

Abstract
Molecular mechanisms of apoptosis may participate in motor neuron degeneration produced by mutant superoxide dismutase-1 (mSOD1), the only proven cause of amyotrophic lateral sclerosis (ALS). Consistent with this, here we show that the proapoptotic protein Bax translocates from the cytosol to the mitochondria, whereas cytochrome c translocates from the mitochondria to the cytosol in spinal cords of transgenic mSOD1 mice during the progression of the disease. Concomitantly, caspase-9 is activated in the spinal cord of transgenic mSOD1 mice. Only in end-stage transgenic mSOD1 mice is the downstream caspase-7 activated and the inhibitor of apoptosis, XIAP, cleaved. These results indicate a sequential recruitment of molecular elements of the mitochondrial-dependent apoptotic pathway in transgenic mSOD1 mice. We also provide immunohistochemical evidence that cytochrome c translocation occurs in the spinal cord of sporadic ALS patients. Collectively, these data suggest that the mitochondrial-dependent apoptotic pathway may contribute to the demise of motor neurons in ALS and that targeting key molecules of this cascade may prove to be neuroprotective.
Keywords: amyotrophic lateral sclerosis, apoptosis, Bax, caspase, cytochrome c, mitochondria, superoxide dismutase, neurodegeneration
Amyotrophic lateral sclerosis (ALS) is a fatal paralytic disease characterized by a progressive loss of spinal cord motor neurons (Rowland, 1995). Important insights into its pathogenesis come from the discovery that missense mutations in superoxide dismutase-1 (mSOD1) are linked to familial ALS (Deng et al., 1993; Rosen et al., 1993) and that overexpression of different mSOD1s in mice replicate the clinical and pathological hallmarks of ALS (Brown, 1995). mSOD1 cytotoxicity is not triggered by a loss of enzymatic activity or by a dominant negative mechanism but by a gain of function (Brown, 1995) of unknown nature.
Mounting evidence indicates that mSOD1-induced spinal cord motor neuron death involves, in part, the apoptotic machinery. Indeed, the expression of the anti-apoptotic protein Bcl-2 is decreased, whereas that of the proapoptotic protein Bax is increased in the spinal cords of human ALS cases and of transgenic mSOD1 mice (Martin, 1999;Vukosavic et al., 1999). Furthermore, the overexpression of Bcl-2 delays the disease process in transgenic mSOD1 mice (Kostic et al., 1997). In addition, caspase-1 and caspase-3, which belong to the family of apoptotic effector cysteine proteases (Earnshaw et al., 1999), are sequentially activated in spinal cords of affected transgenic mSOD1 mice (Pasinelli et al., 1998, 2000; Vukosavic et al., 2000). Activation of these caspases likely participates in the neurodegenerative process of ALS, because overexpression of the dominant negative mutant M17Z of caspase-1 (Friedlander et al., 1997) or the chronic infusion of a pan-caspase inhibitor (Li et al., 2000) provide neuroprotection to transgenic mSOD1 mice.
The release of cytochrome c from the mitochondria to the cytosol is pivotal in the activation of caspases and the ensuing cell death (Kroemer and Reed, 2000). After a death stimulus, cytosolic Bax translocates to mitochondria (Wolter et al., 1997; Gross et al., 1998) in which it can promote the release of cytochrome c (Gross et al., 1998; Jürgensmeier et al., 1998). Once released, cytosolic cytochrome c interacts with apoptotic protease-activating factor-1 (Apaf-1) in the presence of dATP, which stimulates the processing of pro-caspase-9 to its active form, which in turn can then activate the executioner caspase-3 and caspase-7 (Li et al., 1997; Pettmann and Henderson, 1998). Translocation of cytochrome c occurs in several experimental models of acute neurological disorders, such as strokes and encephalitis (Gillardon et al., 1997; Yakovlev et al., 1997; Krajewski et al., 1999;Guégan and Sola, 2000; Vereker et al., 2000). However, whether mitochondrial cytochrome c is released in a chronic neurodegenerative process such as in ALS is not yet known.
Here we show that Bax and cytochrome c translocation occurs in the spinal cord of transgenic mSOD1 mice in parallel with the neurodegenerative process. Concurrently, caspase-9 is activated followed by the activation of caspase-7 and the cleavage of the X chromosome-linked inhibitor of apoptosis protein (XIAP). We also present immunohistochemical evidence supporting the occurrence of cytochrome c translocation in motor neurons of spinal cords from sporadic ALS patients. These findings support the recruitment of the mitochondrial-dependent apoptotic pathway in mice and human ALS cases.
MATERIALS AND METHODS
Time course of behavioral abnormalities in transgenic mSOD1 mice. As described previously (Vukosavic et al., 2000), the first behavioral abnormality appeared at ∼3 months of age in transgenic mSOD1 mice and consisted of a fine tremor and posturing of at least one limb when the animal was held in the air by the tail. Then, progressively, the animals became paralyzed; they were then killed at ∼5 months of age, corresponding to the end stage.
Animals. Two lines of hemizygous transgenic mice were used: (1) line G1H (The Jackson Laboratory, Bar Harbor, ME), which carried a substitution of glycine by alanine at codon 93 of the human SOD1 protein and expressed ∼18 copies of human mutant SOD1 gene (Gurney et al., 1994); and (2) line N1029 (The Jackson Laboratory), which expressed >10 copies of human wild-type (wt) SOD1 gene (Gurney et al., 1994).
Isolation of cytosolic and mitochondrial fractions. Protein extraction of both mitochondrial and cytosolic fractions was performed from fresh spinal cords (n = 4–6 per group) obtained from transgenic wtSOD mice (3 months of age) and from transgenic mSOD1 mice and their nontransgenic littermates of different ages (1, 2, 3, and 5 months). Tissues were gently homogenized with a glass–glass homogenizer in 10 vol (w/v) of cold buffer consisting of (in mm): 250 sucrose, 10 KCl, 1.5 MgCl2, 2 EDTA, 20 HEPES, and protease inhibitor cocktail (Complete mini; Boehringer Mannheim, Indianapolis, IN). Homogenates were centrifuged (500 × g, 5 min, 25°C), and supernatants were collected and centrifuged (13,000 ×g, 20 min, 4°C). Resulting pellets were designated mitochondrial fractions, whereas supernatants were further centrifuged (100,000 × g, 60 min, 4°C). Resulting supernatants were designated cytosolic fractions. To verify the relative mitochondrial purification, each fraction was subjected to Western blotting for β-actin as a cytosolic marker using a mouse monoclonal antibody anti-β-actin (clone AC15; Sigma, St. Louis, MO) and cytochrome c oxidase (COX) as a mitochondrial marker using a mouse monoclonal antibody anti-COX subunit IV (Molecular Probes, Eugene, OR).
Western blot analysis. Concentrations of proteins were determined by BCA protein assay (Pierce, Rockford, IL), and immunoblots were processed as described previously (Vukosavic et al., 2000). For analyzing the subcellular localization of cytochrome c, 30 μg of protein from the cytosolic fraction and 4 μg of protein from the mitochondrial fraction were electrophoresed on a 15% SDS-polyacrylamide gel. For Bax, caspases, and XIAP analysis, 30–70 μg were used. Gels were blotted to nitrocellulose membrane. The primary antibodies used were as follows: a mouse monoclonal cytochrome c antibody (1:1500 and 1:10,000 final dilutions for cytosolic and mitochondrial fractions, respectively; clone 7H8.2C12; PharMingen, San Diego, CA), a mouse monoclonal anti-Bax antibody (1:250 final dilution for both fractions; SC-7480; Santa Cruz Biotechnology, Santa Cruz, CA), a rabbit polyclonal anti-XIAP antibody (1:150 final dilution; catalog #2042; Cell Signaling Technology, Beverly, MA), a rabbit polyclonal anti-caspase-7 antibody recognizing the pro- and cleaved forms (1:250 final dilution; catalog #9492; Cell Signaling Technology), and a rabbit polyclonal anti-caspase-9 antibody recognizing the pro- and cleaved forms (1:1000 final dilution; catalog #AAP-109; StressGen Biotechnologies, Victoria, Canada). Bands were visualized by using enhanced chemiluminescent substrate (SuperSignal Ultra; Pierce). Films (Kodak BioMax MS; Eastman Kodak, Rochester, NY) were scanned, and bands were quantified by using the NIH Image 1.62 software.
Immunohistochemistry. The immunohistochemical analyses of the experimental mice were performed using our standard protocol on cryostat-cut sections (30 μm) obtained from cervical and lumbar segments from spinal cord of transgenic mSOD1 mice at the beginning of symptoms (∼3 months of age) and the end stage (∼5 months of age) (Kostic et al., 1997). The immunostaining for cytochrome c was performed using the Vector M.O.M. immunodetection kit (PK-2200; Vector Laboratories, Burlingame, CA) for eliminating the nonspecific background. Sections were incubated overnight at 4°C with the following primary antibodies: anti-cytochrome c mouse monoclonal antibody (1:1000; PharMingen), anti-cleaved caspase-7 rabbit polyclonal antibody (1:100; catalog #9491; Cell Signaling Technology), and anti-cleaved caspase-9 rabbit polyclonal antibody (1:50; catalog #9501; Cell Signaling Technology). Then the sections were incubated with the biotinylated anti-rabbit IgG (1:200; Vector Laboratories) for 1 hr at room temperature and processed with the ABC system. The peroxidase reaction was revealed with the diaminobenzidine substrate.
The immunohistochemical staining of human spinal cord was applied to 6-μm-thick paraffin sections mounted on coated glass slides. The sections were cleared, rehydrated, and subjected to microwave retrieval before staining them for cytochrome c using the same method as for mice.
Human samples. The tissues used originated from six spinal cord samples from patients with ALS and seven from neurological patients with no spinal cord pathology; samples were obtained from the Brain Bank, Department of Pathology at Columbia University. In the ALS group, the cause of death was pneumonia (n = 3) and respiratory failure (n = 3); in the control group, it was pneumonia (n = 2), respiratory failure (n = 3), pulmonary embolism (n = 1), and septic shock (n = 1). At autopsy, brains and spinal cords were removed and processed for banking as described previously (Przedborski et al., 1996). Half of the brain and selected blocks of each level of the spinal cord were immediately placed in 10% buffered formalin, embedded in paraffin, and subjected to neuropathological examination. There was no significant difference between the time from death to autopsy between the two groups (mean ± SD; ALS, 10.7 ± 5.9 hr; control, 14.6 ± 3.6 hr; p = 0.17; Student's t test). The clinical diagnosis of ALS was confirmed pathologically in all six ALS patients. In these spinal cord specimens, dramatic neuronal loss was observed in the anterior horn with mild to moderate gliosis, whereas no remarkable pathological changes were noted in spinal cords from controls. For the ALS patients, the mean age at onset was 64.3 ± 13.2 years, with a mean duration of disease of 3.3 ± 2.7 years. None of the ALS patients had a family history for the illness.
RESULTS
Release of mitochondrial cytochrome c to the cytosol in ALS
Cytochrome c immunoreactivity was detected by Western blot analysis as a single specific band of 15 kDa (Fig.1A,C), consistent with the gel resolution of the commercially purified cytochrome c protein (data not shown). In nontransgenic mice, the levels of cytochrome c in the cytosolic and mitochondrial fractions did not change over a period corresponding to the lifespan of the transgenic mSOD1 counterparts (Fig. 1). Conversely, in spinal cord of early symptomatic (∼3 months old) and end stage (∼5 months old) and, to a lesser extent, of asymptomatic transgenic mSOD1 mice (1–2 months old), levels of cytochrome c were increased in the cytosolic fraction and decreased in the mitochondrial fraction compared with age-matched nontransgenic controls (Fig. 1). Transgenic wtSOD mice age-matched with early symptomatic transgenic mSOD1 mice showed no alteration of cytosolic and mitochondrial cytochrome c levels (Fig. 1). Likewise, no significant change in cytochrome c level was seen in unaffected brain regions of transgenic mSOD1 mice, such as the cerebellum (data not shown).
Fig. 1.
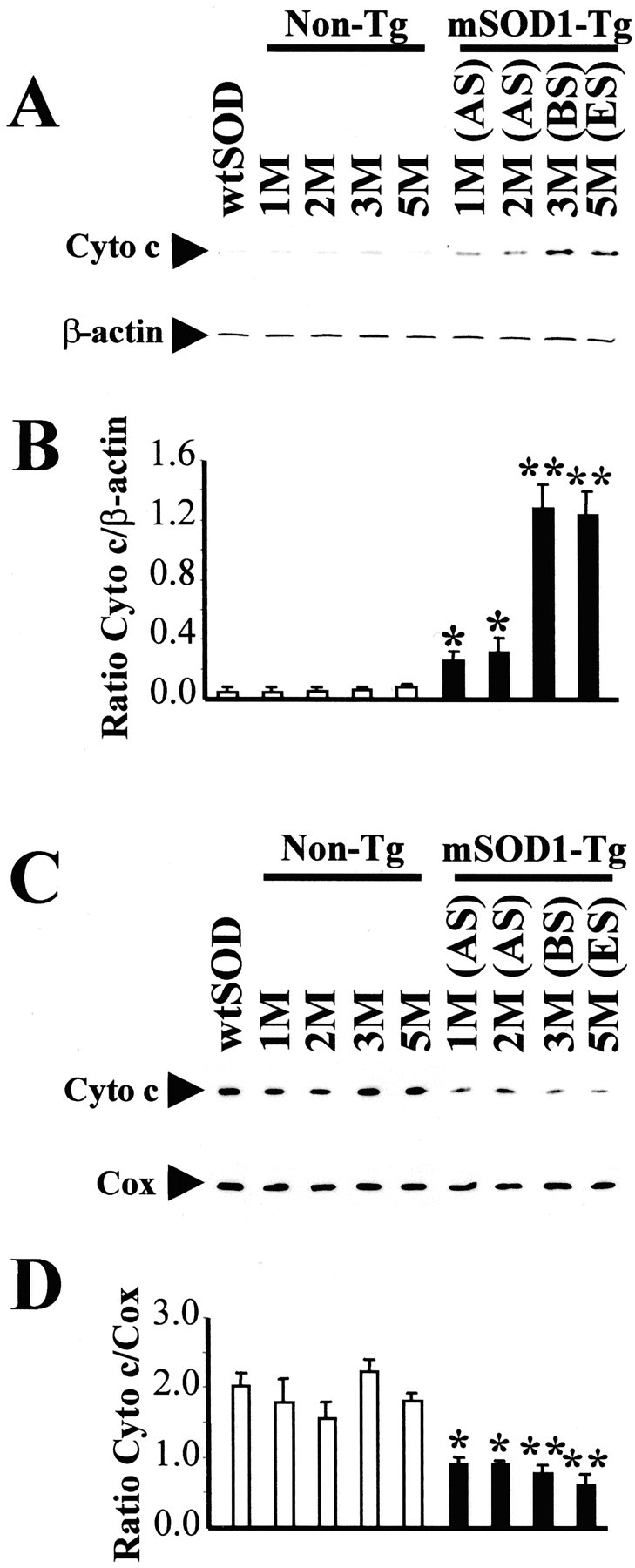
Western blot analysis of cytochrome c. Protein extracts from cytosol (A, B) and mitochondria (C, D) were obtained from spinal cords of wtSOD1 mice (3 months old), of transgenic mSOD1 mice at asymptomatic stage (AS; 1 and 2 months old), at the beginning of symptoms (BS; 3 months old), at end stage (ES; 5 months old), and of their age-matched nontransgenic mice littermates (Non-Tg). β-Actin and COX were used as internal controls for the cytosolic and mitochondrial fractions, respectively. The results of densitometric analysis are shown in B and D. The mean values (n = 4–6 per group; mean ± SEM) obtained for transgenic mSOD1 mice (black columns) were compared with the values of their age-matched littermates (white columns). *p < 0.05; **p < 0.01; Student's ttest.
Cytochrome c immunostaining
To provide more detailed information regarding the cellular localization of released cytosolic cytochrome c, we immunostained spinal cord sections from nontransgenic and symptomatic transgenic mSOD1 mice, both at the beginning of symptoms and at end stage. In nontransgenic mice, numerous neurons immunoreactive for cytochrome c were observed throughout the gray matter of the spinal cord (Fig.2A). Notably, all of these cytochrome c-positive neurons showed immunostaining confined to the cell body and with a fine punctate appearance (Fig.2B,C). In symptomatic mice, many remaining spinal cord neurons in the anterior horn showed a more robust immunoreactivity (Fig. 2D,E) like that described previously in an ischemic model (Fujimura et al., 1999,2000). In those motor neurons, the fine punctate cytochrome c immunoreactivity appeared markedly reduced, giving way to a more diffuse immunostaining (Fig.2E,F), consistent with the redistribution of cytochrome c from mitochondria to cytosol in motor neurons of symptomatic mice.
Fig. 2.

Cytochrome c immunostaining in spinal cord from age-matched littermate nontransgenic mice (A–C) and mSOD1 transgenic mice at end stage (D–F). In nontransgenic controls, the staining was faint and punctate (A–C), whereas in end-stage transgenic mSOD1 mice, it was robust and diffuse (D–F).
mSOD1-related neurodegeneration stimulates Bax translocation
Because translocation of Bax to mitochondria promotes cytochrome c release (Wolter et al., 1997; Jürgensmeier et al., 1998), we determined Bax content in both cytosolic and mitochondrial fractions of the spinal cord of different groups of mice at different time points. In transgenic mSOD1 mice, cytosolic Bax levels were unchanged in the spinal cords of asymptomatic animals compared with that of nontransgenic controls (Fig.3A,B). In contrast, cytosolic Bax levels were reduced in the spinal cord of both early symptomatic and end-stage mice compared with age-matched nontransgenic (Fig.3A,B) and transgenic wtSOD animals (data not shown). In the same transgenic mSOD1 mice, mitochondrial Bax levels steadily augmented in the spinal cords compared with nontransgenic controls during the progression of the disease (Fig. 3C,D), reaching at end stage more than a twofold increase.
Fig. 3.

Subcellular localization of Bax. Bax expression was analyzed by Western blot in cytosolic (A,B) and mitochondrial (C,D) fractions of spinal cords from transgenic mSOD1 mice (black columns) at the asymptomatic stage (AS), at the beginning of symptoms (BS), and at the end stage (ES) and was compared with their age-matched nontransgenic littermates (Non-Tg; white columns). Represented values (n = 4–6 per group) correspond to mean ± SEM. *p < 0.05; **p < 0.01; Student's t test.
Activation of caspase-9 and caspase-7 in transgenic mSOD1 mice
During apoptotic cell death, pro-caspase-9 (52 kDa) is processed into a large active subunit (35 kDa) and a small unit (17 kDa), whereas pro-caspase-7 (37 kDa) is processed into proteolytic fragments of 28 and 20 kDa. In agreement with this, pro-caspase-9 and pro-caspase-7 were detected, respectively, as 52 and 37 kDa full-length inactive proteins in cytosolic fractions of spinal cords obtained from nontransgenic age-matched with end-stage, asymptomatic, and symptomatic transgenic mSOD1 mice (Figs.4A,5A). However, levels of pro-caspase-9 and pro-caspase-7 were decreased in symptomatic transgenic mSOD1 mice compared with nontransgenic controls (Figs.4A, 5A). Although neither active caspase-9 or caspase-7 could be detected in nontransgenic mice, they were both found in symptomatic transgenic mSOD1 (Figs. 4A,5A); note that active caspase-9 was detected sooner than active caspase-7 (Figs. 4A, 5A).
Fig. 4.

Activation of caspase-9 in spinal cords of transgenic mSOD1 mice. A, Using an antibody recognizing both forms of caspase-9, the expression of pro-caspase-9 (52 kDa) and its active fragment (35 kDa) was analyzed by Western blot in cytosolic fractions of spinal cords from transgenic mSOD1 mice at the asymptomatic stage (AS), at the beginning of symptoms (BS), and at the end stage (ES) and was compared with their age-matched nontransgenic littermates (Non-Tg). Each lane corresponds to a different animal. B, The immunolocalization of active caspase-9 was performed on spinal cord sections from transgenic (Tg) mSOD1 mice at end stage (ES) and their age-matched nontransgenic (Non-Tg) littermates.
Fig. 5.

Activation of caspase-7 in spinal cords of transgenic mSOD1 mice. A, Using an antibody recognizing both forms of caspase-7, the expression of pro-caspase-7 (37 kDa) and its cleaved fragment (28 kDa) was analyzed by Western blot in cytosolic fractions of spinal cords from transgenic mSOD1 mice at the asymptomatic stage (AS), at the beginning of symptoms (BS), and at the end stage (ES) and was compared with their age-matched nontransgenic littermates (Non-Tg). The top panel corresponds to β-actin. Each lane corresponds to a different animal and is representative of four to six animals per group.B, The immunolocalization of active caspase-7 was performed on spinal cord sections from transgenic (Tg) mSOD1 mice at end stage (ES) and their age-matched nontransgenic (Non-Tg) littermates.
To determine the cellular localization of activated caspases, we immunostained spinal cord sections from age-matched nontransgenic and transgenic mSOD1 mice at end stage with antibodies directed against cleaved caspase-9 and caspase-7. In nontransgenic controls, neither of the two antibodies generated any significant cellular immunostaining for caspase-9 or caspase-7 fragments (Figs.4B, 5B). On the contrary, in transgenic mSOD1 mice, numerous large motor neurons of anterior horn showed a strong immunostaining for active caspase-9 and caspase-7 (Figs.4B, 5B).
Cleavage of XIAP reflects caspase activation
The member of the inhibitor of apoptosis family, XIAP, antagonizes the caspase cascade through a direct inhibition of caspase-3, caspase-7, and caspase-9 (Deveraux and Reed, 1999). Cleavage of XIAP reflects an excess of activated caspases and produces a 30 kDa fragment with reduced ability of inhibiting caspases (Deveraux et al., 1999). We used an anti-XIAP antibody recognizing the amino acid residues (∼240) corresponding to the cleavage site of XIAP (Deveraux et al., 1999). The full-length protein was detected as a band of 53 kDa in nontransgenic and transgenic mice (Fig. 6). However, at the end stage of the disease, the level of XIAP was dramatically depleted and a ∼30 kDa fragment was concomitantly generated (Fig. 6). A nonspecific band migrating slightly larger than XIAP was also detected as described previously (Deveraux et al., 1999).
Fig. 6.

Cleavage of XIAP in spinal cords of transgenic mSOD1 mice. XIAP expression was analyzed by Western blot in cytosolic fractions of spinal cords from transgenic mSOD1 mice at the asymptomatic stage (AS), at the beginning of symptoms (BS), and at the end stage (ES) and was compared with their age-matched nontransgenic littermates (Non-Tg). The top panel corresponds to β-actin. Each lane corresponds to a different animal and is representative of four to six animals per group.
Immunostaining of cytochrome c of human spinal cord from ALS patient
As indicated above, translocation of cytochrome c is pivotal in the mitochondrial-dependent caspase activation. Therefore, to determine whether this pathway, as in transgenic mSOD1 mice, is in play in human patients with ALS, we assessed cytochrome c immunostaining in spinal cord specimens from sporadic ALS patients and controls. Very similar to the situation seen in mice, neurons with punctate cytochrome c immunostaining were seen in both controls and ALS (Fig.7). As in mice, in ALS specimens but not in controls, several anterior horn neurons exhibited a strikingly more robust and more diffuse cytosolic cytochrome c immunostaining (Fig.7).
Fig. 7.

Immunostaining for cytochrome c of spinal cord from ALS patients and controls. The immunostaining for cytochrome c was processed on spinal cord sections obtained from control (A–D; n = 7) and from ALS (E–H; n = 6) patients. In controls, cytochrome c-positive neurons were faintly stained and punctuate (A–D). In ALS cases, some neurons exhibited a control-like appearance (H), whereas most others were much more intensely and evenly immunostained (F, G).
DISCUSSION
We studied selected key molecular factors of the mitochondrial-dependent apoptotic pathway in the spinal cord of transgenic mSOD1 mice and human ALS because of its major potential clinical and therapeutic significance. Herein, we found that cytochrome c contents rose in the cytosolic fraction of transgenic mSOD1 mice, whereas, concomitantly, it dropped in the mitochondrial fraction over the course of the disease. This finding indicates that, during the neurodegenerative process, the spinal cord of transgenic mSOD1 mice is the site of a translocation of cytochrome c from the mitochondria to the cytosol, which is a critical event in the mitochondrial-dependent apoptotic pathway (Kroemer and Reed, 2000). However, mitochondria in the spinal cord of transgenic mSOD1 mice develop, over the course of the disease, major structural alterations, including cristae distortions, vacuolization, and variable degrees of swelling (Kong and Xu, 1998). Therefore, the appearance of cytochrome c immunostaining in some areas of motor neurons in affected transgenic mSOD1 mice could correspond to distended mitochondria (Fig.2E,F), which could possibly account for some of the cytosolic-like cytochrome c immunostaining. Furthermore, cytochrome c may have leaked out from these damaged organelles. Against these hypotheses is the fact that cytochrome c release was detected in transgenic mSOD1 mice before the appearance of those mitochondria alterations (Kong and Xu, 1998). Supporting the pathological significance of cytochrome c translocation in the transgenic mouse model of ALS are the following observations. First, the redistribution of cytochrome c appeared specific for affected brain regions, because cerebellum, which is devoid of neuropathological changes in this model (Dal Canto and Gurney, 1995), did not show evidence of cytochrome c translocation. Second, the cytochrome c release was related to the cytotoxic effects of the mutant protein and not to increased SOD1 activity, because age-matched transgenic wtSOD1 mice, with also approximately fourfold increased SOD1 activity (Mena et al., 1997), did not show spinal cord cytochrome c translocation. Third, cytochrome c translocation culminates in 3-month-old transgenic mSOD1 mice, the age at which the most active wave of motor neuron death occurs in these animals (Kong and Xu, 1998).
The transgenic mSOD1 mouse is a faithful model of the mSOD1-linked familial form of ALS (Bruijn and Cleveland, 1996), but how good this experimental model is for sporadic ALS is uncertain. Relevant to this issue is the observation that cytochrome c is, as in the transgenic mice, translocated in human postmortem spinal cord samples from sporadic ALS cases. This finding not only provides further credibility to the transgenic mSOD1 mouse model of ALS but also demonstrates that cytochrome c translocation is probably a consistent neuropathological feature of ALS, regardless of whether or not the disease is linked to mSOD1. Of note, as in ALS cases, several controls died from respiratory failure and yet they showed no evidence of spinal cord cytochrome c translocation. It can thus be suggested that the redistribution of cytochrome c in ALS results from the disease process and not from the cause of death.
Proteins of the Bcl-2 family regulate cell death in part by affecting mitochondrial cytochrome c redistribution (Vander Heiden and Thompson, 1999). In symptomatic transgenic mSOD1 mice, expression of Bcl-2 and Bcl-xL, which inhibit apoptosis, is reduced, whereas expression of Bad and Bax, which promote apoptosis, is increased (Vukosavic et al., 1999). In neuronal death induced by trophic factor deprivation or in Fas-inducing cell death, the redistribution of Bax from the cytosol to the mitochondria is a critical event for exerting its proapoptotic activity (Putcha et al., 1999). Given this, it is important to indicate that Bax does translocate from the cytosol to the mitochondria in spinal cords of affected transgenic mSOD1 mice. This finding agrees with the compartmental redistribution of selected Bcl-2 members in vulnerable regions in ALS patients (Martin, 1999). Here we found that the redistribution of Bax to the mitochondria occurs either just before or at the same time as the release of cytochrome c to the cytosol, suggesting that Bax in this model of ALS, as in other cell death settings (Finucane et al., 1999; Putcha et al., 1999), participates in triggering cytochrome c release.
Once released from the mitochondria, cytochrome c interacts in the cytosol with Apaf-1, forming an ATP-dependent complex that activates caspase-9 (Liu et al., 1996; Li et al., 1997; Zou et al., 1997; Hu et al., 1999; Saleh et al., 1999), which is instrumental in the mitochondrial-dependent activation of downstream effector caspases such as caspase-3 and caspase-7 (Slee et al., 1999). In the spinal cord of transgenic mSOD1 mice, a weak activation of caspase-9 is detected in early symptomatic animals, which coincides with the peak of cytosolic cytochrome c, and becomes conspicuous in end-stage animals. In contrast, activation of caspase-7, like that of caspase-3 (Pasinelli et al., 2000; Vukosavic et al., 2000), appears later over the course of the disease in these mice. This sequence of events is consistent with our current knowledge of the hierarchical organization of these different molecules within this apoptotic cell death-related machinery (Pettmann and Henderson, 1998). Accordingly, it is likely that activated caspase-9 probably initiates the processing of caspase-7 (Fig. 5) and of caspase-3 (Pasinelli et al., 2000; Vukosavic et al., 2000). Activated caspase-3 and caspase-7 can cleave other caspases, including caspase-9 (Slee et al., 1999). Consequently, the late and enhanced surge in caspase-9 activation could result in part from a feedback effect of activated caspase-3 and caspase-7.
Aside from the dramatic loss of motor neurons, spinal cord specimens from both human ALS cases and transgenic mSOD1 mice exhibit a strong glial reaction (Adams et al., 1984; Almer et al., 1999; Levine et al., 1999). Because glial cells can produce potent proapoptotic molecules, including tumor necrosis factor-α and interleukin-1-β, it is plausible that gliosis triggers and/or enhances the apoptotic demise of motor neurons in ALS. Alternatively, the described spinal cord apoptotic markers could have originated, at least in part, from glial cells as in Alzheimer's disease (Shimohama, 2000). In the spinal cord of transgenic mSOD1 mice, however, it appears that glial apoptosis is either absent or minimal and that most, if not all, apoptotic cells are reminiscent of neurons (Pasinelli et al., 2000; Vukosavic et al., 2000). Consistent with these previous findings, the present study shows that activated caspase-9 and caspase-7 were found in cells exhibiting a morphology of neurons and not of glial cells. Moreover, the neurons immunopositive for caspase-9, caspase-7, and for diffuse cytochrome c are likely engaged in the dying process, because they appear, in almost all cases, condensed and shrunken. Our observation is consistent with that made by Martin (1999), indicating in human ALS postmortem samples that most of the remaining motor neurons are at an “attritional stage” and thus are suffering–diseased neurons.
The IAPs family members are very well conserved evolutionary proteins that are characterized by the presence of baculoviral-inhibitor of apoptosis repeat (BIR) domains. The overexpression of each member or just the BIR domains is sufficient to block apoptosis in different paradigms of cell death (Liston et al., 1996; Deveraux et al., 1997;Roy et al., 1997). For instance, overexpression of XIAP attenuates cellular and behavioral deficits produced by an ischemic injury to the rat hippocampus, and adenovirus-mediated transgene expression of XIAP blocks the death of dopaminergic neurons in the substantia nigra after the administration of the parkinsonian toxin1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (Xu et al., 1999; Eberhardt et al., 2000). Moreover, some IAP family members can delay motor neuronal death after axotomy (Perrelet et al., 2000). Here we found that XIAP, the most potent inhibitor of apoptosis in the IAP family (Deveraux and Reed, 1999), was cleaved, and thus inactivated, in end-stage transgenic mSOD1 mice. This data, in association with our previous demonstration of a downregulation of Bcl-2 in transgenic mSOD1 (Vukosavic et al., 1999), suggests that not only does the activity of effectors of apoptosis rise over the course of the disease but also that the activity of inhibitors of apoptosis drops. Our demonstration of XIAP cleavage in affected transgenic mSOD1 mice provides meaningful functional information, which allows one to conclude that caspase activation may have real pathological consequences in this model of ALS.
The present study shows that some common apoptotic steps, namely mitochondrial translocation of Bax, cytosolic translocation of cytochrome c, and activation of caspase-9 and caspase-7 are recruited during the degeneration of motor neurons in the spinal cord of transgenic mSOD1 mice. All of these events arise in a sequential manner and are associated with the cleavage of the inhibitor of apoptosis XIAP. Previously, we have demonstrated that caspase inhibition (Li et al., 2000) and Bcl-2 overexpression (Kostic et al., 1997) succeed in prolonging survival but not in stopping the disease in transgenic mSOD1 mice. This suggests that, although the mitochondrial apoptotic machinery may be pivotal in the demise of motor neurons in ALS, its multifactorial nature may require combining several anti-apoptotic strategies to achieve optimal neuroprotection. In keeping with this possibility, the present study, by unraveling key apoptotic factors and the sequence in which they intervene, should help in the development of more effective therapeutic approaches for ALS.
Footnotes
This study is supported by the Muscular Dystrophy Association, the Amyotrophic Lateral Sclerosis Association, Project Amyotrophic Lateral Sclerosis, National Institute of Neurological Disorders and Stroke Grants R01 NS38586, R29 NS37345, and P50 NS38370, United States Department of Defense Grant DAMD 17-99-1-9471, the Lowenstein Foundation, the Smart Foundation, and the Parkinson's Disease Foundation. M.V. is a recipient of a fellowship from the Human Frontier Science Program Organization. We are grateful to Norma Romero for technical help, Dr. Caiping Chen for genotyping, Dr. Brigitte Sola and Dr. Brigitte Onténiente for their continual support, and Dr. Vernice Jackson-Lewis for critical reading of this manuscript.
Correspondence should be address to Dr. Serge Przedborski, Departments of Neurology and Pathology, BB-307, Columbia University, 650 West 168th Street, New York, NY 10032. E-mail: SP30@columbia.edu.
REFERENCES
- 1.Adams JH, Corsellis JAN, Duchen LW. Greenfield's neuropathology. Butler and Tanner; London: 1984. [Google Scholar]
- 2.Almer G, Vukosavic S, Romero N, Przedborski S. Inducible nitric oxide synthase upregulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;72:2415–2425. doi: 10.1046/j.1471-4159.1999.0722415.x. [DOI] [PubMed] [Google Scholar]
- 3.Brown RH., Jr Superoxide dismutase in familial amyotrophic lateral sclerosis: models for gain of function. Curr Opin Neurobiol. 1995;5:841–846. doi: 10.1016/0959-4388(95)80114-6. [DOI] [PubMed] [Google Scholar]
- 4.Bruijn LI, Cleveland DW. Mechanisms of selective motor neuron death in ALS: insights from transgenic mouse models of motor neuron disease. Neuropathol Appl Neurobiol. 1996;22:373–387. doi: 10.1111/j.1365-2990.1996.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 5.Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- 6.Deng H-X, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung W-Y, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 7.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 8.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 9.Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–5251. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 11.Eberhardt O, Coelln RV, Kugler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E, Haid S, Isenmann S, Gravel C, Srinivasan A, Bahr M, Weller M, Dichgans J, Schulz JB. Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci. 2000;20:9126–9134. doi: 10.1523/JNEUROSCI.20-24-09126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 13.Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan J. Inhibition of ICE slows ALS in mice. Nature. 1997;388:31. doi: 10.1038/40299. [DOI] [PubMed] [Google Scholar]
- 14.Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan PH. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J Neurosci. 2000;20:2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillardon F, Bottiger B, Schmitz B, Zimmermann M, Hossmann KA. Activation of CPP-32 protease in hippocampal neurons following ischemia and epilepsy. Brain Res Mol Brain Res. 1997;50:16–22. doi: 10.1016/s0169-328x(97)00162-9. [DOI] [PubMed] [Google Scholar]
- 17.Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guégan C, Sola B. Early and sequential recruitment of apoptotic effectors after focal permanent ischemia in mice. Brain Res. 2000;856:93–100. doi: 10.1016/s0006-8993(99)02347-1. [DOI] [PubMed] [Google Scholar]
- 19.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 20.Hu Y, Benedict MA, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 1999;18:3586–3595. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jürgensmeier JM, Xie ZH, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong JM, Xu ZS. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostic V, Jackson-Lewis V, De Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: Prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- 24.Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 26.Levine JB, Kong J, Nadler M, Xu Z. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS). Glia. 1999;28:215–224. [PubMed] [Google Scholar]
- 27.Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 28.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang XD. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 29.Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda JE, MacKenzie A, Korneluk RG. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature. 1996;379:349–353. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 30.Liu XS, Kim CN, Yang J, Jemmerson R, Wang XD. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 31.Martin LJ. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol. 1999;58:459–471. doi: 10.1097/00005072-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Mena MA, Khan U, Togasaki DM, Sulzer D, Epstein CJ, Przedborski S. Effects of wild-type and mutated copper/zinc superoxide dismutase on neuronal survival and L-DOPA-induced toxicity in postnatal midbrain culture. J Neurochem. 1997;69:21–33. doi: 10.1046/j.1471-4159.1997.69010021.x. [DOI] [PubMed] [Google Scholar]
- 33.Pasinelli P, Borchelt DR, Houseweart MK, Cleveland DW, Brown RHJ. Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc Natl Acad Sci USA. 1998;95:15763–15768. doi: 10.1073/pnas.95.26.15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2000;97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perrelet D, Ferri A, MacKenzie AE, Smith GM, Korneluk RG, Liston P, Sagot Y, Terrado J, Monnier D, Kato AC. IAP family proteins delay motoneuron cell death in vivo. Eur J Neurosci. 2000;12:2059–2067. doi: 10.1046/j.1460-9568.2000.00098.x. [DOI] [PubMed] [Google Scholar]
- 36.Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 37.Przedborski S, Donaldson D, Jakowec M, Kish SJ, Guttman M, Rosoklija G, Hays AP. Brain superoxide dismutase, catalase and glutathione peroxidase activites in amyotrophic lateral sclerosis . Ann Neurol. 1996;39:158–165. doi: 10.1002/ana.410390204. [DOI] [PubMed] [Google Scholar]
- 38.Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 40.Rowland LP. Hereditary and acquired motor neuron diseases. In: Rowland LP, editor. Merritt's textbook of neurology. Williams & Wilkins; Philadelphia: 1995. pp. 742–749. [Google Scholar]
- 41.Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem. 1999;274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- 43.Shimohama S. Apoptosis in Alzheimer's disease—an update. Apoptosis. 2000;5:9–16. doi: 10.1023/a:1009625323388. [DOI] [PubMed] [Google Scholar]
- 44.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vander Heiden MG, Thompson CB. Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- 46.Vereker E, Campbell V, Roche E, McEntee E, Lynch MA. Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J Biol Chem. 2000;275:26252–26258. doi: 10.1074/jbc.M002226200. [DOI] [PubMed] [Google Scholar]
- 47.Vukosavic S, Dubois-Dauphin M, Romero N, Przedborski S. Bax and Bcl-2 interaction in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;73:2460–2468. doi: 10.1046/j.1471-4159.1999.0732460.x. [DOI] [PubMed] [Google Scholar]
- 48.Vukosavic S, Stefanis L, Jackson-Lewis V, Guégan C, Romero N, Chen C, Dubois-Dauphin M, Przedborski S. Delaying caspase activation by Bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2000;20:9119–9125. doi: 10.1523/JNEUROSCI.20-24-09119.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu D, Bureau Y, McIntyre DC, Nicholson DW, Liston P, Zhu Y, Fong WG, Crocker SJ, Korneluk RG, Robertson GS. Attenuation of ischemia-induced cellular and behavioral deficits by X chromosome-linked inhibitor of apoptosis protein overexpression in the rat hippocampus. J Neurosci. 1999;19:5026–5033. doi: 10.1523/JNEUROSCI.19-12-05026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]