

Abstract
Several distinct components of voltage-gated sodium current have been recorded from native dorsal root ganglion (DRG) neurons that display differences in gating and pharmacology. This study compares the electrophysiological properties of two peripheral nerve sodium channels that are expressed selectively in DRG neurons (Nav1.7 and Nav1.8). Recombinant Nav1.7 and Nav1.8 sodium channels were coexpressed with the auxiliary β1 subunit inXenopus oocytes. In this system coexpression of the β1 subunit with Nav1.7 and Nav1.8 channels results in more rapid inactivation, a shift in midpoints of steady-state activation and inactivation to more hyperpolarizing potentials, and an acceleration of recovery from inactivation. The coinjection of β1 subunit also significantly increases the expression of Nav1.8 by sixfold but has no effect on the expression of Nav1.7. In addition, a great percentage of Nav1.8+β1 channels is observed to enter rapidly into the slow inactivated states, in contrast to Nav1.7+β1 channels. Consequently, the rapid entry into slow inactivation is believed to cause a frequency-dependent reduction of Nav1.8+β1 channel amplitudes, seen during repetitive pulsing between 1 and 2 Hz. However, at higher frequencies (>20 Hz) Nav1.8+β1 channels reach a steady state to ∼42% of total current. The presence of this steady-state sodium channel activity, coupled with the high activation threshold (V0.5 = −3.3 mV) of Nav1.8+β1, could enable the nociceptive fibers to fire spontaneously after nerve injury.
Keywords: Nav1.7, Nav1.8, peripheral nerve sodium channels, expression, dorsal root ganglion, nociception, Xenopus oocytes
Voltage-gated sodium channels play an important role in the generation and propagation of action potentials in excitable tissues. At least 10 distinct isoforms of the sodium channel have been identified in brain and neuronal and striated muscle that differ in primary structure, pharmacology, permeation, and gating (Goldin et al., 2000). A major determinant of the functional difference among these isoforms is inherent in the α subunit that determines the selectivity and gating properties of these channels (Goldin et al., 1986). However, in vivo, most sodium channels are associated with auxiliary subunits (β1–β3) that are known to modulate gating and levels of expression (Isom et al., 1992; Morgan et al., 2000). For instance, the inactivation of rat brain sodium channels (Nav1.2) is accelerated and the gating is shifted toward more hyperpolarized voltages when coexpressed with the β1 subunit (O'Leary, 1998). Similar findings have been reported for the skeletal muscle sodium channels, indicating that these auxiliary subunits play an important role in determining the gating properties of these tetrodotoxin-sensitive sodium channels (Bennett et al., 1993; Wallner et al., 1993; Yang et al., 1993). In contrast, the tetrodotoxin-resistant cardiac sodium channels display only subtle changes in gating when coexpressed with the β1 subunit (Makita et al., 1994; Nuss et al., 1995; Makielski et al., 1996).
In addition to changes in gating, the β1subunit is known to enhance the expression of many sodium channels (Chahine et al., 1994; Nuss et al., 1995). The expression of functional Nav1.2 brain sodium channels increases by 2.5-fold when coexpressed with the β1 subunit (Isom et al., 1992). The β1 subunit also is believed to contribute to clustering and remodeling of sodium channels at the neuromuscular junction (Blackburn-Munro and Fleetwood-Walker, 1999; Caldwell, 2000). Such regulations may have important consequences for neuronal tissues such as dorsal root ganglion cells (DRG) in which multiple isoforms of the sodium channels are expressed in the same cell (Akopian et al., 1996; Black et al., 1996; Sangameswaran et al., 1996,1997; Toledo-Aral et al., 1997).
At least five distinct components of voltage-gated sodium channels have been recorded from DRG neurons (Rush et al., 1998). Two components predominate in the small nociceptive neurons: a rapidly inactivating TTX-S current and a slowly inactivating TTX-R current (Kostyuk et al., 1981; Roy and Narahashi, 1992; Elliott and Elliott, 1993; Black et al., 1996). Recently, two peripheral nerve sodium channels have been isolated from the human and rat DRG (Sangameswaran et al., 1996;Toledo-Aral et al., 1997). Nav1.7 (PN1) is a TTX-S rapidly inactivating channel that is expressed widely in DRG neurons (Sangameswaran et al., 1997; Toledo-Aral et al., 1997). Nav1.8 (PN3) is a TTX-R slowly inactivating channel that is expressed predominantly in the small nociceptive C-type pain fibers (Akopian et al., 1996; Sangameswaran et al., 1996). Recently, a novel sodium channel Nav1.9 (NaN) was cloned and also may contribute to the TTX-R current of small DRG neurons (Dib-Hajj et al., 1998). Differential expression of the Nav1.7, Nav1.8, Nav1.9, and several of the brain sodium channels (Nav1.1, Nav1.2, Nav1.3) contributes to the unique electrical excitability of nociceptive neurons (Porreca et al., 1999). Changes in the expression levels of these channels have been implicated in the alterations of neuronal excitability associated with acute and chronic pain syndromes (Rizzo et al., 1995; Gold et al., 1996).
The transcript encoding for the β1 subunit is present in both large and small DRG neurons (Oh et al., 1995; Coward et al., 2001). However, previous studies indicate that the β1 subunit does not alter the gating of Nav1.7 and Nav1.8 sodium channels (Sangameswaran et al., 1996, 1997). This finding is inconsistent with studies showing that many, if not all, sodium channels are modulated by the β1 subunit (Chahine et al., 1994; Isom et al., 1995). In addition, a recent study has shown that the inactivation of heterologously expressed Nav1.7 channels is accelerated when coexpressed with the β1 subunit (Shcherbatko et al., 1999). Currently, little is known about the modulatory effects of the β1 subunit on Nav1.8 channels. In this study the α subunits of Nav1.7 and Nav1.8 sodium channels were expressed in Xenopus oocytes, and the kinetics and voltage sensitivity were compared with and without the coexpressed β1 subunit. The Xenopus oocyte system of expression was used in this study especially because the Nav1.8 sodium channels expressed, with or without the β1 subunit, poorly in the mammalian cell systems (tsA201 and CHO cell line). Coexpression of Nav1.7 with the β1subunit in Xenopus oocytes causes a hyperpolarizing shift in gating and increases the rates of inactivation and recovery from inactivation. For Nav1.8 channels the β1 subunit produces similar changes in the voltage sensitivity and kinetics of gating but, in addition, significantly increases the expression levels of these channels. The β1 subunit modulates the gating of both the Nav1.7 and Nav1.8 sodium channels. Differences in the gating and expression of these sodium channels are likely to have important consequences for the generation and propagation of action potentials in nociceptive neurons.
MATERIALS AND METHODS
Molecular biology: Construction of full-length rat α subunit Nav1.8 cDNA. Total RNA was isolated from Sprague Dawley rat DRG by using the Trizol reagent (Life Technologies, Burlington, Ontario, Canada). Rat DRG RNA was reverse transcribed (RT) with the use of Superscript (Life Technologies) and random primers to create a cDNA library. Total DRG RNA (5 μg) was heat denatured at 70°C for 10 min, followed by rapid cooling on ice. Reverse transcription was performed with 10 mm deoxynucleotide triphosphate (dNTPs), 0.1m dithiothreitol, and 50 ng/μl random primers in a total volume of 20 μl. The mixture was added to the denatured total RNA and incubated at 25°C for 5 min. Superscript (1 U; Life Technologies) was added, and the reactions were incubated at 25°C for 10 min and then at 42°C for 1 hr. Incubating the sample at 70°C for 15 min then terminated the reaction. Finally, the sample was incubated at 37°C with RNaseH for 20 min to remove the total RNA. The RT product was used directly for PCR.
Rat Nav1.8 α subunit-specific primers were designed to amplify 2–4 kb segments within the coding region of the gene. Primer sequences were based on the published sequence (ACC numberU53833). The rat Nav1.8 α subunit gene was amplified from the first ATG start codon; the 5′ untranslated region (UTR) sequence was not included in the clone.
Primer set 1 (1–4247 bp) consisted of the following: primer position 1, sense, GAAGAATGAGAAGATGGAGCTCCCC; primer position 4247, antisense, GAGATTCAGCGTGAAGAAGCCACC. Primer set 2 (3875–5940 bp) consisted of the following: primer position 3875, sense, GTCCTCCTCGTCTGCCTCATCTTC; primer position 5940, antisense, GCCTGAGTGCCTTCACTGAGGTCCAG.
PCR was performed in a 50 μl reaction mixture containing PCR buffer, 10 mm dNTPs, 100 ng/μl of the specific set of primers, 2 μl of the rat DRG cDNA, and 1 U of Pfu-turbo Polymerase (Stratagene, La Jolla, CA). Amplification was performed with the following cycling: 3 min at 94°C, 1 min at 94°C, 1 min at 60°C, and then 2 min/kb at 55°C repeated for a total of 30 cycles. PCR amplicons were subcloned, and the full-length Nav1.8 was constructed in the pCRII–Topo vector (Life Technologies). Full-length Nav1.8 coding sequence was confirmed by fluorescent dideoxy terminator sequencing at the automated sequencing facility of Laval University, Sainte-Foy, Québec. The final Nav1.8 construct was subcloned into pSP64T (β-globin), suitable for high-yield transcription of complementary RNA (cRNA).
The rat Nav1.7 α subunit voltage-gated sodium channel, cloned into the pCDNA3a vector, was kindly donated by Gail Mandel (Department of Neurobiology, State University of New York, NY). cRNA was prepared by the T7 (pCDNA3a) or SP6 (pSP64T) mMessage mMachine kit (Ambion, TX).
Expression and electrophysiology in Xenopusoocytes. Xenopus laevis females were anesthetized with 1.5 mg/ml tricaine (Sigma, Oakville, Ontario, Canada), and two or three ovarian lobes were removed surgically. Follicular cells surrounding the oocytes were removed by incubation at 22°C for 2.5 hr in calcium-free oocyte medium [containing (in mm) 82.5 NaCl, 2.5 KCl, 1 MgCl2, and 5 HEPES, pH 7.6] containing 2 mg/ml collagenase (Sigma). The oocytes were washed first in calcium-free medium and then with a 50% Leibovitz's L-15 medium (Life Technologies) enriched with 15 mm HEPES and 5 mml-glutamine, supplemented with 10 mg/ml gentamycin, pH 7.6. The oocytes were stored in this medium until further use. Stage VI–V oocytes were selected and microinjected with 50 nl of cRNA encoding for the α subunit of Nav1.7 or Nav1.8. The amounts of Nav1.7 cRNA injected in the oocytes were lesser compared with the amounts injected for Nav1.8 channels. This is because the Nav1.7 channels express more readily compared with Nav1.8 channels. Sets of oocytes also were coinjected in parallel with equal ratios of Nav1.7 and β1 subunit or Nav1.8 α subunit and β1subunit.
Oocytes were stored at 18°C and used for experiments depending on the level of expression of each channel type. Parallel sets of experiments with the rat skeletal muscle sodium channel (μ1) were used to confirm the functional association of the α and β1subunits in oocytes (data not shown).
The whole-cell sodium current from cRNA-injected oocytes was measured via two-microelectrode voltage clamp at room temperature ≈22°C. The oocytes were impaled with <2 MΩ electrodes containing 3m KCl and were voltage clamped with an OC-725 oocyte clamp (Warner Instruments, Hamden, CT). The bath Ringer's solution contained (in mm) 90 NaCl, 2 KCl, 1.8 CaCl2, 2 MgCl2, and 5 HEPES, pH 7.6. Currents were filtered at 1.5 kHz with an eight-pole Bessel filter and were sampled at 10 kHz. Data were acquired and analyzed with pClamp software v7 (Axon Instruments, Foster City, CA).
The voltage dependence of activation was determined by eliciting depolarizing pulses from a holding potential of −100 mV to potentials ranging from −80 to +60 mV in 10 mV increments. Current activation curves of the channels were plotted via the following Boltzmann equation: GNa/GNa max = 1/(1 + exp((V +V0.5)/k)), for which the GNa (conductance) values for each clamped oocyte were determined by dividing the peak sodium current by the driving force (Vm −ENa). The reversal potential (ENa) for each oocyte expressing the channels was estimated by extrapolating the linear ascending segment of decay gradient, between 0 and +20 mV for Nav1.7 and between +20 and +40 mV for Nav1.8, of anI–V curve to the voltage axis. V is equal to the test voltage, V0.5 is the voltage at which the channels are half-maximally activated, and k is the slope factor. Conductance versus voltage data were fit with a two-state Boltzmann equation.
Statistical analysis. Results of representative measures were expressed by means ± SEM. The currents of paired groups of oocytes injected with the α subunit of Nav1.7 or Nav1.8 were compared directly with those of oocytes coinjected with the α and β1subunits; a repeated measurement ANOVA was performed. The homogeneity of correlation between repeated measures was tested with the sphericity test. The results were considered significant ifp values were ≤0.05. The data were analyzed by the statistical package program SAS (SAS Institute, Cary, NC).
RESULTS
Effects of β1 subunit on the expression of Nav1.7 and Nav1.8 sodium channels
The α subunit of the peripheral nerve sodium channel Nav1.8 was cloned from the DRG of Sprague Dawley rats via RT-PCR (see Materials and Methods). The fidelity of the Nav1.8 clone was verified by comparing its sequence with the published sequence (ACC number U53833). cRNA of Nav1.7 and Nav1.8 clones was microinjected into stage IV–V Xenopus oocytes. The whole-cell sodium currents of oocytes injected with Nav1.7 and Nav1.8 channels were compared with and without the β1 subunit. The currents were evoked by applying a series of depolarizing voltage steps between −50 and +65 mV in 5 mV increments (Fig.1). Nav1.7 and Nav1.8 channels have distinct expression behaviors in the Xenopus oocytes. The Nav1.7 currents activate at −40 mV and peak at −20 mV, whereas Nav1.8 currents activate and peak at −15 and +20 mV, respectively. For voltage pulses to −20 mV, oocytes expressing the Nav1.7 channels displayed large sodium currents (−3087.8 ± 435.4 nA, n = 6) after <24 hr of incubation (Figs. 1A,2A). In contrast, oocytes injected with Nav1.8 expressed small currents at +20 mV (−557.8 ± 26.3 nA) after >5 d after injection (Figs.1B, 2B), indicating that the α subunit of Nav1.8 is expressed inefficiently in oocytes.
Fig. 1.
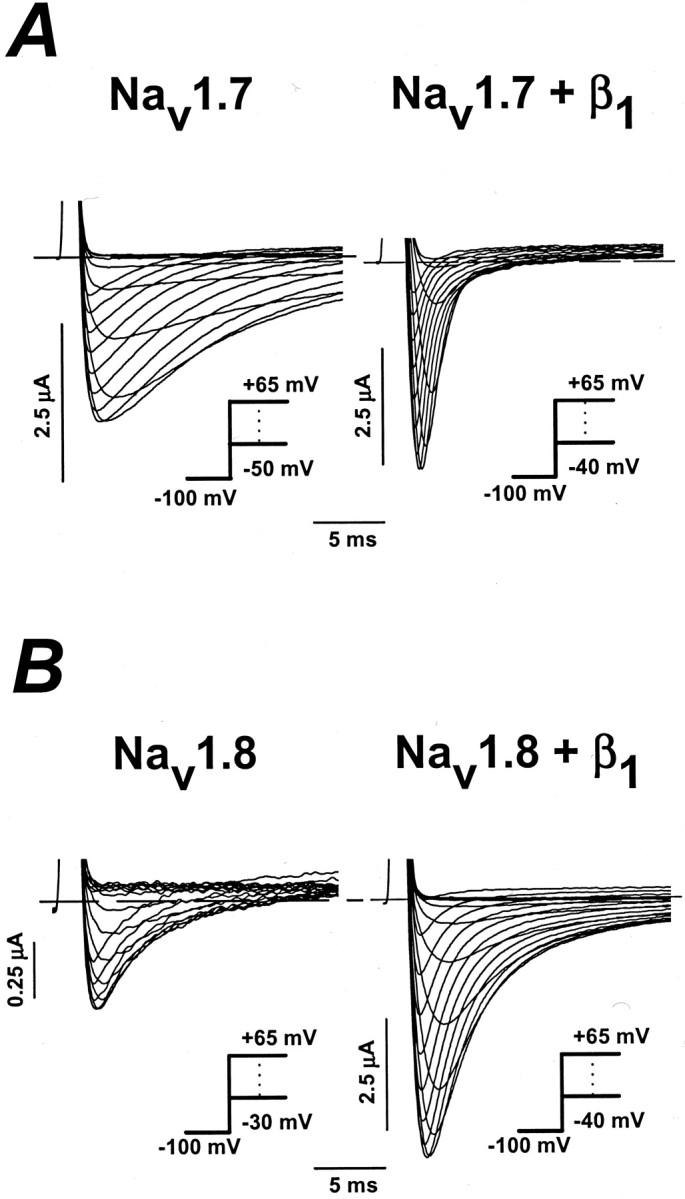
Effects of the β1 subunit on Nav1.7 and Nav1.8 sodium channels heterologously expressed in Xenopus oocytes. The data show the whole-cell sodium currents of oocytes expressing either the Nav1.7 or Nav1.8 sodium channel with and without the β1 subunit. Currents were elicited by depolarizing steps between −50 and +65 mV in 5 mV increments from a holding potential of −100 mV (see inset).A, Whole-cell Nav1.7 currents measured in the absence (left) and presence (right) of the β1 subunit. B, Nav1.8 sodium currents expressed without (left) and with (right) the β1 subunit. Dashed lines are the zero current levels.
Fig. 2.
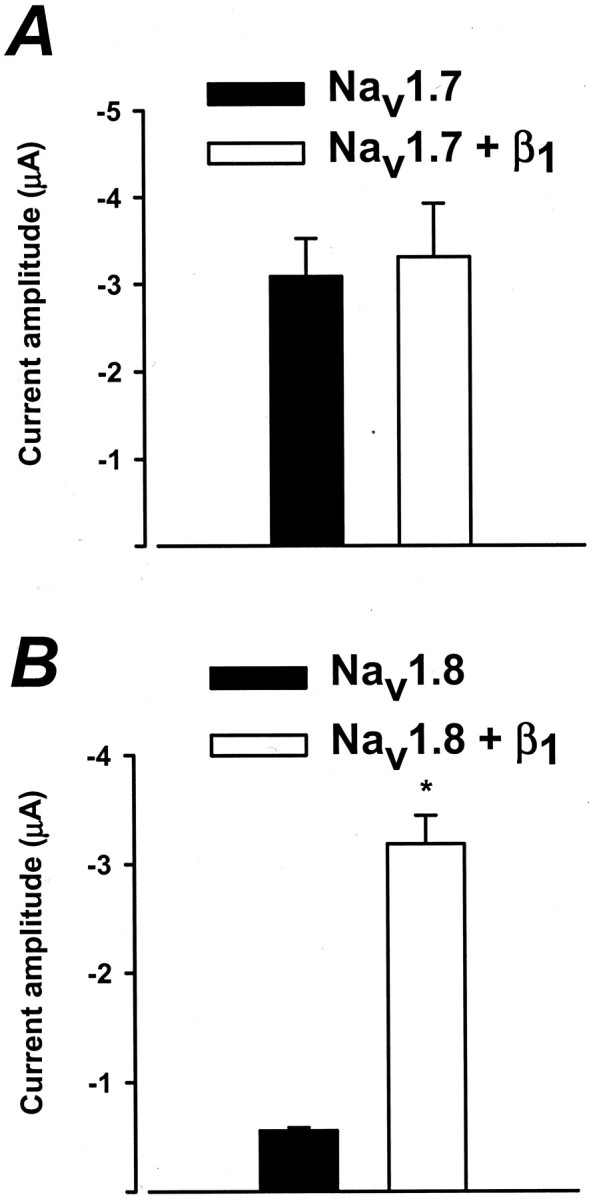
Effects of the β1 subunit on the expression of Nav1.7 and Nav1.8 sodium channels. Shown are the whole-cell sodium currents of paired groups of oocytes expressing either the Nav1.7 or Nav1.8 channels with or without the β1 subunit. Nav1.7 peak currents at −20 mV were measured from oocytes expressing Nav1.7 or Nav1.7+β1after 24 hr of incubation. There was no significant difference in the peak amplitude of current between Nav1.7 and Nav1.7+β1 even at 3 d after injection (data not shown). For Nav1.8 channels the peak currents measured at +20 mV were compared 6 d after cRNA injection. Peak amplitude recorded 3 d after injection for Nav1.8 channels was small (38.1 ± 2.8 nA; n = 3), whereas the coexpression increased expression by 17-fold (695 ± 117 nA;n = 5; data not shown). The β1subunit significantly increased (p < 0.05) the currents of Nav1.8 (n = 7), but not the Nav1.7 (n = 6), sodium channels (p < 0.05). The holding potential was −100 mV.
Figure 2 directly compares the currents of paired groups of oocytes expressing either Nav1.7 or Nav1.8 with and without the β1 subunit. The sodium current of oocytes injected with Nav1.8 substantially increases when coexpressed with the β1 subunit (Fig. 2B). At +20 mV, coexpression with the β1 subunit increases the sodium current of oocytes expressing Nav1.8 by 5.7-fold (−3183 ± 262 nA) (Fig. 2B). Although the mechanism underlying this increase in current is not known, these data are similar to those of previous studies showing that the β1 subunit increases the expression of a number of sodium channel isoforms (Nuss et al., 1995). In contrast, the amplitude of currents of oocytes expressing Nav1.7 is not altered by the β1 subunit (Fig. 2A). The β1 subunit selectively enhances the expression of the Nav1.8 sodium channels. Both Nav1.7 and Nav1.8 sodium channels cDNAs also were transfected into different mammalian cell expression systems (tsA201 and CHO) in the absence and presence of the β1 subunit. However, mammalian cells transfected with Nav1.8 channels, in the absence or presence of the β1 subunit, expressed small to negligible amounts of current (≤1 nA, n = 6; data not shown). In contrast, tsA201 cells transfected with Nav1.7 cDNA expressed ∼10 nA of current (n = 4; data not shown). Because of the poor expression of Nav1.8 channels in the different mammalian systems, Xenopus oocytes were used preferentially in this study.
Effects of the β1 subunit on kinetics of current decay of Nav1.7 and Nav1.8 channels
In addition to changes in expression levels, the β1 subunit also alters the gating properties of these channels. In the absence of the β1subunit, the inactivation of Nav1.7 is slow, resulting in considerable residual current near the end of the 20 msec depolarizing pulse (Figs. 1A,3A). When coexpressed with the β1 subunit, the inactivation is more rapid and the currents are inactivated completely during the 20 msec depolarization (Figs. 1A, 3A). The β1 subunit also accelerates the inactivation of Nav1.8 but to a much lesser extent than that observed for Nav1.7 (Fig. 3B). To quantitate the changes in decay rate, we fit the currents with single exponentials. At −20 mV the decay of Nav1.7 currents has a time constant (τh) of 19.8 ± 3.6 msec (n = 6) and 1.8 ± 0.2 msec (n = 6) for Nav1.7+β1. Coexpression with the β1 subunit significantly accelerates the inactivation of Nav1.7 current (p < 0.05). Coexpression of Nav1.8 with the β1subunit also significantly reduces τh(p < 0.05). At +20 mV, Nav1.8 and Nav1.8+β1 currents decay with time constants of 4.3 ± 0.2 msec (n = 6) versus 2.6 ± 0.1 msec (n = 6), respectively (Fig.3B). The β1 subunit reduces the τh of Nav1.7 and Nav1.8 sodium currents over a wide range of voltages (Fig. 4A,B). Overall, the data indicate that coexpression with the β1 subunit accelerates the inactivation of both Nav1.7 and Nav1.8 sodium channels.
Fig. 3.
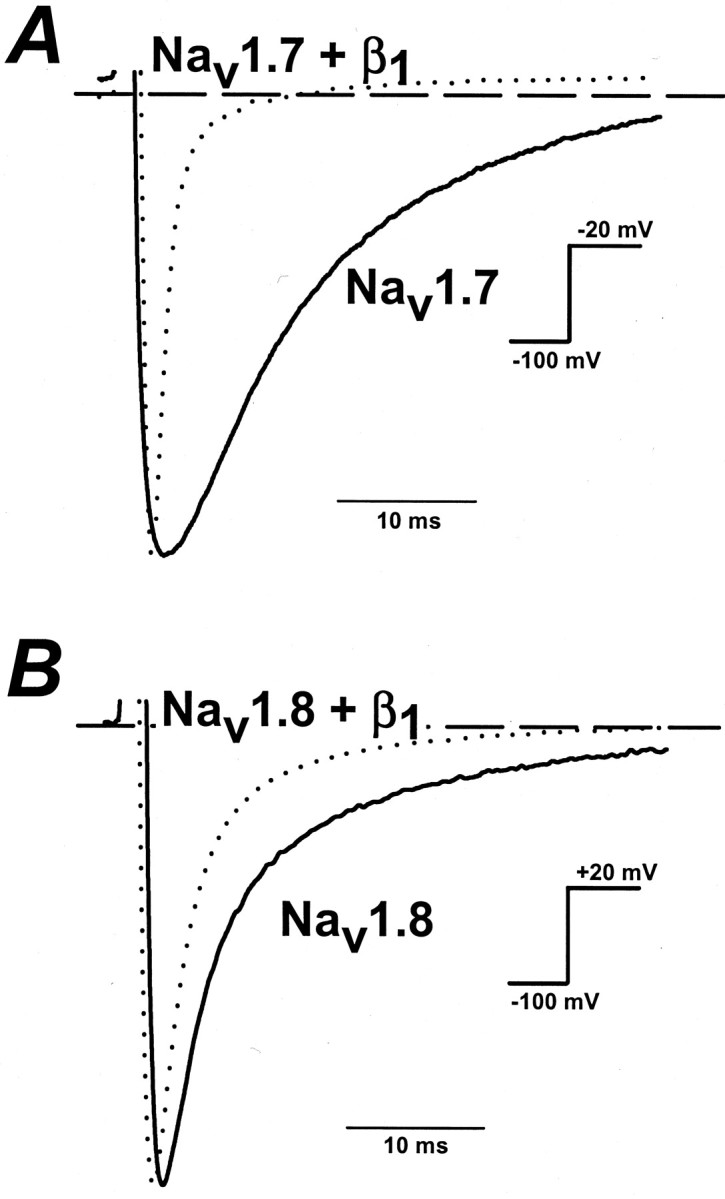
Effects of the β1 subunit on the kinetics of Nav1.7 and Nav1.8 inactivation. Whole-cell sodium currents of Nav1.7 and Nav1.7+β1 sodium channels were elicited by a depolarizing step from −100 to −20 or +20 mV. Currents were normalized to facilitate comparison of the kinetics. A, At −20 mV the time constants of current decay were 19.8 ± 3.6 msec (n = 6) and 1.8 ± 0.2 msec (n = 6) for Nav1.7 and Nav1.7+β1, respectively.B, Nav1.8 and Nav1.8+β1 currents were elicited by a step depolarization to +20 mV and had decay time constants of 4.3 ± 0.2 msec (n = 6) and 2.6 ± 0.1 msec (n = 6), respectively. Dashed linesare the zero current levels.
Fig. 4.

The β1 subunit accelerates the inactivation of Nav1.7 and Nav1.8 channels. The decay of the Nav1.7 and Nav1.8 sodium currents (Fig. 1) was fit to an exponential function, and the time constants were plotted versus the test voltage: I =AI · exp (−t/τi) + C, where I is the current,Ai is the percentage of channels inactivating with time constant τi,t is time, and C is the steady-state asymptote. The data are the means ± SEM of n= 6 for Nav1.7 and Nav1.7+β1 andn = 6 for Nav1.8 and Nav1.8+β1 channels. A, The inactivation time constants of Nav1.7 (filled squares) and Nav1.7+β1 (open squares) plotted versus voltage. B, The time constants of Nav1.8 (filled circles) and Nav1.8+β1 (open circles) plotted versus voltage.
Effects of the β1 subunit on the gating of Nav1.7 and Nav1.8 channels
The effect of the β1 subunit on the voltage sensitivity of activation was investigated also. The relative conductance was determined from families of sodium currents similar to those shown in Figure 1 (see Materials and Methods). The normalized conductance of Nav1.7 and Nav1.8 channels with and without the β1 subunit was plotted versus voltage (Fig.5A,B). The smooth curves are fits to a Boltzmann function with midpoints (V0.5) and slope factors (k) of −22.4 ± 2.7 and 5.4 ± 0.4 mV (n = 10) for Nav1.7 and of −27.7 ± 1.3 mV (V0.5) and 3.7 ± 0.2 mV (k; n = 11) for Nav1.7+β1 (Fig.5A). Coexpression with the β1subunit causes a significant −5.3 mV shift in the midpoint of steady-state activation (p < 0.05). For Nav1.8 the V0.5and k values are 4.7 ± 0.7 and 6.8 ± 0.1 mV (n = 8) and −3.3 ± 0.9 mV (V0.5) and 5.5 ± 0.1 mV (k) for Nav1.8+β1(n = 9) (Fig. 5B). The coexpression with the β1 subunit causes a significant −8 mV shift in midpoint (p < 0.05) and reduces the slope factor, consistent with an increase in the voltage sensitivity of the Nav1.8 sodium channels. Overall, the β1 subunit causes hyperpolarizing shifts in the midpoints of activation and increases the voltage sensitivity of both Nav1.7 and Nav1.8 channels.
Fig. 5.

Effects of the β1 subunit on the activation, inactivation, and recovery of Nav1.7 and Nav1.8 channels. Activation was measured by applying a series of depolarizing voltage pulses between −80 and +60 mV from a holding potential of −100 mV. The peak currents were measured, and the relative conductance was calculated by using the standard procedures (see Materials and Methods). Also plotted is the steady-state availability curve that was determined by using 500 msec conditioning pulses to voltages between −110 and +30 mV and a standard test pulse to either −20 mV (Nav1.7 and Nav1.7+β1) or +20 mV (Nav1.8 and Nav1.8+β1). Test currents were normalized and plotted versus conditioning voltage.A, The normalized conductance versus voltage and steady-state inactivation plots of Nav1.7 (filled squares) and Nav1.7+β1 (open squares) channels. The smooth curves are Boltzmann fits:G = 1/(1 + exp ((V −V0.5)/−k)), with midpoints (V0.5) and slope factors (k) of activation of −22 ± 2.7 and 5.4 ± 0.4 mV for Nav1.7 (n = 10) and −27.7 ± 1.3 and 3.7 ± 0.2 mV for Nav1.7+β1 (n = 11). For inactivation the V0.5 and k values are −68.2 ± 0.43 and 6.4 ± 0.45 mV for Nav1.7 (n = 4) and −69.8 ± 0.3 and 3.9 ± 0.2 mV for Nav1.7+β1 (n = 4).B, Steady-state activation and inactivation of Nav1.8 channels. The smooth curves haveV0.5 and k values for activation of 4.7 ± 0.7 and 6.8 ± 0.1 mV for Nav1.8 (filled circles;n = 8) and −3.3 ± 1.0 and 5.5 ± 0.1 mV for Nav1.8+β1 (open circles;n = 9). The V0.5 andk values for inactivation are −54.8 ± 1.7 and 8.4 ± 0.2 mV for Nav1.8 (n = 3) and −62.6 ± 2.3 and 6.3 ± 0.7 mV for Nav1.8+β1 (n = 6).C, D, The time course of recovery from inactivation of Nav1.7 and Nav1.8 channels. Inactivation was induced by depolarizing to −20 mV (Nav1.7 and Nav1.7+β1) or +20 mV (Nav1.8 and Nav1.8+β1) for 50 msec before returning to −100 mV for intervals between 1 msec and 5 sec. A standard test pulse was used to monitor recovery, and the normalized test currents were plotted versus the recovery interval.C, Recovery from inactivation of Nav1.7 (filled squares) and Nav1.7+β1 (open squares) channels. The smooth curves are fits to the sum of two exponentials:I/IO =AF · (1 − exp(−t/τF)) +AS · (1 − exp(−t/τS)), with time constants (τ) and weighting factors (A) of 19.6 ± 0.8 msec (τF;AF = 0.46 ± 0.02) and 933.4 ± 54.6 msec (τS;AS = 0.54 ± 0.02) for Nav1.7 (n = 7) and 6.6 ± 0.6 msec (τF; AF = 0.89 ± 0.02) and 53.2 ± 12.7 msec (τS;AS = 0.11 ± 0.02) for Nav1.7+β1 (n = 7).D, The recovery of Nav1.8 (filled circles) and Nav1.8+β1 (open circles) channels is described best by the sum of three exponentials:I/IO =AF · (1 − exp(−t/τF)) +AI · (1 − exp(−t/τI)) +AS · (1 − exp(−t/τS)), where τF, τI, and τSare the fast, intermediate, and slow recovery time constants, andAF,AI, and ASare the relative weighting factors. t is the interpulse duration, and I/Io is the normalized current amplitude. Data are the means ± SEM. The time constants of Nav1.8 are 9.9 ± 1.8 msec (τF; AF = 0.41 ± 0.04), 168.6 ± 52.2 msec (τI;AI = 0.28 ± 0.04), and 787.6 ± 112.6 msec (τS;AS = 0.28 ± 0.04) (filled circles; n = 5). Recovery time constants of Nav1.8+β1 are 2.0 ± 0.3 msec (τF;AF = 0.32 ± 0.05), 243.8 ± 85.4 msec (τI; AI= 0.34 ± 0.05), and 1070.1 ± 59.0 msec (τS; AS = 0.34 ± 0.02) (open circles; n= 4).
The effect of the β1 subunit on the steady-state inactivation was investigated also. Steady-state inactivation was measured by using 500 msec conditioning pulses to voltages between −110 and +30 mV. The fraction of available current was determined by using standard test pulses, and the normalized currents were plotted versus the conditioning voltage (Fig.5A,B). The smooth curves are Boltzmann fits with midpoints (V0.5) and slope factors (k) of −68.2 ± 0.4 and 6.4 ± 0.5 mV (n = 4) for Nav1.7 and of −69.8 ± 0.3 mV (V0.5) and 3.9 ± 0.2 mV (k; n = 4) for Nav1.7+β1 (Fig.5A). The β1 subunit only slightly alters the midpoint but significantly increases the voltage sensitivity of Nav1.7 inactivation (p< 0.05). Steady-state inactivation of Nav1.8 sodium channels is shifted significantly toward hyperpolarizing voltages, with V0.5 and kvalues of −54.8 ± 1.7 and 8.5 ± 0.2 mV (n= 3) for Nav1.8 (p < 0.05) and −62.6 ± 2.3 mV (V0.5) and 6.3 ± 0.7 mV (k; n = 6) for Nav1.8+β1 (Fig.5B). Coexpression of Nav1.8 with the β1 subunit significantly shifts the midpoint of steady-state inactivation by −7.8 mV and increases the voltage sensitivity. Hence coexpression with the β1subunit also causes hyperpolarizing shifts in the midpoints and increases the voltage sensitivity of steady-state inactivation of both the Nav1.7 and Nav1.8 sodium channels.
Effects of β1 subunit on recovery from fast inactivation
Effects of the β1 subunit on the recovery from fast inactivation were examined with a standard two-pulse protocol consisting of a depolarizing pulse to −20 mV (Nav1.7) or +20 mV (Nav1.8) for 40 msec to inactivate the channels, followed by a variable duration (1 msec to 5 sec) step to −100 mV to promote recovery. The availability of the channels after the end of the recovery interval was assessed with a standard test pulse, and the normalized currents were plotted versus the recovery interval. Figure 5, C andD, illustrates the time dependence of recovery from fast inactivation of Nav1.7 and Nav1.8 channels. In the absence of the β1 subunit the recovery of Nav1.7 is bi-exponential, with fast and slow time constants of 19.6 ± 0.8 msec (τF) and 933.4 ± 54.6 msec (τS; n= 7) (Fig. 5C). Coexpression with the β1 subunit substantially increases the recovery kinetics and causes the channels to recover fully from inactivation. The fast and slow recovery time constants of Nav1.7+β1 are 6.6 ± 0.6 msec (τF) and 53.2 ± 12.7 msec (τS; n = 7). The more complete recovery of Nav1.7+β1channels can be attributed primarily to a 2.9-fold increase in the fraction of channels recovering with the rapid time constant.
In contrast, the recovery from fast inactivation of Nav1.8 is slow in comparison with Nav1.7, and fitting the data required the sum of three exponentials to describe the time course accurately. The recovery time constants of Nav1.8 channels are 9.9 ± 1.8 msec (τF), 168.6 ± 52.2 msec (τI), and 787.6 ± 112.6 msec (τS; n = 5) (Fig.5D). The recovery time constants of Nav1.8+β1 are 2.0 ± 0.3 msec (τF), 243.8 ± 85.4 msec (τI), and 1070.1 ± 59.0 msec (τS; n = 4) (Fig.5D). The β1 subunit reduces τF but increases τS, consistent with differential effects on the fast and slow components of Nav1.8 recovery from inactivation. Interestingly, the slow component of recovery (τS) is observed only with Nav1.8 channels and is enhanced with the β1 subunit. This suggests that the Nav1.8+β1 channels may enter readily into a slow inactivated state during the short (40 msec) depolarizing prepulses to +20 mV that are used to induce inactivation.
Development of slow inactivation of Nav1.7+β1 and Nav1.8+β1 channels
The recovery from inactivation of Nav1.8+β1 displays a slow component that is not observed with Nav1.7+β1 (Fig.5C,D). The data suggest that, in addition to the fast and intermediate components of inactivation observed for both channels, Nav1.8+β1 channels enter into a slow inactivated state during the short depolarizing prepulses used to inactivate the channels in these experiments. To test this hypothesis, we compared the development of slow inactivation of Nav1.7+β1 and Nav1.8+β1 channels (Fig.6). The onset of slow inactivation was measured by depolarizing the oocytes to either −20 mV (Nav1.7+β1) or +20 mV (Nav1.8+β1) for a variable interval (0 msec to 10 sec) to induce inactivation. Then the voltage was returned to −100 mV for 20 msec to allow for the recovery of fast-inactivated channels (τF = 6.6 msec for Nav1.7+β1; τF = 2 msec for Nav1.8+β1) before a standard test pulse to assay availability was applied. The amplitudes of the test currents were normalized to controls and plotted versus the prepulse interval. In these experiments the progressive decay of the currents observed with increasing prepulse duration reflects the entry of the channels into slow inactivated states from which the channels do not recover readily during the short hyperpolarization (−100 mV for 20 msec) that precedes the test pulse (Fig. 6). The onset of slow inactivation of the Nav1.7+β1 channels is fit with the sum of three exponentials with time constants of 32.6 ± 1.5 msec (τF), 556.5 ± 104.4 msec (τI), and 4071.9 ± 155.1 msec (τS; n = 6) (Fig. 6). For Nav1.8+β1 channels the onset of slow inactivation has time constants of 8.4 ± 0.2 msec (τF), 200.0 ± 30.0 msec (τI), and 8880.0 ± 1150.0 msec (τS; n = 5) (Fig. 6). These data indicate that the onset of slow inactivation (τF) is nearly fourfold faster for the Nav1.8+β1 channels than for Nav1.7+β1 channels. The rapid development of slow inactivation of Nav1.8+β1 may account for the unusually slow component of recovery from inactivation observed for this isoform. In contrast, the onset of slow inactivation of Nav1.7+β1 channels is delayed in comparison with Nav1.8+β1, and these channels consequently lack the slow component of recovery from inactivation (Fig. 5C).
Fig. 6.

Development of slow inactivation of Nav1.7+β1 and Nav1.8+β1 sodium channels. Time course of entry into the slow inactivated state was measured by using a triple-pulse protocol consisting of a variable duration conditioning pulse (2 msec to 10 sec) to −20 mV (Nav1.7+β1; open squares) or to +20 mV (Nav1.8+β1; open circles) to inactivate the channels. A 20 msec pulse to −100 mV was applied to promote the rapid recovery of inactivated channels, and a standard test pulse was used to assay availability. The test currents were normalized and plotted versus the conditioning pulse interval. The decay of the currents is fit best by the sum of three exponentials: I/IO= AF · (1 − exp(−t/τF)) +AI · (1 − exp(−t/τI)) +AS · (1 − exp(−t/τS)), where τF, τI, and τSare the time constants, and AF,AI, and ASare the relative weighting factors. t is the conditioning pulse duration, andI/Io is the normalized current. The data are the means ± SEM. The time constants of Nav1.7+β1 (open squares) are τF = 32.6 ± 1.5 msec (AF = 0.25 ± 0.03), τI = 556.5 ± 104.4 msec (AI = 0.33 ± 0.02), and τS = 4071.9 ± 155.1 msec (AS = 0.42 ± 0.03) (n = 6). For Nav1.8+β1(open circles) the time constants are τF = 8.4 ± 1.5 msec (AF = 0.57 ± 0.07), τI = 200.0 ± 30.0 msec (AI = 0.16 ± 0.07), and τS = 8880.0 ± 1150.0 msec (AS = 0.27 ± 0.04) (n = 5).
Frequency-dependent inhibition of Nav1.7+β1 and Nav1.8+β1 channels
The unusually rapid onset of slow inactivation of Nav1.8+β1 suggests that during short depolarizations (<10 msec) some of the channels are likely to enter into a slow inactivated state. This could have important consequences for the rapid cycling of channels during episodes of repetitive stimulation. The effects of rapid pulsing were tested by applying a series of 50 short (8 msec) depolarizing pulses (−20 mV for Nav1.7+β1 or +20 mV for Nav1.8+β1) at frequencies of between 0.5 and 100 Hz (Fig.7). The 8 msec depolarizing pulse used in the experiments is close to the somatic action potential duration of C fibers (0.6–7.4 msec) previously reported by Harper and Lawson (1985). The currents elicited by the individual test pulses were normalized to control currents and plotted versus the pulse number. For Nav1.7+β1 channels, pulsing frequencies up to 20 Hz have small effects on the amplitude of the currents (Fig. 7A,C; n = 10). The majority of the channels is capable of efficiently cycling via the open, closed, and inactivated conformations at these pulsing frequencies. Increasing the stimulation frequency to 50 or 100 Hz dramatically reduces the currents of Nav1.7+β1, indicating that the channels no longer fully recover during the short intervals between pulses. The decrease in amplitude for pulsing frequencies >25 Hz may reflect the trapping of some channels in slow inactivated states (Fig. 5C).
Fig. 7.

Frequency-dependent inhibition of Nav1.7+β1 and Nav1.8+β1 sodium currents. A,B, A train of 50 pulses was applied to −20 mV (Nav1.7+β1; n = 11) or +20 mV (Nav1.8+β1;n = 8) at frequencies between 0.5 and 100 Hz. The peak currents elicited by each test pulse were normalized to the current of the first pulse (Pn/P1, where n = 1–50) and were plotted versus pulse number. The pulse duration was 8 msec for frequencies between 0.5 and 50 Hz but was reduced to 5 msec for the 100 Hz experiments. The holding and interpulse potential was −100 mV. C, Plotted is the ratio of the currents elicited by the 50th and first pulses (P50/P1) versus the pulsing frequency. Insets are representative raw current traces of Nav1.7+β1 (open squares) and Nav1.8+β1 (open circles) stimulated at 20 Hz.
The response of Nav1.7+β1channels to rapid repetitive stimulation sharply contrasts with that of Nav1.8+β1 channels in which the pulsing frequencies of 1–2 Hz cause a significant reduction in current amplitude (Fig. 7B,C; n = 8). At frequencies >20 Hz the current amplitude reaches a steady-state level of ∼42% of the control current (Fig. 7B,C). The dissimilarity of Nav1.8+β1 and Nav1.7+β1 channels in their sensitivity to rapid repetitive stimulation could have important consequences on the firing frequency of cells predominantly expressing these types of sodium channels.
DISCUSSION
Despite a high degree of sequence homology, the α subunits of voltage-gated sodium channels exhibit substantial heterogeneity in their electrophysiological properties (Goldin et al., 2000). Subtle changes in the primary amino acid sequences can have significant effects in the gating and pharmacology of these channels. In addition to the functional differences encoded in the α subunits, most sodium channels are multimeric proteins that are associated with accessory β subunits (β1, β2, and β3) (Isom et al., 1992; McClatchey et al., 1993; Nuss et al., 1995). The β1 subunit is known to accelerate the kinetics of activation, inactivation, and recovery from inactivation of brain and skeletal muscle sodium channels (Auld et al., 1988). Modulation of these channels by the β1 subunit also contributes to the functional diversity of neuronal and muscle sodium channels.
In this study we examined the effects of the β1subunit on the functional properties of the Nav1.7 (PN1) and Nav1.8 (PN3) sodium channels heterologously expressed in Xenopusoocytes. Our data indicate that the β1 subunit alters the gating and voltage sensitivity of both channels and selectively increases the expression of the Nav1.8 channels.
The β1 subunit modulates levels of sodium currents of Nav1.7 and Nav1.8
The β1 subunit selectively enhances the expression of Nav1.8 channels, but not Nav1.7 channels (Fig. 2). Comparing the average sodium current from paired groups of oocytes indicates that the expression of Nav1.8 increases by almost sixfold in the presence of the β1 subunit versus 1.1-fold for Nav1.7. The selective regulation of Nav1.8 sodium channel expression by the β1 subunit may play an important role in regulating TTX-R current in vivo (Cummins and Waxman, 1997;Novakovic et al., 1998; Gold, 1999). A similar modulation of Nav1.8 sodium channel expression was observed recently with another auxiliary (β3) subunit (Shah et al., 2000).
The β1 subunit alters gating and voltage sensitivity of Nav1.7 and Nav1.8 sodium channels
Coexpression with the β1 subunit significantly alters the kinetics and voltage sensitivity of Nav1.7 and Nav1.8 sodium currents. In the absence of β1 the inactivation of Nav1.7 is slow and incomplete. The β1 subunit accelerates the current decay consistent with a more rapid rate of inactivation. At −20 mV the inactivation of Nav1.7 increases nearly 10-fold when the channels are coexpressed with the β1subunit. The β1 subunit also alters the voltage sensitivity of the channels, shifting the midpoints of steady-state activation and inactivation by −5.3 and −1.6 mV, respectively.
Similar changes in current kinetics and voltage sensitivity are observed when the Nav1.8 channels are coexpressed with the β1 subunit. Coexpression with the β1 subunit decreases the time constant of current decay at +20 mV by 1.6-fold, consistent with more rapid inactivation. In the presence of the β1subunit the midpoints of steady-state activation and inactivation of Nav1.8 channels are shifted significantly by −8 and −7.8 mV, respectively.
The β1 subunit alters the recovery from fast inactivation
The β1 subunit also significantly affects the time course of recovery from inactivation of the Nav1.7 and Nav1.8 sodium channels (Fig. 5C,D). The recovery from inactivation of Nav1.7 is well fit by two exponentials with time constants of 19 and 933 msec for Nav1.7 and time constants of 6.6 and 53 msec for Nav1.7+β1. The recovery of the Nav1.8 channels is also considerably slower and requires three exponentials to describe the time course adequately. Coexpression of Nav1.8 with the β1 subunit accelerates the fast component of recovery from inactivation but delays the intermediate and slow components of recovery from fast inactivation. The mechanism underlying this differential effect on the fast and slow components of inactivation is currently under investigation.
Our data indicate that the β1 subunit has significant effects on both the Nav1.7 and Nav1.8 channels, particularly on the kinetics of inactivation. A similar increase in inactivation rate with β1 coexpression recently has been reported for Nav1.7 channels (Shcherbatko et al., 1999). These findings are inconsistent with previous studies showing that the functional properties of Nav1.7 and Nav1.8 channels expressed in oocytes are not altered by the β1 subunit (Sangameswaran et al., 1996, 1997). Although we have no clear explanation for the differences between our data and those of these previous investigators, our data are consistent with studies showing that the β1 subunit modulates the gating and expression of many neuronal sodium channels (Isom et al., 1995; Shcherbatko et al., 1999).
Comparison with TTX-S and TTX-R sodium channels of DRG neurons
Multiple components of sodium current have been recorded from native DRG neurons that exhibit significant differences in gating and toxin sensitivity (Kostyuk et al., 1981; Roy and Narahashi, 1992; Ogata and Tatebayashi, 1993; Rush et al., 1998). The kinetics of the TTX-S currents are generally faster, and the steady-state activation and inactivation are shifted toward more hyperpolarized voltages in comparison with the TTX-R currents. These properties are qualitatively consistent with those of the Nav1.7 (TTX-S) and Nav1.8 (TTX-R) sodium channels observed in this study. For the Nav1.7+β1channels the midpoints of steady-state activation, inactivation, and the kinetics of inactivation are in reasonable agreement with the properties of TTX-S currents measured from acutely dissociated DRG neurons (Kostyuk et al., 1981; Roy and Narahashi, 1992; Ogata and Tatebayashi, 1993; Rush et al., 1998). The Nav1.7 channel, along with its associated β1 subunit, is likely to contribute to TTX-S sodium currents of both large and small DRG neurons (Ogata and Tatebayashi, 1993; Rush et al., 1998).
In contrast, the midpoint steady-state inactivation of Nav1.8+β1 (−62.6 mV) is more hyperpolarized than the native TTX-R sodium currents (−34 to −52 mV). The reasons for this discrepancy are unclear; however, a recent study has shown that at least three distinct components of TTX-R sodium current are present in the native cells (Rush et al., 1998). Variation in the relative expression levels of these different TTX-R sodium channels may explain the wide range of electrophysiological properties reported for the TTX-R currents of DRG neurons. In addition, the presence of additional β subunits (β2, β3) or a variation in second messenger regulation may contribute further to the differences in the inactivation of heterologously expressed Nav1.8 and the TTX-R current of DRG neurons. In addition, our data show that the development of slow inactivation of the Nav1.8+β1 channels is unusually rapid. This can influence the properties of steady-state inactivation by shifting the midpoint toward more hyperpolarized voltages (Ogata and Tatebayashi, 1992). Interestingly, a TTX-R3 component of sodium current of type D DRG neurons has been identified recently that has a midpoint of steady-state inactivation of −63 mV, identical to what we observed for the Nav1.8+β1 channels (Rush et al., 1998). Despite some quantitative differences the data indicate that coexpression of Nav1.8 and the β1 subunits in oocytes produces currents that have kinetics and voltage sensitivity similar to the TTX-R sodium currents of small DRG neurons.
Several studies have shown that the amplitudes of DRG neuron sodium currents are highly sensitive to the frequency of the applied voltage pulses (Rush et al., 1998; Scholz et al., 1998). In general, the TTX-R currents are reported to be more sensitive to rapid repetitive pulsing than the TTX-S sodium currents. These data suggest important differences in the repriming of these channels after a depolarizing voltage pulse. In this study the Nav1.8+β1 channels display a significant reduction in current amplitude during repetitive stimulation at frequencies between 1 and 2 Hz (Fig. 7). This contrasts with Nav1.7+β1 channels, which are considerably less sensitive to such low-frequency stimulation. The data suggest that during low-frequency repetitive stimulation (1–2 Hz) inactivated Nav1.8+β1 channels fail to recover fully during the interval between pulses. At 2 Hz the rest interval between pulses (492 msec) is sufficient to permit full recovery of fast inactivated channels (τF = 2 msec). The entry and recovery from fast inactivation cannot account for the observed frequency-dependent reduction of Nav1.8+β1 currents. Rather, the data suggest that a fraction of Nav1.8+β1 channels may enter into a slow inactivated state during the brief depolarizations. The time course of entry into the slow inactivated state has been measured directly by a double-pulse protocol (Fig. 6). The onset of slow inactivation of Nav1.8+β1 channels (τF = 8.4 msec) is considerably faster than that of the Nav1.7+β1channels (τF = 33 msec). During the short depolarizations used in the repetitive pulsing protocol (8 msec), a significant fraction of the Nav1.8+β1 channels, but not Nav1.7+β1 channels, is predicted to undergo slow inactivation. Few of these slow inactivated channels recover (τS = 1070 msec) during the short interval (492 msec) between pulses. The data indicate that the high sensitivity of the Nav1.8+β1 channels to low-frequency repetitive stimulation results from the unusually rapid entry of these channels into the slow inactivated state. A similar mechanism has been proposed for the TTX-R sodium current of DRG neurons (Scholz et al., 1998). Nav1.7+β1 channels are more resistant to slow inactivation and are significantly less sensitive to low-frequency repetitive stimulation.
Physiological relevance
Previous work suggests that the rapid repriming and high threshold of TTX-R sodium currents may play an important role in the sustained firing of C fibers after nerve injury (Elliott and Elliott, 1993;Jeftinija, 1994; Schild and Kunze, 1997). Although the majority of the Nav1.8+β1 channels rapidly recovers from inactivation, these channels are also more likely to entering slow inactivated states. During sustained repetitive firing a significant fraction of the Nav1.8+β1 channels is likely to accumulate in this slow inactivated state. However, at very high frequencies (>20 Hz) the amplitudes of Nav1.8+β1 currents reach steady state, being reduced 58% in comparison with the initial current level (Fig. 7). This is nearly equivalent to the fraction of channels (58.3%) that rapidly enters into the slow repriming state in response to sustained depolarization (τF = 8 msec) (Fig.7). The data suggest that during repetitive pulsing at high frequency (>20 Hz), or in response to sustained depolarization, 60% of the Nav1.8+β1 channels rapidly enter into slow inactivated states; however, the other 40% of active channels will contribute to action potential firing.
In addition, the atypical kinetics and voltage sensitivity of the Nav1.8+β1 sodium channels may contribute to the unusual electrical excitability of the small nociceptive neurons in which these channels are expressed preferentially (Caffrey et al., 1992; Arbuckle and Docherty, 1995;Novakovic et al., 1998). The relative contribution of the Nav1.8+β1 channels to the total sodium current of nociceptive neurons may contribute to the high threshold (MeLean et al., 1988) and slow firing frequency of C fibers (Harper and Lawson, 1985). The slow inactivation and recovery kinetics of the Nav1.8+β1 channels would tend to broaden the action potential and reduce the firing frequency of these neurons. These unique properties of the Nav1.8+β1 channels may play a role in the adaptation of nociceptive nerve impulses during low firing frequency. On the other hand, the resistance of Nav1.8+β1 channels to enter fully into the slow inactivated state during high-frequency (>20 Hz) stimulation, coupled with the high threshold for activation (V0.5 = −3.3 mV), could maintain a minimal level of sodium channel activity in rapidly firing or chronically depolarized neurons during sustain noxious stimuli. This may enable pain fibers to continue generating action potentials after peripheral nerve damage.
In conclusion, the present study shows that the modulatory effects of the β1 subunit are likely to have important consequences for the electrical excitability of the DRG neurons expressing these channels. However, the existence of other auxiliary subunits (β2 and β3) in these nociceptive C fibers (Shah et al., 2000; Coward et al., 2001) could have complementary regulatory effects on Nav1.7+β1 and Nav1.8+β1 function. The possible roles of the other β subunits need to be tested.
Footnotes
This study was supported by Grant MOP-49502 from the Canadian Institutes of Health Research (CIHR) and by National Institute of General Medical Sciences Grant GM58058. M.C. is an Edwards Senior Investigator (Joseph C. Edwards Foundation), and K.V. is a recipient of a doctoral research award of the CIHR. We thank Dr. Richard Horn and Dr. Yasushi Okamura for their comments on this manuscript and Dr. G. Mandel for providing the NaV1.7 construct.
Correspondence should be addressed to Dr. M. Chahine, Laval Hospital Research Center, 2725 Chemin Sainte-Foy, Sainte-Foy, Québec, Canada G1V 4G5. E-mail: mohamed.chahine@phc.ulaval.ca.
REFERENCES
- 1.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 2.Arbuckle JB, Docherty RJ. Expression of tetrodotoxin-resistant sodium channels in capsaicin-sensitive dorsal root ganglion neurons of adult rats. Neurosci Lett. 1995;185:70–73. doi: 10.1016/0304-3940(94)11227-a. [DOI] [PubMed] [Google Scholar]
- 3.Auld VJ, Goldin AL, Krafte DS, Marshall J, Dunn JM, Catterall WA, Lester HA, Davidson N, Dunn RJ. A rat brain Na+ channel α subunit with novel gating properties. Neuron. 1988;1:449–461. doi: 10.1016/0896-6273(88)90176-6. [DOI] [PubMed] [Google Scholar]
- 4.Bennett PB, Jr, Makita N, George AL., Jr A molecular basis for gating mode transitions in human skeletal muscle Na+ channels. FEBS Lett. 1993;326:21–24. doi: 10.1016/0014-5793(93)81752-l. [DOI] [PubMed] [Google Scholar]
- 5.Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG. Spinal sensory neurons express multiple sodium channel α subunit mRNAs. Brain Res Mol Brain Res. 1996;43:117–131. doi: 10.1016/s0169-328x(96)00163-5. [DOI] [PubMed] [Google Scholar]
- 6.Blackburn-Munro G, Fleetwood-Walker SM. The sodium channel auxiliary subunits β1 and β2 are differentially expressed in the spinal cord of neuropathic rats. Neuroscience. 1999;90:153–164. doi: 10.1016/s0306-4522(98)00415-1. [DOI] [PubMed] [Google Scholar]
- 7.Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD. Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res. 1992;592:283–297. doi: 10.1016/0006-8993(92)91687-a. [DOI] [PubMed] [Google Scholar]
- 8.Caldwell JH. Clustering of sodium channels at the neuromuscular junction. Microsc Res Tech. 2000;49:84–89. doi: 10.1002/(SICI)1097-0029(20000401)49:1<84::AID-JEMT9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 9.Chahine M, Bennett PB, George AL, Jr, Horn R. Functional expression and properties of the human skeletal muscle sodium channel. Pflügers Arch. 1994;427:136–142. doi: 10.1007/BF00585952. [DOI] [PubMed] [Google Scholar]
- 10.Coward K, Jowett A, Plumpton C, Powell A, Birch R, Tate S, Bountra C, Anand P. Sodium channel β1 and β2 subunits parallel SNS/PN3 α subunit changes in injured human sensory neurons. NeuroReport. 2001;12:483–488. doi: 10.1097/00001756-200103050-00012. [DOI] [PubMed] [Google Scholar]
- 11.Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci USA. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott AA, Elliott JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 1993;463:39–56. doi: 10.1113/jphysiol.1993.sp019583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gold MS. Tetrodotoxin-resistant Na+ currents and inflammatory hyperalgesia. Proc Natl Acad Sci USA. 1999;96:7645–7649. doi: 10.1073/pnas.96.14.7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldin AL, Snutch T, Lübbert H, Dowsett A, Marshall J, Auld V, Downey W, Fritz LC, Lester HA, Dunn R, Catterall WA, Davidson N. Messenger RNA coding for only the α subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes. Proc Natl Acad Sci USA. 1986;83:7503–7507. doi: 10.1073/pnas.83.19.7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Berwald-Netter Y, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 18.Harper AA, Lawson SN. Electrical properties of rat dorsal root ganglion neurones with different peripheral nerve conduction velocities. J Physiol (Lond) 1985;359:47–63. doi: 10.1113/jphysiol.1985.sp015574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 20.Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA. Functional coexpression of the β1 and type IIAα subunits of sodium channels in a mammalian cell line. J Biol Chem. 1995;270:3306–3312. doi: 10.1074/jbc.270.7.3306. [DOI] [PubMed] [Google Scholar]
- 21.Jeftinija S. The role of tetrodotoxin-resistant sodium channels of small primary afferent fibers. Brain Res. 1994;639:125–134. doi: 10.1016/0006-8993(94)91772-8. [DOI] [PubMed] [Google Scholar]
- 22.Kostyuk PG, Veselovsky NS, Tsyndrenko AY. Ionic currents in the somatic membrane of rat dorsal root ganglion neurons. I. Sodium currents. Neuroscience. 1981;6:2423–2430. doi: 10.1016/0306-4522(81)90088-9. [DOI] [PubMed] [Google Scholar]
- 23.Makielski JC, Limberis JT, Chang SY, Fan Z, Kyle JW. Coexpression of β1 with cardiac sodium channel α subunits in oocytes decreases lidocaine block. Mol Pharmacol. 1996;49:30–39. [PubMed] [Google Scholar]
- 24.Makita N, Bennett PB, Jr, George AL., Jr Voltage-gated Na+ channel β1 subunit mRNA expressed in adult human skeletal muscle, heart, and brain is encoded by a single gene. J Biol Chem. 1994;269:7571–7578. [PubMed] [Google Scholar]
- 25.McClatchey AI, Cannon SC, Slaugenhaupt SA, Gusella JF. The cloning and expression of a sodium channel β1 subunit cDNA from human brain. Hum Mol Genet. 1993;2:745–749. doi: 10.1093/hmg/2.6.745. [DOI] [PubMed] [Google Scholar]
- 26.MeLean MJ, Bennett PB, Thomas RM. Subtypes of dorsal root ganglion neurons based on different inward currents as measured by whole-cell voltage clamp. Mol Cell Biochem. 1988;80:95–107. doi: 10.1007/BF00231008. [DOI] [PubMed] [Google Scholar]
- 27.Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. Beta 3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci USA. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR, Eglen RM, Hunter JC. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18:2174–2187. doi: 10.1523/JNEUROSCI.18-06-02174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nuss HB, Chiamvimonvat N, Pérez-García MT, Tomaselli GF, Marbán E. Functional association of the β1 subunit with human cardiac (hH1) and rat skeletal muscle (μ1) sodium channel α subunits expressed in Xenopus oocytes. J Gen Physiol. 1995;106:1171–1191. doi: 10.1085/jgp.106.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogata N, Tatebayashi H. Slow inactivation of tetrodotoxin-insensitive Na+ channels in neurons of rat dorsal root ganglia. J Membr Biol. 1992;129:71–80. doi: 10.1007/BF00232056. [DOI] [PubMed] [Google Scholar]
- 31.Ogata N, Tatebayashi H. Kinetic analysis of two types of Na+ channels in rat dorsal root ganglia. J Physiol (Lond) 1993;466:9–37. [PMC free article] [PubMed] [Google Scholar]
- 32.Oh Y, Sashihara S, Black JA, Waxman SG. Na+ channel β1 subunit mRNA: differential expression in rat spinal sensory neurons. Brain Res Mol Brain Res. 1995;30:357–361. doi: 10.1016/0169-328x(95)00052-t. [DOI] [PubMed] [Google Scholar]
- 33.O'Leary ME. Characterization of the isoform-specific differences in the gating of neuronal and muscle sodium channels. Can J Physiol Pharmacol. 1998;76:1041–1050. doi: 10.1139/cjpp-76-10-11-1041. [DOI] [PubMed] [Google Scholar]
- 34.Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci USA. 1999;96:7640–7644. doi: 10.1073/pnas.96.14.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rizzo MA, Kocsis JD, Waxman SG. Selective loss of slow and enhancement of fast Na+ currents in cutaneous afferent dorsal root ganglion neurones following axotomy. Neurobiol Dis. 1995;2:87–96. doi: 10.1006/nbdi.1995.0009. [DOI] [PubMed] [Google Scholar]
- 36.Roy ML, Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci. 1992;12:2104–2111. doi: 10.1523/JNEUROSCI.12-06-02104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rush AM, Bräu ME, Elliott AA, Elliott JR. Electrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 1998;511:771–789. doi: 10.1111/j.1469-7793.1998.771bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sangameswaran L, Delgado SG, Fish LM, Koch BD, Jakeman LB, Stewart GR, Sze P, Hunter JC, Eglen RM, Herman RC. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J Biol Chem. 1996;271:5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- 39.Sangameswaran L, Fish LM, Koch BD, Rabert DK, Delgado SG, Ilnicka M, Jakeman LB, Novakovic S, Wong K, Sze P, Tzoumaka E, Stewart GR, Herman RC, Chan H, Eglen RM, Hunter JC. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem. 1997;272:14805–14809. doi: 10.1074/jbc.272.23.14805. [DOI] [PubMed] [Google Scholar]
- 40.Schild JH, Kunze DL. Experimental and modeling study of Na+ current heterogeneity in rat nodose neurons and its impact on neuronal discharge. J Neurophysiol. 1997;78:3198–3209. doi: 10.1152/jn.1997.78.6.3198. [DOI] [PubMed] [Google Scholar]
- 41.Scholz A, Kuboyama N, Hempelmann G, Vogel W. Complex blockades of TTX-resistant Na+ currents by lidocaine and bupivacaine reduce firing frequency in DRG neurons. J Neurophysiol. 1998;79:1746–1754. doi: 10.1152/jn.1998.79.4.1746. [DOI] [PubMed] [Google Scholar]
- 42.Shah BS, Stevens EB, Gonzalez MI, Bramwell S, Pinnock RD, Lee K, Dixon AK. Beta 3, a novel auxiliary subunit for the voltage-gated sodium channel, is expressed preferentially in sensory neurons and is upregulated in the chronic constriction injury model of neuropathic pain. Eur J Neurosci. 2000;12:3985–3990. doi: 10.1046/j.1460-9568.2000.00294.x. [DOI] [PubMed] [Google Scholar]
- 43.Shcherbatko A, Ono F, Mandel G, Brehm P. Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys J. 1999;77:1945–1959. doi: 10.1016/S0006-3495(99)77036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toledo-Aral JJ, Moss BL, He Z-J, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, Mandel G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA. 1997;94:1527–1532. doi: 10.1073/pnas.94.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallner M, Weigl L, Meera P, Lotan I. Modulation of the skeletal muscle sodium channel α subunit by the β1 subunit. FEBS Lett. 1993;336:535–539. doi: 10.1016/0014-5793(93)80871-q. [DOI] [PubMed] [Google Scholar]
- 46.Yang JS, Bennett PB, Makita N, George AL, Jr, Barchi RL. Expression of the sodium channel β1 subunit in rat skeletal muscle is selectively associated with the tetrodotoxin-sensitive α subunit isoform. Neuron. 1993;11:915–922. doi: 10.1016/0896-6273(93)90121-7. [DOI] [PubMed] [Google Scholar]