

Abstract
The manner in which white adipose tissue (WAT) expands and remodels directly impacts the risk of developing metabolic syndrome in obesity. Preferential accumulation of visceral WAT is associated with increased risk for insulin resistance, whereas subcutaneous WAT expansion is protective. Moreover, pathologic WAT remodeling, typically characterized by adipocyte hypertrophy, chronic inflammation, and fibrosis, is associated with insulin resistance. Healthy WAT expansion, observed in the “metabolically healthy” obese, is generally associated with the presence of smaller and more numerous adipocytes, along with lower degrees of inflammation and fibrosis. Here, we highlight recent human and rodent studies that support the notion that the ability to recruit new fat cells through adipogenesis is a critical determinant of healthy adipose tissue distribution and remodeling in obesity. Furthermore, we discuss recent advances in our understanding of the identity of tissue-resident progenitor populations in WAT made possible through single-cell RNA sequencing analysis. A better understanding of adipose stem cell biology and adipogenesis may lead to novel strategies to uncouple obesity from metabolic disease.
White adipose tissue as the principal site for energy storage
Proper control of nutrient homeostasis depends on safe and efficient energy storage. In mammals, long-term energy storage is achieved through production of intracellular triglycerides, stored within specialized cells called white adipocytes. White adipocytes are capable of synthesizing triglycerides for long-term storage and liberating free fatty acids from triglycerides in times of energy demand. Beyond their role as an energy bank, adipocytes are also active endocrine cells. Adipocyte secretory products (“adipokines”), such as leptin and adiponectin, regulate organismal energy homeostasis (1–3).
White adipocytes are found throughout the body and are generally organized into anatomically distinct “depots.” Most white adipose tissue (WAT) depots are broadly characterized as either intra-abdominal or subcutaneous (4). WAT’s precise developmental origin during fetal development is still unclear; however, lineage tracing studies in mice indicate that subcutaneous and intra-abdominal depots emanate from distinct lineages (5). Human WAT expands from birth through adolescence via the expansion of cell size and number (6, 7). In adulthood, adipocytes turn over at a rate of 10% per year, and adipocyte number remains relatively stable regardless of BMI or weight loss (6).
WAT’s importance is quite clear from individuals or animal models lacking functional adipose tissue (termed lipodystrophy). Lipodystrophy results from impaired adipocyte development or inability to synthesize triglyceride (8). Congenital generalized lipodystrophy (CGL) is a rare autosomal-recessive disorder usually recognized in neonates. Inactivating mutations in AGPAT2, a key enzyme in triglyceride synthesis, are associated with the development of one type of CGL. Familial partial lipodystrophy (FPL) is a genetic disorder characterized by variable loss of body fat from the extremities and/or truncal region occurring in childhood or puberty. Acquired lipodystrophies also occur, including the loss of subcutaneous fat observed in individuals taking protease inhibitors for treatment of HIV (8, 9). During lipodystrophy, fatty acids overflow into nonadipose tissues (steatosis), including skeletal muscle, heart, pancreas, and liver. Ectopic lipid deposition is deleterious, as accumulated lipid species (e.g., ceramides and diacylglycerols) can interfere with insulin signaling and other tissue functions (termed lipotoxicity) (10–12). Hepatic steatosis, hypertriglyceridemia, and insulin resistance/diabetes all occur early in individuals with lipodystrophy (Figure 1). Leptin, owing to its ability to enhance lipid metabolism, is an effective treatment for managing metabolic abnormalities in lipodystrophic patients (13). As such, both the energy-storing capacity and endocrine functions of white adipocytes play a critical role in controlling energy homeostasis.
Figure 1. Inadequate energy storage in adipose tissue underlies insulin resistance.

Left: Lipodystrophy is characterized by an absolute or partial adipocyte deficiency, triggered by impaired adipocyte differentiation or triglyceride synthesis. This leads to ectopic lipid accumulation in other tissues, including the liver, skeletal muscle, heart, and pancreas. Toxic accumulation of lipid intermediates within these tissues leads to the development of insulin resistance. Middle: Pathologic obesity is often characterized by a relative adipocyte deficiency: limited expandability of the subcutaneous fat tissue, preferential expansion of visceral adipose tissue depots, and unhealthy adipose tissue remodeling (inflammation, fibrosis, limited adipogenesis). This is associated with ectopic lipid accumulation and insulin resistance, similar to what is observed in the lipodystrophy. Right: Metabolically healthy obesity is characterized by adequate expansion of protective subcutaneous depots, healthy adipose tissue remodeling (adipocyte hyperplasia), and limited ectopic lipid deposition.
Studies of WAT development have largely focused on understanding the cellular basis of adipocyte differentiation, termed adipogenesis. The nuclear hormone receptor PPARγ is a bona fide master regulator of adipocyte differentiation. PPARG is expressed in committed adipocyte precursor cells (APCs), or “preadipocytes,” and is further activated upon differentiation into adipocytes. PPARγ is essential for the differentiation of all adipocytes: humans with FPL and engineered mouse strains lacking functional PPARG are severely lipodystrophic and insulin resistant (14, 15). Moreover, in vitro, PPARγ can drive adipocyte differentiation from fibroblasts or cells of mesenchymal origin (16). Great progress has been made in understanding the transcriptional mechanisms controlling adipogenesis (17–19); however, the physiological mechanisms regulating adipocyte number in adulthood are less clear. In particular, the identity of APCs and the importance of adipogenesis in adulthood are still being defined. Here, we highlight recent studies suggesting that the ability to recruit new fat cells through adipogenesis is a critical determinant of both healthy adipose tissue expansion and metabolic health in the setting of overnutrition. We summarize recent single-cell sequencing efforts that have substantially advanced our understanding of adipose tissue–resident progenitor identity and the potential for APC heterogeneity. A better understanding of adipose stem cells and adipogenesis may lead to novel strategies to improve metabolic health in obesity.
Healthy versus unhealthy adipose tissue expansion in obesity
The location of adipose tissue expansion matters.
WAT has an unparalleled capacity to expand under physiological conditions. WAT expansion in response to caloric excess is a physiologically appropriate adaptation; however, the condition of obesity is associated with increased risk for diabetes and cardiovascular disease. Interestingly, many obese individuals are relatively resistant to developing features of metabolic syndrome (20, 21). This implies that factors outside of BMI are driving metabolic syndrome.
Emphasis has been placed on identifying better clinical correlates between obesity and its associated diseases. Such efforts revealed that regional WAT distribution is a strong predictor of metabolic health in obesity (22, 23). Obese individuals who preferentially expand intra-abdominal WAT (i.e., “apple-shaped obesity”) are at greater risk for metabolic syndrome than those who accumulate subcutaneous WAT (i.e., “pear-shaped obesity”) (Figure 1). One reason for the detrimental effects of visceral adipose accumulation may lie in the location of the depot itself. Metabolites and free fatty acids can drain directly from visceral WAT into the portal circulation and disrupt liver function (24). Moreover, the proximity to the gut makes the visceral fat more receptive to gut microbiota–derived products (25–27). Nevertheless, factors beyond visceral WAT mass per se impact the development of insulin resistance (28, 29). Another possibility is that subcutaneous and visceral WAT depots are intrinsically distinct “mini-organs,” and that developmental, functional, and molecular differences may underlie their differential contributions to nutrient homeostasis (30). Several studies using either rodent or human adipocytes collectively demonstrate that anatomically distinct adipocytes, in isolation, are functionally unique, differing in their ability to undergo lipolysis and lipogenesis and activate thermogenic programs (23, 31–33). Further complicating matters, adipocyte heterogeneity within individual WAT depots exists, with specific subpopulations of adipocytes exhibiting more proinflammatory phenotypes (34). Thus, intra-depot adipocyte heterogeneity may influence overall adipose tissue health and susceptibility to metabolic disease.
Understanding how anatomically distinct depots develop may lead to therapeutic strategies to alter body fat distribution or change the functional properties of depot-specific adipocytes. WAT distribution is sexually dimorphic (35, 36). Estrogens and androgens are undoubtedly key regulators of this patterning (35, 37); however, it is likely that genetic variance also plays an important role in determining body fat distribution. GWAS studies revealed a strong association between genetic variance, visceral versus subcutaneous WAT distribution, and insulin resistance (38, 39). Lotta et al. identified 53 independent loci for which genetic variance is associated with insulin resistance and lower levels of peripheral subcutaneous adiposity. Variance at these loci was also linked to familial lipodystrophy. The mechanisms by which individual SNPs influence body fat distribution remain unknown; however, several notable observations were collectively made from these types of studies. First, inability to expand subcutaneous WAT may be a more important determinant of insulin resistance in obesity than accumulation of visceral WAT per se (40). Second, many genes associated with impaired subcutaneous WAT expansion are functionally associated with adipocytes/adipogenesis (38, 39, 41). The latter is meaningful, as it suggests that genetic variance may impact adipogenesis/APCs in a region-specific manner and may underlie variance in body fat distribution and risk for metabolic disease.
The mechanism of adipose tissue expansion matters.
WAT expansion triggered by caloric excess coincides with a dramatic shift in the cellular composition of adipose tissue; this process is often referred to as tissue remodeling. The manner in which WAT depots remodel was also implicated as a critical determinant of insulin sensitivity in obesity (42–47). Individual WAT depots expand by enlarging existing adipocytes (adipocyte hypertrophy) or forming new adipocytes (adipocyte hyperplasia) (48). Analyses of WAT from obese individuals reveal that adipocyte size and number correlate well with risk for metabolic syndrome, independent of BMI (42–44). WAT from patients with metabolic syndrome often exhibits a striking pathologic phenotype: adipose depots are characterized by hypertrophic adipocytes, hypoxia, fibrosis, and accumulation of proinflammatory macrophages (42, 49–52). This phenotype also correlates well with lower serum adiponectin levels and ectopic lipid deposition into nonadipocytes (44). In contrast, WAT depots from metabolically healthy individuals contain numerous and relatively smaller adipocytes, as well as a relatively greater blood vessel density. Such studies support a “limited expandability hypothesis,” whereby the inability of WAT depots to adequately expand to meet the energy storage demands results in adipose tissue dysfunction, ectopic lipid deposition, and insulin resistance (53, 54).
Insight into the importance of “healthy” fat tissue comes from the development of a growing number of animal models. One notable model is the Col6 (collagen VI) knockout mouse (55). Collagen production by adipocytes heavily influences adipose tissue expandability. Collagen VI deficiency leads to a weakening of adipocytes’ extracellular scaffold, enabling seemingly unrestricted WAT expansion in the setting of positive energy balance. Although obese, these mice exhibit an improved inflammatory profile and maintain insulin sensitivity. More evidence comes from transgenic animals expressing Slc2a4 (GLUT4), Adipoq (adiponectin), mitoNEET, or Tnmd (tenomodulin). These engineered animals are more or equally obese compared with their control littermates, but metabolically healthy (56–59). WAT depots in these models are characterized by smaller and seemingly more numerous adipocytes, relatively low inflammation and ectopic lipid accumulation, and relatively better insulin sensitivity than those in control animals. In all of the models using the Adipoq promoter to drive transgenesis, gene expression is induced constitutively within mature adipocytes. Thus, whether the healthy local and systemic phenotypes are due to improved adipocyte function or rather due to the secondary increases in adipocyte number is not entirely clear in many models. Nevertheless, these animal models have provided compelling evidence that adipose tissue health, rather than abundance, is a critical determinant of metabolic health in obesity.
Tracking adipocyte hyperplasia in adult mice.
It has long been appreciated that de novo adipogenesis occurs in adulthood; however, genetic methods to assess new adipocyte formation have been lacking until only recently. Pulse-chase genetic lineage tracing methods have shed considerable insight into how individual adipose depots expand in mice in association with diet-induced obesity (60–64). Importantly, WAT expansion in mice with diet-induced obesity occurs in a depot-specific manner (46, 60, 62–64). Epididymal WAT, representing an intra-abdominal depot, expands in high-fat diet–fed (HFD-fed) adult male mice through both adipocyte hypertrophy and hyperplasia, while the inguinal (subcutaneous) depot in these same animals expands almost exclusively through cellular hypertrophy. Jeffery et al.’s lineage tracing studies in female mice revealed that adipocyte hyperplasia occurs in both perigonadal WAT and, to some variable extent, in inguinal WAT (61). These data support the notion of sex-based differences in regional adipogenesis. We and others have now demonstrated that visceral adipocytes emerging in association with HFD feeding originate, at least in part, from perivascular precursors expressing Pdgfrb (encoding the receptor PDGFRβ) (62, 64). We have derived a doxycycline-inducible pulse-chase lineage tracing system (PdgfrbrtTA;TRE-Cre;Rosa26RmT/mG, or MuralChaser, mice) that allows fate mapping of Pdgfrb-expressing cells in response to various physiological stimuli (62). Pdgfrb-expressing adipocyte precursors undergo adipogenesis in response to HFD feeding (gonadal and retroperitoneal depots), cold exposure (inguinal beige adipocytes), and treatment with PPAR-activating thiazolidinediones (TZDs) (inguinal and gonadal depots) (62, 65). All together, these data highlight the presence of depot- and sex-selective mechanisms for adipose tissue expansion in mice. Importantly, these genetic tools provide novel approaches to track adipogenesis and adipose tissue remodeling in vivo.
Adipogenesis supports healthy adipose tissue remodeling in obesity
Impaired adipogenesis in obesity: cause or consequence?
Adipose tissue remodeling triggered by caloric excess involves dynamic and complex changes in cellular composition of the adipose tissue. Various immune cell populations infiltrate or exit the tissue (66). The precise triggers of inflammation, fibrosis, hypoxia, and adipogenesis are unclear. Consequently, the exact cell types naturally driving healthy versus unhealthy WAT expansion remain uncertain. Selective manipulations of mature adipocytes, immune cells, endothelial cells, and mesenchymal stromal cells can all impact adipose tissue remodeling in mice with diet-induced obesity. Coordinated efforts between adipocytes and the various cell types of the adipose tissue microenvironment are likely required to maintain tissue health and systemic metabolic homeostasis.
A prevailing, albeit simplistic, model proposes that a depot’s inability to expand through adipocyte hyperplasia, combined with inadequate angiogenesis, leads to overloaded and hypoxic adipocytes (17, 51). As adipocytes reach their storage capacity, cell death occurs, leading to the activation of inflammation and fibrosis. The subsequent decline in WAT function leads to detrimental accumulation of lipid species in nonadipose organs, similar to that observed in the setting of lipodystrophy (10, 11). In this model, impaired APC differentiation underlies unhealthy adipose tissue expansion. Accordingly, several cross-sectional or overfeeding studies provided evidence of impaired preadipocyte differentiation in association with hypertrophic adipose tissue (42, 67–71). Moreover, regional differences in preadipocyte characteristics are readily apparent and include depot-specific differences in preadipocyte proliferation, apoptosis, and adipogenic capacity (68). The tremendous challenge is in determining whether such defects in adipocyte differentiation drive pathologic adipose tissue remodeling, or rather represent a consequence of fibrosis, immune cell activity, or any of the multitude of metabolic changes that occur alongside metabolic syndrome.
Promoting healthy adipose tissue expansion in obesity.
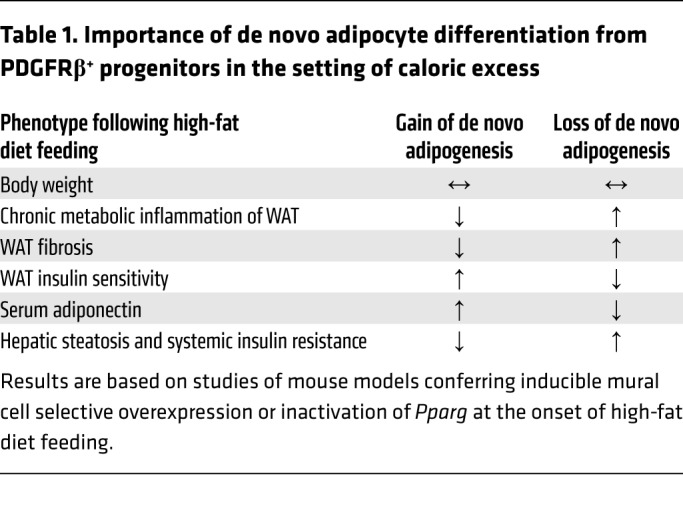
Patients treated with the TZD class of antidiabetic drugs often experience weight gain, associated with the expansion of subcutaneous WAT. Weight gain is likely undesirable to most patients; however, subcutaneous adipocyte hyperplasia and healthy remodeling of adipose tissue appear metabolically favorable and contribute to systemic insulin sensitization. New mouse models conferring inducible gene expression in adipocyte precursors, but not in adipocytes themselves, introduce the opportunity to modulate adipocyte differentiation in adult mice, and assess its importance to healthy WAT expansion in different settings. We recently developed doxycycline-inducible genetic models to ablate or induce Pparg expression in Pdgfrb-expressing precursors of adult mice undergoing HFD feeding and/or TZD treatment (65). Loss of mural cell Pparg, and thereby adipogenesis, led to many of the clinical features of pathologic WAT remodeling (inflammation, fibrosis, insulin resistance) upon HFD feeding. Moreover, TZDs’ ability to promote healthy visceral WAT remodeling (i.e., adipocyte hyperplasia and reduced inflammation) requires mural cell Pparg. Driving adipogenesis from PDGFRβ+ precursors through Pparg overexpression (Mural-PpargTG mice) led to a doubling of the de novo adipogenesis that normally occurs in visceral WAT depots in association with HFD feeding, without increasing body weight or adiposity per se. Instead, intra-abdominal WAT depots accumulated smaller and more numerous fat cells and few proinflammatory macrophages, and maintained local insulin sensitivity. Therefore, stimulating adipogenesis in the setting of caloric excess can drive healthy WAT remodeling (Table 1). Notably, obese Mural-PpargTG mice also maintain the serum adiponectin levels of lean animals. These data suggest that healthy WAT remodeling is a driving force behind the maintenance of adiponectin levels in obesity. How healthy WAT remodeling ultimately promotes systemic insulin sensitization remains unclear. Is it the sequestering of glucose and lipids from circulation? Is the reduction in chronic proinflammatory responses and fibrosis the key driver? Such mechanisms are not necessarily mutually exclusive, and the net metabolic benefit is likely mediated by multiple factors.
Table 1. Importance of de novo adipocyte differentiation from PDGFRβ+ progenitors in the setting of caloric excess.
Adipose tissue progenitor heterogeneity: insights from single-cell sequencing
Growing appreciation of the contribution of de novo adipogenesis to metabolic health has sparked renewed interest in the biology of adipose tissue stem cells. Cell selection strategies based on expression of CD34 and SCA-1 have been used routinely for several years to isolate hierarchical APC populations from stromal vascular fractions (SVFs) of inguinal and gonadal WAT of mice (72). The CD24+ subfraction of CD34+SCA-1+ cells represents a stem cell–like population, whereas the CD24– fraction represents a pool of more committed APCs. Subsequent studies demonstrated that such cells express the fibroblast/mesenchymal cell marker PDGFRα (73, 74). A selection strategy using commercially available antibodies is advantageous; cells can be isolated from any wild-type or engineered mouse strain using validated approaches. Genetic reporter lines have also been used to isolate and localize APCs (62, 75–77). The highly adipogenic subfraction of PDGFRβ+ cells can be isolated via genetic reporters on the basis of expression of Pparg or its upstream regulatory factor Zfp423 (62, 75, 77). An advantage of such genetic reporters is that cell populations of interest can be localized in vivo without dependence on finding suitable antibodies. These Pparg/Zfp423-expressing PDGFRβ+ cells express several mural cell (pericyte/smooth muscle) markers and reside directly adjacent to the endothelium in WAT blood vessels.
A number of APC populations have been described, each isolated through selection with distinct markers (78). As such, it has remained unclear what degree of heterogeneity exists among APCs. The rapid development of single-cell sequencing platforms fueled a recent burst in single-cell RNA sequencing studies of the adipose SVF (79–83). Despite differences in approaches, there seems to be congruence among data sets derived from independent laboratories. Importantly, several important themes/concepts are emerging. First and foremost, far more functional heterogeneity exists within the stromal compartment of adipose tissue than previously appreciated. Isolation of cells based on expression of PDGFRα, PDGFRβ, or CD34/SCA-1 yields molecularly and functionally heterogeneous populations. Second, subpopulations of cells once called APCs can actually be inhibitory to adipogenesis, and may directly impact other aspects of adipose tissue remodeling, e.g., fibrosis and inflammation. Third, the molecular and functional heterogeneity appears to be depot specific. As detailed below, new markers for isolating depot-specific adipose tissue–resident progenitor subpopulations are now emerging.
Stromal cell diversity within intra-abdominal WAT depots.
Recently, Granneman and colleagues explored the cellular landscape of the adipose SVF using single-cell RNA sequencing (83). In this study, Burl et al.’s analysis generated a cellular atlas that included molecularly heterogeneous stromal cell populations as well an array of immune cell types (83). Within gonadal WAT, they identified two prominent PDGFRα+ populations, termed adipose stem cell (ASC) 1 and 2. In addition, they identified two ASC subpopulations that were considered “differentiating” and “proliferating,” respectively, based on their gene expression patterns. The authors postulate that these subpopulations represent different states of adipocyte differentiation, whereby signals emanating from the microenvironment dictate their progression toward an adipogenic phenotype. Two analogous ASC populations were also observed in inguinal depots; however, when comparing ASC populations between depots, ASC1 aligned more closely to ASC2 within the same depot than to ASC1 in the opposing depot. As such, much of the molecular differences between depot-specific ASCs (e.g., gonadal ASC1 versus inguinal ASC1) reflects their anatomical origin.
Our own single-cell sequencing efforts focused specifically on the pool of genetically labeled PDGFRβ+ cells within gonadal WAT of the MuralChaser model, with the rationale that marked cells contribute considerably to adipocyte hyperplasia associated with HFD feeding in this depot. We performed single-cell RNA sequencing of Pdgfrb-expressing cells from epididymal WAT and identified molecularly distinct cell subpopulations (ref. 82 and Figure 2A). LY6C–CD9–PDGFRβ+ cells represent APCs. These cells differentiate spontaneously upon reaching confluence in culture, and are capable of forming an ectopic fat pad upon transplantation into lipodystrophic mice. These cells bear close resemblance to the ASC1 population defined by Burl et al. This subpopulation of PDGFRβ+ cells activates Pparg expression following the onset of HFD feeding, further suggesting that they are the subpopulation of PDGFRβ+ cells that contributes to adipocyte hyperplasia in this depot.
Figure 2. New strategies to isolate adipose tissue–resident progenitor subpopulations from murine gonadal adipose tissue.
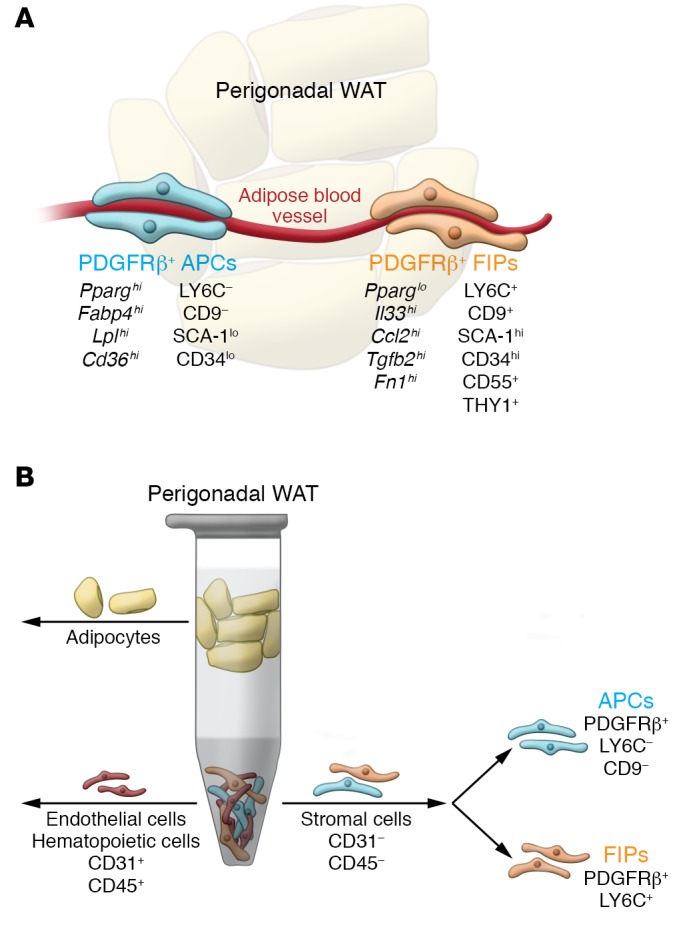
(A) Gonadal WAT depots of adult mice harbor functionally distinct PDGFRβ+ subpopulations. Molecular markers, including cell surface markers, are shown. The PDGFRβ+LY6C–CD9– fraction represents highly adipogenic adipocyte precursor cells (APCs) enriched in the expression of Pparg. These cells differentiate spontaneously upon reaching confluence in vitro when maintained in media containing insulin. The PDGFRβ+LY6C+ fraction represents fibro-inflammatory progenitors (FIPs). These cells are largely refractory to adipogenic stimuli and can actually be anti-adipogenic. (B) FACS strategy to isolate PDGFRβ+ subpopulations from perigonadal WAT of adult mice. Following the depletion of endothelial (CD31+) and hematopoietic cells (CD45+) from the stromal vascular fraction of digested adipose tissue, PDGFRβ+ cells can be subdivided into APCs and FIPs on the basis of LY6C and CD9 expression.
LY6C+PDGFRβ+ cells share a gene expression profile with Burl et al.’s ASC2 population. These cells are enriched in the expression of proinflammatory cytokines and collagens. As such, we refer to these cells as fibro-inflammatory progenitors (FIPs). FIPs express high levels of commonly used APC isolation markers, such as CD34 and SCA-1; however, they lack Pparg expression and do not readily undergo adipocyte differentiation in vitro or in vivo, even in the presence of strong adipogenic stimuli. Moreover, FIPs are also functionally anti-adipogenic and can inhibit differentiation of APCs in vitro through the production of unknown secreted factors. The presence of highly anti-adipogenic stromal cells within the total CD34+SCA-1+ population of gonadal WAT may explain the apparent lack of adipogenic capacity that these cultures possess, despite the presence of APCs (84).
HFD feeding induces collagen expression in FIPs. This suggests that FIPs are fibrogenic progenitors, in accordance with earlier studies indicating that fibrogenic cells residing in mouse and human WAT could be identified by expression of CD9 and PDGFRα (85). FIPs also exert a functional proinflammatory phenotype in response to proinflammatory signals in vitro. FIPs activate mRNA levels of proinflammatory cytokines that are known to promote macrophage infiltration and activation (82). Moreover, FIPs proliferate following the onset of HFD feeding, raising the hypothesis that these cells can modulate local adipose tissue inflammation in obesity.
In vitro studies of FIPs highlight their potential to promote maladaptive adipose tissue remodeling; however, it is certainly possible that FIPs also maintain adipose tissue health in lean animals. Recent independent studies from the Mathis and Artis groups reveal that cells bearing close resemblance to FIPs express IL-33, a key cytokine in maintaining an antiinflammatory state in WAT (79, 86). IL-33 regulates group 2 innate lymphoid cells and Tregs, which maintain eosinophils and promote an antiinflammatory phenotype in macrophages (87). Spallanzani et al. performed single-cell RNA sequencing of PDGFRα+SCA-1+ cells from gonadal WAT of adult mice (79). Functionally distinct populations, closely resembling APCs and FIPs, were identified, along with a population of mesothelial cells. IL-33 expression is largely enriched within FIPs and mesothelial cells. Importantly, deleting Il33 using Pdgfra-Cre led to a sharp reduction in whole-tissue IL-33 expression and eosinophil numbers. These studies provide evidence that specific stromal subpopulations regulate normal immune cell homeostasis in adipose tissue. Additional studies of these cells are needed to determine their precise contribution to WAT inflammation and remodeling in various settings in vivo. Genetic lineage tracing of specific subpopulations is also needed to better define their lineage relationships and phenotypic plasticity. Importantly, whether human WAT harbors cells analogous to FIPs remains unknown. Nevertheless, these studies highlight the vastly underappreciated functional heterogeneity of the adipose stromal compartment. Studies of murine gonadal WAT “adipocyte progenitors” that consist of cells sorted only on the basis of expression of either PDGFRα, PDGFRβ, or CD34/SCA-1 need to account for this functional heterogeneity.
Stromal cell diversity within subcutaneous WAT depots.
Schwalie et al. reported the first single-cell sequencing study of murine WAT (81). The authors used two independent single-cell sequencing approaches to explore the landscape of the murine inguinal WAT SVF, and characterized three stromal cell populations. Populations 1 and 2 (termed P1 and P2) are adipogenic populations that appear to differ molecularly in their level of commitment to the adipocyte lineage. P1 is enriched in expression of CD34 and SCA-1. P2 is enriched in Pparg expression and likely represents committed preadipocytes. Most notably, they defined a previously unrecognized population of cells, termed Aregs, that were refractory to adipogenic signals and instead were anti-adipogenic. These data provided the first evidence of anti-adipogenic stromal cells and highlight a potentially important brake on adipogenesis in vivo. Cells bearing the markers of Aregs were also found in murine gonadal WAT; however, these cells appear to be distinct from anti-adipogenic FIPs. Importantly, Aregs bearing these same markers are present in human WAT. The importance of these cells in controlling adipose tissue hyperplasia under physiological conditions still needs to be evaluated. Moreover, it is of interest to determine whether such cells reside within other tissues (e.g., bone marrow or skeletal muscle) to regulate local adipocyte accumulation.
More recently, Merrick et al. reported their own single-cell sequencing analysis of the inguinal WAT SVF of 12-day-old (postnatal day 12) mice (80). Their study identified three hierarchical APC populations (Figure 3A). One population, termed interstitial progenitor cells (IPCs), molecularly resembles inguinal WAT ASC2 and P1 defined by Burl et al. and Schwalie et al., respectively. Functionally, IPCs are highly proliferative and can differentiate into multiple mesenchymal lineages. The two additional populations are more committed to the adipocyte lineage. ICAM1+ cells represent committed preadipocytes and are enriched in Pparg expression. These cells molecularly resemble the adipogenic cells identified in other studies (inguinal WAT ASC1 and P2). Importantly, both IPCs and ICAM1+ cells exist in human WAT. The third population of cells in mouse inguinal WAT, termed CD142+ preadipocytes, represent a unique adipogenic population. IPCs have the ability to give rise to both the CD142+ and ICAM1+ preadipocytes, in route to becoming adipocytes, suggesting a novel progenitor hierarchy in adipose tissue. Importantly, unlike committed preadipocytes that reside in the perivascular region within WAT, the more primitive IPCs reside in an anatomically distinct region surrounding the adipose tissue, referred to as the reticular interstitium. This defines a new anatomical niche for APCs and introduces opportunities to define local signals that control their activation.
Figure 3. New strategies to isolate adipose tissue–resident progenitor subpopulations from murine inguinal adipose tissue.

(A) Merrick et al. (80) and Schwalie et al. (81) identify functionally distinct subpopulations of adipose precursors in inguinal WAT of adult mice. Molecular markers, including cell surface markers, are shown. Using the nomenclature from Merrick et al., interstitial progenitor cells (IPCs) represent multipotent cells that can give rise to more committed, highly adipogenic, ICAM1+ preadipocytes, along with a previously unrecognized CD142+ adipocyte precursor population. (B) FACS strategy to isolate functionally distinct adipogenic and anti-adipogenic subpopulations from inguinal WAT of adult mice. CD142–DPP4+ IPCs represent a primitive stem cell population. CD142–ICAM1+ cells represent committed preadipocytes. Importantly, these two populations can be identified in human subcutaneous adipose tissue using the same markers, and appear similar to the SCA-1+CD55+ and SCA-1+VAP1+ populations identified by Schwalie et al. Merrick et al. identified an additional preadipocyte population that is among the CD142+ cells (termed group 3 cells). IPCs give rise to ICAM1+ and CD142+ preadipocytes. Anti-adipogenic Aregs can be isolated on the basis of CD142 and ABCG1 expression (CD142+ABCG1+).
Going forward, it will be essential to establish the relative importance of these distinct APC subpopulations under physiological conditions. Prior studies indicate that very little de novo white adipogenesis occurs postnatally in inguinal WAT with age, or in response to HFD feeding (63). During development, inguinal adipogenesis begins during the late stages of fetal development. Most inguinal adipocytes are differentiated by postnatal day 10, with lipid accumulation being the mechanism of tissue expansion thereafter (63). Multiple studies now suggest that the properties of fetal inguinal APCs differ from those present in adulthood (88–90). Genetic lineage tracing should help establish the hierarchical relationships of these cell populations during development and their relative contribution in adulthood. Given that the inguinal WAT depot is a site of beige adipogenesis, it will be of interest to determine which, if any, of these adipogenic populations serve as the precursors of metabolically active beige adipocytes.
New depot-specific markers for isolating APC subpopulations.
Single-cell sequencing has not only shed considerable light on the cellular landscape of the adipose tissue stroma, but also has led to new strategies that can be used to isolate APC populations from different depots by FACS (Figure 2B and Figure 3B). Historically, unpurified adipose SVF cultures have served as a model system for studying adipogenesis in vitro (91). In most studies involving murine cells, inguinal WAT is the predominant source for primary cells. SVF cultures from this depot differentiate robustly and can be used to explore molecular pathways regulating adipogenesis or adipocyte function in vitro. The Merrick et al. and Schwalie et al. sorting strategies enable hierarchical subpopulations to be isolated from native inguinal WAT (Figure 3B). This may enable further dissection of the mechanisms governing preadipocyte commitment. A considerable strength of the Merrick et al. sorting strategy is its adaptability to human WAT. Nevertheless, as noted above, the physiological context in which these individual cell populations contribute to adipogenesis remains unidentified. Generally, the APC populations in inguinal WAT are not activated to undergo adipocyte differentiation in vivo following the onset of HFD feeding. Studying these APC subpopulations, along with Aregs, may shed light on this phenomenon.
It is widely appreciated in the field that primary SVF cells obtained from mouse gonadal WAT lack appreciable differentiation capacity unless used in three-dimensional culture systems (31, 92). This represents somewhat of a paradox since gonadal APCs, but not inguinal APCs, are activated to undergo adipogenesis in vivo in association with HFD feeding. The isolation of PDGFRβ+LY6C–CD9– cells from intra-abdominal WAT yields highly adipogenic cells that can be used in vitro or in transplantation assays (Figure 3B). Importantly, lineage tracing and functional analysis support the notion that these cells represent a major APC population that contributes to adipogenesis in vivo.
Importantly, the heterogeneity of the adipose progenitor pool is, at least in some cases, depot specific and perhaps age dependent. PDGFRβ+ APCs can be isolated from various intra-abdominal WAT depots of adult male and female mice on the basis of LY6C and CD9 expression; however, these markers are indiscriminate in the inguinal WAT depot (82). FIPs may exist in inguinal WAT but express a different cell surface expression profile. Inguinal IPCs are molecularly similar to gonadal WAT FIPs; however, the two populations appear functionally distinct. Unlike inguinal IPCs, FIPs are not highly adipogenic in vitro or in vivo and do not appear to give rise to APCs. These observations serve as a caution that similarities in global gene expression patterns may not necessarily imply similarities in function, and that strategies to isolate cell populations from one depot are not necessarily transferable to other depots.
Concluding remarks
Is pathologic obesity a form of relative lipodystrophy?
Studies of mouse and human adipose tissue provide strong evidence that the inability to expand subcutaneous WAT depots (versus visceral WAT depots) when faced with caloric excess is detrimental to metabolic health in the obese state. Moreover, the inability to recruit new adipocytes to support the demand for increased energy storage can lead to pathologic WAT remodeling. In both cases, the ensuing WAT dysfunction leads to systemic effects reminiscent of lipodystrophy; ectopic lipid deposition occurs and insulin resistance ensues. Much remains unclear regarding the genetic variance identified in recent GWAS; however, a link between insulin resistance and genetic variance at genes potentially involved in adipocyte function and/or adipocyte differentiation suggests that a relative or absolute adipocyte deficiency may be a substantial driver of insulin resistance. As such, it is tempting to think of pathologic obesity as a form of relative lipodystrophy.
Challenges and opportunities.
Many critical questions regarding the regulation of adipogenesis remain unresolved. The exact signals associated with HFD feeding that initiate adipogenesis have not been identified. Whether APCs respond to dietary components themselves or changes in hormone/cytokine levels remains unclear. Rydén and colleagues recently provided evidence that TGF-β locally influences precursor proliferation (93). Such signals may emanate from any number of cells within the adipose tissue. Perhaps it is equally important to identify signals that restrain adipogenesis in vivo. Surprisingly, mouse inguinal APC populations do not readily undergo adipogenesis in response to HFD feeding, despite their robust differentiation potential following removal from the tissue. Thus, the inguinal WAT microenvironment harbors strong inhibitory signals to suppress differentiation (61). Whether this is mediated by Aregs is still unclear. It is notable that transgenic PPARγ expression in PDGFRβ+ cells, combined with HFD feeding, is not sufficient to trigger adipocyte hyperplasia in the inguinal depot of male mice (65). This suggests that such inhibitory mechanisms may include those that target PPARγ at the protein level. Unraveling these inhibitory mechanisms may shed light on the molecular control of body fat distribution.
Identifying the mechanisms underlying progenitor subtype specification is also a challenge. It is not yet clear how and when the various progenitor populations described here emerge during development. The transcriptional mechanisms leading to Pparg activation in subcutaneous white APCs are at least somewhat distinct from those driving Pparg activation in visceral white APCs (90, 94, 95). Further complicating matters, molecular mechanisms controlling adipogenesis during development appear distinct from those controlling adipogenesis in adulthood (60, 96).
Efforts to unravel the functional importance of cellular heterogeneity in adipose tissue will depend highly on new genetic approaches to target cell subpopulations in vivo. To date, a preadipocyte-specific Cre line has not been developed. Inducible Cre lines under the control of Pdgfra, Pdgfrb, or Acta2 (α-SMA) have been quite useful in assaying and manipulating adipogenesis and WAT remodeling in vivo. Nevertheless, these models remain limiting. First, stromal cells bearing these markers are present in multiple tissues. Depending on the targeted gene of interest, this can produce phenotypes unrelated to WAT and confound interpretations of results. Second, these promoters will target heterogeneous cell populations; Cre lines to specifically manipulate subpopulations of adipose progenitors and adipocytes are not yet developed. Moreover, nearly all engineered strains used in the field are generated on a C57BL/6 background. Efforts to explore adipose tissue remodeling across different strains may facilitate the identification of novel regulatory mechanisms.
The heterogeneity and plasticity of the adipose tissue lineage are undoubtedly more complex than previously imagined. Opportunities to selectively target the adipose lineage may emerge from this complexity. Promoting adipogenesis in the setting of nutrient excess, particularly in subcutaneous regions, is metabolically beneficial; however, stimulating ectopic adipogenesis within bone marrow or skeletal muscle is likely deleterious. A deeper understanding of APC regulation in vivo may lead to novel therapeutic strategies to selectively regulate adipogenesis. Promoting healthy energy storage in the face of caloric excess may lead to an uncoupling of obesity from metabolic syndrome.
Acknowledgments
We apologize to our colleagues in the field for not being able to discuss all of the outstanding primary studies related to the topics discussed here. We thank members of the Touchstone Diabetes Center at the University of Texas Southwestern Medical Center for useful discussion. RKG is supported by National Institute of Diabetes and Digestive and Kidney Diseases grants R01 DK104789 and R01 DK119163.
Version 1. 10/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(10):4022–4031.https://doi.org/10.1172/JCI129191.
Contributor Information
Lavanya Vishvanath, Email: lavanya.vishvanath@utsouthwestern.edu.
Rana K. Gupta, Email: Rana.Gupta@utsouthwestern.edu.
References
- 1.Neumann E, Junker S, Schett G, Frommer K, Müller-Ladner U. Adipokines in bone disease. Nat Rev Rheumatol. 2016;12(5):296–302. doi: 10.1038/nrrheum.2016.49. [DOI] [PubMed] [Google Scholar]
- 2.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156(1–2):20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen W, et al. Adipose tissue quantification by imaging methods: a proposed classification. Obes Res. 2003;11(1):5–16. doi: 10.1038/oby.2003.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chau YY, et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat Cell Biol. 2014;16(4):367–375. doi: 10.1038/ncb2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spalding KL, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 7.Knittle JL, Timmers K, Ginsberg-Fellner F, Brown RE, Katz DP. The growth of adipose tissue in children and adolescents. Cross-sectional and longitudinal studies of adipose cell number and size. J Clin Invest. 1979;63(2):239–246. doi: 10.1172/JCI109295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussain I, Garg A. Lipodystrophy syndromes. Endocrinol Metab Clin North Am. 2016;45(4):783–797. doi: 10.1016/j.ecl.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garg A. Clinical review: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313–3325. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaurasia B, Summers SA. Ceramides — lipotoxic inducers of metabolic disorders. Trends Endocrinol Metab. 2015;26(10):538–550. doi: 10.1016/j.tem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510(7503):84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Unger RH. Longevity, lipotoxicity and leptin: the adipocyte defense against feasting and famine. Biochimie. 2005;87(1):57–64. doi: 10.1016/j.biochi.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Oral EA, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 14.Barroso I, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402(6764):880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 15.Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc Natl Acad Sci U S A. 2013;110(46):18656–18661. doi: 10.1073/pnas.1314863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-X. [DOI] [PubMed] [Google Scholar]
- 17.Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20(4):242–258. doi: 10.1038/s41580-018-0093-z. [DOI] [PubMed] [Google Scholar]
- 18.Lee JE, Schmidt H, Lai B, Ge K. Transcriptional and epigenomic regulation of adipogenesis. Mol Cell Biol. 2019;39(11):e00601-18. doi: 10.1128/MCB.00601-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mota de Sá P, Richard AJ, Hang H, Stephens JM. Transcriptional regulation of adipogenesis. Compr Physiol. 2017;7(2):635–674. doi: 10.1002/cphy.c160022. [DOI] [PubMed] [Google Scholar]
- 20.Appleton SL, et al. Diabetes and cardiovascular disease outcomes in the metabolically healthy obese phenotype: a cohort study. Diabetes Care. 2013;36(8):2388–2394. doi: 10.2337/dc12-1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denis GV, Obin MS. ‘Metabolically healthy obesity’: origins and implications. Mol Aspects Med. 2013;34(1):59–70. doi: 10.1016/j.mam.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karpe F, Pinnick KE. Biology of upper-body and lower-body adipose tissue — link to whole-body phenotypes. Nat Rev Endocrinol. 2015;11(2):90–100. doi: 10.1038/nrendo.2014.185. [DOI] [PubMed] [Google Scholar]
- 23.Lee MJ, Wu Y, Fried SK. Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Aspects Med. 2013;34(1):1–11. doi: 10.1016/j.mam.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rytka JM, Wueest S, Schoenle EJ, Konrad D. The portal theory supported by venous drainage-selective fat transplantation. Diabetes. 2011;60(1):56–63. doi: 10.2337/db10-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geurts L, Neyrinck AM, Delzenne NM, Knauf C, Cani PD. Gut microbiota controls adipose tissue expansion, gut barrier and glucose metabolism: novel insights into molecular targets and interventions using prebiotics. Benef Microbes. 2014;5(1):3–17. doi: 10.3920/BM2012.0065. [DOI] [PubMed] [Google Scholar]
- 26.Hersoug LG, Møller P, Loft S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: implications for inflammation and obesity. Obes Rev. 2016;17(4):297–312. doi: 10.1111/obr.12370. [DOI] [PubMed] [Google Scholar]
- 27.Hersoug LG, Møller P, Loft S. Role of microbiota-derived lipopolysaccharide in adipose tissue inflammation, adipocyte size and pyroptosis during obesity. Nutr Res Rev. 2018;31(2):153–163. doi: 10.1017/S0954422417000269. [DOI] [PubMed] [Google Scholar]
- 28.Fabbrini E, et al. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A. 2009;106(36):15430–15435. doi: 10.1073/pnas.0904944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein S. Is visceral fat responsible for the metabolic abnormalities associated with obesity?: implications of omentectomy. Diabetes Care. 2010;33(7):1693–1694. doi: 10.2337/dc10-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7(5):410–420. doi: 10.1016/j.cmet.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macotela Y, et al. Intrinsic differences in adipocyte precursor cells from different white fat depots. Diabetes. 2012;61(7):1691–1699. doi: 10.2337/db11-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan-Bathke M, Chen L, Oberschneider E, Harteneck D, Jensen MD. Sex and depot differences in ex vivo adipose tissue fatty acid storage and glycerol-3-phosphate acyltransferase activity. Am J Physiol Endocrinol Metab. 2015;308(9):E830–E846. doi: 10.1152/ajpendo.00424.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150(2):366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee KY, Luong Q, Sharma R, Dreyfuss JM, Ussar S, Kahn CR. Developmental and functional heterogeneity of white adipocytes within a single fat depot. EMBO J. 2019;38(3):e99291. doi: 10.15252/embj.201899291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank AP, de Souza Santos R, Palmer BF, Clegg DJ. Determinants of body fat distribution in humans may provide insight about obesity-related health risks. J Lipid Res. doi: 10.1194/jlr.R086975. doi: 10.1194/jlr.R086975. [published online ahead of print August 28, 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palmer BF, Clegg DJ. The sexual dimorphism of obesity. Mol Cell Endocrinol. 2015;402:113–119. doi: 10.1016/j.mce.2014.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis KE, et al. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol Metab. 2013;2(3):227–242. doi: 10.1016/j.molmet.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lotta LA, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. doi: 10.1038/ng.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu AY, et al. Multiethnic genome-wide meta-analysis of ectopic fat depots identifies loci associated with adipocyte development and differentiation. Nat Genet. 2017;49(1):125–130. doi: 10.1038/ng.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lotta LA, et al. Association of genetic variants related to gluteofemoral vs abdominal fat distribution with type 2 diabetes, coronary disease, and cardiovascular risk factors. JAMA. 2018;320(24):2553–2563. doi: 10.1001/jama.2018.19329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Justice AE, et al. Protein-coding variants implicate novel genes related to lipid homeostasis contributing to body-fat distribution. Nat Genet. 2019;51(3):452–469. doi: 10.1038/s41588-018-0334-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gustafson B, Hedjazifar S, Gogg S, Hammarstedt A, Smith U. Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab. 2015;26(4):193–200. doi: 10.1016/j.tem.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Hardy OT, et al. Body mass index-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg Obes Relat Dis. 2011;7(1):60–67. doi: 10.1016/j.soard.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klöting N, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299(3):E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- 45.Hoffstedt J, et al. Regional impact of adipose tissue morphology on the metabolic profile in morbid obesity. Diabetologia. 2010;53(12):2496–2503. doi: 10.1007/s00125-010-1889-3. [DOI] [PubMed] [Google Scholar]
- 46.Kim SM, et al. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014;20(6):1049–1058. doi: 10.1016/j.cmet.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hepler C, Gupta RK. The expanding problem of adipose depot remodeling and postnatal adipocyte progenitor recruitment. Mol Cell Endocrinol. 2017;445:95–108. doi: 10.1016/j.mce.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirsch J, Han PW. Cellularity of rat adipose tissue: effects of growth, starvation, and obesity. J Lipid Res. 1969;10(1):77–82. [PubMed] [Google Scholar]
- 49.Klöting N, Blüher M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord. 2014;15(4):277–287. doi: 10.1007/s11154-014-9301-0. [DOI] [PubMed] [Google Scholar]
- 50.Lee MJ, Wu Y, Fried SK. Adipose tissue remodeling in pathophysiology of obesity. Curr Opin Clin Nutr Metab Care. 2010;13(4):371–376. doi: 10.1097/MCO.0b013e32833aabef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121(6):2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun K, Tordjman J, Clément K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18(4):470–477. doi: 10.1016/j.cmet.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Danforth E. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26(1):13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 54.Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome. Endocrinol Nutr. 2013;60(suppl 1):39–43. doi: 10.1016/s1575-0922(13)70026-3. [DOI] [PubMed] [Google Scholar]
- 55.Khan T, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29(6):1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim JY, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117(9):2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kusminski CM, et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med. 2012;18(10):1539–1549. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shepherd PR, Gnudi L, Tozzo E, Yang H, Leach F, Kahn BB. Adipose cell hyperplasia and enhanced glucose disposal in transgenic mice overexpressing GLUT4 selectively in adipose tissue. J Biol Chem. 1993;268(30):22243–22246. [PubMed] [Google Scholar]
- 59.Senol-Cosar O, et al. Tenomodulin promotes human adipocyte differentiation and beneficial visceral adipose tissue expansion. Nat Commun. 2016;7:10686. doi: 10.1038/ncomms10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol. 2015;17(4):376–385. doi: 10.1038/ncb3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeffery E, et al. The adipose tissue microenvironment regulates depot-specific adipogenesis in obesity. Cell Metab. 2016;24(1):142–150. doi: 10.1016/j.cmet.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vishvanath L, et al. Pdgfrβ+ mural preadipocytes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab. 2016;23(2):350–359. doi: 10.1016/j.cmet.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19(10):1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao Z, Daquinag AC, Su F, Snyder B, Kolonin MG. PDGFRα/PDGFRβ signaling balance modulates progenitor cell differentiation into white and beige adipocytes. Development. 2018;145(1):dev155861. doi: 10.1242/dev.155861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shao M, et al. De novo adipocyte differentiation from Pdgfrβ+ preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat Commun. 2018;9(1):890. doi: 10.1038/s41467-018-03196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013;17(6):851–859. doi: 10.1016/j.cmet.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tchoukalova Y, Koutsari C, Jensen M. Committed subcutaneous preadipocytes are reduced in human obesity. Diabetologia. 2007;50(1):151–157. doi: 10.1007/s00125-006-0496-9. [DOI] [PubMed] [Google Scholar]
- 68.Tchoukalova YD, Votruba SB, Tchkonia T, Giorgadze N, Kirkland JL, Jensen MD. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc Natl Acad Sci U S A. 2010;107(42):18226–18231. doi: 10.1073/pnas.1005259107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gustafson B, Gogg S, Hedjazifar S, Jenndahl L, Hammarstedt A, Smith U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am J Physiol Endocrinol Metab. 2009;297(5):E999–E1003. doi: 10.1152/ajpendo.00377.2009. [DOI] [PubMed] [Google Scholar]
- 70.Gustafson B, Hammarstedt A, Hedjazifar S, Smith U. Restricted adipogenesis in hypertrophic obesity: the role of WISP2, WNT, and BMP4. Diabetes. 2013;62(9):2997–3004. doi: 10.2337/db13-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Isakson P, Hammarstedt A, Gustafson B, Smith U. Impaired preadipocyte differentiation in human abdominal obesity: role of Wnt, tumor necrosis factor-α, and inflammation. Diabetes. 2009;58(7):1550–1557. doi: 10.2337/db08-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135(2):240–249. doi: 10.1016/j.cell.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 73.Berry R, Rodeheffer MS. Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol. 2013;15(3):302–308. doi: 10.1038/ncb2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013;18(3):355–367. doi: 10.1016/j.cmet.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gupta RK, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012;15(2):230–239. doi: 10.1016/j.cmet.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hudak CS, et al. Pref-1 marks very early mesenchymal precursors required for adipose tissue development and expansion. Cell Rep. 2014;8(3):678–687. doi: 10.1016/j.celrep.2014.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang W, et al. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322(5901):583–586. doi: 10.1126/science.1156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hepler C, Vishvanath L, Gupta RK. Sorting out adipocyte precursors and their role in physiology and disease. Genes Dev. 2017;31(2):127–140. doi: 10.1101/gad.293704.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spallanzani RG, et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol. 2019;4(35):eaaw3658. doi: 10.1126/sciimmunol.aaw3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Merrick D, et al. Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science. 2019;364(6438):eaav2501. doi: 10.1126/science.aav2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwalie PC, et al. A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature. 2018;559(7712):103–108. doi: 10.1038/s41586-018-0226-8. [DOI] [PubMed] [Google Scholar]
- 82.Hepler C, et al. Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife. 2018;7:e39636. doi: 10.7554/eLife.39636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burl RB, Ramseyer VD, Rondini EA, Pique-Regi R, Lee YH, Granneman JG. Deconstructing adipogenesis induced by β3-adrenergic receptor activation with single-cell expression profiling. Cell Metab. 2018;28(2):300–309.e4. doi: 10.1016/j.cmet.2018.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Church CD, Berry R, Rodeheffer MS. Isolation and study of adipocyte precursors. Meth Enzymol. 2014;537:31–46. doi: 10.1016/B978-0-12-411619-1.00003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marcelin G, et al. A PDGFRα-mediated switch toward CD9high adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab. 2017;25(3):673–685. doi: 10.1016/j.cmet.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 86.Mahlakõiv T, et al. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci Immunol. 2019;4(35):eaax0416. doi: 10.1126/sciimmunol.aax0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cautivo KM, Molofsky AB. Regulation of metabolic health and adipose tissue function by group 2 innate lymphoid cells. Eur J Immunol. 2016;46(6):1315–1325. doi: 10.1002/eji.201545562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hong KY, et al. Perilipin+ embryonic preadipocytes actively proliferate along growing vasculatures for adipose expansion. Development. 2015;142(15):2623–2632. doi: 10.1242/dev.125336. [DOI] [PubMed] [Google Scholar]
- 89.Jiang Y, Berry DC, Tang W, Graff JM. Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis. Cell Rep. 2014;9(3):1007–1022. doi: 10.1016/j.celrep.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shao M, Hepler C, Vishvanath L, MacPherson KA, Busbuso NC, Gupta RK. Fetal development of subcutaneous white adipose tissue is dependent on Zfp423. Mol Metab. 2017;6(1):111–124. doi: 10.1016/j.molmet.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang QA, Scherer PE, Gupta RK. Improved methodologies for the study of adipose biology: insights gained and opportunities ahead. J Lipid Res. 2014;55(4):605–624. doi: 10.1194/jlr.R046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Emont MP, et al. Using a 3D culture system to differentiate visceral adipocytes in vitro. Endocrinology. 2015;156(12):4761–4768. doi: 10.1210/en.2015-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Petrus P, et al. Transforming growth factor-β3 regulates adipocyte number in subcutaneous white adipose tissue. Cell Rep. 2018;25(3):551–560. doi: 10.1016/j.celrep.2018.09.069. [DOI] [PubMed] [Google Scholar]
- 94.Hepler C, et al. Directing visceral white adipocyte precursors to a thermogenic adipocyte fate improves insulin sensitivity in obese mice. Elife. 2017;6:e27669. doi: 10.7554/eLife.27669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu L, et al. The winged helix transcription factor Foxa3 regulates adipocyte differentiation and depot-selective fat tissue expansion. Mol Cell Biol. 2013;33(17):3392–3399. doi: 10.1128/MCB.00244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang QA, et al. Distinct regulatory mechanisms governing embryonic versus adult adipocyte maturation. Nat Cell Biol. 2015;17(9):1099–1111. doi: 10.1038/ncb3217. [DOI] [PMC free article] [PubMed] [Google Scholar]