included rabbit anti-active-caspase-3, MF 397 (Black et al., 1998) (and PharMingen 67341A), rabbit anti-Δ C neoepitope serum (Gervais et al., 1999), rabbit anti-caspase-9 (Bur 49), anti-synaptophysin (Boehringer Mannheim, Mannheim, Germany), and anti-neuron specific (β III) tubulin (TuJ1; Chemicon, Temecula, CA). TUNEL was performed as previously described (Pieper et al., 1999). Briefly, slides were incubated with digoxigenin (DIG)-conjugated UTP and terminal transferase enzyme, incubated with anti-DIG antibodies, and visualized using nitroblue tetrazolium chloride x-phosphate (NBT) and 5-bromo-4-chloro-3-indolyl-phosphate (BCIP) substrate for the antibody-coupled alkaline phosphatase.
Evaluation of caudorostral progression of apoptosis. For temporal evaluation of olfactory neuron loss, we evaluated the number of TUNEL+ neurons in the OE of the mediolateral turbinate 2.5 (±0.45) mm from the cribiform plate at 12–72 hr after bulbectomy. Adjacent sections were assayed for the expression or olfactory marker protein (OMP) (by in situ hybridization).
All cell counts were tabulated and analyzed statistically for mean ± SD using Microsoft Excel. For spatial evaluation of apoptotic patterns, sections were evaluated for the number of TUNEL+ cells in the superior (S), mediolateral (ML) and inferior (I) turbinates at 24 hr after bulbectomy from three sites (1, 2, 3) at 1, 2.5, and 4 mm from the cribiform plate.
ISH was performed as previously described (Blackshaw and Snyder, 1997) using DIG (Boehringer Mannheim) -labeled full-length sense and antisense probes (2 Kb) to the OMP (gift of F. Margolis, University of Maryland, Baltimore, MD). The signal was detected by 12 hr incubation with anti-DIG antibody, followed by overnight color development with NBT/BCIP as described above.
Image capture. All nonconfocal images were captured using a Zeiss Axioskop 2 MOT SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI) and Northern Exposure software on a Pentium II personal computer (PC). Then, they were imported into Adobe Photoshop 5/6 for final compilation. Confocal microscopy was performed using a Zeiss Axiovert S100 TV microscope fitted with Bio-Rad Radiance Plus confocal hardware and LaserSharp software running on a Dell Pentium II PC. Confocal Z-series were processed using NIH Image software version 1.62 (Wayne Rasband, National Institutes of Health) and imported into Adobe PhotoShop 5.0 for colorization and determination of signal colocalization.
RESULTS
Mature olfactory neurons undergo a caudorostral gradient of apoptosis after bulbectomy
To define a spatiotemporal pattern for ORN apoptosis after bulbectomy, we used several techniques to follow the loss of the mature neuron population. Agarose gel electrophoresis of OE-derived DNA first revealed DNA laddering characteristic of apoptosis at 12 hr after bulbectomy, peaking from 24–36 hr after bulbectomy (data not shown), a profile slightly earlier than previously reported (Michel et al., 1994). We used this profile for subsequent immunocytochemical and immunoblotting experiments in which the use of unilateral bulbectomy (Fig. 1G) allows us to directly compare expression patterns of lesioned and unlesioned neurons within each animal.
Fig. 1.

Temporal analysis of olfactory neuron apoptosis by TUNEL labeling and in situ hybridization. Olfactory epithelia were analyzed after unilateral bulbectomy (site of olfactory bulb removal marked by red arrow) by TUNEL staining to detect nicked DNA (dark nuclei). A, TUNEL+ ORNs in the OE (black arrows) at 24 hr after bulbectomy on sagittal. Arrowhead marks the turbinate shown in high-power coronal section (B), in which the contralateral unlesioned side is shown across the nasal septum (Sep). C, By 36 hr, higher densities of TUNEL-labeled cells in the neuronal layer are seen extending further from the lesioned bulb throughout each turbinate (small arrows), and density of TUNEL+ ORNs on coronal section (D) has increased compared with B. From 24 hr (E) to 48 hr (F), the density of OMP-expressing neurons (detected by in situ hybridization for OMP) decreases significantly on the lesioned (L) side of the nasal septum. For temporal evaluation of apoptosis, three independent sites within the mediolateral turbinates on coronal section were assessed for TUNEL+ neurons per linear millimeter. G, Mouse head indicating site of bulbectomy and planes of coronal section. Assessments of TUNEL+ and OMP+ at a single distance (2.5 ± 0.45 mm; position 2 on mouse head) from the cribiform plate (n = 3 animals per group) were plotted (H). H, Numbersshown on y-axes are overall mean ± SD for each group. OMP+ cells are displayed as a percentage of the total cells above the basal lamina per linear millimeter of OE. Tu+, TUNEL+. Scale bars:A, C, 500 μm;B, D–F, 20 μm.
TUNEL staining identified that cells responsible for the DNA laddering pattern lie within the ORN cell layer (Fig. 1). Apoptosis temporally progressed in a caudorostral gradient in which ORNs closest to the olfactory bulb removal site (on sagittal section) underwent apoptosis earlier than more distal ORNs. The same turbinate (Fig.1A,B, arrowhead) is also shown on coronal section at 24 and 36 hr, in which unlesioned OE across the medial septum is essentially devoid of TUNEL+ neurons. TUNEL-labeling of ORNs increases from 24 to 36 hr in all turbinates shown (Fig. 1A–D). ISH confirmed that cells being lost because of apoptosis were expressing OMP, an exclusive marker for mature olfactory neurons (Fig.1E,F). The contralateral OE retains OMP expression and does not become TUNEL-positive. As ORNs undergo apoptosis (between 24 and 48 hr), the thickness of the OE decreases as the number of ORNs expressing OMP mRNA are lost (Fig.1E,F). Adjacent sections, assayed for OMP expression and TUNEL-positivity, demonstrate the peak of loss of the mature ORN population from 24 to 36 hr after bulbectomy and complete loss of mature ORNs by 72 hr (Fig.1H).
Axonal activation of caspase-3 during apoptosis in ORNs
TUNEL positivity reflects, in part, the action of caspase-3-activated deoxyribonuclease (CAD), a unique endonuclease that is liberated in its active form after caspase-3-mediated cleavage of its inhibitor, ICAD (Enari et al., 1998). To assess caspase-3 proenzyme expression and activation during ORN apoptosis, protein extracts from unlesioned and bulbectomized mouse OE were examined by immunoblotting. Using two different antisera to caspase-3, we observed that caspase-3 proenzyme is present (32 kDa) in normal OE (Fig.2A–C, time 0). Over the first 24 hr after bulbectomy, endogenous levels of procaspase-3 increased significantly (Fig. 2A). By 48 hr after deafferentation, proteolytic cleavage of the zymogen to produce active caspase-3 could be readily detected on immunoblot (Fig.2B,C). These results clearly demonstrated an accumulation in procaspase-3 immediately before maximal cleavage and activation but did not provide any information about the site of this process in ORNs. To determine the site of caspase-3 activation, immunohistochemistry was performed using two antisera that specifically recognize only active caspase-3 (Fig.2D–F). Both reagents yielded identical results. Active caspase-3 was clearly detected in some axon bundles and cell bodies of ORNs from which the target had been removed 24 hr earlier (Fig. 2D), but was not detectable in the contralateral (unlesioned) ORNs (Fig. 2E), in which axons and ORN cell bodies still retain expression of OMP (Fig.2G). The immunocytochemical activation profile of caspase-3 for both antisera mirrored the pattern of activation observed on Western blot, i.e., active caspase-3 in some axons and cell bodies at 24 hr after bulbectomy, with more widespread (maximal) immunoreactivity throughout every axon bundle and maximal ORN soma activation at 48 hr after bulbectomy (Fig. 2F).
Fig. 2.
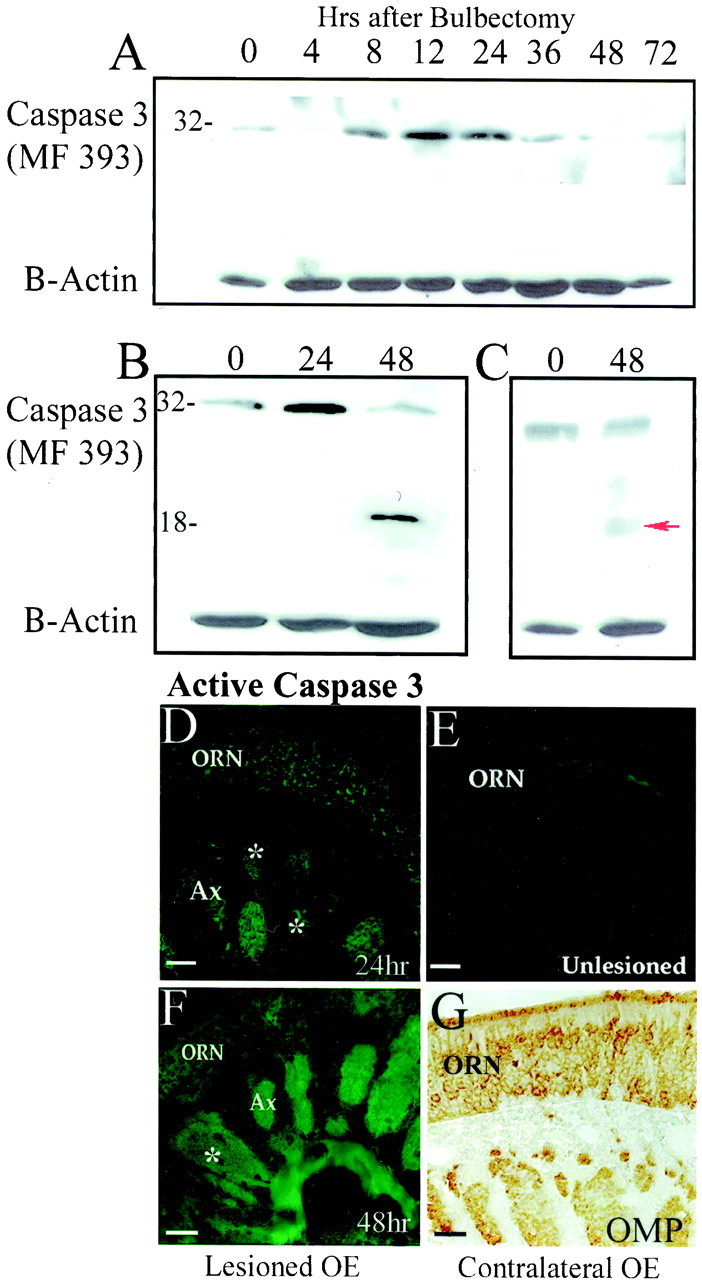
Caspase-3 proenzyme expression and activation after bulbectomy. Caspase-3 proenzyme is detected by Western blot analysis of pooled olfactory epithelium from mice recovering from bilateral bulbectomies (n = 3 per group).A, The endogenous level of caspase-3 proenzyme (detected with MF 393) increases during the first 24 hr after bulbectomy and then falls (because of activation). Using MF 393 (B) and previously described serum raised against the large subunit of human caspase-3 (C), the 32 kDa proenzyme is clearly present in nonlesioned olfactory epithelium (0) and at 24 and 48 hr after bulbectomy. In addition, maximal activation of caspase-3 is demonstrated by the 17 kDa large subunit (←), detected 48 hr after bulbectomy. Blots were reprobed with a β-actin loading control.D, With the more sensitive technique of immunocytochemistry, submaximal levels of active caspase-3 were found in a subpopulation of axons (Ax, partially stained axon bundles indicated with * and some ORN cell bodies (ORN) at 24 hr after unilateral bulbectomy (MF 397 antisera). E, Axons and cell bodies of the side contralateral to the lesion at 24 hr (Unlesioned) do not contain detectable levels of active caspase-3 (MF 397).F, At 48 hr, there is a concomitant increase in axonal caspase-3 activation, in which all axon bundles now contain active caspase-3 (PharMingen 67341A), and in partially labeled axon bundles (*) some breakdown in the integrity of the axon bundle can be seen.G, The unlesioned side still has a stable population of ORNs, expressing OMP throughout their cell bodies and axon bundles. Scale bars, 50 μm.
Caspase-3-dependent cleavage of axonal APLP2
Because the detection of active caspase-3 in axonal bundles was unanticipated, we wanted to examine whether active caspase-3 was being transported retrogradely without cleaving targets, or whether it was actively cleaving downstream targets throughout the neuraxis during ORN apoptosis. Few specific caspase-3 targets have been localized to ORNs and, in particular, their axons. The APP is cleaved by caspase-3 to reveal an epitope (VEVD) recognized by the Δ C-APP antiserum (Gervais et al., 1999). The epitope recognized by the Δ C-APP antiserum is detected by immunocytochemistry in subsets of axons and cell bodies of apoptotic ORNs, but not their unlesioned counterparts, at 24 hr after bulbectomy (Fig.3A–C, low and high magnification). This pattern resembles the cellular distribution of activated caspase-3 (Fig. 2), but is actually more useful, because it allows us to visualize where caspase-3 is being activated and where it has been activated (the cleavage product persists during degeneration, whereas the caspase-3 activation is more transient). At low power, the Δ C-APP epitope first appears most predominantly in subsets of axon bundles and dying ORNs at 24 hr after bulbectomy (Fig. 3A). The epitope is distributed in distinct regions of axon bundles of apoptotic ORNs (Fig. 3B) and, in ORN cell bodies, accumulates in a c-shape deposit surrounding the nucleus (Fig.3C).
Fig. 3.
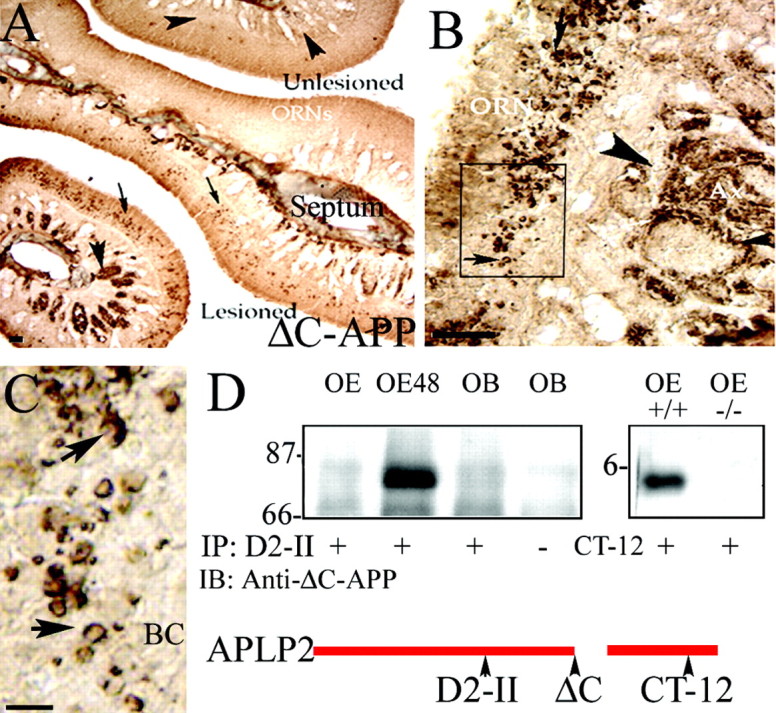
Amyloid precursor-like protein 2 (APLP2) is cleaved in the axons and cell bodies of apoptotic olfactory neurons. A, The ΔC-APP neoepitope antiserum recognizes a caspase-3-dependent epitope found in axons (arrowheads) and cell bodies (small arrows) of ORNs, only on the lesioned side of the septum.B, At higher power, there is a clear and distinct mesaxon subcompartmentalization of the product in each axon bundle (arrowhead), and in the box indicated at higher magnification (C), there is a clear perinuclear distribution of the cleavage product (arrow) in the ORN soma. The basal cell layer (BC) is free of apoptotic cells containing this product. D, Cell lysates from unlesioned OE, OE 48 hr after bulbectomy (OE48), and olfactory bulb (OB) were immunoprecipitated (IP) with an APLP2 affinity-purified N-terminal antiserum (D2-II) and probed (IB) with Δ-C antiserum to confirm that the VEVD epitope is revealed within APLP2 during ORN apoptosis. To confirm that caspase-3 is required for APLP2 cleavage, OE extracts that were similarly prepared from wild-type (+/+) and caspase-3 null (−/−) mice were blotted with an APLP2 C-terminal antiserum, CT-12, which detects the cleavage product in wild-type (+/+) mice but not their caspase-3 null (−/−) littermates. Scale bars:A, B, 100 μm; C, 20 μm.
Because APP is only found in embryonic olfactory neurons, it is not likely to be responsible for the Δ C-APP epitope revealed in apoptotic ORNs. Its homolog, APLP2, is predominantly expressed throughout the axons of ORNs in development and adulthood and also contains the conserved cleavage site (VEVD) for caspase-3-mediated cleavage (Thinakaran et al., 1995). To determine whether APLP2 might be responsible for the observed staining, antisera to the N-terminal (D2-II) and C-terminal (CT12 APLP2) fragments that could be produced by caspase-mediated cleavage of APLP2 were used to immunoprecipitate APLP2. Blotting with the Δ C-APP antisera confirmed that this reagent does recognize an APLP2 cleavage product in apoptotic ORNs (Fig.3D). In addition, the C terminus (CT-12) of APLP2 is also liberated in OE extracts from bulbectomized wild-type mice, but not from mice with a null mutation for caspase-3 (Fig. 3D). Collectively, these results demonstrate that caspase-3-dependent cleavage of APLP2 occurs in a similar spatial pattern to the appearance of activated caspase-3, i.e., predominantly in the axons and later in cell bodies.
Evidence that proteolysis starts at the presynaptic complex
The preceding results suggest that caspase-3 activation may begin in ORN axons and is propagated in a retrograde fashion to ORN cell bodies from the site of lesion. To confirm this hypothesis, we altered our lesion paradigm to that of a partial bulbectomy (removing the cell bodies of the target neurons, but leaving their synaptic complexes intact) in an attempt to encompass the pattern of caspase activation in a complete single slice. After partial bulbectomy, frozen sections of olfactory bulb and epithelium were initially examined at the planes indicated in Figure 4Fby TUNEL, followed by coimmunolocalization of the Δ C-APLP2 cleavage product and the axonal protein type III (neuron-specific) tubulin (Fig.4A,C) (Roskams et al., 1998).
Fig. 4.

Caspase-3-mediated APLP2 cleavage occurs first in the synaptic complexes, then axons and cell bodies of olfactory receptor neurons. Twenty-four hours after partial unilateral removal of the internal layers of the OB, transverse and sagittal sections (site and direction of section indicated on F) of OB (A, B) and OE (C, D) were analyzed for the compartmentalization of the APLP2 C-Δ epitope (green, using the Δ-C antibody) in comparison with TUNEL+ cells (black nuclei in A,C) and neuron-specific tubulin (NST,red in A, C), Synaptophysin (Syn, red inB), and active caspase-3 (aCasp3,red in D) expression. In the lesioned OB, the Δ C epitope is concentrated in the synaptic glomerular complexes (Gl) and axons (Ax) of the nerve fiber layer (NFL), in which the signal becomesyellow where colocalization with axonal NST (A) or glomerular synaptophysin (B) occurs. A, NST+ (red) axons and glomeruli of the unlesioned bulb (the midline between bulbs indicated by white dotted line) do not show any evidence of caspase-3 activation or APLP2 cleavage.C, By 36 hr after bulb lesion, a low-power image of a mediolateral turbinate shows that the Δ C epitope (green) production is highest in NST+ (red) axon bundles (overlap inyellow) closest to the olfactory bulb (marked ←To OB). Neurons (ORN) most distant from the midline were less immunoreactive for Δ C (green) and TUNEL+ (*) and lie adjacent to axons with minimal Δ C/NST overlap (red). D, A retrograde pattern of caspase-3 activation is represented on sagittal section of turbinate immediately adjacent to the septum, in which active caspase-3 (red) is transiently activated in axons and cell bodies (ORN), leaving cleaved Δ C (green) in its axonal wake. OBmarks direction of olfactory bulb location, and arrowaligns the plane of section with that shown on F.E, Sections of successive mediolateral turbinates were sorted into quadrants, based on axonal Δ C/NST labeling on a scale from 0 to 100%. 0% represents NST+ only (red), and 100% represents an entirely yellow axon bundle (complete overlap). ORN soma within each quadrant were then assessed for activation of caspase-3 (green) and TUNEL+ (black nuclei) in correlation with their adjacent axonal caspase-3 activation (mean per quadrant ± SD; three independent sections per animal). Scale bars: A–D, 50 μm;F, 500 μm.
In the olfactory bulb, 24 hr after partial lesion (Fig.4A), the caspase-3-liberated Δ C-APLP2 neoepitope first appears as bright, punctate dots of immunoreactivity in glomerular complexes, each of which contains the synapses of up to 1000 different ORNs. The intensity of the neoepitope also forms a gradient back from the glomeruli into some neuron-specific tubulin (NST)-positive axons of the nerve fiber layer projecting back to the OE (Fig. 4A). The Δ C neoepitope is only seen in axons of lesioned OB and OE, whereas unlesioned axons appear NST-positive (red) only. The synaptic complex localization of the Δ C neoepitope at 24 hr after partial lesion is confirmed by colocalization with synaptophysin in the glomeruli (Fig. 4B). In the OE, 36 hr after bulbectomy, axons displaying the highest degree of caspase-3-mediated cleavage (Δ C+) are most prominent close to where the olfactory nerve (NST+) exits a mediolateral turbinate (Fig.4C). Δ C+/NST (green) and TUNEL+ ORN cell bodies are clustered closest to the axons of highest caspase-3 activation (Fig. 4C, yellow). Axons adjacent to neurons in the part of the turbinate farthest from the bulb, however, are not yet labeled for active caspase-3. To further assess the direction of caspase-3 activation, we examined sagittal slices of OE for the presence of active caspase-3 and coordinate production of the Δ-C neoepitope. Figure 4D shows caspase-3 activation (red) carried through the axon bundle and into ORN cell bodies distant from the olfactory bulb. Caspase-3 activation, which is transient, occurs in advance of the detection of Δ C, for which the neoepitope is seen left behind in the axon (green) in the wake of the active caspase-3 signal.
To assess the relationship between axonal caspase activation and the likelihood of an ORN to undergo apoptosis, we divided successive turbinates, represented by that in Figure 4C, into quadrants in which axon bundles demonstrated Δ–C/NST (yellow) overlap of <20% (red), 20–50% (orange), 50–75% (yellow–orange), and 75–100% (completely yellow) (Fig. 4E). By comparing the number of TUNEL+ neurons and green Δ C+ neuronal soma with adjacent axonal caspase activation, we demonstrate that those axons displaying maximal axonal caspase-3 activation (yellow, 75–100%) are adjacent to regions of OE with maximal cell body caspase activation (green) and maximal TUNEL-labeling (black). Conversely, the areas of OE most distant from the OB display minimal soma caspase-3 activation and minimal TUNEL-positivity and are adjacent to the axons of lowest caspase-3 activation. Collectively, these results suggest that caspase-mediated cleavage begins in the synaptic complexes, is propagated through the axons, and reaches the ORN cell bodies, which then become TUNEL+ last.
Activation of caspase-9 during apoptosis in ORNs
Given that caspase-9 is thought to be the preferential, although not sole, activator of neuronal caspase-3 in vitro andin vivo, we examined caspase-9 expression and activation in ORNs by using multiple different caspase-9 antisera for immunoblotting and immunolocalization. Two different antisera that recognize full-length procaspase-9 demonstrated that levels of this zymogen increase significantly in the OE from 4 to 24 hr after bulbectomy and then drop to barely detectable levels by 72 hr (Figs.5 A, B).
Fig. 5.
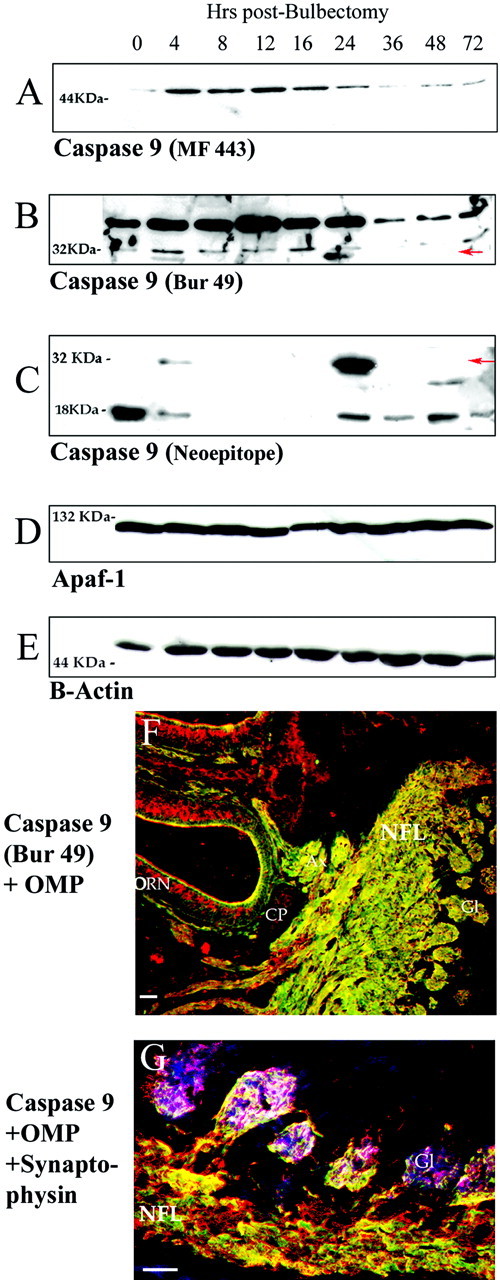
Axonal caspase-9 expression increases before maximal caspase-9 activation during apoptosis in olfactory neurons.A–C, examination of caspase-9 levels in OE after bulbectomy using three different antisera. Caspase-9 zymogen levels rise significantly in OE at 4–24 hr after bulbectomy as assessed using two different sera raised against recombinant human caspase-9 (A, MF 443) and anti-C9/Bur 49 (B). In addition, a cleavage product (C, 221 B) corresponding to the 35 kDa activated form of caspase-9 is detected initially at 4 hr and maximally at 24 hr after bulbectomy. C, A neoepitope antiserum recognizing the cleavage junction between the p19 and p12 subunits of caspase-9 detects the 35 kDa product representing the large subunit and prodomain, as well as a 19 kDa product that could represent the individual large subunit, at 4 and 24 hr (maximal) after bulbectomy.D, E, Control blots used antibodies to Apaf-1 (Stressgen, Victoria, BC, Canada) (D) and mouse anti-β-actin (Sigma) (E).F, Using the antiserum from B, caspase-9 (green) is expressed in axons (Ax) of the normal adult OE, emanating from OMP-positive (red) cell bodies (ORN) and colocalizing with axonal OMP as olfactory axons cross the cribiform plate (CP), course through the nerve fiber layer (NFL) and enter the glomeruli (Gl) of the olfactory bulb. G, Caspase-9 (red) colocalizes at the presynaptic compartment of OMP+ (green) olfactory neurons with synaptophysin (blue) to produce a purple overlap that is highly localized to the inner layers of glomeruli. White areas indicate regions of triple overlap of caspase-9, synaptophysin, and OMP. Scale bars, 100 μm.
An antiserum that recognizes recombinant human caspase-9 (Fig.5B, Bur 49) and another that recognizes a neoepitope that becomes detectable at the C terminus of the large subunit of caspase-9 after proteolytic liberation of the small subunit (Fig. 5C) indicate that multiple species of cleaved caspase-9 were detectable at early (4 hr) and late (24 hr) stages of apoptosis after bulbectomy. These species included the large catalytic subunit linked to the N-terminal prodomain (active caspase-9, 35 kDa) (Fig.5B,C) as well as the p19 large catalytic subunit alone (Fig. 5C). After maximal caspase-9 activation (24 hr), the expression of full-length caspase-9 was significantly reduced (Fig. 5A,B). Blots were reprobed with β-actin and Apaf-1. Note that the levels of Apaf-1 (a known activator of caspase-9 that complexes with it to form an apoptosome) do not change after bulbectomy (Fig.5D,E).
The appearance of maximal caspase-9 activation, immediately before maximal caspase-3 activation and TUNEL-positivity of ORNs, suggests that caspase-9 activation is a significant part of the pro-apoptotic program of deafferented ORNs. For this to be the case, caspase-9 would have to be distributed throughout the neuraxis to activate caspase-3 in response to appropriate extracellular stimuli. Immunofluorescent detection with anti-caspase-9 antiserum (green) combined with OMP (red) shows that caspase-9 is excluded from the ORN cell body, but is highly expressed in ORNs and clearly distributed throughout olfactory axons all the way into olfactory glomeruli (Fig. 5F). The overlap of caspase-9 (Fig.5G, red) with OMP (green) and synaptophysin (blue, magenta on overlap with caspase-9), a protein enriched in presynaptic membranes, confirms that caspase-9 has a synaptic localization. White regions occur where caspase-9, synaptophysin, and OMP all overlap.
Do ORNs develop normally in the absence of caspase-3?
The preceding results suggest a role for the caspase-9–caspase-3 pathway in execution of mature ORNs after deafferentation. Caspase-3 knock-out mice have many neurodevelopmental abnormalities but no reported olfactory impairment (Kuida et al., 1996). To examine whether caspase-3-mediated pathways are used to restrict the number of ORNs during development, we compared sagittal and coronal sections of OE from 1 week-old wild-type and caspase-3 −/− mice by immunohistochemical analysis of sibling groups (n = 3). A measurable increase in the already high levels of caspase-9 was evident throughout the neuraxis of the caspase-3 null (−/−) mice at P7 when equivalent sagittal sections are compared with their wild-type (+/+) littermates (Fig.6A). The olfactory bulb is larger in the null mice, but average glomerular size does not appear to be significantly different (data not shown). The synaptic glomeruli are, however, distributed more evenly around the P7 olfactory bulb, making the glomeruli appear less dense in caspase-3 knock-outs than in littermates (arrowheads). Fluorescence confocal microscopy (Z-series) was used to concurrently visualize and quantify OMP and procaspase-9 expression in these same groups of −/− and +/+ littermates (see Fig. 6B,C for pseudocolored image). OMP expression was almost twofold (1.8×) greater in the caspase-3 null mice than in littermates, and caspase-9 expression was almost fivefold (4.6×) higher. This suggests an expansion of the mature neuron population in the absence of caspase-3, but above that, a specific increase in caspase-9 expression in the axons emanating from each neuron. Because the population density within the OE (in the vertical dimension) is tightly controlled by local factors, we did not initially measure any significant difference in the ORN cell body number per linear millimeter of OE between knock-outs and littermates (data not shown). As a population expansion could be accommodated by additional folding of OE turbinates (instead of increasing the thickness of epithelium), we chose to examine axon bundle number, size, and area at distinct sites in the nasal turbinates of littermates. We selected a series of sites within the turbinates of coronal OE sections selected from position “2” in Figure1G that were subsequently processed for solid phase (peroxidase-VIP) immunohistochemistry for OMP (Fig.6A). Comparing knock-out mice with their littermates (n = 3) allowed us to assess potential changes in the number of olfactory axons projecting from a given turbinate back to the OB. Contrary to what may have been predicted, caspase-3 knock-outs have an overall lower number of axon bundles than their littermates, but each of these axon bundles is significantly larger (two to six times those of littermates) throughout each turbinate examined (Table1). The 1.5 to 2-fold expansion in total axon area seen in the knock-out, coupled with the twofold increase in OMP expression by confocal analysis, suggests that caspase-3 does play a role in controlling the cellular dynamic of the ORN population during development.
Fig. 6.
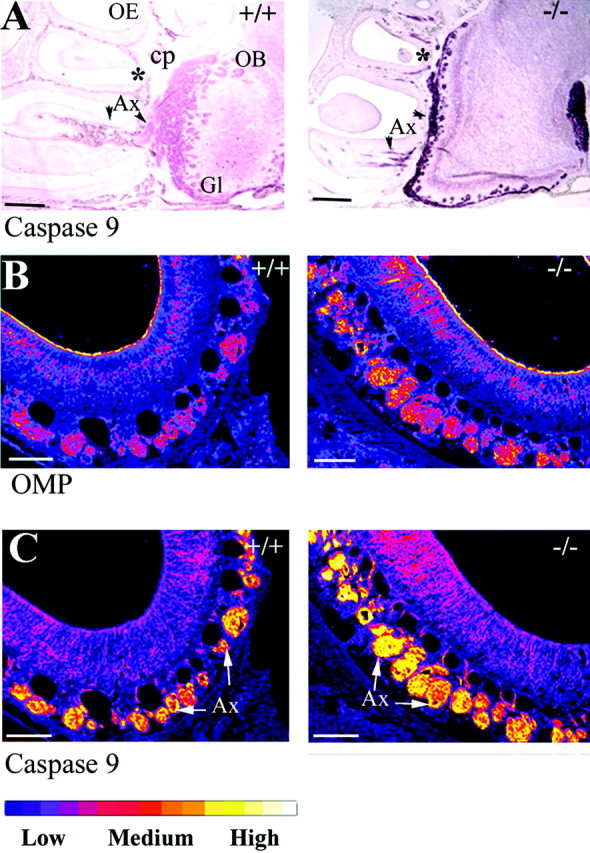
Viable caspase-3 −/− mice have an expanded olfactory neuron population with increased expression of procaspase-9.A, Low-power sagittal sections from 1 week postnatal wild-type (+/+) and caspase-3 null (−/−) mice were simultaneously stained for caspase-9. The caspase-3 null mice have a higher axonal (Ax) expression level of caspase-9 projecting into an enlarged olfactory bulb (OB). Formation of glomeruli (Gl) is also less advanced in the knock-out.B, C, Double immunofluorescence for OMP and caspase-9 was performed on coronal sections (generated from position *) of OE from caspase-3 wild-type and null mice (n = 3), visualized by confocal Z-series and pseudo-colored for relative intensity using NIH Image (see bar for scale). By this scale, OMP expression is twofold greater in cell bodies (ORN) and axons of the −/− than in the +/+, but the increase in intensity of C9 in the enlarged population of axon bundles (Ax) is fivefold higher in the −/− than in the +/+, when compared with OMP. Scale bars: A, 1 mm; B, C, 100 μm.
Table 1.
Changes in axon bundle number and axonal area in caspase-3 knock-outs and heterozygotes
| Superior turbinate | Mediolateral turbinate | Inferior turbinate | ||||
|---|---|---|---|---|---|---|
| +/− | −/− | +/− | −/− | +/− | −/− | |
| Number of axon bundles per millimeter OE (μm) | 21.1 ± 2.54 | 15.8 ± 1.53 | 22.7 ± 2.01 | 11.4 ± 1.00 | 19.1 ± 1.82 | 15.3 ± 0.94 |
| Area of average axon bundle (μm2) | 452 ± 19.1 | 953 ± 27.3 | 506 ± 15.3 | 2999 ± 55.6 | 257 ± 15.0 | 537 ± 17.4 |
| Total area of axons per millimeter OE (×10−3μm2) | 196 ± 0.46 | 223 ± 0.39 | 246 ± 0.42 | 342 ± 0.52 | 97.1 ± 0.35 | 125 ± 0.31 |
Do mature ORNs undergo apoptosis in caspase-3 null mice?
Despite published reports of embryonic and early postnatal lethality in caspase-3 null mutants, we have been able (by backcrossing within our caspase-3 null colony) to generate a caspase-3 −/− mouse that survives for at least 6–9 months. At no time have we detected caspase-3 expression or downstream cleavage (of Δ C or PARP) in the −/− mice, using the antisera reported in this study. To investigate the spatial (in addition to temporal) dynamics of apoptosis in mature wild-type ORNs in the absence of caspase-3, we first evaluated the number of TUNEL+ cells in the S, ML, and I turbinates at 24 hr after bulbectomy from three sites (Fig. 1G,1,2,3) at 1 (±0.2) mm, 2.5 (±0.45) mm, and 4 (±0.56) mm from the cribiform plate. A representative section of a wild-type mouse is shown in Figure7A. Within a given coronal section, the S, ML, and I turbinates contain neurons situated at increasing distances from the olfactory bulb, in which the number of TUNEL+ cells (Fig. 7B) is greater close to the bulb (superior turbinate of sections 1 and 2) and the least at further distances from the bulb (Fig. 7B, inferior turbinate at each position). We then repeated the same experiments on sets of caspase-3 −/− mice and heterozygote littermates subjected to unilateral bulbectomy. At 24 hr after bulbectomy (the beginning of the peak of caspase-3-mediated ORN apoptosis), and 48 hr after bulbectomy (when 90% of olfactory neurons should have completed apoptosis), there were no TUNEL+ ORNs within the OE in any of the knock-out mice tested (Fig.7C,D). TUNEL+ non-neuronal cells in bony tissue of the nasal cavity are, however, observed in these mice (data not shown).
Fig. 7.

Caspase-3 deletion prevents bulbectomy-induced apoptosis of mature ORNs. At 24 hr after bulbectomy, OE coronal sections at defined distances (1, 2.5 ± 0.45, and 4 mm) from the cribiform plate (designated 1, 2,3 on the mouse head in Fig. 1) were assessed to provide spatial patterns of the number of TUNEL-labeled ORNs after bulbectomy in wild-type siblings of caspase-3 null mice. A, Turbinates in a single section (Fig. 1D, position 2) were individually assessed as belonging to superior (close to bulb), mediolateral, and inferior (distant from bulb) turbinates.B, The number of TUNEL+ ORNs were plotted against calculated distance from the olfactory bulb. These data were then used as a baseline for assessing apoptosis in the caspase-3 null mice. The data from the knock-out mice has not been plotted at 24 hr (C) and 48 hr (D), and there is no evidence for TUNEL+ ORNs on the lesioned side in any of the knock-out mice (n = 3 per time point) analyzed.S, Septum. Scale bars, 500 μm.
DISCUSSION
ORNs undergo apoptosis after deafferentation in a spatiotemporal manner in which a chief predictive factor determining the timing of terminal neuronal apoptosis is the length of the ORN axon from the site of lesion (Fig. 1). Because of the three-dimensional structure of the OE, this profile has enabled us to track pro-apoptotic signals all the way from the synapse to the cell body, and even across a single turbinate, to provide data pinpointing where, and at what apoptotic stage, terminal apoptotic pathways become activated. This analysis led to several novel observations. First, endogenous levels of procaspase-3 and -9 increase, beginning as early as 4 hr after bulbectomy. Second, caspase-3 activation is initially recorded at the synapse, then in axon bundles, and only later in the cell bodies. Third, activated caspase-3 is capable of cleaving substrates like APLP2 throughout the olfactory neuraxis. These data have important implications for current models of a role for retrograde signaling in neuronal apoptosis.
In delineating the pathways that drive retrograde ORN apoptosis, caspases 9 and 3 are both expressed by mature olfactory neurons throughout their axons, all the way into the presynaptic compartment (Figs. 4, 5). When the olfactory bulb is lesioned in such a way to remove trophic support from the postsynaptic neuron and deafferent the olfactory nerve, two responses are observed. The endogenous levels of caspase-3 and -9 proenzymes increase, beginning as early as 4 hr after bulbectomy. The peak of expression of caspase-9 (at 24 hr) coincides with its maximal activation, after which proenzyme levels decrease significantly. This peak of caspase-9 activation occurs immediately before the peak of caspase-3 proenzyme expression and cleavage (at 48 hr after bulbectomy). We have not yet been able to generate evidence demonstrating that the elevated proenzyme levels may be a result of transcriptional upregulation, although this could occur marginally within a timeframe allowed by fast retrograde transport. Given the distribution of caspase-3 and -9 throughout the neuraxis before lesion, we believe that it is more likely that the increased levels result from a local upregulation of proenzyme production (at the level of translation) or a change in the constitutive maintenance of caspase-3 and -9. Although further experiments are required to distinguish between these possibilities, these data are the first to show that the commitment to death that occurs early at the initiation of neuronal apoptosis also includes procaspase-9 and –3 accumulation (Putcha et al., 2000). At the time of maximal caspase proenzyme accumulation, caspase activation also becomes evident in these neurons. Strikingly, caspase-3 activation, detected by conformation-sensitive antibodies that recognize only the processed, active form of caspase-3, shows the active enzyme as proximal as the synapse. Active caspase-3 then becomes spatially detectable in axons and only later in ORN cell bodies.
Further support for the synapse to axon to soma propagation of the apoptotic signal comes from use of the Δ-C-APP neoepitope antibody, which provides us with a sensitive way to demonstrate active cleavage of caspase-3 target proteins in ORN synapses and axons in vivo. Although it is conceivable that an alternative caspase (e.g., caspase-6) could also cleave at the same target site, this cleavage event can still be defined as caspase-3-dependent. Cleaved APLP2 cannot be immunoprecipitated or detected immunohistochemically from the OE of lesioned caspase-3 knock-out mice (Fig. 3D). Also, because the APLP2 cleavage product persists in cells and axons during degeneration, whereas active caspase-3 does not, examination of cleaved APLP2 provides a useful map that shows where caspase-3 is currently active and also has been active. The first site at which APLP2 cleavage is detected is within the synaptic complexes of olfactory bulb glomeruli, as evidenced by overlap with synaptophysin, a synaptic protein that, within the glomeruli is primarily found in ORN presynaptic terminals (Kasowski et al., 1999). The active caspase-3 signal is also seen spreading out from the synaptic glomeruli into the nerve fiber layer of the olfactory bulb (Fig.4A,B) and then into the axon bundles of the lamina propria (Fig. 4C,D). The persistence of the Δ–C APP neoepitope in axon bundles after cleavage of APLP2 reveals that there is a level of organization within ORN axon bundles at which synchronously apoptotic ORNs are subtly divided into groups of mesaxons (Fig. 3). Olfactory axons thus appear grouped together on the basis of functional or developmental state, which may underlie their differential vulnerability to apoptosis (Figs. 1,3).
Collectively, the results demonstrating synaptic caspase-3 activation lend strong in vivo support to earlier work targeting the synapse as a focal point for initiation of pro-apoptotic signaling, either in a developing or in a lesion state (Mattson et al., 1998;Mattson and Duan, 1999). The synaptic junction has historically been argued as a site at which positive trophic signals are used to reinforce and stimulate survival of the presynaptic neuron during development and maintenance of the CNS. Our data suggest that, alternatively, the postsynaptic neuron (in this case, the mitral or periglomerular neuron) could control turnover of the presynaptic olfactory neuron population by manipulating the release of pro-survival stimuli, thus shifting the balance of synaptic signaling pathways from pro-survival to pro-apoptotic. The continued expression of caspases 3 and 9 at the highly plastic (NMDA receptor-mediated) olfactory synapse also puts them in an ideal position to participate in the remodeling or local dismantling of inactive synapses via the cleavage of target proteins crucial for synaptic integrity, such as α- and β-spectrin and β-actin (Wang et al., 1998; Chan and Mattson, 1999)
The colocalization of axonal caspase-9 with active caspase-3 (directly mirroring the distribution of ORN mitochondria), coupled with a dependence on Bcl-2 for ORN apoptosis, suggests a significant role for the mitochondrial pathway in mature ORN apoptosis (Krajewski et al., 1999). Procaspases 3 and 9 are also ideally situated to balance pro-apoptotic pathways with those that drive neuronal plasticity, because axonal caspase-9 is detected in many other neuronal axons embryonically (S. Krajewski and C. Cowan, unpublished observations), but is only retained at readily detectable levels postnatally in the olfactory neuraxis. Now that caspase-3 and -9 have been placed at the synapse, it will be important to determine which upstream signaling mechanisms converge to balance synaptic survival and apoptotic pathways in an intact olfactory neuraxis. A number of known pro- and anti-apoptotic signaling proteins (e.g., the tyrosine kinase BDNF receptor, Trk B, and the low-affinity nerve growth factor receptor, P75) are present at the olfactory synapse (Roskams et al., 1996). It is conceivable that differential stimulation of p75 (feeding into mitochondrial apoptotic pathways) and kinase active–inactive Trk B, by bulb-derived neurotrophins could synergistically serve to control caspase-9 activation at the synapse. In addition, nitric oxide, released from periglomerular neurons into a glomerular synapse could directly regulate the activation of presynaptic procaspase-3 or -9 (Tenneti et al., 1997; Tamatani et al., 1998; Li et al., 1999). The upstream signals that may stimulate axonal caspase pathways after physical deafferentation are less obvious and require further investigation. The compartmentalization of caspase-9 in the axon and caspase-3 in axon and cell body introduces the possibility that different pathways could be used for local and distant control of ORN apoptosis, in which caspase-3 not only drives apoptosis of the soma but also retrogradely dismantles proteins that maintain axonal integrity. In contrast, in the mature optic nerve, the process of axonal dismantling and Wallerian degeneration is caspase-3-independent, but neuronal survival is caspase-3-dependent (Finn et al., 2000).
During the course of these studies we also generated caspase-3 null mice that survived beyond the perinatal period. Examination of these mice revealed that the OMP+ ORN population is expanded developmentally, providing evidence that caspase-3 is ordinarily involved in regulating ORN number (Fig. 6). The critical role of caspase-3 in regulating ORN number is also illustrated by the lack of TUNEL+ ORNs in the OE of caspase-3 −/− mice after bulbectomy (Fig. 7). Other cortical neuronal populations are also expanded in long-lived caspase-3 null mice generated by crossing into a black 6 background (G. Robertson, personal communication). The variation in phenotype seen in caspase-3 null mutants, including the upregulation of caspase-9 proenzyme levels, suggests that olfactory neurons use pathways that attempt to compensate for the loss of caspase-3. The identification of alternative compensatory mechanisms in the olfactory system and other areas of the CNS requires further study.
This study highlights the importance of how the location of a caspase within a neuron dictates not only which local pathways may be capable of activating it, but also which substrates are accessible, a key issue when examining candidate neuronal caspase target proteins that could be highly compartmentalized. These data also allow us to place caspase-9 and -3 in a more dynamic role in neuronal apoptosis than previously demonstrated, simultaneously carrying pro-apoptotic signals from a synapse to their final site of action at the neuronal nucleus and actively dismantling axonal proteins. The identification of neuronal caspase activation at the living synapse, coupled with the identification of other apoptotic mediators in the synaptosome (Mattson et al., 1998) now allows us to place caspase-mediated signaling at the epicenter for target-derived trophic support, plasticity, and survival of the olfactory receptor neuron.
Footnotes
This work was supported by Centre for Molecular Medicine and Therapeutics–Merck start-up funds (A.J.R.), a pilot project grant from the British Columbia Health Research Foundation (A.J.R.), and United States Public Health Service Grant NS36821 (S.K.). We thank Harinder Janjua and Joellen Fung for technical assistance, Seth Blackshaw for technical guidance for TUNEL, Murry Gilbert for assistance with confocal microscopy, Frank Margolis for the OMP cDNA and antibody, Gopal Thinakaran for APLP2-specific antibodies, and Phil Hieter for making it possible for our laboratory to do this work.
Correspondence should be addressed to Dr. Jane Roskams, Centre for Molecular Medicine and Therapeutics, 950 W. 28th Avenue, Vancouver, BC, Canada V5Z 4H4. E-mail: roskams@cmmt.ubc.ca.
REFERENCES
- 1.Black SC, Huang JQ, Rezaiefar P, Radinovic S, Eberhart A, Nicholson DW, Rodger IW. Colocalization of the cysteine protease caspase-3 with apoptotic myocytes after in vivo myocardial ischemia and reperfusion in the rat. J Mol Cell Cardiol. 1998;30:733–742. doi: 10.1006/jmcc.1998.0660. [DOI] [PubMed] [Google Scholar]
- 2.Blackshaw S, Snyder SH. Parapinopsin, a novel catfish opsin localized to the parapineal organ. J Neurosci. 1997;17:8083–8092. doi: 10.1523/JNEUROSCI.17-21-08083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burek MJ, Oppenheim RW. Cellular interactions that regulate programmed cell death in the developing vertebrate nervous system. In: Koliatsos VE, Ratan RR, editors. Cell death and diseases of the nervous system. Humana; Ottawa: 1998. pp. 145–179. [Google Scholar]
- 4.Calof AL, Hagiwara N, Holcomb JD, Mumm JS, Shou J. Neurogenesis and cell death in olfactory epithelium. J Neurobiol. 1996;30:67–81. doi: 10.1002/(SICI)1097-4695(199605)30:1<67::AID-NEU7>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 5.Carr VM, Farbman AI. Ablation of the olfactory bulb up-regulates the rate of neurogenesis and induces precocious cell death in olfactory epithelium. Exp Neurol. 1992;115:55–59. doi: 10.1016/0014-4886(92)90221-b. [DOI] [PubMed] [Google Scholar]
- 6.Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res. 1999;58:167–190. [PubMed] [Google Scholar]
- 7.Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- 8.Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, Hamilton RL, Loeffert JE, Graham SH. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human. FASEB J. 1999;13:813–821. doi: 10.1096/fasebj.13.8.813. [DOI] [PubMed] [Google Scholar]
- 9.Davies AM. Neurotrophin switching: where does it stand? Curr Opin Neurobiol. 1997;7:110–118. doi: 10.1016/s0959-4388(97)80128-6. [DOI] [PubMed] [Google Scholar]
- 10.Earnshaw WC, Kaufmann SH. Mammalian caspases: structure, activation, substrates and function during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 11. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391 1998. 43 50 [Erratum (1998) 393:396] [DOI] [PubMed] [Google Scholar]
- 12.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Farbman AI. Olfactory neurogenesis: genetic or environmental controls? Trends Neurosci. 1990;13:362–365. doi: 10.1016/0166-2236(90)90017-5. [DOI] [PubMed] [Google Scholar]
- 14.Farbman AI. Injury-stimulated neurogenesis in sensory systems. Adv Neurol. 1997;72:157–161. [PubMed] [Google Scholar]
- 15.Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 17.Graziadei PP, Graziadei GA. Neurogenesis and neuron regeneration in the olfactory system of mammals. I. Morphological aspects of differentiation and structural organization of the olfactory sensory neurons. J Neurocytol. 1979;8:1–18. doi: 10.1007/BF01206454. [DOI] [PubMed] [Google Scholar]
- 18.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 19.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 20.Huard JM, Youngentob SL, Goldstein BJ, Luskin MB, Schwob JE. Adult olfactory epithelium contains multipotent progenitors that give rise to neurons and non-neural cells. J Comp Neurol. 1998;400:469–486. [PubMed] [Google Scholar]
- 21.Jourdan F, Moyse E, De Bilbao F, Dubois-Dauphin M. Olfactory neurons are protected from apoptosis in adult transgenic mice over-expressing the bcl-2 gene. NeuroReport. 1998;9:921–926. doi: 10.1097/00001756-199803300-00029. [DOI] [PubMed] [Google Scholar]
- 22.Kasowski HJ, Kim H, Greer CA. Compartmental organization of the olfactory bulb glomerulus. J Comp Neurol. 1999;407:261–274. [PubMed] [Google Scholar]
- 23.Keramaris E, Stefanis L, MacLaurin J, Harada N, Takaku K, Ishikawa T, Taketo M, Robertson GS, Nicholson DW, Slack RS, Park DS. Involvement of caspase 3 in apoptotic death of cortical neurons evoked by DNA damage. Mol Cell Neurosci. 2000;15:368–379. doi: 10.1006/mcne.2000.0838. [DOI] [PubMed] [Google Scholar]
- 24.Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 26.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Bombeck CA, Yang S, Kim YM, Billiar TR. Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J Biol Chem. 1999;274:17325–17333. doi: 10.1074/jbc.274.24.17325. [DOI] [PubMed] [Google Scholar]
- 28.Mattson MP, Duan W. “Apoptotic” biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:152–166. [PubMed] [Google Scholar]
- 29.Mattson MP, Keller JN, Begley JG. Evidence for synaptic apoptosis. Exp Neurol. 1998;153:35–48. doi: 10.1006/exnr.1998.6863. [DOI] [PubMed] [Google Scholar]
- 30.Mesner PW, Jr, Bible KC, Martins LM, Kottke TJ, Srinivasula SM, Svingen PA, Chilcote TJ, Basi GS, Tung JS, Krajewski S, Reed JC, Alnemri ES, Earnshaw WC, Kaufmann SH. Characterization of caspase processing and activation in HL-60 cell cytosol under cell-free conditions. Nucleotide requirement and inhibitor profile. J Biol Chem. 1999;274:22635–22645. doi: 10.1074/jbc.274.32.22635. [DOI] [PubMed] [Google Scholar]
- 31.Michel D, Moyse E, Brun G, Jourdan F. Induction of apoptosis in mouse [correction of rat] olfactory neuroepithelium by synaptic target ablation. NeuroReport. 1994;5:1329–1332. [PubMed] [Google Scholar]
- 32.Moulton DG. Dynamics of cell populations in the olfactory epithelium. Ann NY Acad Sci. 1974;237:52–61. doi: 10.1111/j.1749-6632.1974.tb49843.x. [DOI] [PubMed] [Google Scholar]
- 33.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 34.Pieper AA, Brat DJ, Krug DK, Watkins CC, Gupta A, Blackshaw S, Verma A, Wang ZQ, Snyder SH. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Putcha GV, Deshmukh M, Johnson EM., Jr Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J Cell Biol. 2000;149:1011–1018. doi: 10.1083/jcb.149.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 37.Roskams AJ, Bredt DS, Dawson TM, Ronnett GV. Nitric oxide mediates the formation of synaptic connections in developing and regenerating olfactory receptor neurons. Neuron. 1994;13:289–299. doi: 10.1016/0896-6273(94)90347-6. [DOI] [PubMed] [Google Scholar]
- 38.Roskams AJ, Bethel MA, Hurt KJ, Ronnett GV. Sequential expression of Trks A, B, and C in the regenerating olfactory neuroepithelium. J Neurosci. 1996;16:1294–1307. doi: 10.1523/JNEUROSCI.16-04-01294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roskams AJ, Cai X, Ronnett GV. Expression of neuron-specific beta-III tubulin during olfactory neurogenesis in the embryonic and adult rat. Neuroscience. 1998;83:191–200. doi: 10.1016/s0306-4522(97)00344-8. [DOI] [PubMed] [Google Scholar]
- 40.Samali A, Zhivotovsky B, Jones DP, Orrenius S. Detection of pro-caspase-3 in cytosol and mitochondria of various tissues. FEBS Lett. 1998;431:167–169. doi: 10.1016/s0014-5793(98)00740-6. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz Levey M, Chikaraishi DM, Kauer JS. Characterization of potential precursor populations in the mouse olfactory epithelium using immunocytochemistry and autoradiography. J Neurosci. 1991;11:3556–3564. doi: 10.1523/JNEUROSCI.11-11-03556.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Springer JE, Azbill RD, Knapp PE. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord. Nat Med. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- 43.Stefanis L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol. 1997;10:299–305. doi: 10.1097/00019052-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 44.Tamatani M, Ogawa S, Nunez G, Tohyama M. Growth factors prevent changes in Bcl-2 and Bax expression and neuronal apoptosis induced by nitric oxide. Cell Death Differ. 1998;5:911–919. doi: 10.1038/sj.cdd.4400439. [DOI] [PubMed] [Google Scholar]
- 45.Tenneti L, D'Emilia DM, Lipton SA. Suppression of neuronal apoptosis by S-nitrosylation of caspases. Neurosci Lett. 1997;236:139–142. doi: 10.1016/s0304-3940(97)00780-5. [DOI] [PubMed] [Google Scholar]
- 46.Thinakaran G, Kitt CA, Roskams AJ, Slunt HH, Masliah E, von Koch C, Ginsberg SD, Ronnett GV, Reed RR, Price DL, Sisodia SS. Distribution of an APP homolog, APLP2, in the mouse olfactory system: a potential role for APLP2 in axogenesis. J Neurosci. 1995;15:6314–6326. doi: 10.1523/JNEUROSCI.15-10-06314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, Tessier-Lavigne M. En passant neurotrophic action of an intermediate axonal target in the developing mammalian CNS. Nature. 1999;401:765–769. doi: 10.1038/44521. [DOI] [PubMed] [Google Scholar]
- 48.Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS. Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J Biol Chem. 1998;273:22490–22497. doi: 10.1074/jbc.273.35.22490. [DOI] [PubMed] [Google Scholar]