

Abstract
Parkinson's disease (PD) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity are both associated with dopaminergic neuron death in the substantia nigra (SN). Apoptosis has been implicated in this cell loss; however, whether or not it is a major component of disease pathology remains controversial. Caspases are a major class of proteases involved in the apoptotic process. To evaluate the role of caspases in PD, we analyzed caspase activation in MPTP-treated mice, in cultured dopaminergic cells, and in postmortem PD brain tissue. MPTP was found to elicit not only the activation of the effector caspase-3 but also the initiators caspase-8 and caspase-9, mitochondrial cytochromec release, and Bid cleavage in the SN of wild-type mice. These changes were attenuated in transgenic mice neuronally expressing the general caspase inhibitor protein baculoviral p35. These mice also displayed increased resistance to the cytotoxic effects of the drug. MPTP-associated toxicity in culture was found temporally to involve cytochrome c release, activation of caspase-9, caspase-3, and caspase-8, and Bid cleavage. Caspase-9 inhibition prevented the activation of both caspase-3 and caspase-8 and also inhibited Bid cleavage, but not cytochrome c release. Activated caspase-8 and caspase-9 were immunologically detectable within MPP+-treated mesencephalic dopaminergic neurons, dopaminergic nigral neurons from MPTP-treated mice, and autopsied Parkinsonian tissue from late-onset sporadic cases of the disease. These data demonstrate that MPTP-mediated activation of caspase-9 via cytochrome crelease results in the activation of caspase-8 and Bid cleavage, which we speculate may be involved in the amplification of caspase-mediated dopaminergic cell death. These data suggest that caspase inhibitors constitute a plausible therapeutic for PD.
Keywords: caspases, substantia nigra, Parkinson's disease, MPTP, mesencephalic cultures, PC12
Parkinson's disease (PD) is characterized by a progressive degeneration of dopaminergic neurons of the substantia nigra (SN). In postmortem Parkinsonian brain dying neurons are present that have been reported to display morphological characteristics of apoptosis, including cell shrinkage, chromatin condensation, and DNA fragmentation (Mochizuki et al., 1996;Hajimohamadreza and Treherne, 1997; Tatton et al., 1998). In addition, the expression of known effectors of neuronal apoptosis, including the major downstream executioner caspase, caspase-3, has been reported in autopsied SN tissue isolated from PD patients (Anglade et al., 1997; Hartmann et al., 2000). In vivo andin vitro models of PD also have suggested a role for apoptosis in the related human disease pathology (for review, seeAndersen, 2001). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration in mice, for example, produces selective degeneration of dopaminergic neurons of the SN, and the presence of DNA fragmentation has been reported after its administration along with the induction of caspase-3 activity (Tatton and Kish, 1997; Hartmann et al., 2000). In addition, proapoptotic Bax expression is found to be elevated after MPTP administration, and mice in which Bax has been ablated or the expression of the anti-apoptotic protein Bcl-2 has been elevated have been shown to be resistant to this agent (Hassouna et al., 1996; Offen et al., 1998; Yang et al., 1998; Choi et al., 2001). 1-Methyl-4-phenylpyridinium ion (MPP+), a metabolite of MPTP, has been found at low dosages to cause apoptotic cell death in dopaminergic PC12 cells and mesencephalic cultures via the activation of caspase-3 (Hartley et al., 1994; Mochizuki et al., 1996; Dodel et al., 1998). Caspase-3, therefore, has been suggested to be the final effector of apoptotic cell death in Parkinson's-associated neurodegeneration, but the exact biochemical pathways involved in its activation are unclear.
Previously, we have reported the creation of transgenic mice that neuronally express the baculoviral protein p35 (Viswanath et al., 2000). This protein acts as a potent irreversible caspase inhibitor with broad effectiveness against all classes of these proteases (Bump et al., 1995; Zhou et al., 1998; Fisher et al., 1999). Expression of the p35 protein in vivo has been shown to confer functional caspase inhibitory activity and to attenuate apoptosis in a variety of different paradigms (Hay et al., 1994; Sugimoto et al., 1994; Davidson and Steller, 1998; Izquierdo et al., 1999; Hisahara et al., 2000;Viswanath et al., 2000). Unlike animals deficient in the expression of specific caspases (i.e., caspase-3, caspase-8, and caspase-9; Kuida et al., 1996, 1998; Varfolomeev et al., 1998; Colussi and Kumar, 1999;Zheng et al., 1999), p35 transgenics appear phenotypically normal and are viable, and embryonic development does not appear to be affected in these animals (Viswanath et al., 2000). These animals were studied along with the examination of dopaminergic cell lines, primary mesencephalic cultures, and PD brain tissue to assess the possible regulatory role of other caspases in the activation of caspase-3 in the molecular pathway leading to apoptotic cell death in MPTP toxicity and sporadic late-onset PD.
MATERIALS AND METHODS
MPTP treatment of mice. MPTP-HCl (Sigma Aldrich, St. Louis, MO) in 0.9% NaCl was administered to 10- to 16-week-old wild-type and p35 transgenic mice, using an acute dosing regimen of 15 mg/kg intraperitoneally every 2 hr for four doses (n = 5–7 mice in each group) as described previously (Yang et al., 1998). Control animals in both paradigms were treated with equal volumes of saline.
Stereological counts of tyrosine hydroxylase-immunopositive neurons. MPTP and saline-treated animals (n = 5–7) were perfused transcardially with 4% paraformaldehyde. Brain tissue containing SN was sectioned coronally at 40 μm on a sliding microtome. Immunohistochemistry was performed with a rabbit polyclonal anti-tyrosine hydroxylase antibody (Chemicon, Temecula, CA). Briefly, fixed tissue was preincubated with 1% hydrogen peroxide/methanol to reduce background staining, and then the tissue was exposed to a 1:500 dilution of primary antibody in appropriate buffer overnight at 4°C. Sections were washed with PBS, incubated with biotinylated anti-rabbit IgG secondary antibody (Vector Laboratories, Burlingame, CA), rinsed, placed for 1 hr in avidin–biotin peroxidase solution (Vectastain ABC kit, Vector Laboratories), and then developed in 0.01% hydrogen peroxide, 0.01% diaminobenzidine tetrahydrochloride (DAB; Sigma Aldrich) for 5 min. Sections were rinsed, mounted on glass slides in 50% glycerol, and coverslipped. Slides were coded, and calculations of the cell numbers were performed by using the unbiased dissector method (West et al., 1991). Hydroxylase-immunopositive (TH+) cells were counted from a total of 14–18 sections in each field per brain (i.e., every second section) at a magnification of 100× in a 0.2 mm2area, using the optical fractionator approach.
Measurement of levels of dopamine and homovanillic acid.MPTP and saline-treated animals (n = 5–7) were killed 7 d after the last MPTP injection. For each mouse the striatum was dissected, immediately frozen in dry ice, and then stored at −80°C for the measurement of dopamine and its metabolites. Dissected striatal tissues were sonicated and centrifuged in chilled 0.1 m perchloric acid (PCA; 30 μl/mg tissue). The supernatants were analyzed for levels of dopamine and homovanillic acid (HVA) by using 16-electrode electrochemical detection as described previously (Beal et al., 1990). Concentrations of dopamine and HVA are expressed as picomoles per milligram of protein.
Measurement of caspase activities. Caspase activities were measured in PC12 cells and murine SN tissues at the indicated time points after toxin treatment by using specific fluorogenic tetrapeptide substrates. Caspase-3 activity was measured with a fluorace apopain assay kit (Bio-Rad, Hercules, CA) that used the substrate DEVD-AFC according to the manufacturer's directions. Caspase-8 and caspase-1 activities were measured by using the substrates IETD-AFC and YVAD-AFC (Calbiochem, La Jolla, CA), respectively. Caspase-9 activity was measured with an assay kit (Oncogene, Cambridge, MA), using the specific substrate LEHD-AFC. Cells or tissues were lysed in 100–250 μl of lysis buffer [containing (in mm) 10 HEPES-KOH, pH 7.2, 2 EDTA, 5 dithiothreitol (DTT), and 1 phenylmethylsulfonyl fluoride (PMSF) plus 0.1% CHAPS, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, 20 μg/ml leupeptin], vortexed gently, and freeze-thawed four to five times. Lysates were centrifuged at 13,000 ×g for 30 min at 4°C, and the supernatants were collected. Protein concentrations were estimated by using Bradford reagent (Bio-Rad). Supernatant aliquots were incubated with the fluorescent substrates at 37°C for 1–2 hr. Free AFC accumulation resulting from cleavage of the aspartate–AFC bond was measured with a Cytofluor II fluorometer at 360 nm excitation and 515 nm emission wavelengths.
Western blot analysis of cytochrome c levels and Bid cleavage. For analysis of Bid cleavage, tissue extracts were prepared as described above for the measurement of caspase activities. To analyze cytochrome c release, we performed protein extraction of both the mitochondrial and cytosolic fractions as described previously (Kirsch et al., 1999). Cells or tissues were rinsed twice with cold PBS and lysed in cold MSHE buffer [0.21m mannitol (and in mm) 70 sucrose, 10 HEPES-KOH, pH 7.2, 1 EGTA, 1 EDTA, 0.15 spermine, 0.75 spermidine, and 5 DTT plus 2 μg/ml leupeptin, 2 μm benzamidine-HCl, 1 μg/ml pepstatin]. Cells were homogenized with Dounce on ice. Nuclei and unlysed cells were removed by centrifugation at 500 × g at 4°C for 12 min. The supernatant was centrifuged at 9500 × gfor 9 min at 4°C to pellet mitochondria. The supernatant contained the cytosolic fraction; the mitochondrial pellet was resuspended in MSHE buffer and was also used for immunoblot analysis. Protein (5 μg) from the cytosolic fraction and 2.2 μg from the mitochondrial fraction were loaded per lane. The primary antibodies were either a 1:1000 dilution of polyclonal anti-human/mouse Bid (R&D Systems, Minneapolis, MN) or a 1:1000 dilution of monoclonal cytochromec (PharMingen, San Diego, CA), followed by horseradish peroxidase-conjugated secondary antibody (Vector Laboratories) and autoradiography with enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Arlington Heights, IL). Cytosolic extracts also were analyzed with Cox IV antibody, which serves as an indicator of mitochondrial contamination of cytosolic extracts (Clontech, Palo Alto, CA).
PC12 cell culture and treatment with MPP+. PC12 cells were grown in DMEM supplemented with 10% heat-inactivated horse serum, 5% fetal bovine serum, and 2% penicillin/streptomycin (Life Technologies, Gaithersburg, MD). Typically, the cells were plated in 100 × 200 mm culture dishes or six-well plates at a confluency of 50–80% and grown at 37°C in 5% CO2. Cultures were used for no more than 20 passages. Cells were treated with 150 μm MPP+(Sigma Aldrich) for 4–24 hr; untreated cells were used as controls. MPP+ stock was made freshly before its addition to DMEM. For each set of experiments the samples were run in triplicate and repeated three times. For experiments that used the caspase-8 and caspase-9 inhibitors (25 μm in DMSO; Calbiochem-Novabiochem, San Diego, CA and PharMingen, respectively), the cells were preincubated with the inhibitors for 1 hr before treatment with MPP+.
Mesencephalic cultures and MPP+treatment. All experiments were performed by following an institutionally approved protocol in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals. Ventral mesencephalon was dissected from embryonic gestation day 14 (E14) wild-type and p35 transgenic mice. Neurons were dissociated mechanically and incubated for 5 min at room temperature (RT) in 0.05% trypsin-EDTA (Life Technologies) in HBSS without calcium and magnesium (Life Technologies). The digestion reaction was stopped by the addition of 10% fetal bovine serum (FBS; Life Technologies) in high-glucose DMEM (Life Technologies). Cells were plated onto poly-d-lysine-coated eight-well culture slides (Becton Dickinson Labware, Bedford, MA) at a density of 3.5 × 105 cells/well. Neurons were grown in neurobasal medium (NBM) supplemented with 2% B-27, 0.5 mm glutamine, and 1% antibiotic–antimycotic solution (all from Life Technologies). All cultures were incubated at 37°C at 5% CO2. After 4 d one-half of the medium was removed and replaced with an equal volume of medium. Cells were grown an additional 2 d and then treated with 5 μm MPP+ (Sigma Aldrich) for 6, 12, 18, 24, and 48 hr. The caspase-9 inhibitor Z-LEHD-FMK (25 μm; PharMingen) was added 1 hr before MPP+ treatment. Cells were fixed with 4% paraformaldehyde in PBS for 30 min.
Immunocytochemistry for activated caspase-3, caspase-9, and caspase-8 and TH and DAPI staining in cultured TH+ SN neurons. To address the time course of MPP+-induced activation of caspase-3, caspase-9, and caspase-8 in apoptotic TH+ SN neurons, we used double immunolabeling for both the activated caspases and TH and DAPI staining in this study. After fixation the cells were blocked with 10% normal goat serum (NGS) containing 0.3% H2O2 and 0.3% Triton X-100 for 1 hr at RT. After being washed three times in PBS, the cells were incubated with rabbit polyclonal antibodies that recognize the active forms of caspase-3 (1:50; New England Biolabs, Beverly, MA), caspase-9 (1:100; New England Biolabs), and caspase-8 (1:500; SK440, gift from SmithKline Beecham, Philadelphia, PA) at 4°C overnight. After being washed with PBS, the cells were incubated with BODIPY FL goat anti-rabbit IgG conjugates (1:200; Molecular Probes, Eugene, OR) at RT for 1 hr. Then the cells were washed with PBS and incubated with sheep anti-tyrosine hydroxylase polyclonal antibody (1:100; Chemicon) at RT for 1 hr. After being washed with PBS, the cells were incubated with Cy-3-conjugated anti-sheep IgG (1:500; Jackson ImmunoResearch, West Grove, PA) at RT for 1 hr. Finally, the cells were wash with PBS and mounted with VectaShield mounting medium with DAPI (Vector Laboratories).
Cell counts in cultured TH+ SN neurons from p35 transgenic versus wild-type mice treated with MPP+. The total number of TH+ neurons in mesencephalic cultures from both p35 transgenic and wild-type animals was counted in every well in at least three wells per time point (0, 6, 12, and 24 hr after MPP+ treatment). The percentage of TH+ neurons compared with that of the untreated wild type was used to evaluate MPP+ toxicity. Experiments were repeated three times with cultures isolated from independent dissections.
Immunohistochemistry of activated caspase-8 and caspase-9 and TH+ SN neurons in vivo. For mouse tissue, free-floating 40-μm-thick sections were treated first with sheep polyclonal anti-tyrosine hydroxylase antibody (1:500;Chemicon), followed by biotinylated anti-sheep IgG secondary antibody and cy3 streptavidin (Jackson ImmunoResearch). TH+ neurons were localized, and then the sections were reincubated with either rabbit polyclonal anti-activated caspase-9 antibody (1:100; New England Biolabs) or rabbit polyclonal anti-activated caspase-8 antibody (1:2500; gift from SmithKline Beecham), followed by biotinylated anti-rabbit IgG and cy2 streptavidin (Jackson ImmunoResearch); the sections were visualized under fluorescence microscopy. Control experiments were performed in which one or the other of the primary antisera was omitted. No staining was observed under these conditions. Immunostaining was performed similarly with human brain sections (25 μm thick) from both late-onset PD patients with mild-to-moderate focal loss of melanized neurons in ventral and caudal parts of the SN pars compacta (SNpc) and age-matched controls fixed in formaldehyde (n = 3 for each; average postmortem period, 7.25 ± 5 hr; average age, 69.7 ± 9 years). All PD cases were diagnosed clinically and neuropathologically confirmed, whereas controls had no clinical or neuropathological signs of PD or dementia. Caspase expression was not found to be affected by postmortem delay (Stadelmann et al., 1999).
Statistical analysis. Results were expressed as means ± SD of the difference between wild type and p35 transgenics as assessed by ANOVA. Values of p < 0.01 were taken as being statistically significant.
RESULTS
Transgenic mice expressing the general caspase inhibitory protein p35 are resistant to the toxicity associated with the Parkinson's-inducing agent MPTP
Apoptosis has been shown to play a role in MPTP-induced neurotoxicity (Hassouna et al., 1996; Offen et al., 1998). Because caspase-3 has been demonstrated previously to be activated during this process both in vitro and in vivo, we examined whether expression of the general caspase inhibitor p35 in our transgenic animals would act to inhibit the death of dopaminergic SN neurons induced by MPTP treatment. Stereological cell counts of TH+ dopaminergic neurons were performed on coronal sections isolated from the SN of p35 transgenics versus wild-type animals injected with either MPTP or saline (n = 5–7). TH+ cells were counted in every alternate section via the optical fractionator method that combines the optical dissector method and systematic uniform random sampling (West et al., 1991). The number of TH+ neurons in the SNpc of wild-type animals was found to be decreased by ∼37 ± 6.2% after MPTP administration compared with animals injected with saline alone (p < 0.01; Fig.1A). In contrast, a decrease of only 15 ± 4.8% in the number of TH+ cells was observed in the SNpc of p35 transgenics that were injected with MPTP compared with saline-injected animals (p < 0.01).
Fig. 1.

Toxicity induced by MPTP is attenuated in transgenic mice neuronally expressing the general caspase inhibitor protein baculoviral p35. A, Stereological counts of TH+ neurons from wild-type (WT) versus p35 transgenic mice after MPTP administration. Both wild-type and p35 transgenics were killed 7 d after MPTP injection, and SN tissue was immunostained with TH antibody. The cell number was assessed stereologically in every alternate section. Data are means ± SD of five to seven animals per group; *p < 0.01. B, Effects of MPTP administration on dopamine and HVA levels in WT versus p35. For all animals the striatum was dissected for measurement of dopamine and HVA 7 d after MPTP administration. Data are means ± SD from three to four mice per group; *p < 0.01.
The effects of acute administration of MPTP on striatal dopamine and HVA levels also was assessed in wild-type versus p35 transgenic mice (Fig. 1B). No significant differences were noted in striatal dopamine or HVA concentrations after the administration of saline in wild-type versus p35 transgenic animals (p = 0.10; Fig. 1B). However, a large reduction in concentrations of both dopamine and its metabolite was observed in the wild-type animals after drug administration. In the p35 transgenics these depletions were attenuated significantly for both dopamine or HVA (p < 0.01).
MPTP elicits cytochrome c release, caspase-3, caspase-9, and caspase-8 activation, and Bid cleavage in the SN of wild-type mice that are attenuated in p35 transgenics
The inhibition of cell death elicited by MPTP administration in p35 transgenic mice suggests a role for caspases in this process.In vitro kinetic analysis demonstrates that p35 inhibits caspase-1, caspase-3, caspase-6, caspase-7, caspase-8, caspase-9, and caspase-10 most potently (Zhou et al., 1998). The attenuation of MPTP-induced cell death in p35 transgenics implies that either upstream activator or downstream effector caspases or both may be involved in this process.
To determine which caspases are activated in MPTP-induced neuronal degeneration, we injected wild-type and p35 transgenic mice with either MPTP or saline; the SNs were dissected 24 hr later, and cell lysates were prepared to assess caspase activities with the use of specific tetrapeptide fluorogenic substrates. This particular time point was chosen because, according to previous work, this is when the maximum number of apoptotic cells can be observed after MPTP administration, and this time point precedes that of maximal cell death (Hartmann et al., 2001).
Caspase-3 appears to be an essential component of the apoptotic machinery in many cell types and a key player in many types of neuronal apoptosis (Salvesen and Dixit, 1997). We analyzed the ability of MPTP to trigger the activation of caspase-3 activity in SN dissected from p35 transgenics versus wild-type animals. In keeping with previous results (Hartmann et al., 2000), we observed significant activation of caspase-3 in wild-type animals after MPTP injection, approximately threefold (Fig. 2A). Expression of p35 was found to attenuate the MPTP-induced activation of caspase-3 activity significantly (p < 0.01).
Fig. 2.
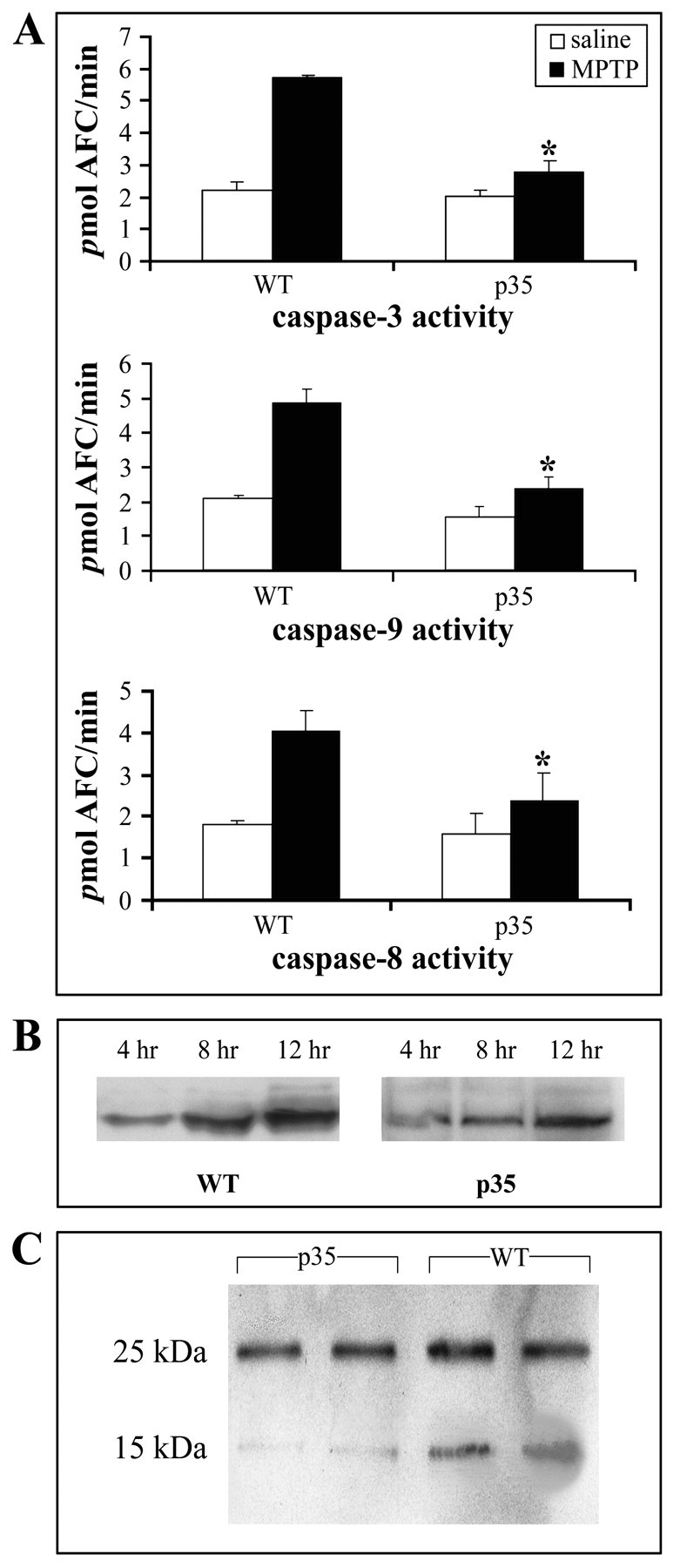
Caspase-3, caspase-8, and caspase-9 activation, cytochrome c release, and Bid cleavage after MPTP administration. A, Caspase-3, caspase-9, and caspase-8 activities. Substantia nigra (SN) dissected from MPTP versus saline-treated wild-type and p35 transgenic animals was used to measure caspase activities with specific fluorogenic tetrapeptide substrates (n = 5–7 animals per group for all assays). The caspase activities were measured 24 hr after MPTP injection. Values represent the means ± SD from three individual experiments; *p < 0.01. B, MPTP-induced cytochrome c release from the mitochondria. Cytosolic extracts were prepared at the indicated times, and cytochromec was evaluated by Western blot analysis with the use of a monoclonal antibody. C, In vivo Bid cleavage 24 hr after MPTP administration in WT and p35 mice. SN was dissected, and total cell lysates were subjected to immunoblotting. Molecular weights are shown on the left. The 25 kDa band represents full-length Bid; the 15 kDa fragment represents the cleaved form.
MPTP is a known mitochondrial toxin (Hartley et al., 1994). Damage to the mitochondria in other paradigms has been demonstrated to result in the release of mitochondrial cytochrome c into the cytoplasm and subsequent activation of caspase-9, which in turn can elicit the activation of caspase-3 (Li et al., 1997). To evaluate whether mitochondrial release of cytochrome c is involved in MPTP-induced cell death, we prepared cytosolic and mitochondrial extracts from SN of wild types and p35 transgenics at various time points after MPTP treatment, and cytochrome c protein levels were measured by immunoblot analysis. Cytosol from tissues isolated from either saline-treated wild-type or p35 animals did not contain any detectable cytochrome c protein (data not shown). In contrast, cytosolic cytochrome c accumulated significantly in wild-type animals treated with MPTP and to a lesser degree in tissue from p35 transgenics (Fig. 2B). The absence of Cox IV in cytosolic extracts confirmed that our preparations were free of mitochondrial contamination (data not shown). Along with mitochondrial cytochrome c release, SN caspase-9 activity was increased by approximately twofold at 24 hr after the induction of apoptosis byin vivo MPTP administration in the wild-type animals; this increase in activity was found to be inhibited greatly in the presence of p35 (p < 0.01; Fig.2A).
Caspase-8 is another caspase that, like caspase-9, appears to be activated upstream of caspase-3 and in addition recently has been implicated possibly to play a role in the cell death associated with neurodegenerative paradigms, including PD (Sanchez et al., 1999; Velier et al., 1999; Hartmann et al., 2001). In wild-type animals caspase-8 activity was found to be induced twofold after treatment with MPTP; this activity increase was attenuated greatly in the p35 transgenics (p < 0.01; Fig.2A). Bid, a Bcl-2 family member, has been shown to be a specific substrate of caspase-8 and to play a role in caspase 8-mediated mitochondrial damage and cell death (Li et al., 1998). To investigate the possible involvement of Bid in MPTP-mediated neurodegeneration, we analyzed Bid cleavage in SN tissue lysates from wild-type and p35 mice after MPTP versus saline injection. Bid was found to be cleaved to a 15 kDa fragment in SN tissue homogenates isolated from MPTP-treated wild-type animals, and this cleavage was attenuated partially in tissues from p35 transgenic animals (Fig. 2C).
MPTP-associated toxicity in dopaminergic cells in culture sequentially involves cytochrome c release, activation of caspase-9, caspase-3, and caspase-8, and Bid cleavage
To determine further the pathway of activation of caspase-3, caspase-9, and caspase-8 in MPTP-induced neurodegeneration, we temporally examined the sequential order of their inductions in vitro by administration of MPP+, the active metabolite of MPTP, to dopaminergic PC12 cells. PC12 cells are a noradrenergic line derived from the rat adrenal medulla that secrete, store, synthesize, and uptake dopamine and are therefore a commonly used model for studying catecholaminergic neurons (Greene and Tischler, 1976). PC12 cells were treated at a dosage of 150 μm MPP+, as described previously (Hartley et al., 1994), and caspase activities were analyzed at various time points (0, 2, 4, 6, 12, and 24 hr after MPP+). At 150 μmMPP+ caspase-9 was activated significantly between 0 and 2 hr and caspase-3 activation between 2 and 4 hr after MPP+ treatment, respectively (p < 0.01; Fig.3A). In contrast, caspase-8 activation did not show significant activation until between 4 and 6 hr after 150 μm MPP+treatment (p < 0.01). This implies that caspase-9 is likely the apical caspase in MPP+-induced toxicity and may be responsible for caspase-8 activation. In agreement with this conclusion, cytochrome c release from the mitochondria was observed by 2 hr after MPP+ treatment (Fig. 3B). Bid cleavage, in contrast, was not seen in MPP+-treated cells until at least 4–6 hr after drug administration (Fig. 3C).
Fig. 3.

MPP+ induces time-dependent activation of various caspases, cytochrome c release, and Bid cleavage in PC12 cells. Values represent the means ± SD from three experiments; p < 0.01.A, PC12 cells were incubated with 150 μmMPP+ for the indicated times. Then the cell lysates were analyzed for caspase activities with the use of specific fluorogenic substrates. B, MPP+-induced cytochrome c release from the mitochondria. Mitochondrial and cytosolic extracts were prepared as described, and cytochrome c release was evaluated by using a monoclonal antibody at 2, 4, and 8 hr after MPP+ treatment. C, Cleavage of Bid in PC12 cells at various times after treatment with MPP+; the 15 kDa fragment was not observed at either 0 or 2 hr after MPP+ application (data not shown).
Caspase-9 activation is necessary for caspase-8 activation and Bid cleavage
To elucidate further the order of caspase activation, we used a specific inhibitor against caspase-9 and examined its effect on caspase-8 activation and Bid cleavage after MPP+ treatment of PC12 cells. LEHD-FMK, a cell-permeable caspase-9 specific inhibitor, was applied 1 hr before the exposure of PC12 cells to 150 μmMPP+ for various periods of time. The dose of the inhibitor used here was predetermined by a set of dose–response experiments in which the ability of LEHD-FMK to inhibit the activity of caspase-9 and the effects of this inhibitor on cell viability were examined (data not shown). Because previous work has implicated a nonspecific caspase inhibition at doses higher than a 50 μm concentration of the inhibitor (Thornberry and Lazebnik, 1998) and because lower dosages (5–20 μm) failed to inhibit caspase-9 activity effectively, a dosage of 25 μm was chosen. Pharmacological inhibition of caspase-9 before MPP+ treatment resulted in inhibition not only of caspase-9 activity but also of both caspase-8 and caspase-3 activities, indicating that the activation of caspase-9 is necessary for the activation of both (Fig.4A). Inhibition of caspase-9 also prevented Bid cleavage (Fig. 4C). However, it did not prevent the release of cytochrome c into the cytosol, an event normally considered to occur upstream of caspase-9 activation (Fig. 4C). Pretreatment with a 5 μm concentration of the cell-permeable caspase-8 specific inhibitor IETD-CHO led to the inhibition of caspase-8 activity and small but significant inhibitions in activities of both caspase-9 and caspase-3 by 6–12 hr after MPP+ treatment (p< 0.01; compare Figs. 4B and 3A).
Fig. 4.

Effects of treatment of PC 12 cells with specific cell-permeable inhibitors of caspase-9 and caspase-8 before treatment with MPP+ on the activation of caspase-9, caspase-3, and caspase-8, cytochrome c release, and Bid cleavage. Values for all assays represent the means ± SD from three experiments; p < 0.01. A, Effects of pretreatment with a caspase-9 specific inhibitor on the activation of caspase-8, caspase-9, and caspase-3. PC12 cells were incubated with 25 μm LEHD-CHO 1 hr before treatment with MPP+. B, Effects of pretreatment with the specific peptide inhibitor to caspase-8 on the activation of caspase-8, caspase-3, and caspase-9. PC12 cells were incubated with 25 μm IETD-CHO at 1 hr before treatment with MPP+. C, Bid cleavage, but not cytochrome c release, is attenuated in PC12 cells after preincubation with LEHD-CHO. Data represent three individual experiments.
Primary mesencephalic cultures isolated from p35 transgenics demonstrate decreased MPP+-mediated loss of primary TH+ neurons
PC12 is a transformed cell line that may have survival mechanisms that are absent in normal primary dopaminergic neurons. Therefore, to verify the role of caspase activation in toxin-induced dopaminergic cell death on a cellular level in primary TH+ neurons, we explored the effects of MPP+ treatment in primary mesencephalic cultures isolated from both p35 transgenic and wild-type animals. A concentration of 5 μm MPP+was chosen for these studies because this dosage is in the range previously reported to elicit primarily apoptotic versus necrotic cell death in dopaminergic cells in these cultures (Hartmann et al., 2001). Preliminary dosage experiments conducted in our own laboratory at MPP+ concentrations of 5, 10, and 50 μm verified this phenomenon. Cells were stained for TH at 0, 6, 12, and 24 hr after MPP+ treatment, and TH+ cells were counted. The percentage of TH+ neurons present versus untreated cultures was 62.0 ± 16.6% for wild-type versus 77.0 ± 28.9% for p35 cultures at 6 hr, 52.7 ± 12.7% wild-type versus 78.6 ± 11.3% p35 at 12 hr, and 28.3 ± 4.8% wild-type versus 38.3 ± 2.7% p35 at 24 hr (p < 0.01; Fig. 5A). As previously observed by Hartmann et al. (2001), this cell loss was found to be accompanied by a noticeable loss in neuritic extensions, especially by the 24 hr time point in the remaining TH+neurons (Fig. 5B). However, unlike the results of Hartmann and colleagues that used the broad pharmacological caspase inhibitor zVAD-FMK, expression of the broad caspase inhibitor p35 was found to attenuate rather than to exacerbate this cell loss, in keeping with ourin vivo data. MPP+-treated wild-type cultures also were pretreated with a 25 μm concentration of the caspase-9 specific inhibitor LEHD-FMK. Although preliminary, results from treatment of wild-type cultures with caspase-9 specific inhibitor suggest that cell loss is attenuated after MPP+ treatment compared with untreated cultures at both 24 and 48 hr after MPP+ (28.0 ± 4.80% wild-type versus 35.3 ± 3.9% wild-type plus inhibitor at 24 hr; 12.7 ± 4.5% wild-type versus 26.0 ± 4.1% wild-type plus inhibitor at 48 hr).
Fig. 5.

TH+ cell counts in MPP+-treated mesencephalic cultures from p35 transgenics (Tg) versus wild-type (WT) animals. A, Percentage of TH+ neurons in p35 transgenic versus wild-type mice mesencephalic cultures after 6, 12, and 24 hr of MPP+ (5 μm) treatment compared with untreated WT; *p < 0.01. B, Representative morphology of TH+ neurons in MPP+-treated wild-type cultures after 0, 6, 12, and 24 hr. Magnification, 40×. Data represent three independent experiments.
Pattern of caspase inductions in mesencephalic cultures recapitulates that observed in the dopaminergic PC12 cell line
To verify the temporal pattern of caspase induction after MPP+ on a cellular level in apoptotic primary dopaminergic neurons, we monitored the induction of caspase-9, caspase-3, and caspase-8 in primary mesencephalic cultures after treatment for 0, 2, 4, 6, 8, 12, 18, 24, and 48 hr with 5 μm MPP+ via immunofluorescence with antibodies specific for both TH+ and the activated forms of each of the enzymes, coupled with DAPI staining. As with the PC12 cells, caspase-9 induction as monitored by immunofluorescence in the TH+ cells occurred first at 2 hr, followed by caspase-3 induction at 6 hr, which in turn preceded caspase-8 induction at 12 hr, demonstrating that MPP+-mediated cell death temporally involves the activation of caspase-9, caspase-3, and then caspase-8 (Fig. 6). The number of TH+ cells at any given time point displaying caspase activation was limited (1–10% or 2–5 per every 50 cells); however, this is in keeping with the findings of others and reflects the fact that only a small subset of dopaminergic neurons is undergoing apoptosis at any given time point (Hartmann et al., 2001). Caspase activation was accompanied by morphological changes in these cells compatible with apoptosis, including chromatin condensation as observed by DAPI staining, apoptotic bodies, and shrunken soma. Although treatment of cultures with caspase-9 specific inhibitor appeared to show a trend toward attenuation of caspase-9, caspase-3, and caspase-8 activation as monitored by immunofluorescence, it was not possible to access this statistically, given the low numbers of caspase-positive TH+ cells present at any of the examined time points (data not shown).
Fig. 6.

Temporal MPP+-induced activation of caspase-9, caspase-3, and caspase-8 in TH+ neurons in mesencephalic cultures. Triple labeling shows immunostaining for the active forms of caspase-9, caspase-3, and caspase-8 in apoptotic (DAPI-stained) TH+ neurons at the time of first induction, i.e., 2, 6, and 12 hr, respectively. Magnification, 60×. Data represent three independent experiments.
Activated caspase-8 and caspase-9 are found within dopaminergic neurons of the substantia nigra in both MPTP-treated mice and Parkinson's patients
To examine caspase activation after MPTP administration on a cellular level in vivo, we used antibodies specific to activated caspase-8 and caspase-9 in conjunction with anti-TH antibodies in MPTP-treated mice. Cellular expression of MPTP-induced activated caspase-3 and caspase-8 has been shown previously to occur in TH+ SN neurons of MPTP-treated mice as well as in PD patients (Hartmann et al., 2000, 2001). In the present study we found that several TH+ SN neurons in the MPTP-treated animals were also positive for immunostaining with both activated anti-caspase-8 and anti-caspase-9 antibodies (Fig.7A).
Fig. 7.

Presence of activated caspase-8 and caspase-9 in TH+ neurons of the substantia nigra of MPTP-treated mice and Parkinsonian brain. A, Wild-type mice were treated with MPTP, and 40 μm sections were double immunolabeled for antibody against TH and the activated form of either caspase-9 or caspase-8. B, Postmortem human brain samples from Parkinson's patients were double immunostained for TH and activated caspase-9 or activated caspase-8. Magnification, 40×.
To determine whether activated caspase-8 and caspase-9 were present in dopaminergic neurons of the Parkinsonian SN and therefore may play a role in the disease pathology, we performed similar experiments on tissues from late-onset sporadic PD cases versus age-matched controls. TH+ SN neurons that also were stained positively for antibodies against activated caspase-8 and caspase-9 were found in the SN isolated from autopsied PD patients (Fig.7B). Although some caspase-8- and caspase-9-positive SN dopaminergic neurons also were detected in tissue from age-matched controls, this was observed less frequently (data not shown).
DISCUSSION
In this study we have demonstrated that neuronal expression of the general caspase inhibitor protein p35 in transgenic mice results in significant reduction in the effects of MPP+/MPTP-induced Parkinsonism bothin vitro and in vivo. Given that neuronal expression of p35 is not completely protective against TH+ SN neuronal cell loss either in vitro or in vivo raises the possibility that other caspase-independent pathways also may be involved in toxin-induced cell death. An alternative explanation is that levels of p35 may not have been elevated sufficiently in our transgenic model to counteract all of the MPP+/MPTP-induced caspase induction and subsequent cell death.
MPTP was found to elicit cytochrome c release, activation of caspase-3, caspase-9, and caspase-8, and Bid cleavage in the SN of wild-type mice, and these events were found to be attenuated in the p35 transgenics in vivo. Studies in dopaminergic PC12 cells and primary mesencephalic cultures revealed that this toxicity appears to involve, sequentially, cytochrome c release, activation of caspase-9, caspase-3, and caspase-8, and Bid cleavage. Furthermore, the inhibition of MPTP-mediated caspase-9 activation appears to prevent caspase-3 and caspase-8 activation and Bid cleavage, but not cytochromec release. On a cellular level the activation of caspase-9 and caspase-3 in TH+ cells in mesencephalic cultures was found to precede that of caspase-8, and either general caspase inhibition via p35 expression or specific pharmacological inhibition of caspase-9 resulted in attenuated MPP+-mediated TH+ cell loss. Both active caspase-8 and caspase-9 were found to be present within dopaminergic SN neurons of MPP+-treated mesencephalic cultures, MPTP-treated mice, and in late-onset Parkinson's patients to a greater extent than in controls. Taken together, these data suggest that caspase-8 activation in dopaminergic neurons occurs downstream of activation of both caspase-9 and caspase-3. Furthermore, caspase-9 activation appears to be required for both caspase-8 activation and Bid cleavage as well as for MPTP-mediated dopaminergic cell death.
MPTP is a mitochondrial toxin that elicits its actions first via monoamine oxidase B-catalyzed conversion to MPP+, which is taken up selectively into nigral dopaminergic neurons by the dopamine transporter. Here it acts to kill these cells by specifically inhibiting mitochondrial complex I activity (for review, see Przedborski and Jackson-Lewis, 1998). Cell death associated with PD also appears to involve a decrease in mitochondrial function via inhibition of the activity of mitochondrial complex I (Mizuno et al., 1998; Schapira et al., 1998). There is evidence both after MPTP administration in mice and in PD that mitochondrial dysfunction results in apoptotic cell death in dopaminergic neurons of the SN (Mochizuki et al., 1996; Hajimohamadreza and Treherne, 1997; Tatton and Kish, 1997; Tatton et al., 1998;Hartmann et al., 2000, 2001; Andersen, 2001). Mitochondrial injury can elicit apoptosis by disruption of mitochondrial membrane potential, resulting in the release of mitochondrial cytochrome c into the cytoplasm where it can complex with apoptosis-activating factor 1 (Apaf-1) and caspase-9, causing the activation of this initiator caspase (Li et al., 1997). Caspase-9 in turn can cleave and activate the downstream executioner caspases, including caspase-3. This leads to cleavage of additional cellular substrates, resulting in the morphological changes associated with apoptosis, including DNA fragmentation and cytoskeletal disruption (for review, see Nuñez et al., 1998; Stennicke and Salvesen, 2000).
Recent evidence from cell-free and in vitro expression systems has suggested that, besides being a final effector in neuronal apoptosis, caspase-3 is also capable of eliciting cleavage and activation of the upstream initiator caspase-8 (Slee et al., 1999; Wolf and Green, 1999; Tang et al., 2000). Caspase-8 traditionally is associated with Fas receptor-induced apoptosis. In this system caspase-8 activation results in the cleavage of Bid, a proapoptotic BH3 domain-containing member of the Bcl-2 family, to produce a truncated form of the protein. Truncated Bid translocates from the cytoplasm to the mitochondria, where it appears to interact with and antagonize the actions of anti-apoptotic members of the Bcl-2 family, thereby causing an efflux of cytochrome c from the mitochondria (Kuwana et al., 1998; Li et al., 1998; Luo et al., 1998; Schendel et al., 1999; Wei et al., 2001). This in turn can result in the activation of caspase-9. Therefore, theoretically, caspase-8 acting via the translocation of cleaved Bid to the mitochondria could amplify apoptotic signals via the continued release of cytochrome cand subsequent activation of caspase-9 and caspase-3. Coupled with our data, this leads to the speculation that the initial activation of mitochondrially associated caspase-9 by MPTP may be potentiated via a feedback amplification loop involving the caspase 8/Bid pathway (Fig.8). In our studies caspase-8 inhibition appeared to have a small but significant effect on activities of caspase-9 or caspase-3 in MPP+-treated dopaminergic cells in culture by 6–12 hr after MPP+ treatment (Fig. 4). It is conceivable that periods of >24 hr are required for more significant effects of caspase-8 inhibition, i.e., for the inhibition of Bid cleavage and cytochrome c release, thereby inhibiting additional activation of caspase-9 and caspase-3. Bid cleavage itself does not occur until ∼4–6 hr after MPP+application.
Fig. 8.

Possible pathway of caspase activation in MPTP-induced dopaminergic cell death. MPTP administration results in the release of mitochondrial cytochrome c and the activation of procaspase-9, leading to the subsequent activation of procaspase-3. Active caspase-3 can cleave downstream substrates, resulting in apoptosis. Active caspase-3 also can activate procaspase-8. Active caspase-8 in turn can cleave Bid, leading to cytochrome c release and setting up a self-amplification loop. Caspase-8 also has been reported to lead directly to the cleavage of caspase-3 (Kumar, 1999). A similar pattern may be at work in the SN of PD patients; alternatively, protein aggregates may lead to caspase-8 oligomerization and activation.
Caspase-8 recently has been shown to be involved in cell death induced by expanded polyglutamine repeats associated with such conditions as Huntington's disease (HD) and spinocerebellar ataxia, possibly by production of protein fragments that form toxic aggregates in affected neurons, although this is somewhat controversial (Kuemmerle et al., 1999; Sanchez et al., 1999; Wellington and Hayden, 2000). Intracellular aggregates also occur in PD in the form of Lewy bodies (Hughes, 1997; Olanow and Tatton, 1999). It is possible in PD that Lewy body aggregation itself may contribute to the recruitment and activation of caspase-8 similar to that which occurs in HD. Indeed, activated caspase-8 recently has been reported to be present in neuromelanin-containing SN neurons in autopsied tissues from PD patients (Hartmann et al., 2001). There also have been reports that both the cell death receptor-associated Fas and FADD are expressed in cells in the adult human SN that undergo degeneration in PD (de la Monte et al., 1998; Hartmann et al., 1998; Michel et al., 1999), although this has been disputed (Jellinger, 2000). This can initiate downstream apoptotic events including the activation of caspase-3 via direct proteolytic cleavage as well as via caspase-9 (Kumar, 1999).
Parkinson's disease develops over a period of several years; however, caspase-mediated apoptosis has been shown to occur within hours or days of initiation. The initiating event in neurodegeneration that is associated with PD appears to be mitochondrial dysfunction. In the MPTP model of PD in which the mitochondrial effects are acute and rapid, apoptosis occurs within 24 hr of drug administration. The delayed development of apoptosis in PD may reflect a slower accumulation of age-related mitochondrial damage over decades. Histological analysis of SN tissue from late-onset PD patients reveals a shrunken, condensed appearance in the remaining neurons and a lack of inflammation (Beal et al., 1993; Ziv et al., 1997). This is in contrast to the type of morphology expected if the associated cell death were necrotic in nature, i.e., cell swelling, rupture, and spillage of cell contents into the extracellular space eliciting significant local immune response. This suggests that cell death is likely apoptotic. Furthermore, the ability of dopaminergic cells of the substantia nigra to undergo PD-related apoptosis is likely dependent on the availability and concentration of activatable caspases in the affected neurons that may be altered over time (Velier et al., 1999). Recent publications have reported that neither the addition of pharmacological caspase inhibitors nor the expression of baculoviral p35 in either cultured primary dopaminergic neurons or dopaminergic cell lines protected them against MPP+-induced cell death (Choi et al., 2001; Hartmann et al., 2001). Indeed, Hartmann and colleagues have reported that both general caspase inhibition and inhibition selective for caspase-8 result in an increase in MPP+-mediated toxicity in rat mesencephalic cultures 24 hr after the addition of toxin, which they have attributed to a switch from apoptotic to necrotic cell death. We found, in contrast, that both the expression of baculoviral p35 and the administration of the caspase-9 specific inhibitor LEHD-FMK resulted in an attenuation of TH+ cell death in murine mesencephalic cultures in vitro up to 24 hr after MPP+ addition. In addition and in agreement with these data, we found that neuronal expression of p35in vivo resulted in significant attenuation of dopaminergic SN cell loss and striatal dopamine/HVA levels up to 7 d after MPTP administration. Our studies clearly imply that caspase inhibition is protective against MPTP-induced cell death and suggest that strategies involving specific caspase inhibition could have utility in the treatment of Parkinson's disease.
Footnotes
This work was funded in part by National Institutes of Health Grants AG12141 and AG09793 (J.K.A.). We thank Dr. John O. Archambeau (Loma Linda, CA) for the use of his stereology setup for performing the neuronal cell counts and Dr. Jytte Larsen (University of Aarhus, Aarhus, Denmark) for advice regarding this procedure. We also thank Dr. Carlos Arruda (SmithKline Beecham, Philadelphia, PA) for the gift of the activated caspase-8 antibody and Julie Schneider and Beth Howard at the Brain Bank (Rush University, Chicago, IL) and Carole Miller at the University of Southern California (USC; Los Angeles, CA) Brain Bank for providing us with human postmortem Parkinsonian and age-matched control tissue. In addition, we acknowledge Dr. Junying Yuan (Harvard University, Cambridge, MA) for advice regarding use of the Bid antibody, Dr. Hadi Zanjani (USC) for assistance with mouse brain dissections and immunohistocytochemistry protocols performed on human tissues, and Dr. Christian Pike (USC) for use of his fluorescent scope and advice on fluorescent immunohistocytochemistry.
V.V. and Y.W. contributed equally to this work
Correspondence should be addressed to Dr. Julie K. Andersen, Associate Professor, Buck Institute for Age Research, 8001 Redwood Boulevard, Novato, CA 94948. E-mail: jandersen@buckinstitute.org.
REFERENCES
- 1.Andersen J. Does neuronal loss in Parkinson's disease involve programmed cell death? BioEssays. 2001;23:640–646. doi: 10.1002/bies.1089. [DOI] [PubMed] [Google Scholar]
- 2.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 3.Beal MF, Kowall NW, Swartz KJ, Ferrante R. Homocysteic acid striatal lesions in rats spare somatostatin-neuropeptide Y neurons. Neurosci Lett. 1990;108:36–42. doi: 10.1016/0304-3940(90)90702-b. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Hyman BT, Koroshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci. 1993;16:125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- 5.Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, Licari P, Mankovitch J, Shi L, Greenberg AH, Miller LK, Wong W. Inhibition of ICE family proteases by baculovirus anti-apoptotic protein p35. Science. 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- 6.Choi WS, Lee EH, Chung CW, Jung YK, Jin BK, Kim SU, Oh TH, Saido TC, Oh YJ. Cleavage of Bax is mediated by caspase-dependent or -independent calpain activation in dopaminergic neuronal cells: protective role of Bcl-2. J Neurochem. 2001;77:1531–1541. doi: 10.1046/j.1471-4159.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- 7.Colussi PA, Kumar S. Targeted disruption of caspase genes in mice: what they tell us about the functions of individual caspases in apoptosis. Immunol Cell Biol. 1999;77:58–63. doi: 10.1046/j.1440-1711.1999.00788.x. [DOI] [PubMed] [Google Scholar]
- 8.Davidson FF, Steller H. Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature. 1998;391:587–591. doi: 10.1038/35385. [DOI] [PubMed] [Google Scholar]
- 9.de la Monte SM, Sohn YK, Ganju N, Wands JR. P53- and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest. 1998;78:401–411. [PubMed] [Google Scholar]
- 10.Dodel RC, Du Y, Bales KR, Ling ZD, Carvey PM, Paul SM. Peptide inhibitors of caspase-3-like proteases attenuate 1-methyl-4-phenylpyridinium-induced toxicity of cultured fetal rat mesencephalic dopamine neurons. Neuroscience. 1998;86:701–707. doi: 10.1016/s0306-4522(98)00154-7. [DOI] [PubMed] [Google Scholar]
- 11.Fisher AJ, Dela Cruz W, Zoog SJ, Schneider CL, Friesen PD. Crystal structure of baculovirus p35: role of a novel reactive site loop in apoptotic caspase inhibition. EMBO J. 1999;18:2031–2039. doi: 10.1093/emboj/18.8.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajimohamadreza I, Treherne JM. The role of apoptosis in neurodegenerative disease. Prog Drug Res. 1997;48:55–98. doi: 10.1007/978-3-0348-8861-5_3. [DOI] [PubMed] [Google Scholar]
- 14.Hartley A, Stone JM, Heron C, Cooper JM, Schapira AH. Complex I inhibitors induce dose-dependent apoptosis in PC12 cells: relevance to Parkinson's disease. J Neurochem. 1994;63:1987–1990. doi: 10.1046/j.1471-4159.1994.63051987.x. [DOI] [PubMed] [Google Scholar]
- 15.Hartmann A, Hunot S, Hirsch EC. CD95 (APO-1/Fas) and Parkinson's disease. Ann Neurol. 1998;44:425–426. doi: 10.1002/ana.410440330. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinvasan A, Ruberg M, Evan GI, Agid Y, Hirsch E. Caspase 3: a vulnerability factor and final effector in apoptotic cell death of dopaminergic neurons in Parkinson's disease. Proc Natl Acad Sci USA. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartmann A, Troadec J-D, Hunot S, Kikly K, Faucheux BA, Prigent A-M, Ruberg M, Agid Y, Hirsch EC. Caspase 8 is an effector in apoptotic death of dopaminergic neurons in Parkinson's disease, but pathway inhibition results in neuronal necrosis. J Neurosci. 2001;21:2247–2255. doi: 10.1523/JNEUROSCI.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hassouna I, Wickert H, Zimmermann M, Gillardon F. Increase in Bax expression in substantia nigra following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment of mice. Neurosci Lett. 1996;204:85–88. doi: 10.1016/0304-3940(96)12323-5. [DOI] [PubMed] [Google Scholar]
- 19.Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- 20.Hisahara S, Araki T, Sugiyama F, Yagami KI, Suzuki M, Abe K, Yamamura K, Miyazaki J, Momoi T, Saruta T, Bernard CC, Okano H, Miura M. Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. EMBO J. 2000;19:341–348. doi: 10.1093/emboj/19.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes AJ. Clinicopathological aspects of Parkinson's disease. Eur Neurol. 1997;38[Suppl 2]:13–20. doi: 10.1159/000113471. [DOI] [PubMed] [Google Scholar]
- 22.Izquierdo M, Grandien A, Criado LM, Robles S, Leonardo E, Albar JP, de Buitrago GG, Martínez-AC Blocked negative selection of developing T-cells in mice expressing the baculovirus p35 caspase inhibitor. EMBO J. 1999;18:156–166. doi: 10.1093/emboj/18.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jellinger KA. Cell death mechanisms in Parkinson's disease. J Neural Transm. 2000;107:1–29. doi: 10.1007/s007020050001. [DOI] [PubMed] [Google Scholar]
- 24.Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA, Hardwick JM. Caspase-3-dependent cleavage of Bcl-2 promotes release of cyt c. J Biol Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- 25.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 26.Kuida K, Zhen TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP-32 deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 27.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MSS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 28.Kumar S. Mechanisms mediating caspase activation in cell death. Cell Death Differ. 1999;6:1060–1066. doi: 10.1038/sj.cdd.4400600. [DOI] [PubMed] [Google Scholar]
- 29.Kuwana T, Smith JJ, Muzio M, Dixit V, Newmeyer DD. Apoptosis induction by caspase 8 is amplified through the mitochondrial release of cytochrome c. J Biol Chem. 1998;273:16589–16594. doi: 10.1074/jbc.273.26.16589. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Zhu H, Xu C, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 31.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmed M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of apaf-1/caspase 9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 32.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 33.Michel PP, Lambeng N, Rugerg M. Neuropharmacological aspects of apoptosis: significance for neurodegenerative diseases. Clin Neuropharmacol. 1999;22:137–150. [PubMed] [Google Scholar]
- 34.Mizuno Y, Yoshino H, Ikebe S, Hattori N, Kobayashi T, Shimoda-Matsubayashi S, Matsumine H, Kondo T. Mitochondrial dysfunction in Parkinson's disease. Ann Neurol. 1998;44:99–109. doi: 10.1002/ana.410440715. [DOI] [PubMed] [Google Scholar]
- 35.Mochizuki H, Goto K, Mori H, Mizuno Y. Histochemical detection of apoptosis in Parkinson's disease. J Neurol Sci. 1996;137:120–123. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- 36.Nuñez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- 37.Offen D, Beart PM, Cheung NS, Pascoe CJ, Hochman A, Gorodin S, Melamed E, Bernard R, Bernard O. Transgenic mice expressing human Bcl-2 in their neurons are resistant to 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Proc Natl Acad Sci USA. 1998;95:5789–5794. doi: 10.1073/pnas.95.10.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 39.Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13:35–38. [PubMed] [Google Scholar]
- 40.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase 8 is required for cell death induced by expanding polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 42.Schapira AH, Gu M, Taanman JW, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Ann Neurol. 1998;44:89–98. doi: 10.1002/ana.410440714. [DOI] [PubMed] [Google Scholar]
- 43.Schendel SL, Azimov R, Pawlowski K, Godzik A, Kagan BL, Reed JC. Ion channel activity of the BH3 only Bcl-2 family member, BID. J Biol Chem. 1999;274:21932–21936. doi: 10.1074/jbc.274.31.21932. [DOI] [PubMed] [Google Scholar]
- 44.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8 and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Brück W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stennicke HR, Salvesen GS. Caspases—controlling intracellular signals by protease zymogen activation. Biochim Biophys Acta. 2000;1477:299–306. doi: 10.1016/s0167-4838(99)00281-2. [DOI] [PubMed] [Google Scholar]
- 47.Sugimoto A, Friesen PD, Rothman JH. Baculovirus p35 prevents developmentally programmed cell death and rescues a ced-9 mutant in the nematode Caenorhabditis elegans. EMBO J. 1994;13:2023–2028. doi: 10.1002/j.1460-2075.1994.tb06475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang D, Lahti JM, Kidd VJ. Caspase 8 activation and BID cleavage contribute to MCF7 cellular execution in a caspase 3-dependent manner during staurosporin-mediated apoptosis. J Biol Chem. 2000;275:9303–9307. doi: 10.1074/jbc.275.13.9303. [DOI] [PubMed] [Google Scholar]
- 49.Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labeling and acridine orange staining. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- 50.Tatton NA, Maclean-Fraser A, Tatton WG, Perl DP, Olanow CW. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson's disease. Ann Neurol. 1998;44:142–148. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- 51.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 52.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 53.Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC, Feuerstein GZ. Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J Neurosci. 1999;19:5932–5941. doi: 10.1523/JNEUROSCI.19-14-05932.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viswanath V, Wu Z, Wei Q, Fonck C, Andersen JK. Transgenic mice neuronally expressing baculoviral p35 are resistant to diverse types of induced apoptosis including seizure-associated neurodegeneration. Proc Natl Acad Sci USA. 2000;97:2270–2275. doi: 10.1073/pnas.030365297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei MC, Zong W-X, Cheng EH-Y, Lindsen T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wellington CL, Hayden MR. Caspases and neurodegeneration: on the cutting edge of new therapeutic approaches. Clin Genet. 2000;57:1–10. doi: 10.1034/j.1399-0004.2000.570101.x. [DOI] [PubMed] [Google Scholar]
- 57.West MJ, Slomianka L, Gundersen HJG. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 58.Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem. 1999;29:20049–20052. doi: 10.1074/jbc.274.29.20049. [DOI] [PubMed] [Google Scholar]
- 59.Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou JC, Penney JB, Jr, Hyman BT, Beal MF. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyride neurotoxicity is attenuated in mice overexpressing Bcl-2. J Neurosci. 1998;18:8145–8152. doi: 10.1523/JNEUROSCI.18-20-08145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng TS, Hunot S, Kuida K, Flavell RA. Caspase knockouts: matters of life and death. Cell Death Differ. 1999;6:1043–1053. doi: 10.1038/sj.cdd.4400593. [DOI] [PubMed] [Google Scholar]
- 61.Zhou Q, Krebs JF, Snipas SJ, Price A, Alnemri ES, Tomaselli KJ, Salvesen GS. Interaction of the baculovirus anti-apoptotic protein p35 with caspases. Specificity, kinetics, and characterization of the caspase/p35 complex. Biochemistry. 1998;37:10757–10765. doi: 10.1021/bi980893w. [DOI] [PubMed] [Google Scholar]
- 62.Ziv I, Offen D, Barzilai A, Haviv R, Stein R, Zilkha-Falb R, Shirvan A, Melamed E. Modulation of control mechanisms of dopamine-induced apoptosis—a future approach to the treatment of Parkinson's disease? J Neural Transm [Suppl] 1997;49:195–202. doi: 10.1007/978-3-7091-6844-8_20. [DOI] [PubMed] [Google Scholar]