

Abstract
If, as is widely believed, information is stored in the brain as distributed modifications of synaptic efficacy, it can be argued that the storage capacity of the brain will be maximized if the number of synapses that operate independently is as large as possible. The majority of synapses in the brain are glutamatergic; their independence will be compromised if glutamate released at one synapse can significantly activate receptors at neighboring synapses. There is currently no agreement on whether “spillover” after the liberation of a vesicle will significantly activate receptors at neighboring synapses. To evaluate the independence of central synapses, it is necessary to compare synaptic responses with those generated at neighboring synapses by glutamate spillover. Here, synaptic activation and spillover responses are simulated in a model, based on data for hippocampal synapses, that includes an approximate representation of the extrasynaptic space. Recently-published data on glutamate transporter distribution and properties are incorporated. Factors likely to influence synaptic or spillover responses are investigated. For release of one vesicle, it is estimated that the mean response at the nearest neighboring synapse will be <5% of the synaptic response. It is concluded that synapses can operate independently.
Keywords: glutamate, synapse, AMPA, NMDA, transporter, synaptic transmission, diffusion, tortuosity, volume fraction, porous medium, synaptic cleft, vesicle, neurotransmitter
Spines of excitatory (glutamatergic) synapses have long been thought to be compartments capable of individual behavior, either electrical (Miller et al., 1985) or chemical (Zador and Koch, 1993). However, such compartmentation would be irrelevant if synaptic signaling at each spine was not independent of that at neighboring spines. Because information is stored in the brain as changes of efficacy at such synapses, their independent operation would have an important benefit: maximizing the information storage capacity of the brain. One prerequisite of independent signaling at spine synapses is that neurotransmitter (glutamate) released at one synapse should not significantly influence neighboring synapses, by activating (or desensitizing) their receptors.
Notwithstanding the evidence in favor of spillover or consistent with its occurrence in a number of experimental situations (Isaacson et al., 1993; Trussell et al., 1993; Barbour et al., 1994; Mennerick and Zorumski, 1994; Kullmann et al., 1996; Silver et al., 1996; Bergles et al., 1997; Clark and Barbour, 1997; Scanziani et al., 1997; Dzubay and Jahr, 1999; Carter and Regehr, 2000), little progress has been made in deciding whether glutamate release at a single synapse can activate receptors at a neighboring synapse. It is difficult to conceive of feasible experiments that could test this phenomenon, so modeling is still likely to give us a useful insight into the importance of this mechanism.
To evaluate the significance of spillover, it is necessary to compare receptor responses caused by spillover with those at the releasing synapse. Calculation of the spillover response obviously requires treatment of diffusion in the complex extrasynaptic structure of brain tissue. However, the manner of transmitter diffusion away from the synapse will also influence receptor activation within the synaptic cleft. Almost all previous models of synaptic activation have made no allowance for this, using an infinite-disk model that will dilute released transmitter much more slowly than the three-dimensional extrasynaptic tissue (Barbour and Häusser, 1997). Synaptic and spillover responses have been compared in one previous model that takes into account the extrasynaptic geometry (Rusakov and Kullmann, 1998b). The results of those calculations suggested that the ratio of spillover to synaptic responses could be as high as 75%.
Calculations are presented here that assess synaptic independence by comparing synaptic receptor activation and that caused by spillover, in a model in which the influence of the extrasynaptic space is taken into account. Analysis of the literature on diffusion measurements in brain tissue allows constraint of the glutamate diffusion coefficient. The influence of several factors that could alter synaptic and/or spillover responses is evaluated: glutamate receptors, glutamate binding sites, glutamate uptake, rate and amount of glutamate release, as well as the properties of the synaptic cleft. In contradiction to previous work, the present simulations predict that the effects of spillover are sufficiently minor to enable synapses to operate independently.
MATERIALS AND METHODS
Diffusion and receptor models. A representation of the synaptic and extrasynaptic space was developed that required only one distance dimension, i.e., which had radial symmetry. It is illustrated in the diagram of Figure1A. The synaptic cleft—the volume between the presynaptic and postsynaptic elements—was represented by a flat cylinder (disk) within which transmitter diffused freely. The concentration of diffusate was assumed to be uniform in the direction perpendicular to the cleft. In the model synaptic cleft, two regions were distinguished for the purpose of placing receptors: a central region corresponding to the postsynaptic density (PSD) and a surrounding annulus. The extrasynaptic space was modeled as a porous medium (Nicholson and Phillips, 1981; Nicholson and Sykova, 1998) in which the standard diffusion equations apply, after slight modification. An apparent diffusion coefficient is used that is related to the free diffusion coefficient by a parameter called the tortuosity, represented by λ. In fact,Dpm (for porous medium) =Dfree/λ2. The tortuosity represents the requirement that diffusion must circumnavigate obstacles, following longer paths between two points than would have been the case in the absence of obstacles. It is also necessary to account for the fact that the diffusion in which we are interested is restricted to the extracellular space, which occupies only a fraction of the total brain volume. In a porous medium, this restriction is described by the volume fraction α. Typical values of volume fraction and tortuosity for brain tissue are α = 0.2 and λ = 1.6 (Nicholson and Sykova, 1998). The use here of these particular values will be justified carefully below (Results). How a smooth transition was effected between the disk and porous medium regions will be described shortly.
Fig. 1.
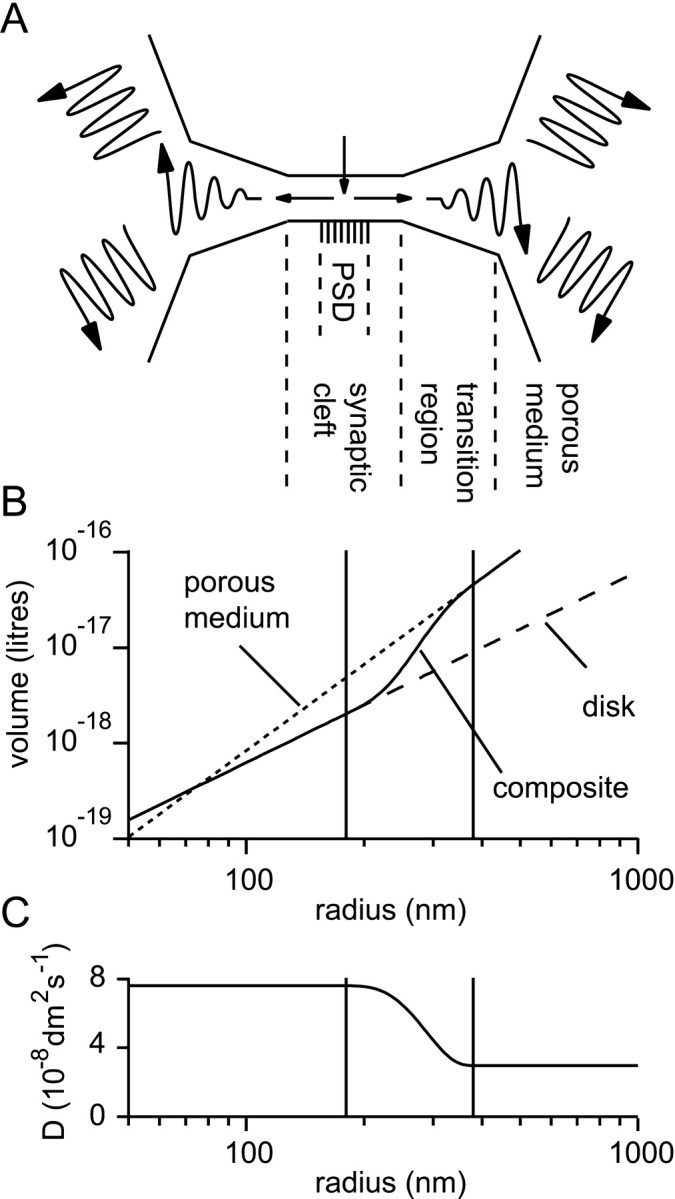
Representation of the synaptic cleft and perisynaptic region. A, Schematic representing the three different regions in the model. A disk-like synaptic cleft formed by the apposition of the presynaptic and postsynaptic elements contains a central PSD. A transition region extends from the cleft edge to a porous medium representing the bulk extracellular tissue.B, The extracellular volume, V, enclosed as a function of the radius, r, from the center of the model. Within the synaptic cleft the volume increases according to the law for the disk (cylindrical) geometry: V = πr2h (dashed line; h = 20 nm). A different law pertains for the three-dimensional porous medium that represents the distant extracellular space: V = α4πr3/3 (dotted line; α = 0.2). In the model a composite volume function (solid line) was constructed by joining an inner disk-like region to an outer porous medium-like region via a smooth transition region of 200 nm between the radii of 180 and 380 nm (indicated by the pair of vertical lines). C, In the porous medium, part of the model an effective diffusion coefficient (D) was used that was in general different to that used for the synaptic cleft (which was in most cases the diffusion coefficient for free medium). A smooth transition, analogous to that above for the volume function, was effected between these two regimes.
Diffusion in the synaptic cleft, porous medium, and transition region between the two was described by one general equation. Following an approach similar to that of Crank (1975), a diffusion equation with one distance variable was derived (although no claim to originality is made). Because the apparent diffusion coefficient in the porous medium will in general differ from that in the synaptic cleft, the form of the equation required for a non-constant diffusion coefficient was obtained:
| Equation 1 |
where V(r) is the volume available for diffusion within radius r. Putting D constant and substituting V = πr2h orV = (4/3)πr3(or even V = kr, k a constant), it is easy to verify that the above equation reduces to the standard forms for cylindrical or spherical (or plane) diffusion with a constant diffusion coefficient.
Little is known about the transition from disk-like synaptic cleft to porous medium extrasynaptic space, except perhaps the diameter of the synaptic cleft. The representation of the transition region is therefore somewhat arbitrary. A smooth transition with adjustable end points was constructed using an interpolating polynomial whose end points corresponded to the radii of the beginning and end of the transition region. In favor of a smooth transition, it can be argued that an abrupt transition would certainly be unrealistic. A smooth transition also helps reduce numerical errors. The interpolating function was chosen to have a value of 0 at its inner end point, of 1 at its outer end point, and zero first and second derivatives at both end points. These conditions ensured that no discontinuities occurred in Equation 1. The interpolating polynomial chosen was thus a quintic whose six coefficients were determined using the above boundary conditions. The volume function was thus:
| Equation 2 |
where f(r) is the interpolating function, and a and b are the radii of the inner and outer end points of the transition region. Also,
| Equation 3 |
(here, h is the cleft height: 20 nm) and
| Equation 4 |
(α is the volume fraction of brain tissue: 0.2). A similar transition of (apparent) diffusion coefficient between the cleft and the extrasynaptic space was also used:
| Equation 5 |
where, unless otherwise stated,Dcleft =Dfree, the diffusion coefficient for unhindered diffusion (7.6 × 10−8dm2/sec = 7.6 × 10−6cm2/sec; in fact the value for glutamine; Longsworth, 1953), and Dpm=Dfree/λ2; λ = 1.6. The choices of diffusion coefficient and tortuosity will be justified in Results. Figure 1B allows a comparison of a typical choice of V withVcleft andVpm. A similar plot, of Dwith Dcleft andDpm is shown in Figure 1C.
The model was populated with AMPA and NMDA-type glutamate receptors, glutamate transporters, and simple binding sites at concentrations described in Results. For each of the receptor types and the binding site, two concentrations were set independently: over the PSD and beyond the PSD. For transporters, two independent concentrations could also be set, but these were for the synaptic cleft (disk-like region) and for the transition region–porous medium. This was because most transporters are expressed in glial cell membranes, which are encountered only outside the region of apposition of the presynaptic and postsynaptic cells. The receptors were modeled according to published kinetic schemes. For AMPA receptors, the “set 1” rate constants of Jonas et al. (1993) were used. Although there is evidence that this model may be mechanistically inexact (Rosenmund et al., 1998), it well describes hippocampal AMPA receptor responses in patches. For NMDA receptors, the model of Lester and Jahr (1992) was used. The kinetics of the simple binding site were characterized by an association rate constant and a dissociation rate constant, the values of which will be given in Results.
A glutamate transporter model with a rapid glutamate-trapping step. In previous simulations of synaptic activation including transporters, only a very simple model was used (Barbour and Häusser, 1997; Rusakov and Kullmann, 1998b), in which transporters were represented as a binding site from which a slow reaction (transporter cycling) could eliminate bound glutamate. Recent work has suggested that an extension to the simple model is necessary. Recordings of transporter currents in the absence of anions that permeate the associated conductance have shown that a very brief transient is induced by rapid applications of glutamate (Bergles and Jahr, 1997; Auger and Attwell, 2000; Otis and Kavanaugh, 2000; Grewer et al., 2000). It is thought that this transient charge movement reflects a conformational change that traps the bound glutamate molecule, probably by translocation across the membrane. The rate of this step (of the order of 1000/sec) far exceeds the much slower overall cycling rate (14–50/sec). It seems likely that transporters with such glutamate-trapping behavior might be particularly effective (Auger and Attwell, 2000), so it is of interest to investigate the effect of such behavior in the present simulations.
A rapid glutamate-trapping step was accordingly incorporated into the simple transporter model. The new reaction scheme is shown in Figure2A. It is described by a second order differential equation. The two characteristic time constants of its solution assume simple forms for saturating and zero glutamate concentrations, allowing the four rate constants to be determined from as many simple experiments. The first piece of information is the apparent steady-state affinity for glutamate,
| Equation 6 |
The kinds of experiments that could be used to obtain the remaining kinetic information are illustrated in Figure 2, Band C, using simulated responses of the model to glutamate applications; the formulas relating the relevant time constants to the model rate constants are given in the figure. The kinetic determinations can to some extent be described by analogy with similar experiments on AMPA receptors. The time constants of “deactivation”, “desensitization”, and “recovery from desensitization” need to be determined (the first two in the absence of permeant anions). Of the four measurements required, only two have been determined accurately for hippocampal glial transporters (EAAT2/GLT1). These are the affinity (13 μm;Bergles and Jahr, 1997) and the recovery time constant (20 msec;Bergles and Jahr, 1998; Otis and Kavanaugh, 2000). The recovery time constant can be used to set k3 to 50/sec forthwith. Based in part on the few published experiments of the types shown in Figure 2, B and C, a range of plausible values was chosen for k+1(Wadiche and Kavanaugh, 1998; Mennerick et al., 1999; Auger and Attwell, 2000) and k2 (Bergles and Jahr, 1997; Otis and Kavanaugh, 2000; Auger and Attwell, 2000), allowing k−1 to be obtained from Equation 6 for the steady-state affinity.
Fig. 2.
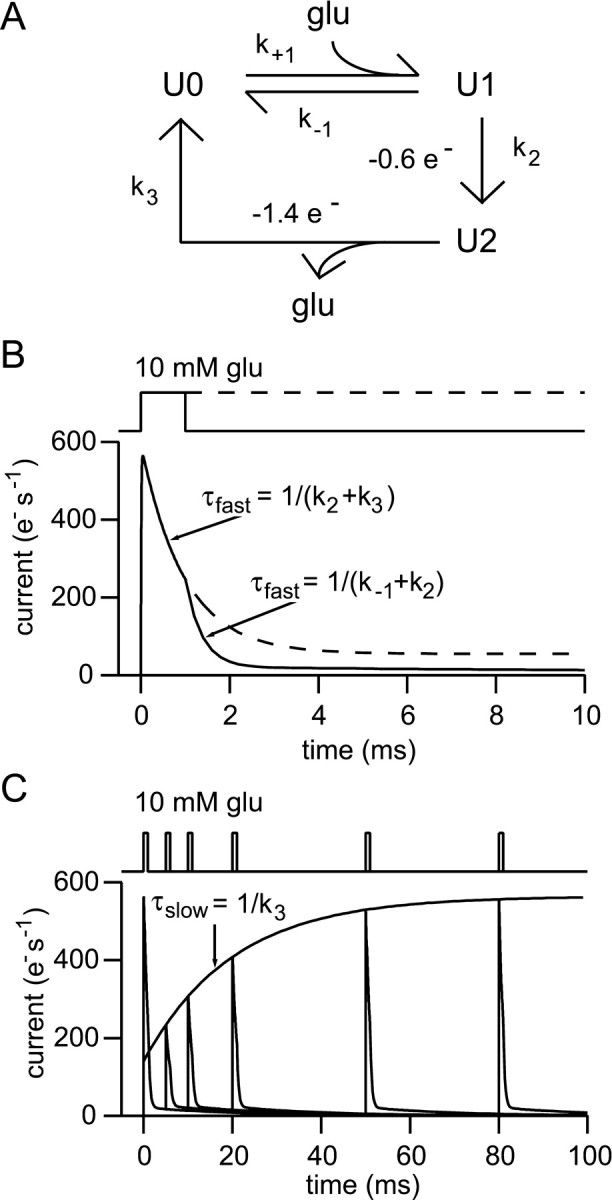
A simple glutamate transporter model including a fast “trapping” reaction. A, Reaction scheme for the transporter model used. The reaction with rate constantk2 represents the rapid trapping (probably by translocation) of bound glutamate. B, Simulated experiment in which rapid glutamate applications and measurement of transporter currents are used to determine the rate constants of the model from the “deactivation” and “desensitization” time constants. The glutamate concentration was 10 mm. The current is given in electronic charges per second per transporter and was calculated by assuming that two charges per cycle were divided between the reactions as shown in A. The choice made has no influence on the subsequent simulations of the effect of uptake on receptor activation. C, Simulated experiment showing determination of the recovery time constant (or cycling time). Pairs of brief applications (10 mm, 1 msec) were applied with different intervals, the first application being att = 0.
Method of solution. The coupled diffusion, receptor, and transporter equations were solved numerically using a space discretization by finite difference methods and integration by the first order explicit (Euler) method. Two spatial grids were used: the first extended from the point of release (r = 0) to a radius of 1 μm at 5 nm intervals; the second extended from there to 16 μm with 50 nm intervals. The first and second spatial derivatives of the concentration, volume, and diffusion coefficient were represented using the standard central difference approximations:
| Equation 7 |
| Equation 8 |
where X is the function to be approximated (e.g., concentration). The derivatives of the volume and diffusion coefficient were calculated with a high accuracy (small δr) at the beginning of each simulation, and the values were stored in a look-up table. At the outermost grid point, the concentration was clamped at zero, but this was sufficiently distant to influence only slightly the diffusion of transmitter in the region and time of interest. This can be shown by the close similarity between the receptor responses to numerically calculated glutamate transients and when the receptors were driven by analytical solutions for geometries where these exist (± <1% at their peaks; data not shown).
The apparent singularity in Equation 1 at r = 0 was dealt with according to Smith (1985). Briefly, noting that aroundr = 0, V = πr2h and the diffusion coefficient was always constant, Equation 1 reduces to:
| Equation 9 |
The last term assumes the indeterminate form 0/0 atr = 0, but application of l'Hôpital's rule shows it to have a limit ofD∂2C/∂r2as r = 0 is approached. Thus, Equation 9 is simplified to:
| Equation 10 |
| Equation 11 |
When release was directly into a porous medium, the factor of 4 in Equation 11 was replaced by a factor of 6 as required for the case of spherical diffusion.
In the model the glutamate released was placed at r = 0 and at the next grid point. To predict the appropriate concentration, it would be necessary to know the effective volume represented by these two grid points. This problem was circumvented by an iterative adjustment of the initial concentrations to fit the model's output to theoretical predictions. When non-instantaneous release was modeled, a steady flux into the first two grid points was maintained for the appropriate time.
It was found convenient during the calculations to convert the distance units of all quantities, in particular the diffusion coefficient, to decimeters (10−1 m; 1 dm3 = 1 liter), since concentrations were then obtained directly.
For synaptic receptor responses, the average activation (Popen) of receptors over the PSD is shown in all figures. This is the average calculated over the area within the radius of the PSD (120 nm), obtained by integrating numerically:
| Equation 12 |
Most of the parameter values were obtained from experiments performed at room temperature, which can therefore be taken as the temperature in the model.
Verification of the model. A time step of 10 nsec was used for the calculations. Halving the distance between space grid points and decreasing the time step by a factor of slightly more than four (which is necessary for stability of the numerical method) had only a very small effect on the solution, as assessed by the peak receptor activation under typical conditions, which was altered by <0.1%. It was concluded that the approximate numerical solution was converging to the true solution.
The full model was solved numerically because no analytical solution is available. It thus poses a problem of verification. It was, however, possible to test the model partially in various ways, against previous models or analytical solutions.
First, the diffusion part of the model was tested. It was verified that the calculated solution for diffusion within an infinite disk reproduced to a reasonable accuracy the solution obtained by analytical methods (Crank, 1975; Barbour & Häusser, 1997). It may be seen from Figure 3A, which shows both the analytical solutions and the calculations, that the calculations give satisfactory results. Very brief discrepancies between the numerical and analytical solutions exist after release; this is presumably the result of the different initial conditions of release. Quite large deviations from the analytical solution are caused by the loss of glutamate at the edge of the model. This becomes significant after 10–20 msec (depending on the geometry) in the periphery of the model. It was shown above that these discrepancies do not greatly influence the receptor responses in the region and time of interest.
Fig. 3.
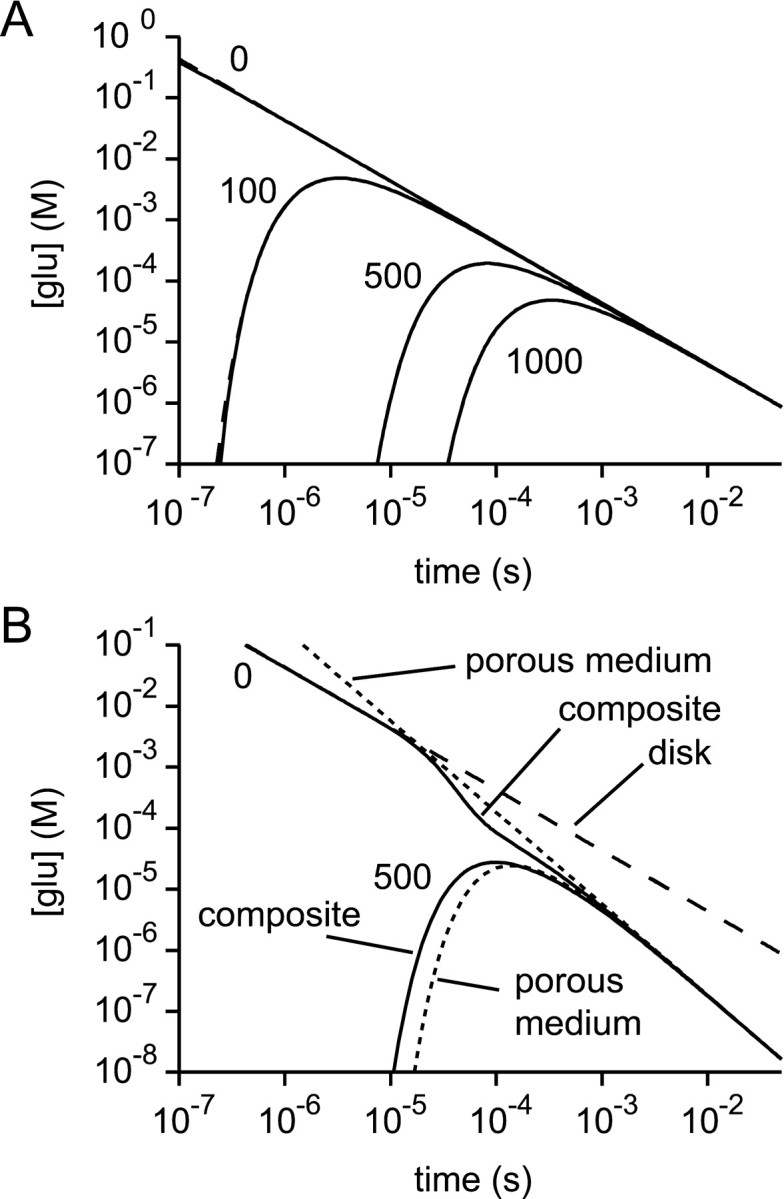
Verification of the diffusion section of the model. A, With a volume function representing an infinite disk, the glutamate concentration time courses at various radii (0, 100, 500, and 1000 nm) predicted by the model (solid lines) gave satisfactory fits to the corresponding analytical solutions (dashed lines, superimposed).B, With the composite volume function and variable diffusion coefficient of Figure 1 the numerical solutions (solid lines) for the glutamate concentration time courses atr = 0 and r = 500 nm eventually approach the analytical solutions for a porous medium (dotted lines). This behavior is expected, because of the tendency of diffusion to “forget” the initial conditions of release. In the composite structure, the time course at r = 0 initially follows the analytical solution for a disk (dashed line) before later approaching the analytical porous medium solution.
A general property of diffusion is that as time advances the initial conditions are “forgotten.” This provides one way of testing the calculations for composite geometries, in which a cylinder joins onto a porous medium (via the smooth transition), because at long times the solution should converge to that of the porous medium. This check is performed in Figure 3B. Although the concentration at the point of release initially follows the analytical solution for the infinite disk, the central concentration and the concentration at 500 nm display the expected behavior by approaching closely the analytical solution for the porous medium toward the end of the calculation.
It is depressingly easy in such calculations to generate or destroy diffusate. The total amount of glutamate present in each simulation was estimated by using Simpson's rule to evaluate the integral:
| Equation 13 |
where C is the concentration of glutamate in all forms. When the transition between disk and porous medium was too abrupt (<100 nm), significant amounts of glutamate were “lost.” For all the simulations shown, the value of the above integral (Eq. 13) did not deviate from the theoretical value by >0.5% between 1 μsec and 10–20 msec (depending on the geometry). The deviation at short times is attributable to the inaccuracy of the integration method when glutamate is present in only a few elements of the model. The deviation at longer times is attributable to the loss of glutamate at the edge of the model.
The implementation of the receptor models was checked by overriding the diffusion calculations of the simulation program and fixing the glutamate concentration as required. For the AMPA receptor model, numerous checks were performed using the values reported by Jonas et al. (1993) for the output of the model. In particular, the deactivation and desensitization time constants were verified, as well as the dose–response curve (EC50, Hill coefficient, and Popen,max). The developers of the NMDA receptor model did not publish such values. The present model does, however, verify their statedPopen,max of ∼0.3. The output of the model conforms to classical NMDA receptor kinetics. In response to a 1 msec application of 1 mm glutamate, the peakPopen was 0.257. The 10–90% rise-time of the response was 9.9 msec, and the decay time constant was 85.4 msec. The responses of the transporter model to various test pulses were verified against the analytical solutions.
RESULTS
Synaptic receptors are not saturated and spillover responses are small
Most models of central synaptic transmission represent the synaptic cleft as a flat cylinder or disk. However, instead of limiting this representation to the true extent of the cleft, it is extended effectively to infinity. This introduces a potential error, because diffusion in the extracellular space around the cleft is probably quite different from that in the cleft. Most obviously, the volume of the extracellular space increases as the cube of the radius, whereas for a disk the volume only increases as the square. This means that eventually the extracellular space is expected to dilute released transmitter much more rapidly than the infinite disk. This behavior is likely to have an important influence both on synaptic responses and on responses caused by spillover at neighboring synapses. Both will be investigated here, because careful assessment of the significance of spillover responses requires comparison with synaptic responses under the same conditions.
A model with a more realistic geometry was developed, in which a disk and a porous medium are combined (see Materials and Methods, Fig. 1). The composite structure is intended to represent a “standard” central synapse—a hippocampal synapse. The model synapse (representing “macular synapses” in the terminology of Ventura and Harris, 1999) had a PSD of radius 120 nm (Takumi et al., 1999), and the disk-like region extended beyond the PSD to a radius of 180 nm (Ventura and Harris, 1999). The disk was joined via a transition region of 200 nm to a porous-medium extracellular space with a volume fraction of 0.2 and a tortuosity of 1.6. The length of the transition region was (arbitrarily) chosen to correspond approximately to the radius of the synaptic cleft. The influence of this choice will be examined below.
Transmitter diffusion and receptor activation were first investigated in the presence of very low concentrations of AMPA and NMDA receptors, to enable their responses to be monitored without altering transmitter diffusion. The diffusion coefficient within the synaptic cleft was set to 7.6 × 10−8dm2/sec, and the effective diffusion coefficient for the porous medium was therefore 3.0 × 10−8dm2/sec. Responses to instantaneous release of 5000 molecules of glutamate (the likely content of a small synaptic vesicle; Bruns and Jahn, 1995) at the center of the synaptic cleft were calculated. Figure 4 shows the mean synaptic Popen (averaged over the PSD) for AMPA and NMDA receptors, as well as their responses at a radius of 500 nm, a typical distance for a neighboring synapse, to give an idea of the likely spillover response.
Fig. 4.

Receptor activation depends on geometry and the diffusion coefficient. A, B, Calculated average synaptic AMPA (A) and NMDA (B) receptor responses to release of one vesicle of glutamate for different combinations of geometry and diffusion coefficient. The composite model (solid line; geometry and D as in Fig. 1) is compared with an infinite disk with two values of diffusion coefficient (D = 7.6 × 10−8 dm2/sec,dashed line; D = 3 × 10−8 dm2/sec, dot-dashed line) and with an open volume surrounding the synapse (D = 7.6 × 10−8dm2/sec; transition region = 200 nm; α = 1; λ = 1; dotted line). C,D, Estimation of the effect of spillover. AMPA (C) and NMDA (D) receptor responses in the composite geometry (solid lines) at distances of 500 nm and 1 μm (the smaller responses). The responses in the composite medium are very similar to those of the receptors driven by the glutamate concentration given by the analytical solution for diffusion from an instantaneous point source in a porous medium with α = 0.2 and λ = 1.6 (dashed lines).
Overall, levels of synaptic receptor activation are quite low (compared with maximum possible Popen values for the AMPA and NMDA receptor models of ∼0.8 and 0.3, respectively). AMPA receptors reach only 15% of the maximumPopen and the figure for NMDA receptors is 20%. The responses caused by spillover are considerably weaker than the synaptic responses, with AMPA receptorPopen reaching 0.8% of the synaptic response, and NMDA receptors attaining a somewhat higher level of activity (3.9% of the synaptic response). We can see immediately that it is difficult to generate significant receptor activation by spillover.
As suggested by the similar time courses of glutamate concentration at radii of the order of 500 nm (and greater) in the composite geometry and for spherical diffusion from a point source in a porous medium (Fig. 3B, solid and dashed lines,respectively), the receptor activities at 500 nm generated by these two glutamate transients are very similar (Fig. 4C). This suggests that the presence of the synaptic cleft per se has little influence on receptor activity at neighboring synapses. This is not the case, however, for synaptic responses, which, as will be shown later, are quite sensitive to the properties of the synaptic cleft.
The difference made by using the present synaptic geometry is illustrated by simulations run for the infinite disk morphology, using two different values of diffusion coefficient (Fig.4A,B). Even with unhindered diffusion, the slower dilution of glutamate in the infinite disk model results in a 1.8-fold increase of AMPA receptor activation and a 3.0-fold increase for NMDA receptors. An even greater increase is observed when a lower diffusion coefficient is used with the infinite disk (a combination found in many previous models). The geometry and diffusion coefficient are therefore critical in determining receptor activation.
In the composite geometry used, the length of the transition region between the disk and porous medium was chosen arbitrarily, so it is important to assess the possible error introduced by this choice. Extreme upper and lower limits on synaptic activation are given by the infinite disk and “open volume” geometries simulated in Figure 4,A and B, (the latter may approximate exposed synapses in culture). Plausible values of the length of the transition region are a few hundred nanometers. Very short transition regions pose technical problems (see Materials and Methods), whereas longer transition regions are constrained by the necessity for the volume fraction of the tissue to be 0.2. If too long a transition region is used, the transition regions from neighboring synapses will overlap frequently, and the tissue volume fraction will fall below the required value. The receptor activation occurring for transition regions ranging from 100 to 300 nm was calculated. The effects on the peak synaptic responses were less than ±5%, respectively for AMPA receptors and less than ±3% for NMDA receptors. The spillover responses were equally insensitive to the length of the transition region in this range (less than ±3%). These tests suggest that large errors are not introduced by the arbitrary choice of the length of the transition region.
It may seem surprising that NMDA and AMPA receptors are activated to a similar degree (∼15–20% of maximumPopen), because NMDA receptors are generally considered to have a much higher affinity for glutamate than AMPA receptors (note however that the definition of affinity for AMPA receptors is problematic because of their desensitization). The explanation of this paradoxical result is that the synaptic glutamate transient is too brief to allow equilibrium binding to be approached. This can be illustrated with the simplest possible reaction of an agonist binding to a single site. If the agonist concentration is initially zero and the sites are unoccupied, the fraction of sites bound following a step change of the agonist concentration (att = 0) follows a time course described by:
| Equation 14 |
where C is the agonist concentration,Kd the dissociation constant (the ratio of the rate constants of dissociation,koff, and association,kon), and τ = 1/(Ckon +koff). The left-hand factor of Equation 14 (in curly braces) reflects the equilibrium binding position, whereas the right-hand term governs the exponential approach to equilibrium. If we set Ckon ≫koff (which is the same asC ≫ Kd), the above equation reduces to:
| Equation 15 |
which is independent of koff and therefore of equilibrium affinity. It is interesting to point out that under these conditions (and assuming the agonist is removed before the sites are saturated) it is perfectly possible for a “low-affinity” site with a fast kon to attain a higher occupancy than a “high-affinity” site with a slowkon. Synaptic receptor activation appears to operate under conditions resembling those for which Equation15 applies, and the similar kon values for AMPA and NMDA receptors lead to the approximately equivalent activation of the two receptor types.
The time-dependent binding behavior illustrated by Equations 14 and 15also explains why NMDA receptors at neighboring synapses are not saturated and are, in fact, hardly activated. Although glutamate concentrations that would saturate NMDA receptors can occur at neighboring synapses (e.g., 28 μm at 500 nm), these concentrations are not maintained for a sufficient length of time for equilibrium to be approached. In fact, the glutamate transient is so brief that receptor activation is unlikely.
The glutamate diffusion coefficient is not a free parameter
The value of the glutamate diffusion coefficient is a critical parameter that ultimately determines the degree of synaptic and spillover receptor activation. If diffusion is slower, receptor binding has more time to approach equilibrium before the glutamate is diluted. Unfortunately, this issue is very confused in the literature, and the present choice will require careful justification. The problem can be divided into two: determining the diffusion coefficient within the synaptic cleft and within the extrasynaptic space. The latter will be treated first.
It is an experimental fact that many tracer molecules restricted to the extracellular space diffuse through brain tissue with effective diffusion coefficients some 2.5-fold smaller than in dilute aqueous solution (Nicholson and Sykova, 1998; Nicholson et al., 2000). This is equivalent to saying that their diffusion can be described using the diffusion equations modified for use with a porous medium with a tortuosity value of 1.6. If such measurements existed for glutamate, the appropriate effective diffusion coefficient could be used without further ado. Unfortunately, the presence in brain tissue of powerful glutamate uptake systems and various glutamate binding activities, coupled with the difficulty of detecting glutamate on a microscopic scale (glutamate-sensitive microelectrodes do not exist and glutamate cannot be oxidized during amperommetry), makes it very unlikely that accurate direct measurements of glutamate diffusion in brain tissue will be obtained. Thus, all modellers are faced with the problem of deducing the properties of glutamate diffusion from measurements made with other tracer molecules.
Tracer molecules of either positive or negative charge and of molecular weights bracketing that of glutamate all diffuse with tortuosity values of ∼1.6. It might therefore seem reasonable to assume that glutamate behaves similarly, but this course of action has rarely been followed, and a number of different arguments have been advanced in justification, invoking viscosity, nonspecific binding, or specific binding. These will be considered in turn.
The measurements of diffusion in brain naturally include the effects on the tracer molecule of any viscosity, so a different tortuosity value for glutamate need only be used if it is affected differently by the viscosity of the extracellular fluid. The effects of solution viscosity can depend on the size of the diffusate molecules (smaller molecules being less sensitive to viscosity), and it has been suggested that the diffusion of glutamate [molecular weight (MW) 146] would be more affected than that of typical tracer molecules such as tetramethylammonium (MW 60) (Rusakov and Kullmann, 1998b). In fact, because tracer molecules both larger and smaller than glutamate diffuse similarly (for review, see Nicholson and Sykova, 1998), it is unlikely that glutamate itself behaves differently. Amazingly, there exist molecules with molecular weights of up to 106 that diffuse in brain tissue with tortuosities of 1.6 (Nicholson et al., 2000). However, some rigid macromolecules do exhibit somewhat higher tortuosities (up to 2.2). The values of tortuosity obtained for brain tissue could plausibly arise on geometrical grounds alone (Gardner-Medwin, 1980), and it is currently thought that the viscosity of the extracellular fluid is little different to that of normal saline (Nicholson and Sykova, 1998). It is concluded that a different tortuosity value for glutamate cannot be justified on the grounds of a different sensitivity to the viscosity of the extracellular fluid.
The principal argument against an important influence of nonspecific binding of glutamate and other molecules is that tracer molecules of different charges diffuse similarly (Nicholson and Sykova, 1998). The relatively low values of tortuosity determined for brain tissue also argue against a significant slowing of the diffusion of all tracer molecules. The absence of any such effect is not particularly surprising, because nonspecific binding sites would probably have low affinities for glutamate and would therefore need to be present in high concentrations to have a noticeable effect.
In contrast, it is certainly the case that glutamate is subject to specific interactions with numerous glutamate binding sites. These include the multitude of glutamate receptors and also several isoforms of glutamate transporters. The binding of glutamate to these binding sites will retard its diffusion. However, it is incorrect in a model to account for this by lowering the diffusion coefficient, because binding sites do not only retard diffusion, they also reduce the free concentration of diffusate. Although the slowed diffusion will tend to increase receptor activation, this effect will be opposed by the concomitant reduction of concentration. In fact, as will be shown in the next section, the introduction of glutamate “buffering” sites never increases receptor activation. Any binding sites considered potentially important should thus be modeled explicitly, without changing the diffusion coefficient of unbound diffusate. This also ensures correct modeling of the temporal and concentration-dependent behavior of the glutamate buffer.
Because empirical measurements of tortuosity are available, it is mostly of theoretical interest to estimate tortuosity by other means. Nevertheless, theoretical values have on occasion been preferred to the experimental determinations (Rusakov and Kullmann, 1998a,b). However, it should be noted that the ab initio calculations of geometrical tortuosity used to obtain those theoretical values (Rusakov and Kullmann, 1998a,b) are controversial and have attracted criticism from Chen and Nicholson (2000). The issue is amplified in a brief to this paper. For the simulations presented here, the empirically determined value of tortuosity has been used.
To summarize, if the extracellular space is to be modeled as a porous medium, which is the case here, the effective diffusion coefficient for glutamate should be obtained by setting the tortuosity equal to the value measured for small tracer molecules in the desired tissue. The influence of any important binding sites should be evaluated by explicit modeling, rather than by altering the diffusion coefficient for glutamate. The values of 1.6 for tortuosity and 0.2 for volume fraction that have been reported for many brain regions (Nicholson and Sykova, 1998) have been used here. Very similar values have been found for the rat hippocampus (Mazel et al., 1998). Significantly lower values of volume fraction for the hippocampus have been reported (α = 0.12; McBain et al., 1990) and used in previous modeling (Rusakov and Kullmann, 1998b). Mazel et al. (1998) suggest that the discrepancy arises because the influence of tissue anisotropy was not recognized in those studies. In any case, the disputed values concern the stratum pyramidale, which contains few excitatory spine synapses.
It is worthwhile to comment briefly on the problem of choosing the extracellular diffusion coefficient for a model in which the extracellular space is not treated as a porous medium but is instead represented explicitly, based on an electron microscopic reconstruction of a volume of neuropil. Diffusion in such models is conveniently investigated by Monte Carlo methods. As argued above, current thinking suggests that the diffusion coefficient for unhindered diffusion will correctly describe the behavior of glutamate within the extracellular space. However, it would in theory be necessary to confirm that the value chosen can reproduce the tortuosity measured in the tissue. This may not be trivial (and has not been reported), because it is not known how the volume and shape of the extracellular space are altered by fixation, although this compartment is suspected of shrinking.
Because synaptic clefts contribute a negligible fraction of the extracellular space, the properties of diffusion within them could be very different from those of the extracellular space without influencing diffusion measurements in bulk tissue. For this reason, little is known about diffusion in the synaptic cleft, and modellers still have some room for maneuver here. It is incorrect, however, to justify slowed diffusion in the synaptic cleft on the basis of the tortuosity of brain tissue, because the obstacles that make the brain tortuous are larger than the synaptic cleft and are outside it. It is the subject of speculation that the cleft may contain microscopic or submicroscopic obstacles to diffusion, and the possible effects of this will be investigated below. However, for most of the simulations here the choice of parsimony is made: it is assumed that glutamate diffuses freely in the synaptic cleft.
Glutamate buffers can only reduce receptor activation
Given the large number of glutamate receptor subtypes and transporters and the paucity of information about the distribution and kinetics of some of these molecules, it will be useful to investigate in general terms the possible effects of binding sites, in particular to assess the consequences of omitting some binding sites from the model. Therefore, before populating the model with realistically modeled receptors and transporters (except NMDA and AMPA receptors at negligible concentrations, to monitor receptor activation in the various conditions), the effects of placing simple binding sites at the synapse and/or in the extracellular space were investigated. A series of simulations were run in which 100 μm (20 μm with respect to tissue volume) binding sites with a kon of 107/(m sec) andkoff values of 103, 102, and 10/sec (giving Kd values of 100, 10, and 1 μm) were placed either in the synapse (over the PSD) and/or in the extracellular space (beyond the PSD). The results are illustrated in Figure 5.
Fig. 5.

Adding “generic” glutamate binding sites improves synaptic independence. Simple binding sites withKd values of 100 μm(dotted lines), 10 μm (dashed lines), or 1 μm (dot-dashed lines) were added throughout the model at a concentration of 100 μm in the extracellular space (20 μm tissue concentration). The synaptic responses (A, B) and spillover responses at 500 nm (C, D) of AMPA (A, C) and NMDA (B, D) receptors are shown, compared with the control situation in the absence of binding sites (solid lines).
In no case did the addition of binding sites lead to increased NMDA or AMPA receptor activation. It is sometimes argued that the slowed diffusion caused by glutamate binding will allow receptor activation to build up to higher levels. This is seen to be mistaken, for the simple reason that binding to the sites also reduces the free concentration of glutamate, which, of course, tends to reduce receptor activation. In fact, the time that a bound molecule spends not diffusing is exactly the time during which interaction with receptors (other than the bound site) is impossible. The reductions of free concentration and effective diffusion coefficient are therefore proportional. For receptor activation, the two effects exactly cancel under certain conditions (very brief glutamate transients). For binding sites with plausible kinetic properties, their effect will be time- and concentration-dependent and this, coupled with the receptor kinetic properties, causes a net reduction of receptor activation.
Several other important features emerged from these simulations. The first is that placing binding sites only in the cleft had remarkably little effect on receptor responses anywhere (data not shown). In addition, synaptic responses were in general rather difficult to alter, however binding sites were distributed. Importantly, in all the conditions modeled, the reduction of spillover responses after the addition of binding sites was greater than the corresponding decrease of the synaptic response, for both AMPA and NMDA receptors. This differential effect is plain in Figure 5.
Reasonable estimates are available for the synaptic and extrasynaptic concentrations of AMPA and NMDA receptors. It is thought that hippocampal synapses will contain no more than ∼200 AMPA receptors (Nusser et al., 1998), which is equivalent to 400 μmwithin the PSD radius of the model synapse. The synaptic NMDA receptor concentration is assumed to be some 15% (60 μm) of this on the basis of well clamped recordings of EPSCs in cerebellar granule cells (Silver et al., 1992). The concentrations of these receptors outside the synapse of interest can be estimated from binding experiments to be 1 μm for AMPA receptors and ∼150 nm for NMDA receptors (Barbour and Häusser, 1997). A simulation was run with these receptor concentrations, and the receptor responses were compared to the control case of negligible receptor concentrations. The differences were minor, for both synaptic and spillover responses. The largest effect was an 8% reduction of the spillover NMDA response (data not shown).
On this basis it is concluded that realistic AMPA and NMDA receptor concentrations have very modest effects on the glutamate transient and receptor activation in and around the synapse and were neglected in the following simulations (negligible concentrations were present to monitor their activation). Other receptors and binding sites (apart from glutamate transporters) will not be modeled. For most of them it is not expected that they will be present in sufficient concentrations to have an important effect on the responses under investigation here. Possible exceptions to this rule may include the kainate-binding protein present at high concentrations in avian and fish cerebella (Gregor et al., 1989; Barbour and Häusser, 1997). Metabotropic receptors may also be present in significant quantities, particularly in the cerebellum. However, the simulations above with the simple binding site allow these other receptors to be ignored in the knowledge that their most likely action would be to reduce spillover responses slightly, while leaving synaptic responses largely unaffected.
Glutamate transporters selectively limit spillover
The simulations above further confirm the widely accepted notion that dilution by diffusion is sufficiently rapid to terminate the synaptic action of glutamate. However, transporter concentrations are so high (Lehre and Danbolt, 1998) that it is natural to ask whether they could nevertheless influence the synaptic glutamate transient. A potentially important role of transporters is to limit the spread of glutamate from its site of release, thereby reducing spillover responses. To evaluate these transporter functions, the model was populated with transporters. The reaction scheme developed in Materials and Methods was used, and a quantitative study of transporter expression in the hippocampus guided transporter placement and the choice of concentration (Lehre and Danbolt, 1998).
Lehre and Danbolt (1998) concluded that the EAAT2 (GLT-1; the predominant form in the hippocampus) and EAAT1 (GLAST) transporter isoforms are present at a total concentration of 25 μm(which corresponds to 125 μm when referred to the extracellular volume). Simulations were therefore performed with 25 μm of glutamate transporters outside the synaptic cleft. In contrast, none were placed in the synaptic cleft, because in the hippocampus they appear to be expressed essentially only in glial cells, which are absent from the cleft (Lehre and Danbolt, 1998). Three values of k+1 (association) were tested: 5 × 106, 107, and 5 × 107(m sec). These were combined with values for k2(trapping) of 1000 and 2000/sec.
Including transporters, whatever their kinetic properties, led only to very modest reductions of synaptic responses (<2% for AMPA receptors, data not shown, and < 8% for NMDA receptors) (Fig.6A). This confirms that dilution by diffusion alone dominates the process of terminating the synaptic action of glutamate, at least at the kind of small isolated synapses modeled here.
Fig. 6.

Glutamate transporters selectively limit spillover. A, B, The effects of zero (solid lines), 25 μm (dashed lines), or 5 μm (dotted lines) transporters on synaptic (A) and spillover (500 nm; B) NMDA receptor responses. Transporters were distributed uniformly outside the synaptic cleft; their concentrations are with respect to tissue volume. C, Release of a single vesicle does not saturate nearby transporters (present at 25 μm). Solid line, 200 nm; dotted line, 300 nm; dashed line, 500 nm; anddot-dashed line, 1 μm. Subsequent simulations, unless otherwise stated, are in the presence of 25 μmtransporters outside the synaptic cleft.
Much more significant reductions of spillover responses were obtained after the addition of transporters to the model. For k+1 = 107and k2 = 1000, AMPA receptor responses at 500 nm were reduced by 32% (data not shown), whereas NMDA responses were reduced by 58% (Fig. 6B). The effect on spillover increased with distance: NMDA responses at 1 μm were reduced by 85%. Paradoxically, the results obtained (particularly for NMDA responses), showed almost no sensitivity to the rate constants chosen (over the combinations tested). The explanation for this counter-intuitive observation is that the dissociation rate constant was always determined by the choices of association and trapping rate constants, to preserve the apparent steady-state affinity. This dependence of the parameters introduces a compensatory action that ultimately leaves the effect of the transporters unchanged, suggesting that the present predictions may be relatively insensitive to uncertainties in the two poorly constrained rate constants.
These results show that transporters selectively limit spillover responses, especially over greater distances, but do not interfere with orthodox synaptic transmission. Although transporters have a significant effect in the above simulations, two uncertainties about the transport mechanism could reduce that effect. First, it is implicit in the model that in the absence of glutamate all the transporters soon accumulate in the state that can bind and rapidly trap glutamate. If the whole transport reaction is considered, it is not obvious that this should be the case, although the benefits of the system working in this way are plain. It is certainly possible, for instance, that at rest a fraction of transporters are in states requiring the binding of one or more sodium ions before glutamate can bind and be translocated, and this might be expected to reduce the effectiveness of the transporters on a synaptic timescale. In fact, the available evidence provides no support for this notion. There are two independent lines of evidence. In recordings of transporters (hEAAT2) expressed in oocytes, Wadiche et al. (1995) were able to resolve sodium-dependent charge movements after voltage steps. These currents were attributed to the binding and unbinding of one or more sodium ions. Inspection of their data show that near the resting potential of a hippocampal glial cell (−93 mV;Bergles and Jahr, 1997), the voltage-sensitive sodium binding would be nearly saturated. Raising the extracellular sodium concentration from the amphibian (∼100 mm) to the mammalian (∼145 mm) value will further decrease the small fraction of transporters without bound sodium.
An alternative point of view is suggested by an explicit model of the complete reaction cycle of a glutamate transporter (Otis and Jahr, 1998). According to their model, although 93% of transporters will have bound at least one sodium ion at rest, only a small fraction of transporters will have bound all three required for glutamate to bind. However, all the rates of sodium binding (and unbinding) are very rapid indeed (the sodium concentration is hardly limiting… ), so the states immediately preceding the glutamate-receptive state can be considered at equilibrium with it under all relevant conditions. The final effect will be to reduce the apparent affinity for glutamate. However, because the parameters of the model are determined by measurement of the apparent affinity, any such effect is automatically taken into account. The model of Otis and Jahr (1998) also leaves open the possibility that protons must bind before glutamate, with a rate that may not allow the approximation of equilibrium under all conditions. However, this issue has been reexamined by Auger and Attwell (2000), who show that proton binding probably occurs in a different part of the transport cycle.
The second problem is that glutamate transporters may form multimers, and it is currently unclear whether the functional unit of transport is a pentamer (Haugueto et al., 1996; Eskandari et al., 2000) or the monomer, which has been the assumption here so far. If a single functional transporter is a pentameric assembly, the true concentration of transporters in the model must be divided by five, which will reduce their effect. The results of a simulation with one-fifth of the previous transporter concentration have accordingly been added to Figure 6, A and B.
If nearby transporters are saturated by release of a vesicle of glutamate, they will become ineffective at limiting spillover for succeeding release events. Recordings of transporter currents in hippocampal glia have indicated that transporters are not saturated in this way (Diamond and Jahr, 2000). This behavior is reproduced in the model (Fig. 6C), which shows that the levels of transporter “occupancy” are in fact quite low, even at the exit from the synaptic cleft.
Duration of glutamate release and timing of receptor activation
Thus far, release of transmitter has been modeled here as an instantaneous event. Obviously, release will in fact be spread out over time, although little precise information is available concerning the time course of release from a small synaptic vesicle. It is therefore worthwhile to evaluate by simulation the influence of the duration of release on receptor activation. The issue of release duration has been lent importance by the suggestion that an acceleration of initially slow release could explain the transition from “silent” synapses (Isaac et al., 1995; Liao et al., 1995), with only an NMDA receptor-mediated response detectable, to “normal” synapses, with an AMPA receptor-mediated component also observable (Choi et al., 2000).
It is instructive to calculate exactly when AMPA and NMDA receptors are activated after instantaneous transmitter release. Here, “activation” is defined as the binding of a second glutamate molecule, because in both receptor models this is the necessary and sufficient condition for the channel to open (with a certain probability). By following the rate at which binding of a second glutamate occurs, it is possible to determine the time course of receptor activation. The rate at which receptor activation occurs is therefore given by the probability of being in the state with one bound glutamate multiplied by the rate for glutamate binding, thus:
| Equation 16 |
In the notation of Jonas et al. (1993) the monoliganded state of the AMPA receptor model is C1, and k[glu] is given by k+2c (wherec is the glutamate concentration). In the AMPA receptor model, a second glutamate could, in theory, also bind to the monoliganded desensitized state (C3). This flux was not included when calculating receptor activation and always negligible under the conditions of the present simulations. In the notation ofLester and Jahr (1992), the monoliganded state of the NMDA receptor is “AR”, and the corresponding rate constant iskon. If the activation rate (Eq. 16) is integrated with respect to time, it is possible to calculate the number of receptor activations occurring before a certain time. This analysis was performed for instantaneous release (in the presence of glutamate transporters), and the “activation integral” was then normalized to its maximum (Fig.7A). Receptor activation is terminated extremely quickly. Some 70 (NMDA) to 80% (AMPA) of activated receptors are activated within ∼35 μsec of release. For AMPA receptors, 95% of all receptor activations occur by 160 μsec, whereas for NMDA receptors the same point is reached in 760 μsec. This reinforces just how quickly dilution of transmitter by diffusion is able to terminate the synaptic action of glutamate. For instantaneous release, very little AMPA receptor activation therefore takes place after the peak of the EPSC (which occurs at 440 μsec). If release takes place over a time longer than a few tens of microseconds, it is clear that the time course of receptor activation will largely follow the time course of release.
Fig. 7.
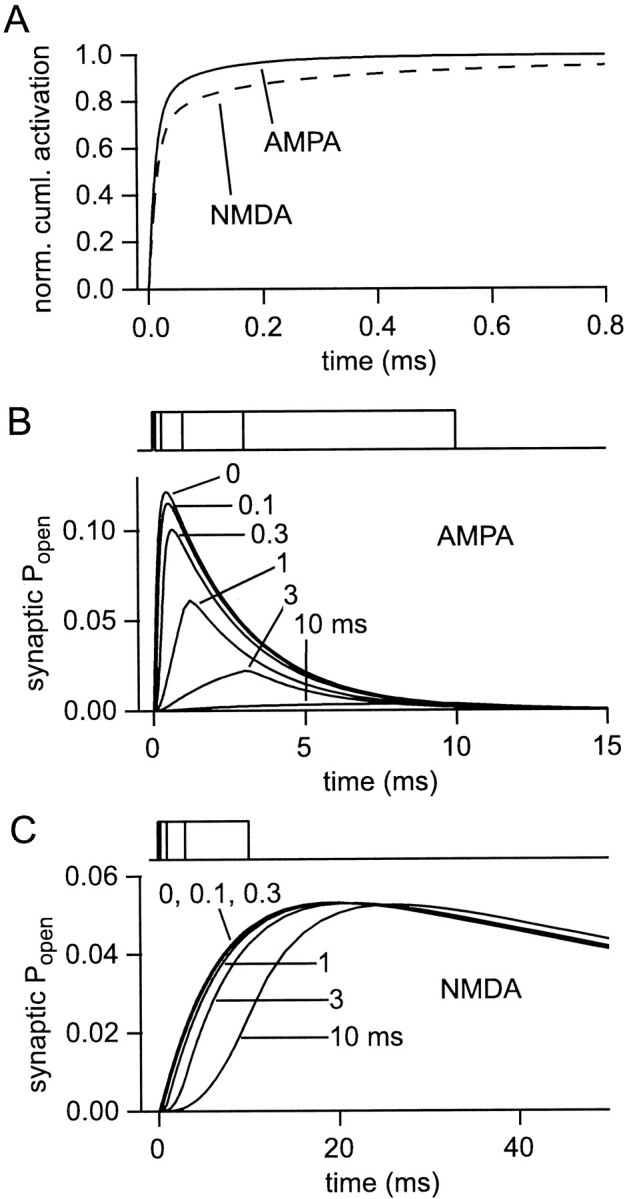
Timing of glutamate release and receptor activation. A, Receptor activation is terminated very rapidly. Curves showing when synaptic AMPA (solid line) and NMDA (dashed line) receptors are activated, defined as binding a second glutamate molecule (see Results). The curves represent the cumulative fraction of all activations (up to 50 msec) occurring after instantaneous release of a single vesicle.B, C, Slow glutamate release reduces AMPA responses and delays NMDA responses. Families of synaptic AMPA (B) and NMDA (C) receptor responses to the instantaneous release of a vesicle (“0”) or to release of the contents of one vesicle at a uniform rate over 0.1, 0.3, 1, 3 or 10 msec (timing indicated above each panel).
To investigate the influence of the time course of transmitter release, the receptor responses to the same amount of glutamate released at various uniform rates were calculated (Fig. 7B,C). For instantaneous release, the ratio of peak NMDA response to peak AMPA response (in terms of fraction of maximalPopen) is 1.21. Releasing the glutamate over a relatively short time, 100 or 300 μsec instead of instantaneously, had little effect on the overall aspect of the AMPA (or NMDA) responses. Nevertheless, the time-to-peak of the AMPA response increased (from 440 μsec) to 500 and 640 μsec, respectively, whereas the amplitude decreased slightly, by 7 and 19%, with respect to the response to instantaneous release. However, as the glutamate release is prolonged, the NMDA/AMPA response ratio indeed increases dramatically, e.g., to 41.3 for release over 10 msec. The mechanism is simply that the slower dissociation of glutamate from the NMDA receptor permits the accumulation of the occupancy of those sites over much longer times than for the AMPA receptor.
These simulations confirm the theoretical validity of the mechanism proposed by Choi et al. (2000). Interestingly, the simulations generate an additional prediction with which to test their hypothesis. A slowing of release sufficient to render the AMPA response undetectable might also be expected to generate an observable delay of the rising phase of the NMDA response. Thus, if induction of LTP causes an acceleration of initially slow glutamate release (Choi et al., 2000), the rising phase of the NMDA component should be advanced in time.
Prolonging release duration can reduce the specificity of synaptic activation, because the spillover/synaptic ratio of AMPA responses falls somewhat (approximately twofold for release lasting 10 msec; data not shown). However, the effect is small, and synaptic activation is reduced to very low levels. The ratio for NMDA responses is hardly altered.
Multivesicular release impairs synapse specificity
It has long been assumed that small central synaptic contacts are not capable of releasing more than one vesicle of transmitter per presynaptic action potential. The inaccessibility of most central synapses and the difficulty of isolating a single contact have made this a remarkably difficult hypothesis to test. Nevertheless, there is some evidence that multivesicular release can occur (Auger et al., 1998). The family of response curves in Figure8 shows the results of multivesicular release (and can also be used to assess the effects of variations in the vesicular content of glutamate). When five vesicles are released instead of one, synaptic AMPA Popenrises from 0.12 to 0.44, and NMDAPopen rises from 0.053 to 0.21. Note that, despite the increasing amplitudes, there are only minor kinetic changes, reflecting the rapidity of transmitter dilution compared with receptor gating kinetics. Spillover responses for both receptor types are increased even more than the synaptic responses. The AMPAPopen at 500 nm rises from 0.00069 to 0.016, whereas the NMDA response at the same distance is increased from 0.00093 to 0.019. Synaptic independence is therefore impaired.
Fig. 8.

Multivesicular release impairs synaptic independence. Synaptic (A, B) and spillover (500 nm;C, D), AMPA (A, C), and NMDA (B, D) receptor responses are shown for the release of 1, 2, 3, 4, and 5 vesicles containing 5000 molecules of glutamate each (more vesicles give larger responses in all cases). The spillover/synaptic response ratio rises as more vesicles are released.
The synaptic responses for multivesicular release are likely to be somewhat underestimated here, because all the vesicles are released at the same point, leading to some mutual occlusion of their responses near the point of release. Multivesicular release at real synapses will usually involve release at different points over the PSD. A limit on this error is provided by a saturated synaptic response. Thus, for five vesicles, the true responses could not be >80% (AMPA) and 38% (NMDA) greater than those shown. If the synaptic responses are underestimated, the relative significance of spillover will be overestimated in proportion.
Obstructing diffusion in the synaptic cleft enhances synaptic independence
It was argued above that the value of the effective diffusion coefficient for the extracellular space, which is modeled as a porous medium, has been tightly constrained by experiment. In contrast, very little is known about the properties of diffusion in the synaptic cleft. It has been suggested that synaptic clefts might contain sufficient material to retard diffusion on a microscopic or submicroscopic scale, much in the same way that brain tissue retards diffusion on a larger scale. Such an effect (approximately twofold) has been deduced for diffusion of acetylcholine at the neuromuscular junction and attributed to the basal lamina (Land et al., 1984). There are also reports of material within central synaptic clefts (Ichimura and Hashimoto, 1988). However, because the arguments in favor of retarded diffusion are rather indirect, especially as concerns central synapses, this must be regarded as an open question. Nevertheless, the effect of retarding diffusion within the cleft is interesting and potentially beneficial to synapse operation. This can be judged from the simulations of Figure 9, where diffusion within the synaptic cleft is modeled as occurring in a porous medium with different values of volume fraction and tortuosity. The decreased volume fraction concentrates the released transmitter while the tortuosity causes retention of transmitter in the cleft. Both these changes lead to increased receptor activation at the synapse. What is particularly interesting is that the extrasynaptic responses are hardly affected. This means that introducing obstacles to diffusion within the cleft would produce more synaptic receptor activation for a given amount of transmitter, an economy of transmitter. It would also improve the spatial selectivity of synaptic transmission, because the ratio of the synaptic to spillover responses is increased.
Fig. 9.

Obstructions to diffusion in the synaptic cleft increase synaptic independence. Possible submicroscopic obstacles to diffusion within the synaptic cleft were modeled by setting the volume fraction and tortuosity of the synaptic cleft to the following pairs of values: αcleft = 1, λcleft = 1 (ctrl, unimpeded diffusion); αcleft = 0.7, λcleft = 1.3; αcleft = 0.3, λcleft = 1.7; αcleft = 0.1, λcleft = 1.9. Increased synaptic AMPA (A), and NMDA (B) responses are observed as α is decreased and λ is increased, but spillover responses (500 nm; C, D) are unchanged.
The combinations of cleft volume fraction and tortuosity chosen are physically plausible combinations, in the sense that approximately equivalent combinations have been measured for anoxic brain tissue (Chen and Nicholson, 2000). Thus, unless the obstacles have a very special structure, very dense packing of obstacles is required to generate high tortuosities. It should be noted that the extreme values of cleft volume fraction and tortuosity modeled above represent an improbably dense packing of obstacles to diffusion, denser even than that, on a larger scale, of brain tissue.
These simulations make clear the sensitivity of synaptic responses to the properties of the synaptic cleft. Some possible variations in the details in the cleft geometry have not been modeled here, because quantitative data on cleft geometry have not been reported: a narrower cleft; a cleft that is wider centrally and narrows toward the PSD edge. Both of these situations would be expected to increase synaptic responses, without affecting spillover responses; synaptic independence would be enhanced.
Receptor desensitization after synaptic activation
AMPA receptors undergo profound desensitization. It is possible that desensitization at the synapse could alter the ratio of spillover to synaptic responses, so the possible influence of this mechanism was investigated. A brief application of glutamate to a patch containing AMPA receptors can induce quite a large reduction of the amplitude of the response to a second application, despite little desensitization appearing to occur while channels are open during the first pulse (Colquhoun et al., 1992). In the AMPA receptor model used here this phenomenon is accounted for by a high probability of the receptor entering the desensitized state when only one glutamate is bound. Such observations have led to the expectation that desensitization should similarly provide a depressive mechanism during paired-pulse synaptic stimulation. Surprisingly, at least in some preparations, little or no desensitization could be observed (Stevens and Wang, 1995; Debanne et al., 1996; Hjelmstad et al., 1999). In contrast, Otis et al. (1996)have shown that at synapses in the chick cochlear nucleus, a compound EPSC will desensitize ∼40% of receptors.
Investigation of synaptic desensitization is often confounded by the profound presynaptic changes that occur during paired-pulse stimulation, so the present model was used to evaluate the expected contribution of receptor desensitization during paired pulse stimulation. The release of two vesicles separated by various intervals was simulated (Fig. 10), and the ratio of the AMPA receptor responses calculated. Desensitization has a modest effect (15% at a 10 msec interval). This is in fact expected, given the low levels of activation induced by one vesicle: receptors that do not bind glutamate cannot desensitize. Moreover, this represents a slight overestimate of the effect of desensitization, because normally a second vesicle would not be released exactly at the same point as the first and would therefore usually face even fewer desensitized receptors. These results provide a possible reconciliation between the desensitization behavior observed in patches and the apparent lack of desensitization at hippocampal synapses, without the need to postulate different kinetic properties for receptors in the two preparations. Nevertheless, even such a small effect might have been detectable in some of the preparations (Stevens and Wang, 1995; Hjelmstad et al., 1999), suggesting that synaptic AMPA receptors may desensitize more slowly than those in patches or that receptor occupancy is even lower than predicted here. In any case, the many different types of AMPA receptors probably have different desensitization behavior (Liu et al., 1999). Naturally, multivesicular release or the obstruction of diffusion would increase desensitization.
Fig. 10.

A single vesicle desensitizes a small fraction of synaptic AMPA receptors. Simulated synaptic paired-pulse experiment, showing the amplitudes of synaptic responses to successive vesicle liberations separated by 10, 20, 50, and 100 msec. The peak effect of desensitization causes a 15% reduction in synaptic AMPA receptorPopen for the second vesicle.
Calculating the average spillover response
To summarize the various factors that can affect synaptic independence, the peak Popen values for AMPA and NMDA receptors are shown in Figure11, A and B, as a function of the distance from the point of release. Several simulations are plotted: a single vesicle in the absence of uptake, showing the effect of diffusion alone; a single vesicle in the presence of uptake; three vesicles in the presence of uptake; and a single vesicle with an obstructed synaptic cleft in the presence of uptake. AMPAPopen falls off very rapidly with distance in all the simulations, and it seems unlikely that activation of AMPA receptors by spillover glutamate will play an important role. The situation for NMDA receptors is less clear, however, and a more careful analysis is required to assess the effects of spillover.
Fig. 11.
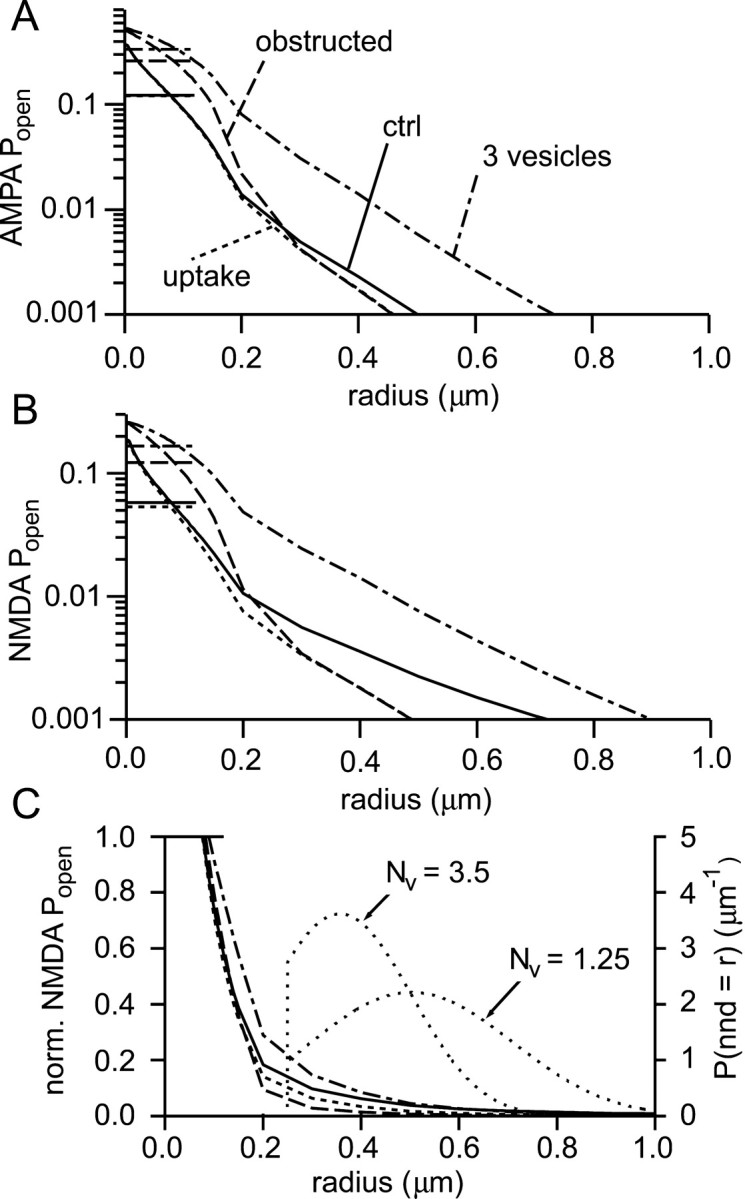
Assessing synaptic independence.A, Peak AMPA receptor Popenas a function of distance under a variety of conditions: a single vesicle with no uptake (solid; ctrl), with uptake (25 μm transporters; dotted,uptake), three vesicles (with 25 μmtransporters; dot-dashed, 3 vesicles), and with obstructions to diffusion in the synaptic cleft (αcleft = 0.7, λcleft = 1.3; one vesicle with 25 μm transporters; dashed,obstructed). The average peak synapticPopen values are represented by thehorizontal line segments in the top leftcorner of the graph. The synaptic responses in descending order are: 3 vesicles, obstructed, ctrl ≈ uptake. B, Analogous plot for NMDA receptors. Same key as in A.C, The Popen curves fromB for NMDA receptors are replotted (left axis; same key as in A) after normalization to the average synaptic Popen in each condition (horizontal line segment top left). Two probability density functions (lightly dotted lines) for the distance to the nearest neighbor synapse are plotted (right axis) and labeled with the corresponding densities:Nv = 3.5 synapses μm−3 or Nv = 1.25 synapses μm−3. nnd, Nearest neighbor distance.
To simplify comparison between different conditions, the curves for NMDA receptor Popen have been normalized to their respective average synaptic values (Fig.11C). To estimate average spillover responses, the calculated Popen values must be combined with knowledge of intersynaptic distances. Unfortunately, although a distribution of distances to nearest-neighbor synapses for hippocampus has been published (Rusakov et al., 1999), quite large discrepancies exist between the synaptic density reported (1.25/μm3) and that observed in another study, in hippocampal slices (∼3.5/μm3; Sorra and Harris, 1998). It will therefore not be possible to obtain a unique result. Estimated distributions of distances to nearest neighbor synapses were obtained as follows. It was first assumed that synapse positions are random (i.e., a Poisson process). However, in this distribution, synapses can be arbitrarily close, which is of course impossible. A region around the liberating synapse was therefore cleared of synapses within a certain radius (without altering the rest of the distribution). For consistency with the size of synapse modeled here, that radius should be 360 nm. However, synapses do vary in size, and shorter intersynaptic distances are found. To allow for this, a compromise boundary of 250 nm was chosen. The probability density function for the distance to the nearest neighbor (nnd) for this distribution is given by:
| Equation 17 |
where r0 = 250 nm as just described, and NV is the density of synapses in the original Poisson distribution. This distribution was calculated for the two reported synapse densities (Fig.11C). Using these distributions, it is now possible to calculate the mean receptor activation for nearest neighbor synapses. In the order (1) one vesicle without uptake, (2) one vesicle with uptake, (3) three vesicles with uptake, and (4) one vesicle with uptake and an obstructed cleft, the mean spillover/synaptic NMDA response ratios were: (1) 0.07, (2) 0.04, (3) 0.10, and (4) 0.02 for the dense synapse packing; (1) 0.04, (2) 0.02, (3) 0.06, and (4) 0.01 for the lesser synapse density. The results show that NMDA receptor activation resulting from spillover can be considered unimportant, even in the absence of glutamate uptake, except perhaps if significant multivesicular release occurs.
DISCUSSION
Synapses can operate independently
The present synaptic model, which takes into account the three-dimensional extrasynaptic space and incorporates the correct diffusion coefficient for glutamate, predicts that synaptic receptors are far from saturated by a single vesicle of glutamate and that spillover responses are small in comparison. This suggests that typical small glutamatergic synapses can operate independently. However, whether such synapses do always operate independently, particularly as regards NMDA receptors, remains a finely balanced question. The above conclusions apply at room temperature (at which most of the parameters of the model were determined) for synaptic transmission that is isolated, both in space and time. The ability of NMDA receptors to integrate activation over tens or even hundreds of milliseconds means that it will be necessary to know more about the density and frequency of synaptic activations in the brain before deciding whether spillover has a role to play in activating NMDA receptors. It is therefore interesting that only a small fraction of the total number of synaptic inputs that a pyramidal cell receives need to be activated to excite it (Sayer et al., 1990; Larkman et al., 1991; Otmakhov et al., 1993). Moreover, in vivo recordings suggest that only a small fraction of hippocampal pyramidal cells are active at any one time (Henze et al., 2000). Together, these observations suggest that most synapses may be inactive most of the time under physiological conditions.
Comparison with experiment
Because no parameters of the model were based on synaptic recordings, it is valid to compare the predictions with synaptic data. That release of one vesicle does not saturate AMPA receptors is now established for several synapses (Silver et al., 1996; Forti et al., 1997; Liu et al., 1999; McAllister and Stevens, 2000), although most studies have been performed in culture. The present predictions for AMPA receptor occupancy are somewhat lower than the 40% reported byLiu et al. (1999) or the upper limit of ∼50% of Silver et al. (1996). If this difference is confirmed, it seems likely that the geometry of the cleft or obstacles in it enhance synaptic responses somewhat. In the model, NMDA receptors are only activated to a slightly greater extent than AMPA receptors. Two-photon imaging of NMDA receptor-mediated spine calcium signals has provided direct evidence that the release of one vesicle is insufficient to saturate NMDA receptors (Mainen et al., 1999), a result also indicated by the variability and correlation of the AMPA and NMDA components of mEPSCs (McAllister and Stevens, 2000). Comparison of the NMDA receptor component of mEPSCs and responses to iontophoretic agonist applications at putative isolated synapses in culture (McAllister and Stevens, 2000) further suggested that an unexpectedly small fraction of NMDA receptors is activated by a single vesicle, a result that is consistent with the present simulations.
The present prediction of a small spillover effect may appear to contradict experimental results consistent with this phenomenon. However, the model is designed to simulate small central synaptic contacts operating in isolation, whereas the available experimental evidence concerns large synaptic contacts or widespread synaptic activity, for which the synergistic interaction of transmitter released from numerous sources is possible. Such effects may play interesting network roles physiologically (Scanziani, 2000). Recordings of glia have provided direct evidence for glutamate activating receptors outside the synaptic cleft (Mennerick and Zorumski, 1994; Bergles et al., 1997; Clark and Barbour; 1997; Dzubay and Jahr, 1999). However, receptor occupancy was not determined in those studies, and all of the observations were made when many vesicles were released simultaneously, making direct comparison with the present model difficult.
A powerful technique for determining the transmitter time course to which receptors are exposed is to measure the inhibition of a synaptic response by a rapidly unbinding (low-affinity) competitive antagonist. This technique was applied in a pioneering study using NMDA receptors (Clements et al., 1992), in which it was deduced that synaptically activated receptors were exposed to a glutamate transient that, when assumed to follow an exponential time course, was best approximated by a peak amplitude of 1.1 mm and a decay time constant of 1.2 msec. The same data were later fitted by a double exponential decay (Clements, 1996). An analogous experiment has since been performed for AMPA receptors (Diamond and Jahr, 1997). The calculated time courses are rather different from the theoretical predictions here (Fig.12A). This is not too surprising, because the expected glutamate concentration time course is not biexponential, but instead approximately proportional tot−3/2. Because up to 99% of glutamate can leave the PSD region within 100 μsec (Fig.12A), before much antagonist has unbound (Clements et al., 1992), the method is also relatively insensitive to events occurring immediately after release. The slow decays that were reported are generally interpreted as showing that diffusion of glutamate out of the synaptic cleft is in some way slowed. However, Wahl et al. (1996)have already pointed out that such slowing would have to be quite significant, even in an infinite disk model. If release is assumed to be instantaneous in the present model, it is not possible to recover the slow time courses with plausible slowing of diffusion within the synaptic cleft (Fig. 12A). Although extrapolation of the present model to culture synapses may not be wholly accurate (synapse size could make a difference), another or an additional explanation is probably required to explain the difference. If release is prolonged, the slow time courses can be approximately recovered, but most of the glutamate released (>60%) must be released slowly (time constant, 300 μsec to 2 msec). These estimates were obtained by convolving the glutamate time course of the model with monoexponential or biexponential release time courses (data not shown). If diffusion is unhindered in the cleft, several vesicles are required for recovery of the published time courses. If diffusion is obstructed (as shown), less than one vesicle is required, but the kinetics of the release function are not faster than stated above. In the case of Clements et al. (1992), one possible alternative source of glutamate at late times could be neighboring synapses.
Fig. 12.

Comparison with reported glutamate concentration time courses. A, Glutamate time courses determined using low-affinity glutamate receptor antagonists are slower than predicted here. The difference is difficult to account for only by slowing diffusion within the synaptic cleft. Time courses reported by: Clements et al. (1992) (CLTJW 1; dashed line); Clements (1996)(CLTJW 2; dashed curve); Diamond and Jahr (1997)(DJ1,2; dotted). The predictions of the present model (solid lines, average concentration over the PSD) are for instantaneous release of one vesicle in the absence of transporters (ctrl), and the same but with an extreme obstruction of the synaptic cleft (obstructed; αcleft = 0.1, λcleft = 1.9). The peak concentrations for ctrl and obstructed time courses are 10.9 and 109 mm, respectively. B, There is a discrepancy between the glutamate time courses reported by Rusakov and Kullmann (1998b) and Rusakov et al. (1999) and the present simulations, which agree with an approximate theoretical prediction. The glutamate concentration at 300 nm reported by Rusakov and Kullmann (RK; dot-dashed line) is approximately twice that produced using very similar parameters in the present simulation (dashed line). The accuracy of the present simulation is confirmed by its close agreement with the analytical solution for diffusion from a point source in a porous medium (dotted line). Some of the (slight) difference between the present simulation and the analytical solution is attributable to the presence of transporters, because a similar simulation without transporters (solid line) agrees even more closely with the analytical solution.
Comparison with other models
Compared with the numerous infinite disk models of synaptic activation, the simulations here predict lower levels of receptor activation. This is expected on the basis of the faster dilution of transmitter in a three-dimensional space, coupled with the choice of diffusion coefficient.
In a recently published model, Rusakov and Kullmann (1998b) and Rusakov et al. (1999) make a very similar choice of geometry to the present simulations. Under certain conditions they predict quite significant extrasynaptic NMDA receptor activation via glutamate spillover after release of a single vesicle (e.g.,Popen of ∼0.03, 25 times the 0.0013 calculated here; Rusakov et al., 1999). A good deal of the discrepancy can be explained by those authors' use of a slower diffusion coefficient, and other different parameter choices also contribute. However, a discrepancy of about a factor of 2 for NMDA receptor activation at 0.5 μm remains even when their parameter values are adopted in the present simulations. Figure 12Breplots a glutamate transient of Rusakov et al. (1999; their Fig. 3) and that produced when their published parameter values are used with the present model. Also plotted is the theoretical curve calculated for diffusion in a porous medium (i.e., without the synaptic cleft and transition regions). The theoretical curve is very close to the transient calculated in the present simulations. The time course ofRusakov et al. (1999) reaches almost double the concentration. A similar excess is observed for all the radii shown in their figure. It is difficult therefore to avoid the conclusion that their model contains >5000 glutamate molecules. It is not possible to identify precisely the source of this additional glutamate, but the description of the conditions imposed at the boundary of the synaptic cleft (Rusakov and Kullmann, 1998b) suggests that an imbalance of centripetal and centrifugal fluxes there might “create” glutamate. The larger glutamate transient will, of course, increase receptor activation and the difference probably accounts for the remaining discrepancy with the present model.
Conclusion
It is extremely rare in biological modeling to be able to determine all the relevant parameters with the accuracy implied by the explicit calculations practiced on them. The author willingly acknowledges that this is still the case here. Critical issues remain: the properties of diffusion in the synaptic cleft; better information about the number of vesicles released, their glutamate content and how it is released; confirmation that synaptic receptors in situbehave as has been determined for extrasynaptic receptors in patches. Because models (such as this one) based on the porous medium approximations can only describe average diffusion behavior, simulations based on explicit tissue reconstructions will provide an important test of the present estimates and yield invaluable information regarding variations between synapses.
Rusakov and Kullmann (1998a,b) report a method for calculating the “geometrical” tortuosity of a porous medium in which diffusion is restricted to a network of thin lamellae. The method has attracted criticism from Chen and Nicholson (2000), who describe the difficulty of such attempts to calculate tortuosity ab initio. In addition, the derivation contains a number of problems that suggest it cannot be accurate. The following arguments will refer to the longer exposition in Rusakov and Kullmann (1998a).
Rusakov and Kullmann (1998a) define an “elementary geometrical tortuosity” (EGT; their Eq. 1) using a plane element of extracellular space oriented at an arbitrary angle to the imposed “direction” of diffusion. The tortuosity (following Chen and Nicholson, I define λRK as the tortuosity calculated by Rusakov and Kullmann) is then to be obtained by averaging the EGTs throughout the porous medium. An obvious difficulty with this first step of the calculation is that the EGT can assume very large or even infinite values for some angles (in their notation, β2 = 90°). Although this behavior does not prevent the subsequent average from remaining bounded, the large values are unrealistic, and the result therefore cannot be exact. A serious omission in the derivation is the absence of any treatment of the behavior of diffusion at branches of the network of lamellae, although the connection properties might be expected to have an important influence on diffusion through the structure.
For a porous medium that is by assumption uniform and isotropic, the unusual claim is made that tortuosity will be “different, depending on whether it is calculated with respect to unidirectional or radial diffusion”. Normally, in a uniform isotropic structure the value of the diffusion coefficient (and therefore the tortuosity) is expected to be constant and independent of both the form of the diffusion source and the coordinate system. The authors make their statement because they average the EGT over the structure in two different ways and obtain two different values. However, one method is erroneous; both methods should give the same result.
The EGT is defined on a plane element whose orientation can be defined by two angles. λRK is obtained by averaging many EGTs. The authors state that all orientations are equally likely in an isotropic medium (although this may be invalid for the orientations encountered along a path) and therefore in the average equal weight should be given to all orientations. This average can be calculated as an integral over the surface of a sphere (divided by its surface area). Accordingly, their Equation 1 is transformed into Equations 3 and 4 and integrated to yield λRK = π/2 (in fact, the integral is most simply evaluated using a surface of revolution). The authors then proceed to transform their Equation 3 to spherical coordinates (Eq. 5) and integrate it again, this time by a Monte Carlo method, to obtain their estimate of tortuosity for radial diffusion, 1.398, which differs from the value above. However, because the transformation to spherical coordinates amounts to nothing more than a change of variable in the integration, the result should be unaffected. In fact, in the Monte Carlo integration, all combinations of the two angles defining the plane element orientations, rather than the orientations themselves, are considered equally likely. This is a different integration that, not surprisingly, gives a different result. Put another way, the surface integral should use an elementary surface area of dS =r2sinθdθdφ , but the Monte Carlo method implicitly uses dS =r2dθdφ. Including the factor of sinθ enables recovery of the original result (λRK = π/2).
Footnotes
This work was supported by the Centre National de la Recherche Scientifique (Unité Mixte de Recherche 8544). I thank Philippe Ascher and the members of the Laboratoire de Neurobiologie for their generous support and Steven Traynelis for helpful discussion. For their comments on this manuscript, I thank Etienne Audinat, Philippe Isope, Jacques Neyton, and Stéphane Supplisson (who also helped operate Mathematica).
Correspondence should be addressed to Boris Barbour, Laboratoire de Neurobiologie, Centre National de la Recherche Scientifique, Unité Mixte de Recherche 8544, Ecole Normale Supérieure, 46 rue d'Ulm, 75230 Paris Cedex 05, France. E-mail: barbour@ens.fr.
REFERENCES
- 1.Auger C, Attwell D. Fast removal of synaptic glutamate by postsynaptic transporters. Neuron. 2000;28:547–558. doi: 10.1016/s0896-6273(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 2.Auger C, Kondo S, Marty A. Multivesicular release at single functional synaptic sites in cerebellar stellate and basket cells. J Neurosci. 1998;18:4532–4547. doi: 10.1523/JNEUROSCI.18-12-04532.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbour B, Häusser M. Intersynaptic diffusion of neurotransmitter. Trends Neurosci. 1997;20:377–384. doi: 10.1016/s0166-2236(96)20050-5. [DOI] [PubMed] [Google Scholar]
- 4.Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- 5.Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- 6.Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergles DE, Dzubay JA, Jahr CE. Glutamate transporter currents in Bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci USA. 1997;94:14821–14825. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruns D, Jahn R. Real-time measurement of transmitter release from single synaptic vesicles. Nature. 1995;377:62–65. doi: 10.1038/377062a0. [DOI] [PubMed] [Google Scholar]
- 9.Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen KC, Nicholson C. Changes in brain cell shape create residual extracellular space volume and explain tortuosity behavior during osmotic challenge. Proc Natl Acad Sci USA. 2000;97:8306–8311. doi: 10.1073/pnas.150338197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses”. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- 12.Clark BA, Barbour B. Currents evoked in Bergmann glial cells by parallel fibre stimulation in rat cerebellar slices. J Physiol (Lond) 1997;502:335–350. doi: 10.1111/j.1469-7793.1997.335bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clements JD. Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–170. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- 14.Clements JD, Lester RAJ, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- 15.Colquhoun D, Jonas P, Sakmann B. Action of brief pulses of glutamate on AMPA/kainate receptors from different neurones of rat hippocampal slices. J Physiol (Lond) 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crank . The mathematics of diffusion, Ed 2. Oxford UP; Oxford: 1975. [Google Scholar]
- 17.Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol (Lond) 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- 20.Dzubay JA, Jahr CE. The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci. 1999;19:5265–5274. doi: 10.1523/JNEUROSCI.19-13-05265.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eskandari S, Kreman M, Kavanaugh MP, Wright EM, Zamphigi G. Pentameric assembly of a neuronal glutamate transporter. Proc Natl Acad Sci USA. 2000;97:8641–8646. doi: 10.1073/pnas.97.15.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forti L, Bossi M, Bergamaschi A, Villa A, Malgaroli A. Loose-patch recordings of single quanta at individual hippocampal synapses. Nature. 1997;388:874–878. doi: 10.1038/42251. [DOI] [PubMed] [Google Scholar]
- 23.Gardner-Medwin AR. Membrane transport and solute migration affecting the brain cell microenvironment. Neurosci Res Progr Bull. 1980;18:208–226. [Google Scholar]
- 24.Gregor P, Mano I, Maoz I, McKeown M, Teichberg VI. Molecular structure of the chick cerebellar kainate-binding subunit of a putative glutamate receptor. Nature. 1989;342:689–692. doi: 10.1038/342689a0. [DOI] [PubMed] [Google Scholar]
- 25.Grewer C, Watzke N, Wiessner M, Rauen T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci USA. 2000;97:9706–9711. doi: 10.1073/pnas.160170397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haugueto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- 27.Henze DA, Borhegyi Z, Csicsvari J, Mamiya A, Harris KD, Buzsaki G. Intracellular features predicted by extracellular recordings in the hippocampus in vivo. J Neurophysiol. 2000;84:390–400. doi: 10.1152/jn.2000.84.1.390. [DOI] [PubMed] [Google Scholar]
- 28.Hjelmstad GO, Isaac JT, Nicoll RA, Malenka RC. Lack of AMPA receptor desensitization during basal synaptic transmission in the hippocampal slice. J Neurophysiol. 1999;81:3096–3099. doi: 10.1152/jn.1999.81.6.3096. [DOI] [PubMed] [Google Scholar]
- 29.Ichimura T, Hashimoto PH. Structural components in the synaptic cleft captured by freeze-substitution and deep etching of directly frozen cerebellar cortex. J Neurocytol. 1988;17:3–12. doi: 10.1007/BF01735373. [DOI] [PubMed] [Google Scholar]
- 30.Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 31.Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- 32.Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol (Lond) 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kullmann DM, Erdemli G, Asztély F. LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 34.Land BR, Harris WV, Salpeter EE, Salpeter MM. Diffusion and binding constants for acetylcholine derived from the falling phase of miniature endplate currents. Proc Natl Acad Sci USA. 1984;81:1594–1598. doi: 10.1073/pnas.81.5.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larkman A, Stratford K, Jack J. Quantal analysis of excitatory synaptic action and depression in hippocampal slices. Nature. 1991;350:344–347. doi: 10.1038/350344a0. [DOI] [PubMed] [Google Scholar]
- 36.Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lester RA, Jahr CE. NMDA channel behavior depends on agonist affinity. J Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 39.Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 40.Longsworth LG. Diffusion measurements at 25° of aqueous solutions of amino acids, peptides and sugars. J Am Chem Soc. 1953;75:5705–5709. [Google Scholar]
- 41.Mainen ZF, Malinow R, Svoboda K. Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature. 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- 42.Mazel T, Simonova Z, Sykova E. Diffusion heterogeneity and anisotropy in rat hippocampus. NeuroReport. 1998;9:1299–1304. doi: 10.1097/00001756-199805110-00008. [DOI] [PubMed] [Google Scholar]
- 43.McAllister AK, Stevens CF. Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc Natl Acad Sci USA. 2000;97:6173–6178. doi: 10.1073/pnas.100126497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McBain CJ, Traynelis SF, Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674–677. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- 45.Mennerick S, Zorumski CF. Glial contributions to excitatory neurotransmission in cultured hippocampal cells. Nature. 1994;368:59–62. doi: 10.1038/368059a0. [DOI] [PubMed] [Google Scholar]
- 46.Mennerick S, Shen W, Xu W, Benz A, Tanaka K, Shimamoto K, Isenberg KE, Krause JE, Zorumski CF. Substrate turnover by transporters curtails synaptic glutamate transients. J Neurosci. 1999;19:9242–9251. doi: 10.1523/JNEUROSCI.19-21-09242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller JP, Rall W, Rinzel J. Synaptic amplification by active membrane in dendritic spines. Brain Res. 1985;325:325–330. doi: 10.1016/0006-8993(85)90333-6. [DOI] [PubMed] [Google Scholar]
- 48.Nicholson C, Phillips JM. Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J Physiol (Lond) 1981;321:225–257. doi: 10.1113/jphysiol.1981.sp013981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- 50.Nicholson C, Chen KC, Hrabetova S, Tao L. Diffusion of molecules in brain extracellular space: theory and experiment. Prog Brain Res. 2000;125:129–154. doi: 10.1016/S0079-6123(00)25007-3. [DOI] [PubMed] [Google Scholar]
- 51.Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- 52.Otis TS, Jahr CE. Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J Neurosci. 1998;18:7099–7110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otis TS, Kavanaugh MP. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci. 2000;20:2749–2757. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otmakhov N, Shirke AM, Malinow R. Measuring the impact of probabilistic transmission on neuronal output. Neuron. 1993;10:1101–1111. doi: 10.1016/0896-6273(93)90058-y. [DOI] [PubMed] [Google Scholar]
- 56.Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- 57.Rusakov DA, Kullmann DM. Geometric and viscous components of the tortuosity of the extracellular space in the brain. Proc Natl Acad Sci USA. 1998a;95:8975–8980. doi: 10.1073/pnas.95.15.8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998b;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rusakov DA, Kullmann DM, Stewart MG. Hippocampal synapses: do they talk to their neighbours? Trends Neurosci. 1999;22:382–388. doi: 10.1016/s0166-2236(99)01425-3. [DOI] [PubMed] [Google Scholar]
- 60.Sayer RJ, Friedlander MJ, Redman SJ. The time course and amplitude of EPSPs evoked at synapses between pairs of CA3/CA1 neurons in the hippocampal slice. J Neurosci. 1990;10:826–836. doi: 10.1523/JNEUROSCI.10-03-00826.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scanziani M. GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 62.Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- 63.Silver RA, Traynelis SF, Cull-Candy SG. Rapid-time-course miniature and evoked excitatory currents at cerebellar synapses in situ. Nature. 1992;355:163–166. doi: 10.1038/355163a0. [DOI] [PubMed] [Google Scholar]
- 64.Silver RA, Cull-Candy SG, Takahashi T. Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol (Lond) 1996;494:231–250. doi: 10.1113/jphysiol.1996.sp021487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith GD. Numerical solutions of partial differential equations: finite difference methods, Ed 3. Oxford UP; Oxford: 1985. [Google Scholar]
- 66.Sorra KE, Harris KM. Stability in synapse number and size at 2 hr after long-term potentiation in hippocampal are CA1. J Neurosci. 1998;18:658–671. doi: 10.1523/JNEUROSCI.18-02-00658.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- 68.Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- 69.Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- 70.Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wadiche JI, Kavanaugh MP. Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci. 1998;18:7650–7661. doi: 10.1523/JNEUROSCI.18-19-07650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP. Kinetics of a human glutamate transporter. Neuron. 1995;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 73.Wahl LM, Pouzat C, Stratford KJ. Monte Carlo simulation of fast excitatory synaptic transmission at a hippocampal synapse. J Neurophysiol. 1996;75:597–608. doi: 10.1152/jn.1996.75.2.597. [DOI] [PubMed] [Google Scholar]
- 74.Zador A, Koch C. The function of dendritic spines: devices subserving biochemical rather than electrical compartmentalization. J Neurosci. 1993;13:413–422. doi: 10.1523/JNEUROSCI.13-02-00413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]