

Abstract
The Shaw-like potassium (K+) channels Kv3.1 and Kv3.3 are widely coexpressed in distinct neuronal populations in the CNS, possibly explaining the relatively “mild” phenotypes of the Kv3.1 and the Kv3.3 single mutant. Kv3.1-deficient mice show increased cortical γ- and decreased δ-oscillations (Joho et al., 1997, 1999); otherwise, the Kv3.1-mutant phenotype is relatively subtle (Ho et al., 1997; Sánchez et al., 2000). Kv3.3-deficient mice display no overt phenotype (Chan, 1997). To investigate whether Kv3.1 and Kv3.3 K+ channels are functionally redundant, we generated the Kv3.1/Kv3.3 double mutant. Kv3.1/Kv3.3-deficient mice were born at the expected Mendelian frequencies indicating that neither Kv3.1 nor Kv3.3 K+ channels are essential for embryonic development. Although there are no obvious changes in gross brain anatomy, adult Kv3.1/Kv3.3-deficient mice display severe ataxia, tremulous movements, myoclonus, and hypersensitivity to ethanol. Mice appear unbalanced when moving, whereas at rest they exhibit whole-body jerks every few seconds. In spite of the severe motor impairment, Kv3.1/Kv3.3-deficient mice are hyperactive, show increased exploratory activity, and display no obvious learning or memory deficit. Myoclonus, tremor, and ethanol hypersensitivity are only seen in the double-homozygous Kv3.1/Kv3.3-deficient mice, whereas increased locomotor and exploratory activity are also present in double-heterozygous mice. The graded penetrance of mutant traits appears to depend on the number of null alleles, suggesting that some of the distinct phenotypic traits visible in the absence of Kv3.1 and Kv3.3 K+ channels are unrelated and may be caused by localized dysfunction in different brain regions.
Keywords: cerebellum, ataxia, tremor, ethanol, double knock-out, fast-spiking, interneurons
Voltage-gated potassium (K+) channels form a large family of ion channels that are involved in establishing the resting membrane potential, in determining the action potential (AP) waveform and duration, in regulating release of neurotransmitter, and in modulating rhythmic firing patterns and pacemaker activity of neurons. Subunits for the Shaw-like K+ channels Kv3.1 and Kv3.3 are extensively coexpressed throughout the CNS (Drewe et al., 1992; Perney et al., 1992; Rettig et al., 1992; Rudy et al., 1992;Goldman-Wohl et al., 1994; Lenz et al, 1994; Weiser et al., 1994, 1995;Du et al., 1996; Perney and Kaczmarek, 1997; Sekirnjak et al., 1997; for review, see Rudy et al., 1999).
In heterologous expression systems, Kv3.1 and Kv3.3 subunits form homotetrameric K+ channels with rapid kinetics of activation and deactivation, high thresholds of activation (more than −20 mV), and relatively large unit conductances (∼30 pS) (Weiser at al., 1994). It is remarkable that Kv3.1 channels deactivate 10 times faster than do all tested Kv1-type (Shaker-type) voltage-gated K+ channels (Grissmer et al., 1994). These properties are probably responsible for the fast afterhyperpolarization (fAHP) required for high-frequency firing observed in Kv3.1-expressing neurons (Massengill et al., 1997; Martina et al., 1998; Wang et al., 1998). Hence, it is reasonable to hypothesize that regulating Kv3.1 activity affects AP duration and influences Ca2+ entry in axonal terminals with dramatic consequences for subsequent neurotransmitter release. Indeed, computer simulations show that a K+channel with the biophysical properties of Kv3.1 keeps the AP narrow and the refractory period short; hence, Kv3.1 and, presumably, Kv3.3 channel-mediated repolarizing currents are likely requirements for sustained high-frequency firing (Kanemasa et al., 1995; Perney and Kaczmarek, 1997).
Mice in which the genes encoding Kv3.1 and Kv3.3 are individually inactivated have been generated (Chan, 1997; Ho et al., 1997). The Kv3.1 single mutant shows increased γ- and decreased δ-oscillations and a motor-skill deficit caused by altered muscle contractility and force generation (Ho et al., 1997; Joho et al., 1997, 1999). The motor-skill deficit is, however, only present in 129/Sv and not in 129/Sv × C57BL/6 F1 mice; i.e., the penetrance of the Kv3.1-null mutation depends strongly on the genetic background (Sánchez et al., 2000). The Kv3.3 single mutant displays no overt phenotype (Chan, 1997). It is possible that the wide coexpression of Kv3.1 and Kv3.3 K+ channel subunits in the same cells in the CNS results in functional redundancy, explaining the lack of stronger phenotypes in both single mutants. To address the possibility that Kcnc1 and Kcnc3 genes (encoding Kv3.1 and Kv3.3) represent a pair of redundant K+channel genes, we generated the double-mutant mouse lacking Kv3.1 and Kv3.3 K+ channels. Adult Kv3.1/Kv3.3-deficient mice display severe ataxia, tremulous movements, myoclonus, and hypersensitivity to ethanol. Mutant mice appear unbalanced when moving, whereas at rest they exhibit whole-body jerks every few seconds. In spite of these debilitating motor deficits, Kv3.1/Kv3.3-deficient mice are hyperactive, show dramatically increased exploratory activity, and display no obvious learning or memory deficit.
Parts of this paper have been published previously (Espinosa et al., 2000).
MATERIALS AND METHODS
Mouse breeding
The generation of the Kv3.1−/−mouse on the 129/Sv, C57BL/6, and 129/Sv × C57BL/6 F1 background has been described previously (Ho et al., 1997; Sánchez et al., 2000). The Kv3.3-encoding gene was inactivated by homologous recombination using a targeting vector in which the region containing the ATG initiation triplet was deleted and replaced with the phosphoglycerate kinase-neomycin resistance cassette inserted in the opposite orientation relative to the direction of transcription of the Kv3.3 gene (Chan, 1997). The loci Kcnc1 and Kcnc3encoding Kv3.1 and Kv3.3 K+ channel subunits are on chromosome 7 in the mouse (Wymore et al., 1994; Kalman et al., 1998) at an estimated genetic distance of 0.5–2.0 centimorgans (cM; Mouse Genomic Database, October 1998; The Jackson Laboratory, Bar Harbor, ME). To obtain double-homozygous mutants, we crossed double-heterozygous Kv3.1+/−Kv3.3−/+males of mixed genetic background (∼25% 129/Sv and ∼75% C57BL/6) carrying the Kv3.1 and Kv3.3 null alleles on the paternal and maternal chromosome 7, respectively, with Kv3.1−/− 129/Sv × C57BL/6 F1 females (see Fig. 1 for breeding scheme). Because of poor breeding habits, we found it impractical to maintain homozygous Kv3.1- and Kv3.3-deficient mice on a pure 129/Sv or C57BL/6 background. As expected, ∼50% of the offspring were Kv3.1+/−, and ∼50% were Kv3.1−/−. To detect a recombination event, only homozygous Kv3.1−/−offspring were then genotyped for Kv3.3. Nearly all of the Kv3.1−/− pups were Kv3.3+/+ (no recombination); however, after ∼100 offspring, we obtained a Kv3.1−/−Kv3.3+/−animal because of a recombination event that had occurred in the male germ line between the Kcnc1 and Kcnc3 locus (Fig.1).
Fig. 1.
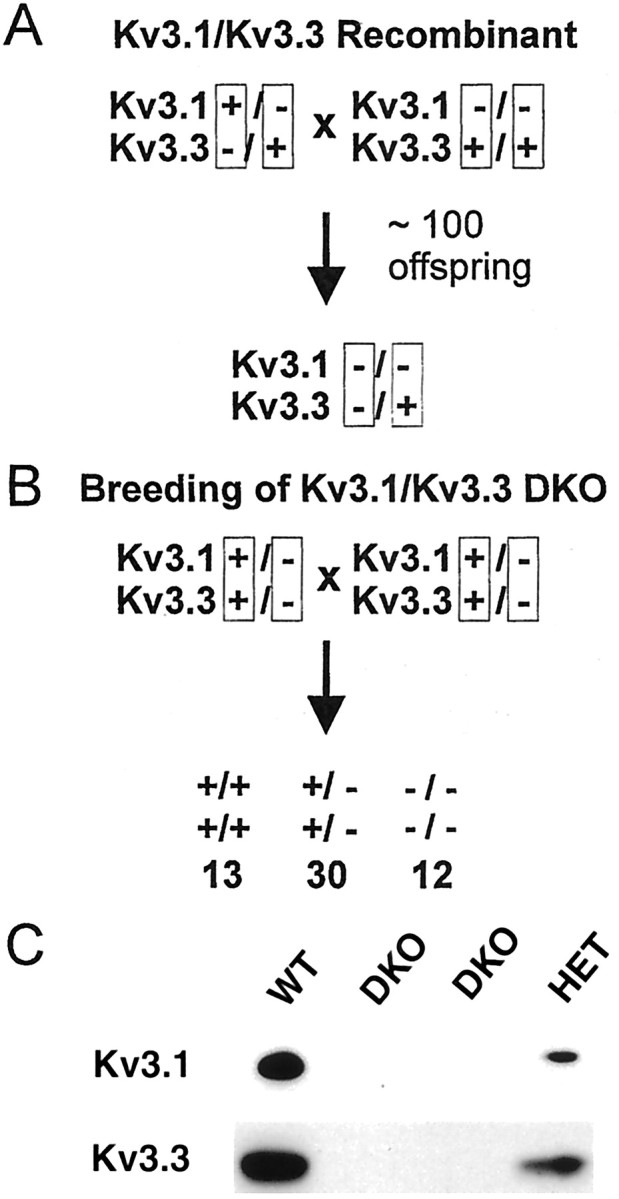
Generation of the Kv3.1/Kv3.3 double mutant by recombination. A, A double-heterozygous male with the null alleles for Kv3.1 and Kv3.3 on two different chromosomes 7 was crossed to a Kv3.1−/− female. Recombination in the male germ line (after ∼100 offspring) linked the two null alleles on the same chromosome. B, By the use of double-heterozygous (+/−,+/−) breeding pairs, the two null alleles segregate together and yield offspring at the expected Mendelian ratios of 1:2:1 (indicated below genotypes), indicating that Kv3.1 and Kv3.3 K+ channels are not required for embryonic development. C, In comparison with wild-type mice (WT), immunoblot analyses of cerebellar protein extracts show reduced levels of Kv3.1 and Kv3.3 in double-heterozygous mice (HET) and no detectable Kv3.1 or Kv3.3 protein in double-homozygous mice (DKO).
Genotyping
Mice were genotyped using tail DNA (∼100–400 ng in 25 μl reaction volume) as the template for amplification by PCR. The oligonucleotides (ONs) used for Kv3.1 genotyping were the following: ON1, GCGCTTCAACCCCATCGTGAACAAGACC (wild-type or mutant forward primer at 300 nm); ON2, GGCCACAAAGTCAATGATATTGAGGGAG (wild-type reverse primer at 150 nm); and ON3, CTACTTCCATTTGTCACGTCCTGCACG (mutant reverse primer at 150 nm). The wild-type allele yields a 216 bp fragment with ON1 and ON2; the mutant allele yields a 420 bp fragment with ON1 and ON3. For Kv3.3 genotyping, the following oligonucleotides were used: ON4, CGGCGACAGCGGTAAGATCGTGATCAACG (wild-type forward primer at 150 nm); ON5, CCTGAAAACACAGACGCTTGAGCTCGCC (wild-type or mutant reverse primer at 150 nm); and ON6, CTTCCATTTGTCACGTCCTGCACGACGCGAGC (mutant forward primer at 150 nm). For Kv3.1 genotyping, PCR amplification for the wild-type and mutant allele was done together; for Kv3.3 genotyping, amplification was done in separate reactions to obtain the 490 bp wild-type fragment (with ON4 and ON5) and the 658 bp mutant fragment (with ON5 and ON6). The PCR protocol for both genes (performed in a PTC-100 Programmable Thermal Controller; MJ Research, Inc.) was as follows: 95°C for 3 min, followed by 34 cycles at 94°C for 1 min, 55°C for 2 min, and 72°C for 3 min, and a final extension at 72°C for 3 min. The DNA products were analyzed by standard gel electrophoresis in 1.6% agarose.
Western blot analysis
Mice were anesthetized deeply with Avertin, and cerebella were dissected out and homogenized in homogenization buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 1% Triton X-100, containing the complete protease inhibitor cocktail from Boehringer Mannheim, Indianapolis, IN). Homogenized samples were left on ice for 15 min and were then centrifuged at 10,000 ×g for 10 min. Supernatant protein was assayed using the BCA protein assay kit (Pierce, Rockford, IL). Ten micrograms of each protein sample were separated by 6% SDS-PAGE and transferred to Immobilon-P membranes. The Kv3.1b protein was detected using a rabbit anti-rat polyclonal antibody (Alomone), and the Kv3.3 protein was detected using a rabbit anti-mouse polyclonal antibody (Chan, 1997); the secondary antibody was a peroxidase-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA). Enhanced chemiluminescence (ECL) was used as the detection system according to the manufacturer's instructions (Pierce).
Behavioral testing
Mice were kept on a 12 hr light/dark cycle. All tests were performed with age-matched males (3–4 months of age unless otherwise indicated). Double-heterozygous breeding pairs (+/−,+/− × +/−,+/−) were used to obtain mice of all three genotypes studied here: Kv3.1+/+Kv3.3+/+(+/+,+/+), Kv3.1+/−Kv3.3+/−(+/−,+/−), and Kv3.1−/−Kv3.3−/−(−/−,−/−). By the use of this breeding scheme, the genetic background varied among individual mice; however, the variability was the same for mice of all three genotypes (∼44% of genes were derived from 129/Sv, and ∼56% were from C57BL/6). All experiments were approved by the Institutional Animal Care and Research Advisory Committee.
Spontaneous myoclonus. Mice were placed in a small Plexiglas cage (a restraining chamber) to minimize spontaneous movements, and they were allowed to habituate to the environment. Individual mice were videotaped for ∼30 min. Myoclonic jerks were operationally defined as spontaneous, brief movements involving the animal's whole body, not just a single limb, the head, or an ear twitch [corresponding to scores 4 and 5 on a scale from 1 to 5 by Kanthasamy et al. (2000)]. These jerks could be counted relatively easily when mice were at rest and their movements were restricted. Wild-type and double-heterozygous mice showed no jerks under these conditions.
Rotarod test. The rotarod test was performed as described previously (Ho et al., 1997; Sánchez et al., 2000). Briefly, male mice were placed on a rod (8.9 cm long and 3.8 cm in diameter), initially rotating at 5 rpm and accelerating at 10 rpm/min (Rotamex System; Columbus Instruments, Columbus, OH). The dependent measure was the length of time on the rod. When the mice fell to the bottom of the cage, they received a brief electrical foot shock (0.2 mA for 1 sec). Five trials were performed with each animal within a 1 hr period, and the means of the median performance times were compared by statistical analysis.
Open field test. Male littermates were individually tested in an open field (44 × 44 cm Plexiglas cage; Opto-Varimex and Auto-Track-System software, Columbus Instruments). Spontaneous activity was recorded for 60 min and plotted in 15 min intervals. To determine center-field occupancy, the open field was divided into 8 × 8 squares, and the mean occupancy (in seconds) in each square was determined (Auto-Track Multiple Zone Motion Monitor). Center-field occupancy is defined as the time spent in the area delineated by the 16 squares (4 × 4) in the center of the open field (in seconds per 60 min period).
Ethanol sensitivity. A male mouse was placed in the open-field setup and allowed to explore the environment while its activity was recorded for 60 min. The mouse was then injected intraperitoneally with ethanol (20–200 mg/ml in 0.9% saline), and the activity was recorded and videotaped for another 60 min. Sensitivity to ethanol was monitored by counting sideways falls for the first 10 min after ethanol injection.
Active avoidance test. A male mouse was placed in the dark chamber of a light/dark shuttle box (each compartment was 29 × 22 × 13.5 cm) with a small opening to allow transitions between the two compartments. After 10 sec, an electrical current (0.2 mA) was applied through a bottom grid for a duration of 20 sec. For the first trial on the first day of training, the opening was kept closed until the foot shock was initiated. Mice were subjected to 10 trials per day on 5 consecutive days. Fourteen days after the fifth day of training, mice were tested again to assess memory. An avoidance response occurred when a mouse actively avoided the foot shock by leaving the dark chamber during the first 10 sec. The avoidance index is the ratio of the number of avoidance reactions to the total number of trials.
Statistical analysis of results. For data presentation, means and SEM were used. For statistical analyses, two-tailed Student's t test and one- or two-factor ANOVA followed by multiple comparisons (Tukey or Dunnett tests) were used as appropriate. Analyses were performed in Microsoft Excel 97.
RESULTS
Kv3.1 and Kv3.3 channels are essential for postnatal but not for embryonic development
Kv3.1 and Kv3.3 K+ channels show regional coexpression in the CNS (for review, see Rudy et al., 1999). We reasoned that these channels might be encoded by a pair of redundant K+ channel genes (Kcnc1 andKcnc3) that could explain the absence of more dramatic phenotypic changes in the Kv3.1 and the Kv3.3 single mutants. Hence, we decided to generate the Kv3.1/Kv3.3 double mutant, expecting a more severe phenotype.
The loci Kcnc1 and Kcnc3 had been mapped to chromosome 7 in the mouse (Wymore et al., 1994; Kalman et al., 1998), and the genetic distance between the two loci had been estimated from 0.5 to 2.0 cM (Mouse Genomic Database, October 1998; The Jackson Laboratory), indicating that the two K+channel genes were not tandemly linked. Because of the genetic distance of 0.5–2.0 cM between Kcnc1 and Kcnc3, we expected to obtain a recombination event between the two loci among a few hundred offspring from appropriate mating pairs. After ∼100 offspring, we obtained a Kv3.1−/−Kv3.3+/−female resulting from a recombination event between theKcnc1 and the Kcnc3 locus in the male germ line (Fig. 1A).
We used this founder animal to generate double-heterozygous Kv3.1+/−Kv3.3+/−(+/−,+/−) mice in which the two null alleles were linked and segregated together on the same chromosome. Using heterozygous breeding pairs (+/−,+/− × +/−,+/−), we obtained wild-type (+/+,+/+), double-heterozygous (+/−,+/−), and double-homozygous (−/−,−/−) mice at a ratio of ∼1:2:1, indicating that neither Kv3.1 nor Kv3.3 channel function was required for embryonic development (Fig.1B). Immunoblot analyses showed that protein levels for Kv3.1 and Kv3.3 were reduced in brain extracts of +/−,+/− mice and completely absent in −/−,−/− mice (Fig. 1C). All subsequent experiments were done with offspring from such double-heterozygous breeding pairs; therefore, although variability of the genetic background was necessarily present among individual mice, the variability was the same for mice of all three genotypes.
Starting at approximately postnatal day 7 (P7), it became apparent that the offspring of heterozygous breeding pairs consisted of two phenotypically different populations (Fig.2A). By P7, some pups were clearly smaller than others of the same litter. The smaller pups gained less weight than did their littermates. At ∼P14, the smaller pups stopped growing, began to loose weight gradually, and died between P19 and P26. In contrast, all pups that were larger at P7 continued to grow and developed into normal-looking adults. Two to 3 d before death, the body temperature of the smaller pups began to drop from ∼38°C to as low as ∼25–28°C on the day of death (Fig.2A, inset). All small pups that died by ∼P26 were of the −/−,−/− genotype, i.e., deficient for Kv3.1 and Kv3.3 K+ channels. The pups that developed normally were of either +/+,+/+ or +/−,+/− genotype. The small −/−,−/− pups showed some type of ataxia and motor incoordination, manifested as poor balance while moving, tremulous movements, and sudden, brief muscle jerks. The motor problems and the “death phenotype” by P26 were fully penetrant; i.e., none of the −/−,−/− mice survived beyond P26.
Fig. 2.

A, Kv3.1 and Kv3.3 K+ channels are required for postnatal development. In this litter of seven pups, two double mutants began to loose weight at ∼P15 and died by P20 and P21. Wild-type and heterozygous littermates gained weight normally and grew to adulthood.Inset, The drop in body temperature of the double mutants is shown. The Kv3.1 and Kv3.3 genotypes and the gender [female (f), male (m)] are shown on the right. B, Adult Kv3.1/Kv3.3-deficient mice are smaller than are wild-type and double-heterozygous mice. At 1.5 months (45 d) and 3.5 months (100 d) of age, Kv3.1−/−Kv3.3−/−(DKO), Kv3.1+/−Kv3.3+/−(HET), and Kv3.1+/+Kv3.3+/+(WT) male mice differ from each other in body weight [mean ± SEM and number of mice (above eachvertical bar) shown; one-factor ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001].
Observing the Kv3.1/Kv3.3-deficient mice, we suspected that the motor deficit might place the double mutants at a competitive disadvantage for feeding (suckling) compared with their littermates. This, in turn, might result in weight loss and death by 3–4 weeks of age. When ataxic double mutants were allowed to feed on semisolid food (rodent chow softened in water) for ∼1 hr/d (from P15 to P30) in the absence of their competing littermates, ∼80–90% of the pups survived to adulthood. At P45, the homozygous −/−,−/− double mutants reached however only ∼50–60% of the body weight of +/+,+/+ or +/−,+/− mice (one-factor ANOVA; p < 0.001); double-heterozygous +/−,+/− mice remained 10–15% smaller than +/+,+/+ wild-type mice (p < 0.05), suggesting a graded effect on body weight depending on the actual number of mutant alleles (two for +/−,+/− vs four for −/−,−/− mice) (Fig.2B). The difference in body weight was still present when the mice were ∼3 months old.
Adult Kv3.1/Kv3.3-deficient mice show severe ataxia, myoclonus, and ethanol hypersensitivity but no changes in gross brain anatomy
We examined the brain of −/−,−/− mice for possible neuroanatomical alterations. Hematoxylin- and eosin-stained parasagittal brain sections of a wild-type and a double-mutant mouse are shown in Figure 3. At this level of analysis, we could not detect any obvious neuroanatomical changes in the brains of Kv3.1/Kv3.3-deficient mice. In particular, the mutant cerebellum shows an unchanged appearance of the molecular, Purkinje cell, and granule cell layers, including normal foliation and cytoarchitecture. This finding is remarkable in light of several phenotypic traits that may reflect cerebellar dysfunction (see below).
Fig. 3.
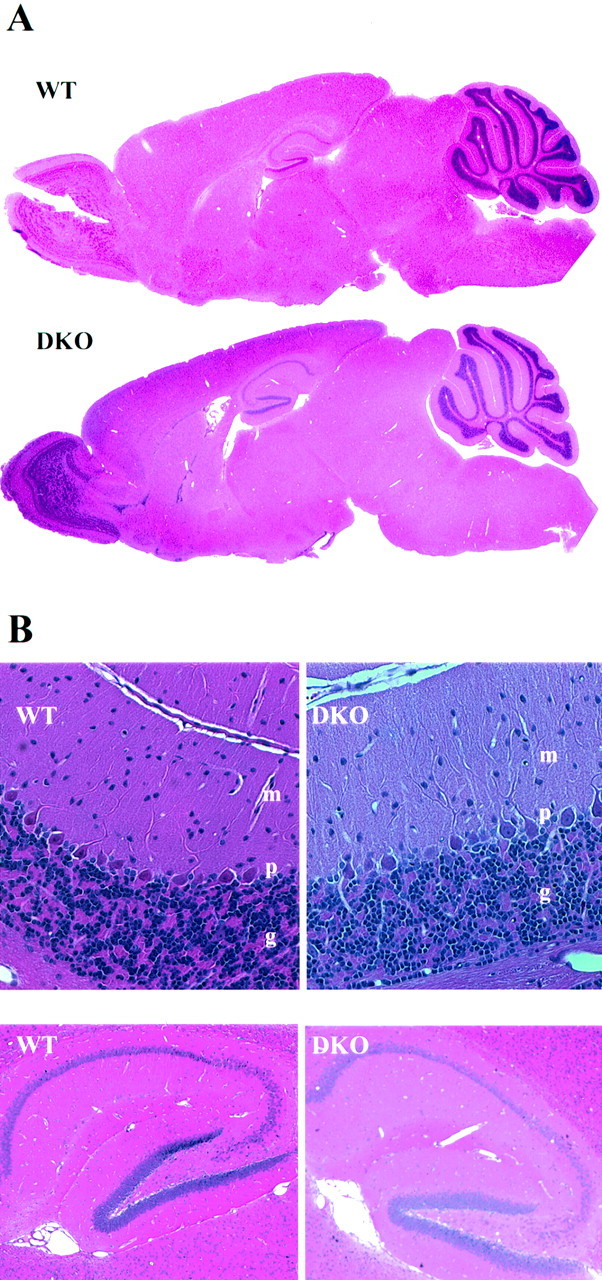
No obvious alterations in gross brain anatomy in the Kv3.1/Kv3.3 double mutant. A, Hematoxylin- and eosin-stained parasagittal sections (4 μm thick) of a wild type and a Kv3.1/Kv3.3 double mutant show no changes in gross brain anatomy. Cerebellar foliation is unchanged in the mutant brains.B, When examined at higher magnification, both brains display the same characteristic layering of the molecular (m), Purkinje (p), and granule (g) cell layers (top). Hippocampal cytoarchitecture also displays no obvious alterations (bottom).
Adult Kv3.1/Kv3.3-deficient mice showed severe ataxia, intermittent tremor-like episodes, and spontaneous, brief, and involuntary muscle contractions (myoclonus). Adult −/−,−/− mice appeared uncoordinated and unbalanced when moving around the cage, and they exhibited whole-body jerks every few seconds (31.7 ± 1.3 jerks/min;n = 4). Some of the jerks were strong enough to lift the mouse off the cage floor. Wild-type +/+,+/+ and heterozygous +/−,+/− mice showed no jerks.
We quantified some aspects of the motor deficit of Kv3.1/Kv3.3 double mutants using the rotating-rod (rotarod) test. When +/+,+/+ and +/−,+/− mice were subjected to the rotarod test, mice of both genotypes stayed on the accelerating, rotating rod for ∼80 sec, and the two groups did not differ from one another (Fig.4). In marked contrast, the −/−,−/− double mutants were unable to stay on either the rotating or stationary rod for more than a few seconds (one-factor ANOVA; p < 0.001). In some instances, it appeared that a double homozygous −/−,−/− mouse fell off the rod because of a sudden jerk that made the animal loose its balance.
Fig. 4.

Kv3.1/Kv3.3-deficient mice have a severe motor-skill deficit. In contrast to wild-type or heterozygous mice, Kv3.1−/−Kv3.3−/− mice cannot stay on a rotating rod (one-factor ANOVA; ***p < 0.001). DKO mice show no difference between the rotating and the stationary rod (paired t test,p = 0.36). Male mice were placed on an accelerating, rotating rod (diameter, 3.8 cm), and the time until fall was measured [mean ± SEM and number of mice (above eachvertical bar) shown]. At time 0, the rod turned at 5 rpm and accelerated at 10 rpm/min. Each animal was subjected to five trials during an ∼1 hr test period.
Ethanol is effective in reducing some types of myoclonus and tremor; therefore, we compared the effects of ethanol on wild-type and Kv3.1/Kv3.3-deficient mice. Ethanol (up to 2 mg/gm of body weight) had almost no effect on wild-type and double-heterozygous mice; in stark contrast, Kv3.1/Kv3.3-deficient mice were highly sensitive to even low doses of ethanol (0.5 mg/gm), indicated by periodically occurring sideways falls when moving (Fig. 5). In contrast to +/+,+/+ and +/−,+/− mice that showed no sideways falls in the absence of ethanol, −/−,−/− mice fell approximately every 3 min (0.30 ± 0.09 falls/min; n = 16). With as little as 0.5 mg/gm ethanol, −/−,−/− mice showed an increase in sideways falls, reaching a plateau at 1.0 mg/gm (7.4 ± 1.8 falls/min;n = 6). Wild-type or double-heterozygous mice displayed no sideways falls at 1.0 mg/gm ethanol; they began to show a few sideways falls at 2.0 mg/gm ethanol at a frequency similar to that of double-homozygous mutants in the absence of ethanol [0.40 ± 0.07 and 0.53 ± 0.21 falls/min for +/+,+/+ and +/−,+/−, respectively (n = 4)] (Fig. 5).
Fig. 5.

Kv3.1/Kv3.3-deficient mice are hypersensitive to ethanol. Ethanol (in 0.9% saline) was injected intraperitoneally in male mice, and sideways falls were counted for the first 10 min after ethanol injection [mean ± SEM and number of mice (above eachvertical bar) shown]. The ethanol effect was fully visible within 2 min of injection and lasted for ∼20 and ∼45 min for 1 and 2 mg/gm, respectively.
The Kv3.1/Kv3.3 double mutant shows increased locomotor and exploratory activity despite a severe motor-skill deficit
We studied spontaneous locomotion and exploratory behavior in the open field. Surprisingly, the double mutant, despite unbalanced movements, was significantly more active during the 60 min test when compared with +/+,+/+ mice (one-factor ANOVA; p < 0.001) (Fig. 6, top left). Double-heterozygous +/−,+/− mice showed spontaneous locomotor activity intermediate between that of wild-type (p < 0.001) and double-mutant (p < 0.05) mice, suggesting again, as for body weight, a null allele dosage effect. This increase in spontaneous locomotion was directly related to a significant increase in ambulatory time during the 60 min observation period [in seconds, +/+,+/+, 288.6 ± 45.9 (n = 8); +/−,+/−, 621.2 ± 42.8 (n = 10); and −/−,−/−, 806.6 ± 66.4 (n = 11)] (Fig. 6, top right); the traveling speed, however, was not changed [in centimeters/second, +/+,+/+, 21.4 ± 0.29 (n = 8); +/−,+/−, 21.7 ± 0.22 (n = 10); and −/−,−/−, 21.0 ± 0.18 (n = 11)] in spite of the dramatic motor-skill deficits that resulted in uncoordinated movements, balance problems, and myoclonus.
Fig. 6.
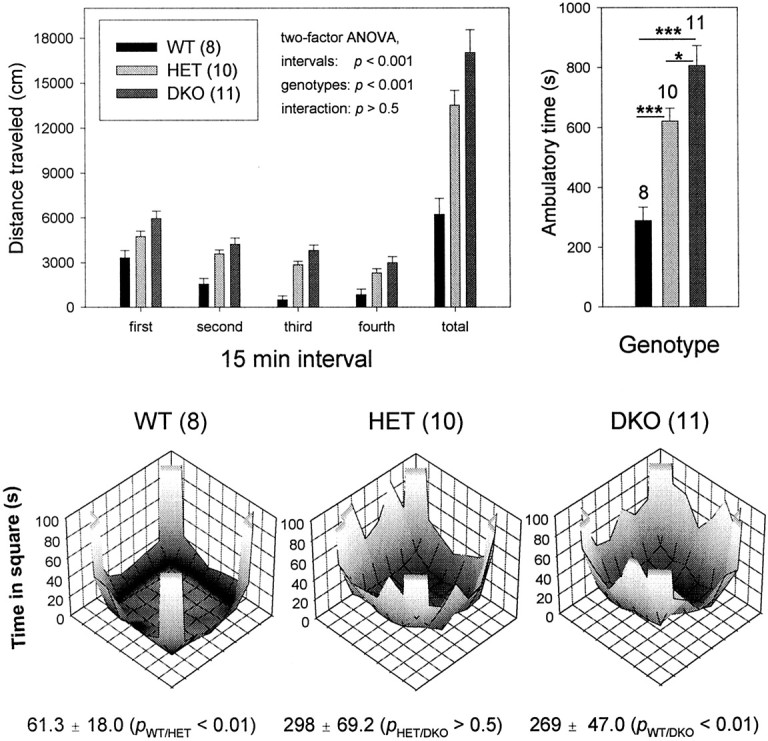
Kv3.1/Kv3.3-deficient mice show increased spontaneous locomotion and center-field occupancy. Top left, Spontaneous locomotor activity of male mice was monitored for 60 min and plotted in 15 min intervals [mean ± SEM and number of mice (in parentheses) shown]. Heterozygous and homozygous Kv3.1 and Kv3.3 mutants were significantly more active than were wild-type mice. For mice of all three genotypes, locomotor activity decreased during the 60 min interval (two-factor ANOVA). Top right, The increased distance traveled (shown on the top left) is caused by increased ambulatory activity (one-factor ANOVA; *p < 0.05; ***p < 0.001). Bottom, The open field was divided into 64 squares (8 × 8), and occupancy in each square was determined. Center occupancy (4 × 4 squares) was significantly increased for +/−,+/− and −/−,−/− mice (one-factor ANOVA). The test was conducted in an open field (44 × 44 cm) bounded by Plexiglas walls where the movement of the mouse was monitored along thex- and y-axes by infrared beams ∼2.5 cm apart (Opto-Varimex and Auto-Track-System software; Columbus Instruments). Low and high occupancy (seconds/60 min) is indicated bydark and light gray shades, respectively.
When mice are exposed to a novel environment, they generally display thigmotaxis; i.e., when moving around the cage, they tend to stay close to the wall of the test chamber. Brief excursions toward the center are initially rare but occur with increased frequency after the mouse has become more familiar with the environment. An increase in center-field activity or occupancy may be interpreted to correlate with a lower anxiety level (Gershenfeld and Paul, 1997). Figure 6,bottom, shows the occupancy patterns of wild-type and of double-heterozygous and double-homozygous Kv3.1/Kv3.3-deficient mice. During the 60 min test, wild-type mice remained for most of the time in the corners of the open field with only brief excursions to the center (61.3 ± 18.0 sec; n = 8). In contrast, both double-heterozygous and double-homozygous mice spent more time along the edges and made longer-lasting excursions to the center of the open field. Both +/−,+/− mice (298 ± 69.2 sec; n = 10; p < 0.01) and −/−,−/− mice (269 ± 47.0 sec; n = 11; p < 0.01) show significant increases in center-field occupancy (one-factor ANOVA). For this phenotypic trait, there is no difference between +/−,+/− and −/−,−/− mice (p > 0.5), in contrast to body weight and spontaneous locomotor activity that show graded penetrance. It is remarkable that the phenotypic trait of increased exploratory activity is fully penetrant in +/−,+/− mice in which only two of four possible K+ channel alleles are nonfunctional. It is tempting to speculate that increased spontaneous locomotion and center-field occupancy may reflect an increase in GABA tone because of increased GABA release as a consequence of prolonged APs in the absence of Kv3.1 and Kv3.1 K+channels (see Discussion).
Kv3.1/Kv3.3-deficient mice show normal learning and memory in an active avoidance task
The double-mutant mice were severely impaired in several motor functions. This finding raised the possibility that the absence of two K+ channels needed for rapid AP repolarization could have had a “globally” perturbing effect on brain function. To test this possibility, we subjected the Kv3.1/Kv3.3-deficient mice to an active avoidance test that assessed their ability to learn and memorize the simple task of avoiding a foot shock (Fig. 7). Although the double-mutant mice showed severe ataxia and myoclonus while performing the active avoidance task, Kv3.1/Kv3.3-deficient mice learned to avoid the foot shock as quickly as did wild-type and double-heterozygous mice, and they remembered the task equally well, at least for the 2 week interval until they were retested (two-factor ANOVA; days,p < 0.001; genotype, p > 0.5). Hence, although Kv3.1/Kv3.3-deficient mice were severely impaired in their motor skills, exhibited myoclonus and tremulous movements, and were hypersensitive to ethanol, the double mutants did not show an obvious deficit in performing a simple learning and memory task. Hence, it appears that the phenotypic changes resulting in the absence of Kv3.1 and Kv3.3 K+ channels do not perturb general CNS function in a nonspecific and global manner.
Fig. 7.

Kv3.1/Kv3.3-deficient mice display normal active avoidance learning. There are no differences between WT,HET, and DKO male mice in learning an avoidance task (days 1–5) and in recalling it 2 weeks later (day 19) (mean ± SEM and number of mice shown).
Gene (null allele) dosage effect on the penetrance of phenotypic traits
In the current study, we studied eight phenotypic traits in wild-type, double-heterozygous, and double-homozygous Kv3.1/Kv3.3-deficient mice (Table 1). Each phenotypic trait could be assigned to one of four different groups. Active avoidance (group 1), a simple form of learning and memory, is not altered in −/−,−/− mice, indicating that the absence of Kv3.1 and Kv3.3 K+ channels does not perturb in a nonspecific or global way cognitive function of the brain. The phenotypic traits of ataxia (poor rotarod performance), tremor, myoclonus, and ethanol sensitivity (group 2) are present in −/−,−/− mice but not in either +/+,+/+ or +/−,+/− mice. In contrast to the phenotypic traits listed in group 2, the traits of reduced body weight and increased spontaneous locomotion (group 3) are present both in +/−,+/− and in −/−,−/− mice; however, these two traits are only partially penetrant in the double-heterozygous +/−,+/− mice compared with the homozygous −/−,−/− mice. In contrast to the group 3 traits, increased center-field occupancy (group 4) is fully penetrant, both in +/−,+/− and in −/−,−/− mice.
Table 1.
Phenotypic traits of Kv3.1/Kv3.3-deficient mice
| Genotype | Kv3.1+/+Kv3.3+/+ | Kv3.1+/−Kv3.3+/− | Kv3.1−/−Kv3.3−/− |
|---|---|---|---|
| Active avoidance | Normal | Normal | Normal |
| Ataxia (rotarod) | None | None | Severe1-a |
| Tremor | None | None | Present |
| Myoclonus | None | None | Every few seconds |
| Ethanol sensitivity | Normal | Normal | Hypersensitive |
| Body weight | 100% | 88%1-b | 50% |
| Spont. locomotion1-c | 100% | 217% | 273% |
| Ctr-field occupancy1-d | 100% | 486% | 439% |
Graded penetrance of phenotypic traits is shown. Depending on the number of mutant alleles (0, 2, or 4), phenotypic traits show different degrees of penetrance and may be classified in four distinct groups.
Full penetrance is shown in bold letters.
Altered phenotypic traits are italicized.
Spont., spontaneous.
Ctr, Center.
DISCUSSION
Many Kv3.1-expressing cells in the hippocampus, neocortex, and striatum contain the calcium-binding protein parvalbumin, a marker for fast-spiking GABAergic interneurons. Some of these neurons fire short-duration APs at frequencies of up to 600 Hz (Kawaguchi et al., 1987; Brew and Forsythe, 1995; Wang et al., 1998). Neurons with fast-spiking activity appear in the cortex during the second postnatal week (Du et al., 1996; Massengill et al., 1997), approximately at a time when Kv3.1 and Kv3.3 mRNA and protein levels begin to increase (Drewe et al., 1992; Perney et al., 1992; Goldman-Wohl et al., 1994;Ho, 1996). It appears that the expression levels of Kv3.1 and Kv3.3 mRNAs are developmentally regulated and determine the firing type of a neuron by promoting rapid AP repolarization and fAHP, leading to the brief refractory periods necessary for sustained high-frequency firing.
The unique biophysical properties of Kv3.1 channels seem indeed to be responsible for fast-spiking neurons. It has been shown that a voltage-gated K+ channel with the kinetic and pharmacological properties resembling those of Kv3.1 participates in postsynaptic signal integration in the medial nucleus of the trapezoid body (MNTB), a brainstem relay nucleus involved in sound-source localization (Brew and Forsythe, 1995). MNTB neurons express Kv3.1 mRNA, and blockade of Kv3.1 channels with tetraethylammonium (TEA) prevents MNTB neurons from faithfully following and firing APs at stimulation frequencies above ∼200 Hz (Wang et al., 1998). In agreement with this finding, in the Kv3.1 single mutant, K+ currents are dramatically reduced in Kv3.1−/− MNTB neurons compared with those of Kv3.1+/+mice, and Kv3.1-deficient MNTB neurons can no longer follow stimulating inputs >200 Hz (Macica et al., 2000).
Kv3.1 and Kv3.3 K+ channels show functional redundancy
The wide coexpression of Kv3.1 and Kv3.3 K+ channels in many of the same neurons in the CNS may explain the lack of strong phenotypes in either single mutant because of the functional redundancy of the Kcnc1 andKcnc3 genes. To investigate this possibility, Kv3.1/Kv3.3 double mutants were generated. Double-homozygous (−/−,−/−) mutant mice were born at the expected Mendelian frequency, indicating that neither Kv3.1 nor Kv3.3 channels are required for embryonic development. Kv3.1/Kv3.3-deficient mice are ataxic and die at ∼3 weeks of age, probably because they cannot compete with their littermates for food. Most −/−,−/− mice survive, however, when they are protected from their littermates and given daily access to soft food. The surviving adult −/−,−/− mice remain significantly smaller than +/+,+/+ or +/−,+/− mice. Homozygous Kv3.1/Kv3.3-deficient mice display severe ataxia, tremulous movements, spontaneous myoclonus, and hypersensitivity to ethanol. They show poor balance when moving, whereas at rest they exhibit whole-body jerks every few seconds. In spite of this severe motor impairment, Kv3.1/Kv3.3-deficient mice are hyperactive and show increased exploratory activity and no obvious learning or memory deficit (Table1).
The phenotype of the Kv3.1/Kv3.3 double mutant is pleiotropic. It is possible that some of the distinct phenotypic traits that emerge in the absence of two voltage-gated K+ channels involved in AP repolarization and fAHP are unrelated and are caused by distinct local dysfunction in different brain regions. This idea is supported by the finding that some phenotypic traits are only visible in double-homozygous −/−,−/− mice (group 2 traits are ataxia, tremor, myoclonus, and ethanol sensitivity), whereas other traits are present with intermediate penetrance in double-heterozygous +/−,+/− mice (group 3 traits are reduced body weight and increased spontaneous locomotor activity). Increased exploratory activity (group 4 trait is increased center-field occupancy) is even fully penetrant in +/−,+/− mice (Table 1). We hypothesize that different phenotypic traits, or groups of traits, are caused by regional dysfunction of distinct neuronal subpopulations in different parts of the brain. Targeted rescue of K+ channel function in areas where Kv3.1 and Kv3.3 channels are normally expressed in wild-type mice may help to determine which brain regions are causally linked to a particular mutant phenotype.
Severe motor impairment in Kv3.1/Kv3.3-deficient mice
Of the phenotypic traits described in Kv3.1/Kv3.3-deficient mice, the motor-related dysfunctions of ataxia, myoclonus, and tremor have frequently been associated with cerebellar pathology, and ethanol sensitivity may also be related to perturbed cerebellar function. Interestingly, these putative cerebellar traits cluster in group 2 (Table 1), and they are penetrant only in double-homozygous but not in double-heterozygous mice.
Ataxia is commonly attributed to altered output from the cerebellar cortex or from deep cerebellar nuclei (DCN) (Klockgether and Evert, 1998). Many mouse mutants with neurological deficits like ataxia show degeneration of the cerebellum, either of granule cells or Purkinje cells, or both. Although Kv3.1/Kv3.3-deficient mice are severely ataxic, we did not find any obvious neuroanatomical changes in the mutant brain. Particularly, the cerebella of Kv3.1/Kv3.3-deficient mice show normal foliation and unaltered cytoarchitecture (Fig. 3). We consider it likely that the dramatic motor impairment and hypersensitivity to ethanol are caused by the altered physiological properties of neurons and result from increased neurotransmitter release (presumably GABA) in the absence of Kv3.1 and Kv3.3. Interestingly, ethanol is known to impart some of its behavioral effects by enhancing GABAA receptor-mediated inhibition in neuronal circuits in the cerebellum (Harris, 1999; Mehta and Ticku, 1999).
If group 2 traits are of cerebellar origin, this raises the question of how the lack of Kv3.1 and Kv3.3 K+channels may result in cerebellar dysfunction. Kv3.1 channels are expressed in the somata of granule cells and along the entire length of their axons, the parallel fibers, including presynaptic boutons but with low or no expression in dendrites and glomeruli (Sekirnjak et al., 1997). There is little or no Kv3.1 expression in Purkinje, basket, stellate, and Golgi cells or in the incoming mossy fibers and climbing fibers (Sekirnjak et al., 1997). The absence of Kv3.1 from parallel fibers may lead to prolonged APs with concomitantly enhanced glutamate release resulting in increased excitation of Purkinje and Golgi cells. Indeed, in cerebellar slices, application of TEA at a concentration that preferentially blocks Kv3.1 channels resulted in parallel fiber AP broadening and an increase in EPSCs at granule→Purkinje cell synapses (Sabatini and Regehr, 1997). Increased Golgi cell activity could lead to enhanced feedback inhibition of granule cells. Interestingly, it has been shown that ataxia results when the feedback inhibition via Golgi cells is altered (Watanabe et al., 1998). In addition, the absence of Kv3.3 channels from Purkinje cells may lead to the broadening of their APs and to increased GABA release on DCN neurons. Perhaps it is the synergistic effect of the lack of Kv3.1 channels from parallel fibers leading to enhanced excitation of Purkinje and Golgi cells and the absence of Kv3.3 channels from Purkinje cells leading to increased GABA release on DCN neurons that results in cerebellar dysfunction.
The phenotypic traits of decreased body weight, increased spontaneous locomotor activity, and center-field occupancy are already penetrant in double-heterozygous +/−,+/− mice (Table 1). There are many reasons why body weight may be lower in the Kv3.1/Kv3.3-deficient mutant mouse, particularly given the dramatic ataxia that keeps infant mutant mice from feeding properly (Fig. 2). The phenotypic traits of enhanced locomotor and center-field activity may reflect a general increase in GABAergic tone because of increased neurotransmitter release in the absence of Kv3.1 and Kv3.3 K+ channels from fast-spiking interneurons.
Here, we have only studied the Kv3.1/Kv3.3 double mutant (four null alleles) and its corresponding controls, the double-heterozygous (two null alleles) and the wild-type (zero null allele) mouse. Animals of these three genotypes were exclusively derived from heterozygous breeding pairs; therefore, the variability of the genetic background was the same (mixed background of 129/Sv and C57BL/6). Because of the graded penetrance of some of the phenotypic traits, it will be interesting to study mice that carry three of the possible four null alleles.
Footnotes
This work was supported by the Howard Hughes Medical Institute and by National Institutes of Health–United States Public Health Service Grant NS30532 (N.H.).
Correspondence should be addressed to Dr. Rolf Joho, Center for Basic Neuroscience, The University of Texas Southwestern Medical Center, Dallas, TX 75390-9111. E-mail: Rolf.Joho@UTSouthwestern.edu.
REFERENCES
- 1.Brew HM, Forsythe ID. Two voltage-dependent K+ conductances with complementary functions in postsynaptic integration at a central auditory synapse. J Neurosci. 1995;15:8011–8022. doi: 10.1523/JNEUROSCI.15-12-08011.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan E. PhD thesis. The Rockefeller University; 1997. Regulation and function of Kv3.3. [Google Scholar]
- 3.Drewe JA, Verma S, Frech G, Joho RH. Distinct spatial and temporal expression patterns of K+ channel mRNAs from different subfamilies. J Neurosci. 1992;12:538–548. doi: 10.1523/JNEUROSCI.12-02-00538.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du J, Zhang L, Weiser M, Rudy B, McBain CJ. Developmental expression and functional characterization of the potassium-channel subunit Kv3.1b in parvalbumin-containing interneurons of the rat hippocampus. J Neurosci. 1996;16:506–518. doi: 10.1523/JNEUROSCI.16-02-00506.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Espinosa F, Ho CS, McMahon A, Chan E, Heintz N, Burns D, Joho RH. Kv3.1/3.3 double-mutant mice display ataxia and myoclonus. Soc Neurosci Abstr. 2000;26:898. [Google Scholar]
- 6.Gershenfeld HK, Paul SM. Mapping quantitative trait loci for fear-like behaviors in mice. Genomics. 1997;46:1–8. doi: 10.1006/geno.1997.5002. [DOI] [PubMed] [Google Scholar]
- 7.Goldman-Wohl DS, Chan E, Baird D, Heintz N. Kv3.3b: a novel Shaw type potassium channel expressed in terminally differentiated cerebellar Purkinje cells and deep cerebellar nuclei. J Neurosci. 1994;14:511–522. doi: 10.1523/JNEUROSCI.14-02-00511.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz J, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- 9.Harris RA. Ethanol actions on multiple ion channels: which are important? Alcohol Clin Exp Res. 1999;23:1563–1570. [PubMed] [Google Scholar]
- 10.Ho CS. PhD thesis. The University of Texas Southwestern Medical Center; 1996. Generation and characterization of a mutant mouse carrying the disrupted gene for the voltage-gated potassium channel Kv3.1. [Google Scholar]
- 11.Ho CS, Grange RW, Joho RH. Pleiotropic effects of a disrupted K+ channel: reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci USA. 1997;94:1533–1538. doi: 10.1073/pnas.94.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joho RH, Ho CS, Armitage R, Marks G. Physiological and behavioral alterations in a mutant mouse deficient for the voltage-gated K+ channel Kv3.1. Pflügers Arch. 1997;434:R90–R91. [Google Scholar]
- 13.Joho RH, Ho CS, Marks GA. Increased γ- and decreased δ-oscillations in a mouse deficient for a potassium channel expressed in fast-spiking interneurons. J Neurophysiol. 1999;82:1855–1864. doi: 10.1152/jn.1999.82.4.1855. [DOI] [PubMed] [Google Scholar]
- 14.Kalman K, Nguyen A, Tseng-Crank J, Dukes ID, Chandy G, Hustad CM, Copeland NG, Jenkins NA, Mohrenweiser H, Brandriff B, Cahalan M, Gutman GA, Chandy KG. Genomic organization, chromosomal localization, tissue distribution, and biophysical characterization of a novel mammalian Shaker-related voltage-gated potassium channel, Kv1.7. J Biol Chem. 1998;273:5851–5857. doi: 10.1074/jbc.273.10.5851. [DOI] [PubMed] [Google Scholar]
- 15.Kanemasa T, Gan L, Perney TM, Wang LY, Kaczmarek LK. Electrophysiological and pharmacological characterization of a mammalian Shaw channel expressed in NIH 3T3 fibroblasts. J Neurophysiol. 1995;74:207–217. doi: 10.1152/jn.1995.74.1.207. [DOI] [PubMed] [Google Scholar]
- 16.Kanthasamy AG, Nguyen BQ, Truong DD. Animal model of posthypoxic myoclonus. II. Neurochemical, pathologic, and pharmacologic characterization. Mov Disord. 2000;15:31–38. doi: 10.1002/mds.870150707. [DOI] [PubMed] [Google Scholar]
- 17.Kawaguchi Y, Katsamuru H, Kosak T, Heizmann CW, Hama K. Fast spiking cells in the rat hippocampus (CA1 region) contain the calcium-binding protein parvalbumin. Brain Res. 1987;416:369–374. doi: 10.1016/0006-8993(87)90921-8. [DOI] [PubMed] [Google Scholar]
- 18.Klockgether T, Evert B. Genes involved in hereditary ataxias. Trends Neurosci. 1998;21:413–418. doi: 10.1016/s0166-2236(98)01315-0. [DOI] [PubMed] [Google Scholar]
- 19.Lenz S, Perney TM, Qin Y, Robbins E, Chesselet MF. GABA-ergic interneurons of the striatum express the Shaw-like potassium channel Kv3.1. Synapse. 1994;18:55–66. doi: 10.1002/syn.890180108. [DOI] [PubMed] [Google Scholar]
- 20.Macica CM, Wang LY, Joho RH, Ho CS, Kaczmarek LK. Knockout of the Kv3.1 gene impairs high frequency firing in auditory neurons. Soc Neurosci Abstr. 2000;26:1705. [Google Scholar]
- 21.Martina M, Schultz JH, Ehmke H, Monyer H, Jonas P. Functional and molecular differences between voltage-gated K+ channels of fast-spiking interneurons and pyramidal neurons of rat hippocampus. J Neurosci. 1998;18:8111–8125. doi: 10.1523/JNEUROSCI.18-20-08111.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massengill JL, Smith MA, Son DI, O'Dowd DK. Differential expression of K4-AP currents and Kv3.1 potassium channel transcript in cortical neurons that develop distinct firing phenotypes. J Neurosci. 1997;17:3136–3147. doi: 10.1523/JNEUROSCI.17-09-03136.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehta AK, Ticku MK. An update on GABAA receptors. Brain Res Rev. 1999;29:196–217. doi: 10.1016/s0165-0173(98)00052-6. [DOI] [PubMed] [Google Scholar]
- 24.Perney T, Marshall J, Martin KA, Hockfield S, Kaczmarek L. Expression of the mRNA for the Kv3.1 potassium channel gene in the adult and developing rat brain. J Neurophysiol. 1992;68:756–766. doi: 10.1152/jn.1992.68.3.756. [DOI] [PubMed] [Google Scholar]
- 25.Perney TM, Kaczmarek LK. Localization of a high threshold potassium channel in the rat cochlear nucleus. J Comp Neurol. 1997;386:178–202. [PubMed] [Google Scholar]
- 26.Rettig J, Wunder F, Stocker M, Lichtinghagen R, Mastiaux F, Beckh S, Kues W, Pedarzani P, Schröter KH, Ruppersberg JP, Veh R, Pongs O. Characterization of a Shaw-related potassium channel family in rat brain. EMBO J. 1992;11:2473–2486. doi: 10.1002/j.1460-2075.1992.tb05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudy B, Kentros C, Weiser M, Fruhling D, Serodio P, Vega-Saenz de Miera E, Ellisman MH, Pollock JA, Baker H. Region-specific expression of a K+ channel gene in brain. Proc Natl Acad Sci USA. 1992;89:4603–4607. doi: 10.1073/pnas.89.10.4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, Moreno H, Nadal MS, Hernández-Pineda R, Hernández-Cruz A, Erisir A, Leonard C, Vega-Saenz de Miera E. Contributions of Kv3 channels to neuronal excitability. Ann NY Acad Sci. 1999;868:304–343. doi: 10.1111/j.1749-6632.1999.tb11295.x. [DOI] [PubMed] [Google Scholar]
- 29.Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sánchez JA, Ho CS, Vaughan DM, Garcia MC, Grange RW, Joho RH. Muscle and motor-skill dysfunction in a K+ channel-deficient mouse are not due to altered muscle excitability or fiber type but depend on the genetic background. Pflügers Arch. 2000;440:34–41. doi: 10.1007/s004240000248. [DOI] [PubMed] [Google Scholar]
- 31.Sekirnjak C, Martone ME, Weiser M, Deerinck T, Bueno E, Rudy B, Ellisman M. Subcellular localization of K+ channel subunit Kv3.1b in selected rat CNS neurons. Brain Res. 1997;766:173–187. doi: 10.1016/s0006-8993(97)00527-1. [DOI] [PubMed] [Google Scholar]
- 32.Wang LY, Gan L, Forsythe ID, Kaczmarek LK. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. J Physiol (Lond) 1998;509:183–194. doi: 10.1111/j.1469-7793.1998.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe D, Inokawa H, Hashimoto K, Suzuki N, Kano M, Shigemoto R, Hirano T, Toyama K, Kaneko S, Yokoi M, Moriyoshi K, Suzuki M, Kobayashi K, Nagatsu T, Kreitman R, Pastan I, Nakanishi S. Ablation of cerebellar Golgi cells disrupts synaptic integration involving GABA inhibition and NMDA receptor activation in motor coordination. Cell. 1998;95:17–27. doi: 10.1016/s0092-8674(00)81779-1. [DOI] [PubMed] [Google Scholar]
- 34.Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiser M, Bueno E, Sekirnjak C, Martone ME, Baker H, Hillman D, Chen S, Thornhill W, Rudy B. The potassium channel subunit Kv3.1b is localized to somatic and axonal membranes of specific populations of CNS neurons. J Neurosci. 1995;15:4298–4314. doi: 10.1523/JNEUROSCI.15-06-04298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wymore RS, Korenberg JR, Kinoshita KD, Aiyar J, Coyne C, Chen XN, Hustad CM, Copeland NG, Gutman GA, Jenkins NA, Chandy KG. Genomic organization, nucleotide sequence, biophysical properties, and localization of the voltage-gated K+ channel gene KCNA4/Kv1.4 to mouse chromosome 2/human 11p14 and mapping of KCNC1/Kv3.1 to mouse 7/human 11p14.3-p15.2 and KCNA1/Kv1.1 to human 12p13. Genomics. 1994;20:191–202. doi: 10.1006/geno.1994.1153. [DOI] [PubMed] [Google Scholar]