

Abstract
Loss-of-function mutations in genes encoding TET DNA dioxygenase occur frequently in hematopoietic malignancy, but rarely in solid tumors, which instead commonly have reduced activity. The impact of decreased TET activity in solid tumors is not known. Here we show that TET2 mediates the IFN-γ/JAK/STAT signaling pathway to control chemokine and PD-L1 expression, lymphocyte infiltration, and cancer immunity. IFN-γ stimulated STAT1 to bind TET2 and recruit TET2 to hydroxymethylate chemokine and PD-L1 genes. Reduced TET activity was associated with decreased Th1-type chemokines and tumor-infiltrating lymphocytes and the progression of human colon cancer. Deletion of Tet2 in murine melanoma and colon tumor cells reduced chemokine expression and tumor-infiltrating lymphocytes, enabling tumors to evade antitumor immunity and to resist anti–PD-L1 therapy. Conversely, stimulating TET activity by systematic injection of its cofactor ascorbate/vitamin C increased chemokines and tumor-infiltrating lymphocytes, leading to enhanced antitumor immunity and anti–PD-L1 efficacy and extended lifespan of tumor-bearing mice. These results suggest an IFN-γ/JAK/STAT/TET signaling pathway that mediates tumor response to anti–PD-L1/PD-1 therapy and is frequently disrupted in solid tumors. Our findings also suggest TET activity as a biomarker for predicting the efficacy of and patient response to anti–PD-1/PD-L1 therapy, and stimulation of TET activity as an adjuvant immunotherapy of solid tumors.
Keywords: Cell Biology, Oncology
Keywords: Cancer immunotherapy, Chemokines, Signal transduction
Introduction
The ten-eleven translocation (TET) family of proteins, which includes TET1, TET2, and TET3 in mammalian cells, belong to the family of dioxygenases that use α-ketoglutarate (α-KG) and reduced iron (Fe2+) as cofactors to oxidize substrates (1, 2). TET DNA dioxygenases catalyze 3 sequential oxidation reactions, converting 5-methylcytosine (5mC) first to 5-hydroxylmethylcytosine (5hmC), then to 5-formylcytosine (5fC), and finally to 5-carboxylcytosine (5caC) (3–5). Replication-dependent dilution of these oxidized forms of 5mC or thymine DNA glycosylase–mediated (TDG-mediated) excision of 5fC and 5caC coupled with base excision repair results in demethylation (6, 7). Human and mouse TET1, TET2, and TET3 proteins are very closely related, sharing 52%, 57%, and 89% identity over the entire sequences and the identical catalytic mechanisms. Physiologically, TET enzymes have important functions in cell fate determination, cell differentiation, and development. Genetic studies in individual and compound Tet mutant mice have revealed the function of Tet genes in zygotic, embryonic, and perinatal development (8, 9), differentiation of hematopoietic cells (3, 10–12), and reprogramming of induced pluripotent stem cells (13, 14). Such diverse and specific roles are consistent with the binding of TET proteins and the distribution of their catalytic products 5hmC, 5fC, and 5caC throughout the genome (15–18). Several DNA sequence–specific transcription factors have been identified as recruiting TET2 to, and activating the expression of, their target genes (19–21), providing mechanistic support for how TET enzymes achieve such diverse and specific functions.
Pathologically, loss-of-function mutations in TET genes, predominantly TET2, occur frequently in hematopoietic malignancy of both myeloid and lymphoid lineages (3, 22, 23). TET2 mutation is believed to represent one of the first genetic alterations in the onset of hematopoietic malignancy and to cause aberrant hematopoietic stem cell self-renewal (24). In a subset of acute myeloid leukemia (AML) with the wild-type TET2 gene, TET2 enzyme is catalytically inactivated by d-2-hydroxyglutarate, an oncometabolite produced by mutation targeting isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2) (25, 26), which occurs in about 20% of AMLs in a mutually exclusive manner with TET2 mutations (27). This further underscores the critical importance of TET2 function in suppressing leukemogenesis. TET2 mutations have also been frequently observed in aged asymptomatic individuals and are associated with clonal hematopoiesis (28–32), which was recently linked to elevated expression of inflammation-associated cytokines and increased risk of atherosclerosis (33, 34). Collectively, these studies support a model that, via interaction with different transcription factors to regulate the expression of potentially many different genes, TET2 controls the proliferation and differentiation of hematopoietic stem or progenitor cells and suppresses hematopoietic malignancies.
Mutations of TET genes are uncommon in solid tumors. Although missense mutations in TET genes have been observed in different tumor types with relatively low frequency, the significance of these changes for the activity and function of TET enzymes is yet to be established (6). Instead, TET activity, as measured by the catalytic product of TET enzyme, 5hmC, has been found to be significantly reduced across different types of human and mouse tumors (35–41). Several mechanisms have been described for the nonmutational loss of TET activity in solid tumors, including deprivation of oxygen in hypoxic tumors (42) and downregulation of IDH2 and TET gene expression (36, 37, 39, 41, 43). The significance of loss of TET activity in solid tumors is not known. The present study is directed toward this issue.
Results
Loss of Tet2 confers tumor resistance to antitumor immunity and immunotherapy.
We explored the function of TET enzyme in solid tumors using a murine B16-OVA melanoma tumor model. We deleted Tet2 (Tet2-KO) using CRISPR/Cas9 genome-editing technology (ref. 44, Figure 1A, and Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI129317DS1). Tet2 is the most highly expressed Tet gene in B16-OVA cells, and deletion of Tet2 did not affect the expression of Tet1 and Tet3 (Supplemental Figure 1B). Tet2-KO B16-OVA cells showed similar rates of proliferation in vitro and of tumor growth in vivo in nude mice compared with the parental Tet2-WT B16-OVA cells (Figure 1, B and C), indicating that Tet2 does not play a significant intrinsic role in B16-OVA cell proliferation in vitro or tumor growth in vivo in immunodeficient mice.
Figure 1. Loss of Tet2 confers tumor resistance to immunotherapy.

(A) Western blot confirmation of Tet2 KO in B16-OVA melanoma cells is shown. (B) Tet2-KO B16-OVA cells proliferated similarly to wild-type cells in culture. Proliferation curves for Tet2-WT and -KO B16-OVA cells were determined by seeding of the same number of cells and counting every day. Error bars represent cell numbers ± SD for triplicate experiments. (C) Tet2-KO B16-OVA cell–derived tumors grow similarly to wild-type cells in nude mice. A quantity of 2 × 105 WT or Tet2-KO B16-OVA cells were injected s.c. into nude mice, and tumor volume and weight were determined and analyzed. Data represent mean ± SEM for 8 tumors. (D) Kaplan-Meier survival curves for mice injected with WT or Tet2-KO B16-OVA cells and treated with adoptive T cell immunotherapy are shown. A quantity of 2 × 105 WT or Tet2-KO B16-OVA cells were injected s.c. into C57BL/6 mice at day 0 and 5 × 106 OT-I cells injected i.v. at day 15. Kaplan-Meier survival curves for these mice are shown (n = 10 mice for groups without OT-I injection and n = 12 mice for groups with OT-I injection). (E) Kaplan-Meier survival curves for mice injected with WT or Tet2-KO B16-OVA cells and treated with anti–PD-L1 therapy are shown (n = 10 mice for each group). A quantity of 2 × 105 WT or Tet2-KO B16-OVA cells were injected s.c., and anti–PD-L1 antibody was injected i.p., into C57BL/6 mice at the indicated time points. The survival curve of mice injected with only WT or Tet2-KO B16-OVA cells without treatment in D is also shown by the dashed gray or pink line for reference. The P values of D and E are shown in the tables at right, determined using log-rank (Mantel-Cox) test comparing each 2 groups; ***P < 0.001.
Next, we carried out 2 experiments to investigate the role of Tet2 in adoptive antitumor immunity and in response to anti–PD-L1 immunotherapy, respectively, in syngeneic immunocompetent C57BL/6 mice. First, we subcutaneously transplanted equal numbers of Tet2-WT or Tet2-KO B16-OVA cells, and 2 weeks later intravenously injected with MHC class I–restricted, ovalbumin-specific CD8+ T (OT-I) cells that recognize the ovalbumin (OVA) antigen expressed by B16-OVA cells (45). Mice in which either Tet2-WT or Tet2-KO B16-OVA cells were transplanted died with comparable mean lifespan of 19.0 and 18.5 days, respectively, in the absence of OT-I cells (Figure 1D and Supplemental Figure 1C). Injection of OT-I cells slowed down the progression of Tet2-WT B16-OVA melanoma and extended the mean lifespan of the tumor-bearing mice to 25.5 days. Notably, OT-I cell transfusion did not affect the growth of Tet2-KO tumors or mouse survival (Figure 1D), indicating that TET2 plays an important role in tumor cells in response to T cell immunity.
Next, to determine the role of TET2 in the response to immunotherapy, we intraperitoneally injected with anti–PD-L1 antibody following subcutaneous transplant of Tet2-WT and Tet2-KO B16-OVA cells. While anti–PD-L1 extended the mean lifespan of the mice bearing Tet2-WT melanoma to 23 days compared with mice injected with PBS, it had no effect on mice bearing Tet2-KO tumors (Figure 1E). These results indicate that Tet2 loss also impaired the efficacy of anti–PD-L1 immunotherapy. To provide additional evidence supporting this, we deleted Tet2 in another syngeneic mouse tumor cell line, MC38 cells derived from C57BL/6 murine colon adenocarcinoma (Supplemental Figure 2, A and B). We confirmed that TET2 deletion did not affect the expression of either Tet1 or Tet3 (Supplemental Figure 2C). Like in B16-OVA cells, deletion of Tet2 in MC38 cells did not appreciably affect cell proliferation in vitro (Supplemental Figure 2D), but almost completely abolished the response to anti–PD-L1 treatment in vivo (Supplemental Figure 2E). Anti–PD-L1 treatment extended the lifespan of C57BL/6 mice bearing Tet2-WT MC38 tumors from average 24.5 days to beyond 45 days of experimental duration but had essentially no effect on C57BL/6 mice bearing Tet2-KO MC38 tumors (average lifespan: 21 days). Hence, loss of Tet2 confers both B16-OVA melanoma and MC38 colon tumors resistance to immune checkpoint blockade.
Loss of Tet2 reduces tumor-infiltrating lymphocytes.
Compared with Tet2-KO B16-OVA tumors, there were significantly more CD3+ and CD8+ T cells in the Tet2-WT tumors in mice without injection of OT-I cells or anti–PD-L1 antibody (4.2- and 3.7-fold, respectively; Figure 2A). Injection of OT-I cells resulted in a significant increase of intratumoral CD8+ (2.6-fold, P < 0.0001) and CD3+ (1.5-fold, P = 0.0003) T cells in Tet2-WT tumors, but only a slight increase in Tet2-KO tumors, resulting in 5.5- and 4.2-fold differences in intratumoral CD8+ and CD3+ cells between Tet2-WT and Tet2-KO tumors, respectively (Figure 2, B, D, and E). Likewise, injection of anti–PD-L1 antibody also resulted in a significant increase of intratumoral CD8+ (2.5-fold, P < 0.0001) and CD3+ (1.6-fold, P = 0.0004) T cells in Tet2-WT tumors, but only a slight increase in Tet2-KO tumors, resulting in 5.1- and 5.3-fold differences in intratumoral CD8+ and CD3+ cells between Tet2-WT and Tet2-KO tumors, respectively (P < 0.0001 for both; Figure 2, C–E). These results suggest that loss of TET2 in B16-OVA cells reduced the infiltration of T cells, leading to decreased antitumor immunity and resistance to anti–PD-L1 immunotherapy.
Figure 2. Loss of Tet2 reduces tumor-infiltrating lymphocytes.

(A–C) Tumor-infiltrating T cells were reduced in Tet2-KO tumors compared with WT tumors. Paraffin sections from WT or Tet2-KO B16-OVA melanomas in Figure 1, D and E, without secondary injection (A), injected with OT-I cells (B), or with anti–PD-L1 antibody (C) were subjected to IHC analysis of CD3 and CD8. Scale bars: 200 μm. (D) Quantification of CD8+ T cells from A, B, and C. Average cell number per high-power field (HPF) is shown; 5 HPFs were calculated per group. Bonferroni-adjusted **P < 0.01, ***P < 0.001, with raw P value derived from unpaired Student’s t test. Error bars represent ± SD. (E) Quantification of CD3+ T cells from A, B, and C. Average cell number per HPF is shown; 5 HPFs were calculated per group. Bonferroni-adjusted **P < 0.01, ***P < 0.001, with raw P value derived from unpaired Student’s t test. Error bars represent ± SD.
Loss of TET2 impairs IFN-γ–induced chemokine and PD-L1 expression.
The above results led us to determine the expression of PD-L1 (CD274/B7H1) and the interferon-γ–induced (IFN-γ–induced) chemokines CXCL9, CXCL10, and CXCL11, which are often referred to as T helper 1 type (Th1 type) and are recognized by CXC chemokine receptor 3 (CXCR3), expressed in several types of antitumor effector T cells, including cytotoxic CD8+ T cells, IFN-γ–expressing Th1 cells, natural killer (NK) cells, and NKT cells (46). We found that the expression of Pdl1, Cxcl9, and Cxcl10 was significantly decreased in Tet2-KO B16-OVA tumors compared with that in Tet2-WT tumors (Figure 3A). The expression of PD-L1 and these chemokine genes has been reported to be stimulated by IFN-γ (47–50). To test whether TET2 mediates IFN-γ–stimulated expression of these 4 genes, we treated B16-OVA and MC38 as well as THP-1 human monocytic cells, commonly used to study the response to IFN signaling, with IFN-γ and performed quantitative reverse transcriptase PCR (RT-qPCR) analyses. We confirmed that deletion of TET2 did not affect the expression of TET1 and TET3 in THP-1 cells (Supplemental Figure 3B). Then we found that IFN-γ indeed potently induced the expression of PD-L1, CXCL9, CXCL10, and CXCL11 genes and deletion of TET2 significantly reduced the IFN-γ induction of these genes in B16-OVA cells (Figure 3B), MC38 cells (Supplemental Figure 3A), and THP-1 cells (Figure 3C). Next, we found that Tet2 deletion reduced protein levels of CXCL9 and CXCL10 (Figure 3D) and PD-L1 (Supplemental Figure 3C) in response to IFN-γ treatment. To further confirm the effect of TET2 deletion on the expression of these 4 genes and exclude off-target effects associated with the CRISPR/Cas9 system, we performed a rescue experiment in TET2-KO THP-1 cells (Supplemental Figure 3D). We found that ectopic expression of wild-type, but not AML-derived, catalytic inactivating mutant (R1896S), TET2 largely restored the induction of CXCL10 and PD-L1 (Figure 3E) as well as CXCL9 and CXCL11 (Supplemental Figure 3E) genes by IFN-γ. These results identify TET2 as an important mediator in the induction of these 3 chemokine and PD-L1 genes by IFN-γ via a catalytic-dependent mechanism.
Figure 3. Loss of TET2 impairs Th1-type chemokines and PD-L1 expression.

(A) Deletion of Tet2 in allograft tumors reduced chemokine Cxcl9, Cxcl10, and Pdl1 expression. Total mRNA was extracted from WT or Tet2-KO tumor (n = 10 for each group), and mRNA levels of genes were determined by qPCR. Data represent mean ± SD. *P < 0.05, **P < 0.01 by unpaired Student’s t test. (B and C) Knocking out Tet2 blocked IFN-γ–induced chemokines and Pdl1 gene expression in B16-OVA (B) and THP-1 (C) cells. WT or Tet2-KO cells were treated with IFN-γ for 20 hours, and total RNA was extracted. The relative mRNA levels were determined by qPCR. Error bars represent ± SD for triplicate experiments. (D) Knocking out Tet2 decreased IFN-γ–induced CXCL9 and CXCL10 protein levels in B16-OVA cells. WT or Tet2-KO B16-OVA cells were treated with IFN-γ for 72 hours; then medium was collected and subjected to ELISA analysis. Error bars represent ± SD for triplicate experiments. **P < 0.01, ***P < 0.001 by unpaired Student’s t test. (E) TET2 catalytic activity was required for IFN-γ–induced CXCL10 and PD-L1 expression. TET2-WT and catalytic mutant R1896S were overexpressed in TET2-KO THP-1 cells; then cells were treated with IFN-γ for 20 hours as indicated, and total RNA was extracted. The relative mRNA levels were determined by qPCR. Error bars represent ± SD for triplicate experiments. (F) Deletion of Tet2 impaired T cell attraction by Transwell assay. WT or Tet2-KO B16-OVA cells were treated with IFN-γ for 48 hours, and CM was collected. Triplicate independent experiments were performed for each group. Error bars represent ± SD. Bonferroni-adjusted **P < 0.01, with raw P value derived from unpaired Student’s t test.
Regulation of Th1-type chemokines by TET2 led us to assess the function of TET2 in T cell migration. A Transwell assay showed that migration of CD8+ T cells was accelerated by conditional medium (CM) derived from IFN-γ–treated Tet2-WT B16-OVA culture but was not affected by CM from IFN-γ–treated Tet2-KO B16-OVA culture (Figure 3F). The preincubation of antibody against CXCR3 with T cells blocked T cell migration to the CM from IFN-γ–treated Tet2-WT culture, and addition of recombinant murine CXCL10 to the CM from IFN-γ–treated Tet2-KO culture restored T cell migration. These results demonstrate that loss of TET2 function decreases IFN-γ–induced chemokine expression and enables tumor cells to avoid attracting local CD8+ T cells.
Loss of TET2 alters IFN-γ transcriptome.
The IFN-γ signaling pathway plays a critical role in tumor’s response to antitumor immunity and immunotherapy (51). The findings that TET2 mediates IFN-γ stimulation of PD-L1 and 3 chemokine genes led us to determine whether TET2 plays a broad role in the IFN-γ signaling. We analyzed the impact of TET2 loss on the transcriptome after IFN-γ treatment in TET2-WT and TET2-KO THP-1 cells. RNA-Seq analysis showed that deletion of TET2 significantly altered the IFN-γ transcriptome (Supplemental Figure 4A). IFN-γ stimulated 677 genes 2-fold or more in TET2-WT THP-1 cells, but only 27 genes in TET2-KO cells (Figure 4A), and 176 genes induced by IFN-γ were downregulated 5-fold or more in TET2-KO THP-1 cells compared with TET2-WT cells (P < 0.05; Figure 4B). Among IFN-γ–stimulated genes whose expression was significantly impaired by TET2 deletion are CXCL9 (7.7-fold), CXCL10 (18.9-fold), and CXCL11 (22.5-fold), as well as PD-L1 (29-fold) in TET2-KO cells as compared with TET2-WT cells (Figure 4, B and C, and Supplemental Figure 4B). These results indicate that a large fraction of IFN-γ–induced genes were suppressed, many severely, by the deletion of TET2, suggesting an important role of TET2 in mediating the IFN-γ signaling pathway. These results are also consistent with the results from targeted RT-qPCR analyses above.
Figure 4. Loss of TET2 alters IFN-γ transcriptome.

WT or TET2-KO THP-1 cells were treated with 100 ng/mL IFN-γ for 20 hours, and total RNA was extracted and subjected to RNA-Seq. (A) Venn diagram of affected genes (stimulated 2-fold or more) in the whole transcriptome is shown. (B) Global gene expression analysis of WT and TET2-KO THP-1 cells stimulated with IFN-γ by RNA-Seq is shown. The region (P < 0.001 and fold change > 5) is highlighted by dashed lines. Red dots represent CXCL9, CXCL10, and CXCL11; green dot represents PD-L1. (C) Heatmap depiction of differentially expressed genes (fold change ≥ 5) between control and TET2-KO THP-1 cells.
TET2 mediates the IFN-γ/JAK2/STAT1 signaling pathway to activate IFN-γ–induced chemokines and PD-L1 gene expression.
The signaling pathways activated downstream from IFN-γ rely primarily on the Janus family of protein tyrosine kinases (JAK) and the signal transducer and activator of transcription factors (STAT) family. Ligation of IFN-γ with its receptor (IFNGR1 and IFNGR2) activates IFNGR-associated JAK1/2, leading to Tyr701 phosphorylation of STAT1 and its translocation to the nucleus, where it binds to specific DNA sequences (51). A weak, but reproducible, TET2-STAT1 binding could be detected in unstimulated THP-1 and B16-OVA cells, and this binding was significantly enhanced by IFN-γ treatment (Figure 5A and Supplemental Figure 5A) and disrupted by a Y701F mutation in STAT1, which blocks STAT1 nuclear translocation (Figure 5B). We also found that TET2 can bind to other STAT family members (Supplemental Figure 5B), suggesting a broad function of TET2 in mediating the STAT signaling pathway. Chromatin immunoprecipitation–qPCR analysis showed that TET2 binds to the CXCL10 (Figure 5C) and PD-L1 (Figure 5D) promoters in an IFN-γ–dependent manner. 5hmC levels of the CXCL10 (Figure 5E) and PD-L1 (Figure 5F) promoters were substantially increased, 15.6-fold and 13.9-fold, respectively, in TET2-WT but not TET2-KO THP-1 cells after IFN-γ treatment. Treatment of cells with CHZ868, a specific JAK2 inhibitor (52, 53), blocked IFN-γ induction of chemokine and PD-L1 genes (Supplemental Figure 5, C and D) and TET2 binding to and upregulation of 5hmC on the CXCL10 (Figure 5, G and H) and PD-L1 (Figure 5, I and J) promoters. Taken together, these results demonstrate that TET2 is an important factor in the IFN-γ/JAK/STAT signaling pathway and is recruited by IFN-γ–activated STAT1 to catalyze 5mC hydroxylation and activate the expression of STAT1 target genes, including Th1-type chemokines and PD-L1.
Figure 5. TET2 mediates the IFN-γ/JAK2/STAT1 signaling pathway to activate Th1-type chemokine and PD-L1 gene expression.

(A) IFN-γ promoted TET2-STAT1 binding. THP-1 cells were treated with or without IFN-γ, and the interaction of TET2 and STAT1 was determined by IP–Western blot. (B) Y701F mutation disrupted TET2-STAT1 binding. STAT1-WT and Y701 mutant plasmids were transfected into THP-1 cells and treated with IFN-γ, and their binding to TET2 was determined by IP–Western blot. (C and D) IFN-γ promoted TET2 binding to the CXCL10 (C) and PD-L1 (D) promoters. THP-1 cells were treated with or without IFN-γ, and TET2 binding to the CXCL10 (C) and PD-L1 (D) promoters was determined by TET2 ChIP-qPCR. Error bars represent ± SD for triplicate experiments. (E and F) IFN-γ increased the 5hmC level of the CXCL10 (E) and PD-L1 (F) promoters. hMeDIP assays were performed in control and TET2-KO THP-1 cells treated or untreated with IFN-γ. 5hmC levels on the CXCL10 (E) and PD-L1 (F) promoters were determined by qPCR. Error bars represent ± SD for triplicate experiments. (G and H) JAK2 inhibitor blocked binding to (G) and demethylation of (H) the CXCL10 promoter by TET2 upon IFN-γ treatment. THP-1 cells were treated with IFN-γ and JAK2 inhibitor as indicated, and the ability of TET2 to bind to (G) and demethylate (H) the CXCL10 promoter was determined by TET2 (G) or 5hmC (H) ChIP-qPCR. Error bars represent ± SD for triplicate experiments. (I and J) JAK2 inhibitor blocked binding to (I) and demethylation of (J) the PD-L1 promoter by TET2 upon IFN-γ treatment. THP-1 cells were treated with IFN-γ and JAK2 inhibitor as indicated, and the ability of TET2 to bind to (I) and demethylate (J) the PD-L1 promoter was determined by TET2 (I) or 5hmC (J) ChIP-qPCR.
Loss of TET activity is associated with decreased IFN-γ–induced chemokines and infiltrating lymphocytes in human colon cancer.
TET activity, as measured by 5hmC level, is significantly reduced in different types of human solid tumors. To corroborate the findings from cultured cells and mice, we examined the levels of 5hmC, the expression of chemokine genes, and infiltrating lymphocytes by immunohistochemistry (IHC) in human colon cancer, which develops through a number of well-defined clinical stages. 5hmC was evidently decreased during colon tumorigenesis, notably during the progression from low-grade to high-grade dysplasia (Table 1 and Supplemental Figure 6A). We selected representative tumors with high or low 5hmC from different grades and performed IHC-immunofluorescence for CD3+ T cells, CD8+ cytotoxic T lymphocytes, and CD56+ NK cells. This study showed that 5hmC level exhibited significant (P < 0.01) positive correlation with the infiltration of each of 3 types of immune cells (Figure 6, A and B, and Supplemental Figure 6B). We also determined these chemokines’ expression in 5hmC-high and -low tumors as well as in normal colon epithelial (Figure 6C and Supplemental Figure 6C). We found that intratumoral CXCL9 (Figure 6D), CXCL10 (Figure 6E), and CXCL11 (Figure 6F) expression also positively correlated with 5hmC levels in colon tumors, independent of tumor stage. Close examination showed that CXCL10 expression was tightly correlated with 5hmC level in the individual cells from different areas within same tumor (Supplemental Figure 6D). These results link TET activity with the expression of IFN-γ–induced, Th1-type chemokines and infiltration of lymphocytes in human primary tumors.
Table 1. 5hmC is substantially reduced during the progression of colon adenomas.
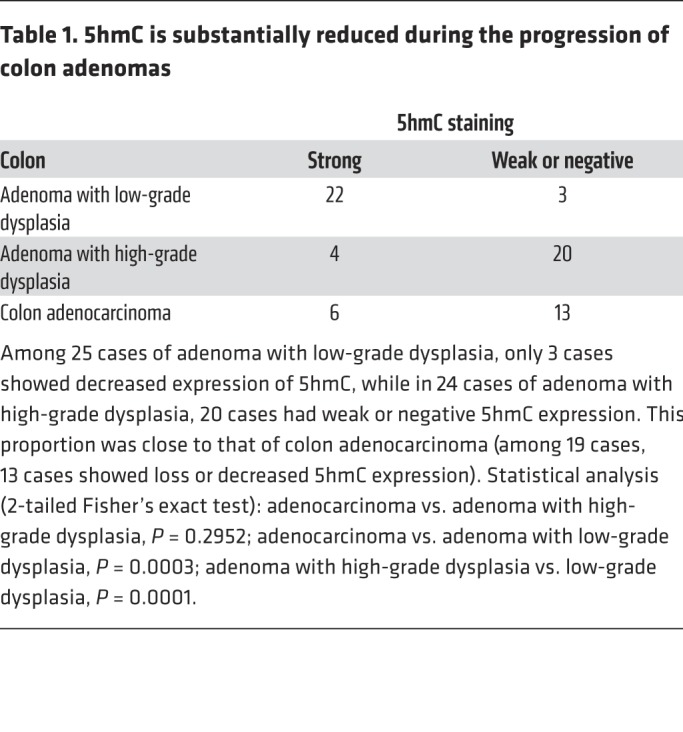
Figure 6. Loss of TET activity is associated with decreased Th1-type chemokines and infiltrating lymphocytes in human colon cancer.

(A) Infiltrating lymphocyte numbers including CD3+ T cells, CD8+ cytotoxic T lymphocytes (CTLs), and CD56+ NK cells were decreased along with the loss of 5hmC in adenoma of colon. Two representative pictures show multicolor, fluorescently labeled inflammatory cells in adenomas expressing high 5hmC levels and adenomas with decreased 5hmC expression, separately. Scale bars: 100 μm. (B) Quantification of CD3+ T cells, CD8+ CTLs, and CD56+ NK cells in colon adenomas classified by high and low 5hmC staining. Four cases of adenoma with high 5hmC expression and 8 cases of adenoma with low 5hmC expression were used to count cytotoxic T cells (CD3+ and CD8+) and NK cells (CD56+). For each case, 3 areas highly infiltrated with inflammatory cells were selected. **P < 0.01 by Student’s t test. Data represent mean ± SEM. (C) Intratumoral CXCL9, CXCL10, and CXCL11 levels were correlated with 5hmC levels in colon adenomas. Representative photographs show the expression of CXCL9, CXCL10, and CXCL11 in samples with high 5hmC and in samples with low 5hmC in the same fields, on serial sections in adenoma with low-grade dysplasia, adenoma with high-grade dysplasia, and adenocarcinoma specimens. Scale bars: 50 μm. (D–F) Quantification of CXCL9 (D), CXCL10 (E), and CXCL11 (F) expression classified by high and low 5hmC staining. Five samples representing cases with low 5hmC expression and 5 with high 5hmC expression were selected separately in adenoma with low-grade dysplasia, adenoma with high-grade dysplasia, and adenocarcinoma. For each case, 5 fields were randomly selected to calculate the integrated staining density by i-Solution image analysis software. *P < 0.05, **P < 0.01, ***P < 0.001 by unpaired Student’s t test. Data represent mean ± SEM.
Ascorbate/vitamin C stimulates TET activity to enhance tumor infiltration of lymphocytes and antitumor immunity.
TET activity, as measured by 5hmC, is significantly decreased in solid tumors of different types, but rarely mutated (35–40). Analysis of publicly available TCGA data sets of 10 different cancer types showed that there is no significant difference in the mRNA levels for combination of 3 TET genes between tumor and matched normal samples (Supplemental Figure 7A). Inactivation of TET activity through a nonmutational mechanism prompted us to explore the possibility of reactivating TET as a means to stimulate the expression of Th1-type chemokines and tumor infiltration of lymphocytes. Vitamin C (VC)/l-ascorbic acid is a cofactor for TET enzymes that promotes recycling of inactive oxidized ferric iron (Fe3+) to the actively reduced ferrous iron (Fe2+) and/or TET protein folding and has been shown to stimulate TET activity in vitro and in vivo (54–58). Addition of VC stimulated TET activity significantly in both TET2-WT THP-1 and B16 OVA cells, but only minimally in TET2-KO (Figure 7A). VC increased IFN-γ–stimulated expression of 3 Th1-type chemokines and PD-L1 genes in a TET2-dependent manner in both THP-1 (Figure 7B) and B16-OVA (Figure 7C) cells, indicating that VC is a rate-limiting factor for TET activity in these cells.
Figure 7. VC stimulates TET activity to promote Th1-type chemokine and PD-L1 expression.

(A) VC increased TET activity in cultured cells. Control and TET2-KO THP-1 cells or B16-OVA cells were treated with l-ascorbic acid (L-AA; 250 μM) as indicated for 24 hours, total genomic DNA was extracted, and 5hmC level was determined by dot blot and quantified. Error bars represent ± SD for triplicate dilutions. (B) VC increased IFN-γ–induced chemokine and PD-L1 gene expression in THP-1 cells. Control and TET2-KO THP-1 cells were treated with IFN-γ and L-AA (250 μM) as indicated, and total RNA was extracted. mRNA levels of chemokines and PD-L1 were determined by qPCR. UD, undetectable. Error bars represent ± SD for triplicate experiments. (C) VC enhanced IFN-γ–induced chemokines and Pdl1 expression in B16-OVA cells. Control and Tet2-KO B16-OVA cells were treated with IFN-γ and L-AA (250 μM) as indicated, and total RNA was extracted. mRNA levels of chemokines and Pdl1 were determined by qPCR. Error bars represent ± SD for triplicate experiments.
We then determined the effect of VC on adoptive T cell antitumor immunity in vivo. Daily i.p. injection of VC at a dose of 4 g/kg for 12 days significantly (P = 0.048) extended the mean lifespan of mice with transplanted Tet2-WT B16-OVA cells from 24 days to 30 days (group 3 [G3] vs. G5; Figure 8A). Even for mice with transplanted Tet2-KO tumor cells, VC injection still displayed a significant (P = 0.011) benefit and extended the mean lifespan from 18.5 days to 20.5 days (G4 vs. G6; Figure 8A), presumably because of either the stimulation of TET1 and/or TET3 activity, other Fe2+-dependent enzymes, or increased hydrogen peroxide–induced oxidative stress (59). Notably, VC injection conferred significant benefits to mice bearing Tet2-WT melanoma as compared with mice with Tet2-KO melanoma (G5 vs. G6, mean lifespan increased by 9.5 days; P = 0.0005), indicating that most antitumor benefits of VC are mediated by TET2. Supporting this notion, VC treatment significantly (P < 0.001) increased tumor-infiltrating CD8+ (Figure 8B) and CD3+ (Supplemental Figure 7B) cells in both Tet2-WT and Tet2-KO tumors, with a much more pronounced effect when Tet2 was expressed (Figure 8C).
Figure 8. VC stimulates TET activity to enhance tumor-infiltrating lymphocytes and efficiency of adoptive T cell therapy.

(A) VC enhanced the adoptive T cell immunotherapy. Kaplan-Meier survival curves for mice injected with WT or Tet2-KO B16-OVA cells and treated with adoptive T cell immunotherapy and VC (sodium ascorbate) as indicated are shown. A quantity of 2 × 105 WT or Tet2-KO B16-OVA cells, 5 × 106 OT-I cells, and sodium ascorbate were injected s.c., i.v., and i.p., respectively, into C57BL/6 mice at the indicated time points. Kaplan-Meier survival curves for these mice are shown (n = 10 mice for each group). The survival curve of mice injected with only WT or Tet2-KO B16-OVA cells without treatment in Figure 1D is also shown by a dashed gray or pink line for reference. The P value was determined using log-rank (Mantel-Cox) test, comparing every 2 groups, shown in the table of the figure. *P < 0.05, **P < 0.01, ***P < 0.001. (B) VC enhanced tumor-infiltrating lymphocytes with adoptive T cell immunotherapy. CD8 immunostaining of Tet2-WT and -KO tumor from A treated with adoptive T cell immunotherapy and VC is shown. Scale bars: 200 μm. (C) Quantification of CD8+ and CD3+ T cells from B and Supplemental Figure 7B. Average cell number per high-power field (HPF) is shown, with 5 HPFs calculated from each group. *P < 0.05, **P < 0.01, ***P < 0.001. The quantification of CD8+ and CD3+ T cells of mice injected with OT-I cells in Figure 2, D and E, is also shown for reference. Error bars represent ± SD.
Finally, we determined the effect of VC as an adjuvant for anti–PD-L1 immunotherapy. Daily i.p. injection of VC for 12 days, together with i.p. injection of anti–PD-L1 (200 μg) for 6 days, significantly extended survival of mice with transplanted Tet2-WT B16-OVA cells by more than 20% (G1 vs. G3, mean lifespan 23 days vs. 28 days, P = 0.043), or of mice with transplanted Tet2-KO cells by nearly 10% (G2 vs. G4, mean lifespan 19 days vs. 21 days, P = 0.049; Figure 9A). Notably, VC injection conferred significantly more benefits to mice bearing Tet2-WT melanoma than to mice with Tet2-KO melanoma (G3 vs. G4, P = 0.0008). This was accompanied by a significant increase of tumor-infiltrating CD8+ (Figure 9B) and CD3+ (Supplemental Figure 7C) cells in both Tet2-WT and Tet2-KO tumors, with a much more pronounced effect when Tet2 was expressed (Figure 9C). Taken together, these results demonstrate that VC is a rate-limiting factor for TET activity in vivo in transplanted melanoma and, when systematically injected into mice, can stimulate tumor infiltration of lymphocytes and enhance the effect of antitumor immunity and efficacy of anti–PD-L1 immunotherapy. These results provide a proof of principle that TET activity can be stimulated in vivo in solid tumors to achieve therapeutic benefit.
Figure 9. VC stimulates TET activity to enhance tumor-infiltrating lymphocytes and anti–PD-L1 immunotherapy.

(A) VC enhanced anti–PD-L1 immunotherapy. Kaplan-Meier survival curves for mice injected with WT or Tet2-KO B16-OVA cells and treated with anti–PD-L1 antibody and VC (sodium ascorbate) as indicated are shown. 2 × 105 WT or Tet2-KO B16-OVA cells, anti–PD-L1 antibody, and sodium ascorbate were injected s.c., i.p., and i.p., respectively, into C57BL/6 mice at the indicated time points. Kaplan-Meier survival curves for these mice are shown (n = 10 mice for each group). The survival curve of mice injected with WT or Tet2-KO B16-OVA cells with anti–PD-L1 treatment in Figure 1E is also shown by a dashed black or red line for reference. The P value was determined using log-rank (Mantel-Cox) test, comparing every 2 groups, and is shown in the table of the figure. *P < 0.05, ***P < 0.001. (B) VC enhanced tumor-infiltrating lymphocytes with anti–PD-L1 immunotherapy. CD8 immunostaining of Tet2-WT and -KO tumors from A treated with anti–PD-L1 antibody and VC is shown. Scale bar: 200 μm. (C) Quantification of CD8+ and CD3+ T cells from B and Supplemental Figure 7C. Average cell number per HPF is shown, with 5 HPFs calculated for each group. **P < 0.01, ***P < 0.001. Quantification of CD8+ and CD3+ T cells of mice injected with anti–PD-L1 in Figure 2, D and E, is also shown for reference. Error bars represent ± SD.
Discussion
The key finding reported here is that loss of TET function enables melanoma cells to evade antitumor immunity and resist anti–PD-L1 therapy. Our results support a model that IFN-γ stimulation, which activates JAK and results in phosphorylation and nuclear translocation of STAT1 transcription factor, leads to STAT1-TET2 association and subsequent recruitment of TET2 to and activation of STAT1 target genes. Our results suggest that potentially many IFN-γ–stimulated genes are kept in silence, in part by DNA methylation, and are activated by TET2-mediated DNA demethylation, identifying TET2 as an important mediator in the IFN-γ/JAK/STAT signaling pathway and providing a mechanistic basis for TET2-mediated antitumor immunity.
Among the genes whose expression are impaired by the deletion of TET2 are PD-L1 and 3 Th1-type chemokine genes, CXCL9, CXCL10, and CXCL11. We demonstrated that IFN-γ stimulates TET2 binding to and increased 5hmC level on the promoters of these genes in a manner that is dependent on the activity of JAK. PD-L1 expression is associated with chemokine expression and immune cell infiltration. This coexpression of PD-L1 and chemokine protects host tissue and limits inflammation and is also linked to positive response to anti–PD-1/PD-L1 immunotherapy. How this coexpression is achieved, however, is not clear. Our study suggests a plausible mechanism for coordinated expression of PD-L1 and chemokines: Both PD-L1 and chemokine genes are silenced by DNA methylation in normal noninflamed cells and activated by TET2-mediated demethylation following inflammation and IFN-γ stimulation.
This study also bears several clinical implications. First, TET2-mediated chemokine expression helps to explain why the genes on the IFN-γ/JAK/STAT pathway are the top hits in multiple experimental screens for the resistance to anti–PD-1 therapy (60–63), but only a small number of anti–PD-1–resistant human tumors (~5%) have been found to harbor loss-of-function mutations targeting genes on the pathway, mostly JAK2 and to a lesser extent JAK1 (64–66). Our findings suggest that, like JAK1/2 mutation, loss of TET activity, seen in many solid tumors of different types, would functionally inactivate the IFN-γ/IFNGR/JAK/STAT/TET/chemokine pathway and confer the resistance to anti–PD-1/PD-L1 therapy.
Second, the results presented here identify a novel biomarker for improving immunotherapy. Despite their extraordinary success, anti–PD-1 or anti–PD-L1 immunotherapy has demonstrated objective responses in only approximately 20%–30% of patients treated (67), emphasizing the critical need to identify biomarkers to select appropriate patients for treatment and to identify the cause and develop strategy for nonrespondents. The current biomarkers include tumor mutational load, the presence of IFN-γ signaling, and the colocalization of PD-1–expressing CD8+ T cells with tumor cells expressing PD-L1 in the tumor microenvironment (67). We show that 5hmC levels correlate closely with the expression of PD-L1 and chemokines in tumor cells, infiltration of lymphocytes to tumors, and response to anti–PD-1 therapy. 5hmC epitope is highly stable, can be sensitively and simply detected by IHC in tumor samples preserved by different methods, and measures accurately the activity of TET. We suggest that 5hmC merits exploration as a new biomarker for predicting the efficacy of and patient response to anti–PD-1/PD-L1 therapy.
Third, this study suggests a new strategy — stimulating TET activity — for improving immunotherapy. The anticancer effect of VC has been investigated for decades, and, despite controversy, it is reemerging as a potential anticancer agent as a result of better mechanistic understanding and improved intravenous delivery (see recent reviews, refs. 59, 68). Two mechanisms of anticancer activity of VC are proposed: hydrogen peroxide–induced oxidative stress and DNA demethylation mediated by TET enzyme activation. Our study provides novel insight into the antitumor effect of VC through stimulation of TET activity and thus TET-mediated chemokine expression and T cell infiltration. Our results demonstrate that VC is a rate-limiting factor for TET activity in vivo and, when systematically injected into mice, can stimulate tumor infiltration of lymphocytes and enhance the effect of antitumor immunity and efficacy of anti–PD-L1 immunotherapy. Restoration of TET2 activity after reversible knockdown blocks aberrant hematopoietic stem cell self-renewal and leukemia progression in mice and can be mimicked by high-dose VC treatment (54, 69). The results presented here extend the potential therapeutic benefit of high-dose VC to solid tumors. Although VC could also stimulate other α-KG/Fe(II)–dependent dioxygenases, its function in blocking self-renewal and leukemogenesis was attributed primarily to the increase of TET activity. We found that deletion of TET2 substantially reduced the activity of VC to stimulate tumor-infiltrating CD8+ and CD3+ cells and the survival of B16-OVA melanoma–bearing mice after injection with either OT-I or anti–PD-L1, suggesting that TET2 is the major target of VC in boosting the antitumor immunity and efficacy of anti–PD-L1 therapy. We suggest that high-dose VC should be considered as an adjuvant to immunotherapy, especially for solid tumors expressing low levels of 5hmC. An important insight from this finding is that TET activity can be stimulated in vivo in solid tumors to achieve therapeutic benefit in response to immunotherapy.
Methods
Cell culture and cell transfection.
THP-1 cells were acquired from the University of North Carolina (UNC) Lineberger Tissue Culture Facility (ATCC TIB-202, passage 25) and were maintained in RPMI 1640 medium containing 10% FBS, 1% penicillin/streptomycin antibiotics, 2 mM glutamine, 10 mM HEPES, and 1× nonessential amino acids (all from GIBCO). B16-OVA cells (B16F10 cells expressing ovalbumin) were cultured in DMEM (GIBCO) supplemented with 10% FBS and 1% penicillin/streptomycin. MC38 cells were purchased from Kerafast (catalog ENH204-FP) and were maintained in DMEM containing 10% FBS, 1% penicillin/streptomycin antibiotics.
Transfection of THP-1 cells was performed using Amaxa Cell Line Nucleofector Kit V according to the manufacturer’s protocol (Lonza VCA-1003). Transfected cells were cultured at 0.5 × 106 per milliliter for 24 hours before further experiments.
Cell lysis, immunoprecipitation, and immunoblotting.
Cells were washed with ice-cold PBS and lysed in NP-40 lysis buffer at 4°C for 30 minutes. Cell lysates were incubated with anti-FLAG beads (MilliporeSigma) or protein A/G-agarose (Thermo Fisher Scientific) and antibodies (indicated in the figures) for 3 hours at 4°C; the beads were washed 3 times with NP-40 buffer and then subjected to SDS-PAGE. Western blotting was performed according to standard protocol.
Antibodies.
Antibodies against FLAG (clone M2, MilliporeSigma), TET2 (Abcam, ab124297; Millipore, MABE462), tubulin (Santa Cruz Biotechnology, sc- 23948), PD-L1 (Cell Signaling Technology, 51296S), STAT1 (Santa Cruz Biotechnology, sc-345), STAT2 (Proteintech, 51075-2-AP), STAT3 (Cell Signaling Technology, 9139S), STAT4 (Abcam, ab68156), STAT5a (Abcam, ab32043), STAT5b (Abcam, ab178941), and STAT6 (Abcam, ab32520) were purchased commercially.
Gene deletion by CRISPR/Cas9 system.
TET2-knockout B16-OVA cells were generated through the CRISPR/Cas9 system by transient CRISPR strategy (44). Cells were transiently transfected with a Cas9 and single-guide RNA (sgRNA) plasmid with EGFP expression (PX458; Addgene plasmid 48138). The gRNA sequence used for targeting TET2 was GAAAGTGCCAACAGATATCC. Following transfection for 2 days, single cells were sorted by FACS based on EGFP expression into 96-well plates. Knockout clones were validated by Western blot with TET2 antibody and DNA sequencing. PCR primers used for amplifying the sgRNA-targeted sequence from genomic DNA were as follows: forward, 5′-CAGATGCTTAGGCCAATCAAG-3′; reverse, 5′-AGAAGCAACACACATGAAGATG-3′.
In vivo tumor progression and immunotherapy models.
B16-OVA cells or MC38 cells (2 × 105) were transplanted s.c. into the back flanks of 5- to 6-week-old C57BL/6 mice (The Jackson Laboratory) or nude mice. Tumor size was measured with a caliper every 2–3 days, and tumor volume was calculated by width2 × length × 0.523. Mice were sacrificed when tumors reached maximum allowed size (20 mm in diameter).
For the adoptive T cell immunotherapy model, OT-I cells were isolated from 6- to 8-week-old C57BL/6-Tg(TcraTcrb)1100Mjb/J mice (The Jackson Laboratory, stock 003831) using CD8a microbeads (Miltenyi Biotec) according to the manual. OT-I CD8+ T cells (5 × 106) were transfused i.v. into tumor-bearing mice at day 12. For the anti–PD-L1 immunotherapy model, mice were injected i.p. with 200 μg anti–PD-L1 (clone 10F.9G2, BP0101, Bio X Cell) 3 times per week for 2 weeks after tumor implantation. For VC combination treatment, mice were injected i.p. with sodium ascorbate (4g/kg) or PBS at indicated days. Mice were monitored for tumor growth every 3 days and sacrificed when tumors reached 20 mm in diameter. Statistical analysis was conducted using GraphPad Prism software. Kaplan-Meier curves and corresponding log-rank (Mantel-Cox) test were used to evaluate the statistical differences between groups in survival studies.
RNA purification, quantitative PCR, and RNA sequencing.
For quantitative PCR (qPCR), total RNA was purified from cells treated as indicated in the figure legends or mouse tumor samples using RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer’s instructions. cDNA was synthesized with 1 μg of RNA using SuperScript III First-Strand Synthesis System (Invitrogen). qPCR was performed in triplicate using cDNA and SYBR Green PCR Master Mix (Applied Biosystems) in a QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). All primers for qPCR are listed in Supplemental Table 1.
For RNA sequencing (RNA-Seq), THP-1 control and TET2-KO cells were treated with or without IFN-γ for 20 hours, and total RNA was isolated. Each condition was prepared in triplicate for each RNA-Seq experiment. Library preparation using Illumina TruSeq mRNA sample preparation kit and sequencing on an Illumina HiSeq4000 machine were performed by the High Throughput Genomic Sequencing Facility at UNC. Paired-end FASTQ sequences were aligned to the human genome (GRCH37/hg19). Differential gene expression analysis was performed with the DESeq2 package (70) using raw gene counts output from Rsubread (71), and a gene was declared as significantly differentially expressed if its adjusted P value (controlling the FDR at 0.05) was less than 0.05. The RNA-Seq data were deposited in the NCBI’s Gene Expression Omnibus database (GEO GSE132408).
TCGA database analysis.
The Cancer Genome Atlas (TCGA) level 3 gene expression (~20,000 genes) data were downloaded from the TCGA-GDC portal using RTCGAToolbox (72). Data on TET1, TET2, and TET3 mRNA expression were then extracted for tumor samples and matched normal samples for all 30 cancer types. We then kept those tumor samples with available matched normal samples, and only kept 10 cancer types that had more than 20 pairs of tumor and matched normal samples. For each of these 10 cancer types, box plots of log2 of the ratio of expression of genes (TET1, TET2, TET3, and sum of TET1/TET2/TET3) between the tumor and matched normal were provided. A 2-sided paired Student’s t test was performed, with a Benjamini-Hochberg adjustment for multiple tests.
ChIP-qPCR.
ChIP assay was performed as described previously (73). DNA was sheared by sonication using Covaris Sonicator for 12 minutes at 4°C. ChIP-enriched DNA was analyzed by qPCR with SYBR Green Master Mix. The ChIP primers are listed in Supplemental Table 1.
Transwell migration assay.
OT-I CD8+ lymphocytes were isolated from 6- to 8-week-old C57BL/6-Tg(TcraTcrb)1100Mjb/J mice by positive selection using CD8a microbeads (Miltenyi Biotec), then were stimulated with anti-CD3/CD28 Dynabeads (Thermo Fisher Scientific, catalog 11456D) at a 1:1 beads/cells ratio and 30 U/mL human IL-2 (PeproTech, catalog 200-02) for 72 hours. 5 × 105 OT-I cells in 100 μL complete media were loaded into the top chamber of Transwell inserts (5.0 μm pore size; Corning). The bottom well was filled with RPMI medium or conditioned medium (CM) derived from B16-OVA control or Tet2-KO cells, with or without 100 ng/mL recombinant murine CXCL10 (R&D Systems, catalog 466-CR-010). For blockade of CXCR3, T cells were preincubated in 10 μg/mL anti-CXCR3 (BioLegend, 126517) for 30 minutes prior to loading into the top chamber. Plates were incubated at 37°C overnight, the contents of the lower chamber were collected, and the viable OT-I CD8+ cells were counted with trypan blue.
Immunohistochemistry.
All colon adenoma and adenocarcinoma samples were acquired from the First Affiliated Hospital of Henan Medical University (Zhengzhou, China). Informed consent for research was given by patients for all samples. The procedures related to human subjects were approved by Ethics Committee at Shanghai Medical College, Fudan University (Shanghai Shi, China).
Tissue sections from colon adenoma and adenocarcinoma samples were deparaffinized twice by xylene and then hydrated. Hydrogen peroxide (0.3%) was used to eliminate endogenous peroxidase activity. For 5hmC staining, the slides were treated with 2N HCl for 15 minutes at room temperature; sections were neutralized with 100 mm Tris-HCl (pH 8.5) for 10 minutes and washed 3 times with PBS. The sections were blocked with goat serum in PBS for 30 minutes. Sections were then incubated with either anti-5hmC (Active Motif; 1:1000 dilution), anti-CXCL10 (abCam Cambridge, UK; 1:100 dilution), or anti-CXCL11 antibody (abCam Cambridge, UK; 1:100 dilution) overnight at 4°C. Secondary antibody was then applied and incubated at 37°C for 1 hour. Sections were developed with DAB kit and stopped with water. Cells showing either cytoplasmic or nuclear signals (brown) were counted as positive. To quantify the positive area of staining in samples, 5 fields from each sample were randomly selected and microscopically examined by 2 pathologists in a double-blind manner. Images were captured using a charge-coupled device (CCD) camera and analyzed using IMT i-Solution Images processing, measurement, and analysis software (IMT i-Solution Inc.). The integral staining density (including the information of the positive area and the staining density) was obtained for further statistical analysis (74) (http://www.imt-digital.com).
Fluorescent multiplex immunofluorescence staining.
For the multiple fluorescence staining, Opal 4-Color Fluorescent IHC Kit (MEL794001kt, PerkinElmer Inc.) was used. According to the protocol, the paraffin-embedded slides were routinely dewaxed (as described above). After antigen retrieval and blocking, the first primary antibody (anti-CD8, 1:100 dilution; R&D Systems) was incubated on the slides for 1.5 hours at room temperature, followed by HRP-conjugated mouse or rabbit secondary antibodies and the Opal working solution (with fluorescence) for 10 minutes at room temperature. Slides were then subjected to antigen retrieval by microwave for 15 minutes to strip the antibodies. Subsequently, the process was repeated using the second primary antibody (anti-CD56, 1:100 dilution; R&D Systems) and the third primary antibody (anti-CD3, 1:300 dilution; R&D Systems) with different opal fluorophore (Cy5 for CD8, FITC for CD56, and Cy3 for CD3). Finally, the slides were stained using DAPI for 5 minutes at room temperature and images acquired by Mantra Quantitative Pathology Workstation (CLS140089, PerkinElmer Inc.). Inflammatory cell numbers were counted from 5 images from each sample, using a multi-spectral slice flow cytometric analysis system (Vectra Polaris Automated Quantitative Pathology Imaging System, PerkinElmer Inc.).
Statistics.
Data analysis was performed using GraphPad Prism software. Normally distributed data were analyzed using an unpaired, 2-tailed Student’s t test, and multiple comparisons were corrected using Bonferroni’s method; a log-rank (Mantel-Cox) test was used for the mouse survival assay. Statistical significance was defined as a P value of less than 0.05. Levels of significance were indicated as *P < 0.05, **P < 0.01, and ***P < 0.001.
Study approval.
All animal experiments were approved by the IACUC at UNC–Chapel Hill. For human samples, all colon adenoma and adenocarcinoma samples were acquired from the First Affiliated Hospital of Henan Medical University. Informed consent for research was given by patients for all samples. The procedures related to human subjects were approved by Ethic Committee of the Institutes of Basic Medical School, Fudan University.
Author contributions
Co-first authorship was determined by overall intellectual and experimental contributions. YPX is named first because her contributions were made over a longer period than those of LL and YL and took a leading role in collecting and organizing the figures provided by all authors. YPX initiated and performed the experimental work, including the animal experiments, RNA-Seq, biochemical and cellular studies, and validation experiments, and analyzed the data. LL found that TET2 regulates PD-L1 expression and performed ChIP experiments and additional cellular and molecular experiments. YL performed immunohistochemistry for human cancer samples and demonstrated the clinical relevance. YPX played major roles in designing the experiments, collecting and analyzing the results, organizing the figures, and writing the paper. WCL provided human colon cancer samples. MDS assisted with animal work. XMT performed RNA-Seq and TCGA bioinformatics analyses. ZL and MC helped to conduct the experiments. MB and JA synthesized the JAK2 inhibitor. YX conceived and designed the experiments and supervised the study. YPX and YX wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Jon Serody, Yisong Wan, and Dan Ye for critical reading of the manuscript, Kun-Liang Guan for providing the B16-OVA cells, and Deborah Ann Lee and Sarah Scarry for administrative assistance. Tail vein (intravenous) injections were performed within the UNC Lineberger Animal Studies Core Facility at UNC, supported in part by a National Cancer Institute (NCI) Center Core Support Grant (CA0016086) to the UNC Lineberger Comprehensive Cancer Center. Animal histopathology services were performed by the UNC Animal Histopathology and Laboratory Medicine Core, which is supported in part by an NCI Center Core Support Grant (CA0016086) to the UNC Lineberger Comprehensive Cancer Center. This study was supported by startup funds from UNC to JA, by a Samuel Waxman Research Foundation Investigator Award, and by NIH grant CA163834 to YX.
Version 1. 07/16/2019
In-Press Preview
Version 2. 09/04/2019
Electronic publication
Version 3. 10/01/2019
Print issue publication
Footnotes
LL’s present address is: Department of Biochemistry and Molecular Biology, Shanghai Medical College, Fudan University, Shanghai, China.
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(10):4316–4331.https://doi.org/10.1172/JCI129317.
Contributor Information
Yan-ping Xu, Email: yanping@email.unc.edu.
Lei Lv, Email: lvlei@fudan.edu.cn.
Ying Liu, Email: yliu@shmu.edu.cn.
Matthew D. Smith, Email: uncmatt@email.unc.edu.
Wen-Cai Li, Email: 13523563997@126.com.
Xian-ming Tan, Email: xianming@email.unc.edu.
Meng Cheng, Email: meng93@email.unc.edu.
Zhijun Li, Email: zhijunl@email.unc.edu.
Michael Bovino, Email: mibovino@davidson.edu.
Jeffrey Aubé, Email: jaube@email.unc.edu.
Yue Xiong, Email: yxiong@email.unc.edu.
References
- 1.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14(6):341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quivoron C, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1):25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30(7):733–750. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18(9):517–534. doi: 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- 8.Gu TP, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477(7366):606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 9.Dawlaty MM, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24(3):310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ko M, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011;108(35):14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moran-Crusio K, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z, et al. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doege CA, et al. Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature. 2012;488(7413):652–655. doi: 10.1038/nature11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costa Y, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature. 2013;495(7441):370–374. doi: 10.1038/nature11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams K, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347):343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473(7347):389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song CX, et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153(3):678–691. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen L, et al. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153(3):692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell. 2015;57(4):662–673. doi: 10.1016/j.molcel.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525(7569):389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma S, et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. J Exp Med. 2017;214(5):1471–1491. doi: 10.1084/jem.20161149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delhommeau F, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 23.Tefferi A, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23(5):905–911. doi: 10.1038/leu.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowman RL, Levine RL. TET2 in normal and malignant hematopoiesis. Cold Spring Harb Perspect Med. 2017;7(8):a026518. doi: 10.1101/cshperspect.a026518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chowdhury R, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Busque L, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolli N, et al. Characterization of gene mutations and copy number changes in acute myeloid leukemia using a rapid target enrichment protocol. Haematologica. 2015;100(2):214–222. doi: 10.3324/haematol.2014.113381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuster JJ, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaiswal S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin SG, et al. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011;71(24):7360–7365. doi: 10.1158/0008-5472.CAN-11-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H, et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32(5):663–669. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kudo Y, et al. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012;103(4):670–676. doi: 10.1111/j.1349-7006.2012.02213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haffner MC, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2(8):627–637. doi: 10.18632/oncotarget.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lian CG, et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150(6):1135–1146. doi: 10.1016/j.cell.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orr BA, Haffner MC, Nelson WG, Yegnasubramanian S, Eberhart CG. Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS One. 2012;7(7):e41036. doi: 10.1371/journal.pone.0041036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu C, et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One. 2013;8(5):e62828. doi: 10.1371/journal.pone.0062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thienpont B, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537(7618):63–68. doi: 10.1038/nature19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collignon E, et al. Immunity drives TET1 regulation in cancer through NF-κB. Sci Adv. 2018;4(6):eaap7309. doi: 10.1126/sciadv.aap7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78(2):110–117. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- 46.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 48.Farber JM. Mig and IP-10: CXC chemokines that target lymphocytes. J Leukoc Biol. 1997;61(3):246–257. doi: 10.1002/jlb.61.3.246. [DOI] [PubMed] [Google Scholar]
- 49.Qian C, An H, Yu Y, Liu S, Cao X. TLR agonists induce regulatory dendritic cells to recruit Th1 cells via preferential IP-10 secretion and inhibit Th1 proliferation. Blood. 2007;109(8):3308–3315. doi: 10.1182/blood-2006-08-040337. [DOI] [PubMed] [Google Scholar]
- 50.Rani MR, Foster GR, Leung S, Leaman D, Stark GR, Ransohoff RM. Characterization of beta-R1, a gene that is selectively induced by interferon beta (IFN-beta) compared with IFN-alpha. J Biol Chem. 1996;271(37):22878–22884. doi: 10.1074/jbc.271.37.22878. [DOI] [PubMed] [Google Scholar]
- 51.Alspach E, Lussier DM, Schreiber RD. Interferon γ and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb Perspect Biol. 2019;11(3):a028480. doi: 10.1101/cshperspect.a028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu SC, et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell. 2015;28(1):29–41. doi: 10.1016/j.ccell.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer SC, et al. CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell. 2015;28(1):15–28. doi: 10.1016/j.ccell.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cimmino L, et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell. 2017;170(6):1079–1095.e20. doi: 10.1016/j.cell.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blaschke K, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500(7461):222–226. doi: 10.1038/nature12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet. 2013;45(12):1504–1509. doi: 10.1038/ng.2807. [DOI] [PubMed] [Google Scholar]
- 57.Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem. 2013;288(19):13669–13674. doi: 10.1074/jbc.C113.464800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yin R, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc. 2013;135(28):10396–10403. doi: 10.1021/ja4028346. [DOI] [PubMed] [Google Scholar]
- 59.Shenoy N, Creagan E, Witzig T, Levine M. Ascorbic acid in cancer treatment: let the phoenix fly. Cancer Cell. 2018;34(5):700–706. doi: 10.1016/j.ccell.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan D, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359(6377):770–775. doi: 10.1126/science.aao1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burr ML, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549(7670):101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mezzadra R, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549(7670):106–110. doi: 10.1038/nature23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manguso RT, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547(7664):413–418. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin DS, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sucker A, et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. 2017;8:15440. doi: 10.1038/ncomms15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klimant E, Wright H, Rubin D, Seely D, Markman M. Intravenous vitamin C in the supportive care of cancer patients: a review and rational approach. Curr Oncol. 2018;25(2):139–148. doi: 10.3747/co.25.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Agathocleous M, et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. 2017;549(7673):476–481. doi: 10.1038/nature23876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41(10):e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Samur MK. RTCGAToolbox: a new tool for exporting TCGA Firehose data. PLoS One. 2014;9(9):e106397. doi: 10.1371/journal.pone.0106397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lv L, et al. Vpr targets TET2 for degradation by CRL4VprBP E3 ligase to sustain IL-6 expression and enhance HIV-1 replication. Mol Cell. 2018;70(5):961–970.e5. doi: 10.1016/j.molcel.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shinde V, et al. Applications of pathology-assisted image analysis of immunohistochemistry-based biomarkers in oncology. Vet Pathol. 2014;51(1):292–303. doi: 10.1177/0300985813511124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.