

SUMMARY
Small proteins are traditionally overlooked due to computational and experimental difficulties in detecting them. To systematically identify small proteins, we carried out a comparative genomics study on 1,773 human-associated metagenomes from four different body sites. We describe >4,000 conserved protein families, the majority of which are novel; ~30% of these protein families are predicted to be secreted or transmembrane. Over 90% of the small protein families have no known domain and almost half are not represented in reference genomes. We identify putative housekeeping, mammalian-specific, defense-related, and protein families that are likely to be horizontally transferred. We provide evidence of transcription and translation for a subset of these families. Our study suggests that small proteins are highly abundant and those of the human microbiome, in particular, may perform diverse functions that have not been previously reported.
Graphical Abstract
In Brief
Computational identification and characterization of thousands of conserved small ORFs from human microbiome sequences spanning multiple anatomical sites suggests a diversity of unknown protein domains and families with diverse functions.
INTRODUCTION
To support the transition of the microbiome field from descriptive science to a more mechanistic one, there is an ongoing shift from 16S ribosomal RNA sequencing to whole-metagenome shotgun (WGS) sequencing projects (Ranjan et al., 2016; Lloyd-Price et al., 2017; Gilbert et al., 2018). While accumulating WGS studies have illuminated the remarkable genetic diversity encoded by human-associated microbes, our ability to link specific genes to phenotypes is still lagging behind (Koppel and Balskus, 2016). One of the challenges in linking genes to phenotypes is that the process of gene annotation overlooks an entire class of potentially important genes.
Small open reading frames (sORFs) and the small proteins they encode, here defined as proteins of ≤50 amino acids in length, have traditionally been ignored (Duval and Cossart, 2017; Storz et al., 2014; Su et al., 2013). It is difficult to distinguish protein coding ORFs from the numerous random in-frame genome fragments, and thus most prediction tools require a minimum ORF length, resulting in incomplete databases. In mutational screens, sORFs are less likely to be targeted and classical biochemical approaches are usually not optimized to detect small proteins. Finally, experiments that rely on databases, such as mass spectrometry, will fail to identify small proteins if their sequences are not present in reference databases.
Despite this bias, recent studies have elucidated interesting functions for small proteins in both eukaryotes and prokaryotes (reviewed in Couso and Patraquim, 2017; Duval and Cossart, 2017; Kemp and Cymer, 2014; Storz et al., 2014; Plaza et al., 2017). Here, we sought to characterize the small proteins encoded by the healthy human microbiome, represented by the NIH Human Microbiome Project (HMP) dataset (Lloyd-Price et al., 2017). We leveraged the concept that protein-coding sORFs likely have protein sequences that are conserved. Our analysis reveals 4,539 candidate small protein families encoded by human-associated microbes, very few of which have been previously described.
For each family, we provide taxonomic classification, prevalence across body sites, predicted cellular localization (secreted/transmembrane), and prediction of antimicrobial function. We provide information about homologs of the families among ~6,000 non-human metagenomes. Finally, because in bacteria, gene context can inform predictions of function, we describe the genes that are encoded in vicinity of the sORF. We highlight several novel small proteins with diverse predicted functions, including house-keeping, cell-cell crosstalk, adaptation, as well as defense against phage or against other bacteria.
For a subset of small protein families that have homologs in metatranscriptomic datasets (Abu-Ali et al., 2018; Tropini et al., 2018) , we show that at least 75% are actively transcribed. For homologs that are found in Bacteroides thetaiotaomicron, we use ribosome-profiling (Ribo-Seq) to show that at least 40% are translated. We contribute to building a more complete understanding of the full coding potential encoded by the human microbiome, including the thus far overlooked sORFs. This is a fundamental step toward understanding of the mechanisms that underlie the role of the microbiome in health and disease.
RESULTS
Only a Small Subset of Well-Characterized Small Proteins Are Relevant to the Human Microbiome
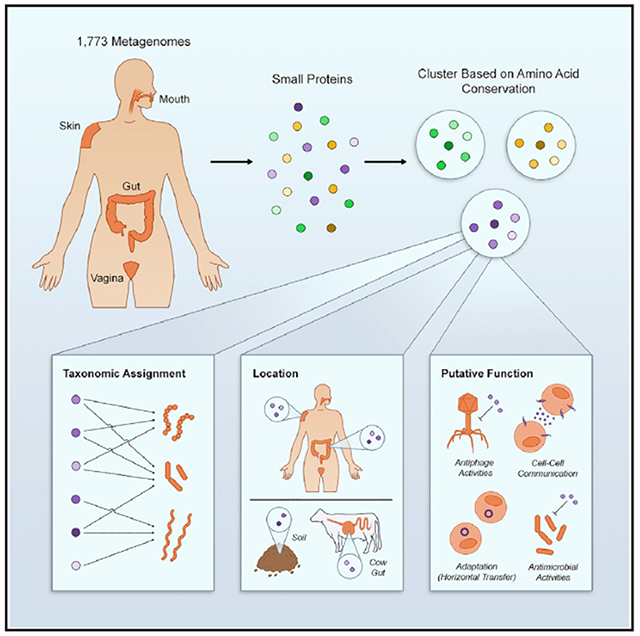
Small proteins that have been studied in depth generally originate from model organisms (for review, see Duval and Cossart, 2017; Storz et al., 2014). To infer their potential relevance to the human microbiome, we sought to identify those that are also found in human-associated microbes. To not limit our search to species that have a reference genome, we undertook a reference-free approach and conducted our analysis on HMPI-II metagenomic sequencing data (Lloyd-Price et al., 2017). We used MetaProdigal (Hyatt et al., 2012) to annotate all open reading frames, as short as 15 base pairs (bp), on 128,368,337 contigs spanning more than 180 billion bp of sequenced DNA from 1,773 metagenomes from 263 healthy individuals (Table S1) sampled from four different major body sites (Figure S1; Table S1). We filtered out ORFs that encode for proteins that are >50 amino acids in length, resulting in 2,514,099 sORFs (Figure 1A).
Figure 1. Small Protein Discovery and Characterization Pipeline Applied to HMPI-II Metagenomic Data.
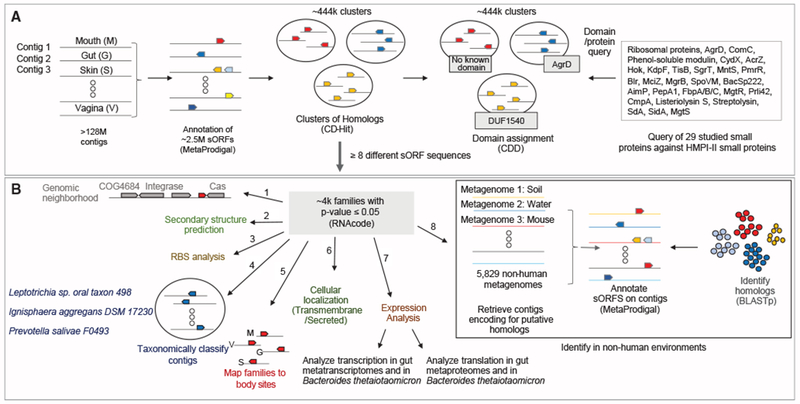
(A) Identification of 29 known small proteins in HMPI-II metagenomes. More than 128 million contigs were annotated using MetaProdigal with a lower size limit of five amino acids. The small proteins were then clustered using CD-Hit based on amino acid similarity and protein length. Representatives of each of the ~444,000 clusters were queried against the Conserved Domain Database (CDD), to assign domains to clusters. The list of CDD domains was then queried for the small known proteins that have an assigned domain. Known small proteins that do not have an assigned domain or that failed the domain search were queried against HMPI-II small proteins using BLASTp.
(B) Identification and characterization of HMPI-II small proteins. RNAcode was used to assign p values to the ~444,000 clusters. The following analyses were conducted on the ~4,000 protein families whose p value was ≤0.05. (1) Identification of neighboring genes on longest contig associated with each family. (2) Prediction of secondary structure. (3) Analysis of ribosomal binding sites (RBS) upstream of the small genes. (4) Taxonomic classification of contigs encoding each of the small protein families. (5) Assignment of small protein families to body sites. M-mouth; V - vagina; G - gut; S - skin. (6) Prediction of signal peptide and transmembrane domains to assign likely cellular localization. (7) Analysis of expression of the small genes using metatranscriptomic, metaproteomic datasets as well as Bacteroides thetaiotaomicron transcriptomics and proteomics. (8) Identification of homologs of small protein families in non-human metagenomes.
See also Figures S1, S2, and S7, Tables S1, S2, S3, and S4, and Data S1 and S2.
We queried a set of 29 known small proteins that have been studied in depth (reviewed by Duval and Cossart, 2017; Storz et al., 2014) (Tables 1 and S2) as well as a set of small ribosomal proteins, to identify homologs of these known small proteins among the predicted ~2,500,000 putative small proteins. Whenever possible, we used a domain-based approach (RPS-BLAST) that would detect even distant homologs (Altschul et al., 1997), and we used a sequence-based approach (BLASTp) for small known proteins that have not been assigned a protein domain.
Table 1.
Representation of Known Small Proteins in HMPI-II Data
| Abundant in HMPI-II Samples | Identified at Low Levels in HMPI-II Samples | Not Identified in HMPI-II Samples |
|---|---|---|
| Ribosomal proteins | CydX (Escherichia coli) | MciZ (Bacillus subtilis) |
| AgrD (Gram+ bacteria) | AcrZ (Escherichia coli) | MgrB (Escherichia coli) |
| ComC (Streptococcus) | Hok (Escherichia coli) | SpoVM (Bacillus subtilis) |
| Phenol soluble modulin (Staphylococcus) | KdpF (Escherichia coli) | BacSp222 (Staphylococcus pseudintermedius) |
| TisB (Escherichia coli) | AimP (Bacillus subtilis phages) | |
| SgrT (Escherichia coli) | FbpA/B/C (Bacillus subtilis) | |
| MntS (Escherichia coli) | MgtR (Salmonella typhimurium) | |
| PmrR (Salmonella enterica) | Prli42 (Listeria monocytogenes) | |
| SidA (Caulobacter crescentus) | CmpA (Bacillus subtilis) | |
| MgtS (Escherichia coli) | PepA1 (Staphylococcus aureus) | |
| Blr (Escherichia coli) | Listeriolysin S (Listeria monocytogenes) | |
| Streptolysin (Streptococcus pyogenes) | ||
| SdaA (Bacillus subtilis) |
Known proteins were queried against CDD-assigned domains of all ~444,054 representatives whenever they had an assigned domain and against all protein sequences of the ~444,054 representatives using BLASTp (Camacho et al., 2009) when the known protein was not assigned a known domain (Table S2). Only 12 of the 29 small proteins have an assigned protein domain (AcrZ, CydX, KdpF, AgrD, ComC, MciZ, MgrB, SpoVM, SgrT, Hok, TisB, phenol-soluble modulins as well as small ribosomal proteins). Approximately 3.5% of small proteins that were assigned a domain (3,930/113,693) were homologous to the extensively studied quorum-sensing small protein, Staphylococcal AgrD. ComC, a quorum-sensing signal that enables Streptococci to regulate DNA uptake and genetic transformation in response to population density as well as environmental queues such as antibiotic stress (Moreno-Gámez et al., 2017), was found in ~2% (2,176/113,693) of small proteins. Homologs of AgrD and ComC were clustered into 153 and 19 clusters, respectively, suggesting rapid evolution of these proteins, in line with what has been previously documented (Hyatt et al., 2012; Allan et al., 2007). CydX (YbgT) is a small protein required for the function of cytochrome bd oxidase (Sun et al., 2012). KdpF is part of the high-affinity ATP-driven potassium transport system (Gassel et al., 1999). Hok (Chukwudi and Good, 2015) and TisB (Steinbrecher et al., 2012) are toxins. AcrZ is a multidrug efflux pump accessory protein (Hobbs et al., 2012). SgrT is a regulator of glucose metabolism (Lloyd et al., 2017). MntS that takes part in manganese chaperoning (Martin et al., 2015). PmrR, is a regulator of a membrane-bound enzyme (Kato et al., 2012). SidAisan inhibitor of cell division (Modell et al., 2011). MgtS (formerly known as YneM) modulates intracellular Mg2+ levels to maintain cellular integrity upon Mg2+ limitation (Wang et al., 2017). Blr is involved in B-lactamase resistance (Karimova et al., 2012). Names of organisms in parentheses indicate the model organism in which small protein was mainly studied.
To reduce computational load associated with analysis of such large amounts of sequences, we first clustered all ~2,500,000 putative small proteins based on sequence and length similarity using CD-Hit (Fu et al., 2012), resulting in 444,054 clusters. We then queried each of the 444,054 families against the Conserved Domain Database (CDD) (Marchler-Bauer et al., 2011, 2017) (Figure 1A). Only ~4.5% (113,693/2,514,099) of the putative small proteins, spanning ~0.5% (2,225/444,054) of the clusters, could be assigned a known domain (Table S3). The most common types of domains identified are of diverse small ribosomal proteins, assigned to ~64% of all domain-assigned small proteins (72,982/113,693). Other well studied proteins that were abundant in our dataset (such as AgrD and ComC) are encoded by commonly studied organisms that are often constituents of the healthy microbiome (such as Staphylococcus and Streptococcus, respectively), making it unsurprising that we identified them in our human-associated microbiome dataset. Otherwise, we found limited overlap between well characterized small proteins and those that are abundant in human microbiomes (Tables 1 and S2).
Identification of ~4,000 Small Protein Families of the Human Microbiome
Intrigued that such a small proportion of previously described small proteins were present in the human-associated microbiomes, we sought to better understand what types of small proteins exist in this unexplored space. First, we revisited the 444,054 clusters (Table S3) of potential small proteins that were generated in the previous step of our analysis (Figure 1A). Most were not assigned a known functional domain, which raised concerns for the potential presence of spurious sORFs. To enrich for families that are more likely to be protein-coding families, we used RNAcode (Washietl et al., 2011), a gene predictor program that distinguishes between coding and non-coding sequences by evaluating evolutionary signatures. We applied RNAcode on the 11,715 clusters that contained ≥8 different DNA sequences. Using a p value threshold of ≤0.05, we identified 4,539 clusters (containing 467,538 small proteins) that are predicted to be bona fide sORFs (Figure 1A; Table S3). A ribosomal binding site (RBS) motif was detected in 91% (426,581/467,538) of all proteins (Figure S2; Table S3). These 4,539 “small protein families” are subjected to further analyses hereafter (Figure 1A; Table S3).
The Majority of the ~4,000 Small Protein Families of the Human Microbiome Are Novel
Reassuringly, the ~4,000 family subset is significantly enriched for small protein families that were assigned a protein domain (p < 1 × 10−5 Fisher exact test): among the 4,539 small protein families, 4% (190/4,539) were assigned a domain (compared to 0.5% of the 444,054 clusters), (Figures 2A and 2B). These families contain 12% of the 467,538 small proteins (compared to 4.5% of the 2,514,099 in the initial database). Interestingly, ~96% (4,349/4,539) of small protein families were not assigned a CDD domain, some of which are actually encoded by a large number of species (Figure 2C; Table S3), emphasizing the incompleteness of knowledge in the small protein domains space. We also asked what proportion of the sORF families are found in reference genome databases such as RefSeq (Pruitt et al., 2007). We performed sequence similarity searches of all 4,539 representative proteins against proteins of ≤50 amino acids annotated in 69,681 RefSeq bacterial reference genomes. Only ~25% of the small protein families (1,149/4,539) in our dataset have annotated homologs in RefSeq genomes (Figure 2A; Table S3). We postulated that at least some of the small proteins in our dataset do have homologs that were not annotated. We therefore re-annotated all 69,681 RefSeq genomes with a permissive size threshold to include all potential sORFs. Indeed, this step revealed an additional set of 1,230 (~27%) small protein families. Still, for 48% (2,164/4,539) of the small protein families, we could not identify any homologs (Figure 2A; Table S3). This confirms that any effort to comprehensively identify candidate novel small proteins of the human microbiome would be very limited if applied only to genomes from reference databases that have, generally speaking, a limited representation of human-associated microbes.
Figure 2. Many of the ~4,000 Families, Some of which Are Very Abundant, Are Not Assigned a Known Protein Domain nor Are They Represented in RefSeq Genomes.
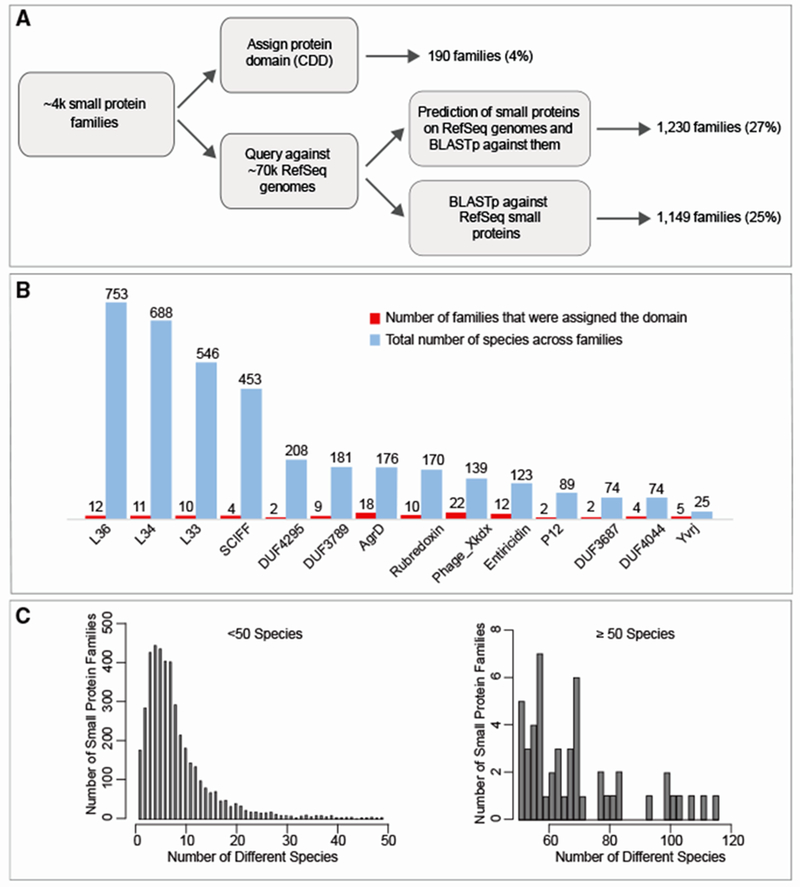
(A) Pipeline to identify families that do not have an assigned domain and families that are not represented in RefSeq genomes. Upper path of the flow diagram: only a small subset of the ~4,000 small protein families were assigned a protein domain (identified by RPS-blast against CDD position specific scoring matrices, PSSMs). Lower path of the flow diagram: representatives of all ~4,000 families were blasted against ~3,000,000 small RefSeq annotated proteins originating from ~70,000 RefSeq genomes and against ~7,000,000 putative small proteins that we annotated using Prodigal with adjusted thresholds. The second step allowed the identification of an additional set of homologs that are encoded but not annotated in RefSeq genomes.
(B) Domains identified among ~4,000 families. Domains that were classified to ≥5 families and/or ≥50 species are shown. A complete list of domains can be found in Table S3.
(C) Number of species encoding small proteins of families with no known domain are shown in histogram.
We next looked for evidence of transcription and translation of the 4,539 small gene families. Analysis of 226 publicly available human fecal metatranscriptomes (Abu-Ali et al., 2018) (Table S4) revealed homologs of 689 of the families. Of these, 518 (75%) have at least one actively transcribed homolog (Figure S3). We then selected two publicly available metaproteomic datasets and re-analyzed the raw data to determine if any of the predicted sORFs are found as detectable proteins (Zhang et al., 2017; Zhang et al., 2018). Altogether, 25 small protein families were detected within the two sets. Finally, we focused on Bacteroides thetaiotaomicron, in which we annotated 35 “high-confidence” sORFs. We find evidence that 19 (54%) are transcribed in a publicly available dataset (Tropini et al., 2018). We performed Ribo-Seq analysis, which shows that 14 (40%) of the 35 sORFs predictions are translated; using mass-spectrometry based proteomics, we find almost 10% of the 35 sORFs (Table S4).
Because most families in our dataset do not have an assigned protein domain nor do they have well-characterized homologs from which we can try to infer function, we subsequently used several approaches to provide insight into the potential functions of these small proteins (Figure 1B). In the next sections, we focus on specific classes of small proteins.
Putative Novel “Housekeeping” Small Protein Families among Human-Associated Microbes
We sought to identify small protein families that could be playing housekeeping roles. We posited that such families would be highly prevalent across species. To characterize the taxonomic distribution of families, we classified each of the contigs that encode small proteins against a set of 83,701 microbial reference genomes, consisting of 53,193 bacteria, 27,020 viruses, 1,892 eukaryota, and 1,756 archaea genomes, using the k-mer based One Codex platform (Minot et al., 2015).
We focused our next analysis on the 14 most prevalent families that are encoded by ≥ 100 species (Figure 3A). Whereas most families in the overall dataset are taxonomically unique to one (2,353, 52%) or two (1,183, 26%) phyla, there is strong enrichment among the 14 most prevalent families for presence in multiple phyla (Figure 3B), suggesting a role that is not clade-specific. In all 14 families, the average percentage of k-mers that could be classified is >10%, implying that classification is likely reliable in these families. Second, we determined whether these families are specific to a particular ecological niche. To do so, we mapped each family to the body site(s) in which homologs of the family were identified. Whereas most small protein families are identified uniquely in mouth (1,188, 26%) or gut (2,220, 48%) (Table S3), 13 of the 14 most prevalent families were identified in ≥3 body sites, suggesting a role that is not niche-specific (Figure 3A). Because the HMP data resource we used for this study has a limited representation of skin and vagina samples (Table S1), it is possible that families that seem absent from one of these body sites are present but not detected.
Figure 3. A Subset of Small Protein Families Is Prevalent across the Tree of Life.
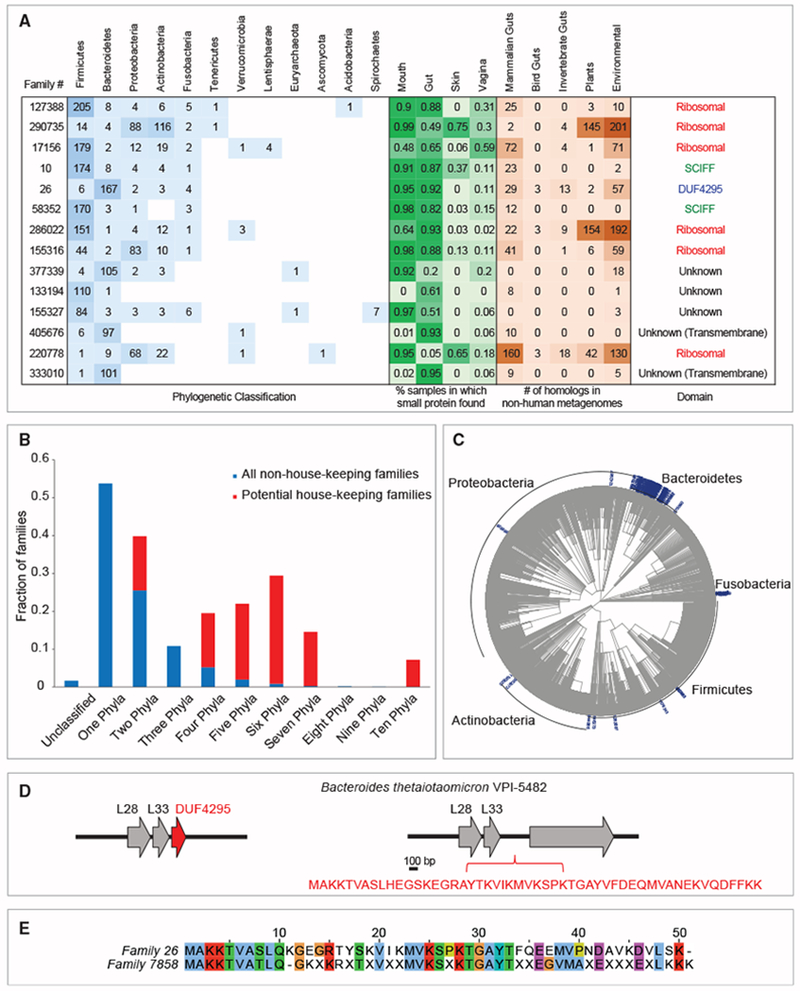
(A) Most abundant families. Each row represents one of the 14 families that were identified in ≥100 species. The taxonomic distribution of the 14 families is presented in the blue table, the prevalence among body sites is presented in the green table and the number of homologs identified in non-human metagenomes is presented in the brown table. Potential novel ribosomal is family 26. When multiple homologs were mapped to the same taxa, it is counted as one event in this table. SCIFF, “six cysteines in forty-five residues.”
(B) The fraction of families assigned to different number of phyla for the 14 potential housekeeping (red) and the 4,525 remaining families (blue) is shown. For example, >50% of the non-housing-keeping families were assigned to one phyla versus zero housekeeping families that were assigned to one phylum.
(C and D) Potential novel ribosomal protein. (C) Phylogenetic tree of family 26. (D) The genomic neighborhood of DUF4295 (family 26) next to two known ribosomal proteins is illustrated. In Bacteroides thetaiotaomicron VPI-5482 it is encoded in the intergenic region downstream of these genes (locus tags BT0914 and BT0915).
(E) Homology between family 26 and family 7858, two potential novel ribosome-associated families of proteins. Family 7858 is encoded by 26 species from 3 different phyla and did not pass the required ‘housekeeping’ threshold (which requires ≥100 species). The family 7858 gene is genomically positioned next to two ribosomal proteins; it is found in 85% of mouth samples (but not in any gut samples) as well as in diverse non-human environments.
See also Figures S3 and S4 and Tables S1 and S3.
Positing that true housekeeping genes are likely to be conserved among a broad range of ecological niches, we tested whether these 14 prevalent families are more likely to have homologs in non-human metagenomes. To do so, we checked for sequence homology of the ~4,000 small proteins within a set of 5,829 non-human metagenomes, including mammalian and bird gut metagenomes, as well as environmental samples of different types (Table S1). While we could not identify homologs in non-human metagenomes for the majority of small protein families (3,551, 78%), we were able to identify homologs in at least one non-human environment for all 14 candidate “house-keeping” families (Figure 3A).
Altogether, the taxonomic abundance and the existence in multiple niches of these 14 “housekeeping” families suggest a role that is not clade- or niche-specific. Indeed, among these 14, six encode different ribosomal proteins. Among the remaining eight families, three were assigned a CDD domain and five were not. Two of the CDD-assigned families were assigned the “SCIFF” domain, which is associated with a small ribosomally synthesized natural product (Haft and Basu, 2011; Haft and Haft, 2017). The biological function of this small protein is unknown. Family 26 was assigned a DUF4295 domain, which we address below. There are five families that were not assigned a protein domain, two of which are predicted to be transmembrane. Analysis of transcription datasets shows that at least 12 of the 14 are actively transcribed (Figure S3). The three families that have homologs in Bacteroides thetaiotaomicron (26, 286022, and 220778) were all detected in our Bacteroides thetaiotaomicron Ribo-Seq (Table S4).
We also asked which small protein families in our dataset could be playing key roles that are associated with a specific body niche(s). To identify the body site(s) with which each family is associated, we mapped all contigs associated with the ~4,000 protein families back to body site from which these contigs were assembled. A total of 458 families (10%, 458/4,539) were identified in ≥ 50% of samples of at least one body site (“core families”). In most cases, “coreness” is associated with a specific body site, suggesting that among the small protein families there are those that may be “housekeeping” in a specific body niche and are probably not essential in other body niches (Figure S4).
Identification of a Putative Novel Ribosome-Associated Protein Prevalent among Human-Associated Microbes
Family 26 is among the 14 families that are very abundant and was assigned a domain of unknown function, DUF4295 (Figures 3A and 3C). This 50-amino acid protein was detected in 182 species originating from four different phyla. We identified homologs of this protein in diverse non-human metagenomes and in a high percentage of gut and mouth samples, as well as in vaginal samples. It drew our attention because the sORF is located in a strongly conserved genomic locus, downstream of two known ribosomal proteins, L28 and L33 (Figure 3D). In light of its wide phylogenetic distribution and genomic localization, we hypothesize that this small protein family encodes a novel small ribosome-associated protein that has thus far escaped detection. In the lab strain Bacteroides thetaiotaomicron VPI-5482, the small gene encoding this protein was not annotated, as is the case for many small proteins, but nevertheless is encoded in the intergenic region downstream these two genes (Figure 3D). In support of the hypothesis that family 26 is probably highly expressed, we could detect it in all expression datasets described above (Figure S3; Table S4). DUF4295 domain is also encoded by family 7858 and displays significant sequence homology to family 26 (Figure 3E).
Small Proteins that Are Potential Mediators of Cell-Cell and Cell-Host Communication
We were particularly interested in small proteins that could be involved in the crosstalk between microbial cells and their environment (host or other microbial cells). Communication is typically mediated through direct cell-cell contact or via small diffusible molecules secreted by cells (Hayes et al., 2010; Moreno-Gámez et al., 2017). We thus postulated that proteins that are at the cell surface or are secreted are more likely to be involved in cell-cell communication.
We looked in our dataset for small protein families that are either transmembrane and/or potentially secreted. To predict transmembrane and signal peptides, we applied two algorithms, TMHMM (Krogh et al., 2001) and SignalP-5.0 (Almagro Armenteros et al., 2019), on all 467,538 small proteins that constitute the 4,539 small protein families. We classified a family as predicted to be transmembrane/secreted if ≥80% of the homologs of the family are predicted to be such. Due to the limitations associated with prediction of secreted proteins, we believe that the number of secreted proteins in our dataset is in fact higher than we predict here.
In addition, we sought to identify small protein families that could display antimicrobial activity. To do so, we used AmPEP (Bhadra et al., 2018), which uses a Random Forest algorithm to identify antimicrobial peptides. By applying the algorithm on the 4,539 representatives, we identified 39 small protein families (Table S3) that are potential novel antimicrobial peptides.
Of the 4,539 small protein families, a total of 1,402 families (30% of the 4,539 families) are predicted to be transmembrane and/or secreted (Figure S1). Specifically, 1,054 (23%) families, consisting of 168,165 small proteins (35% of the total 467,538 small proteins) are predicted to be solely transmembrane, 107 (2%) families, consisting of 19,749 small proteins (4% of the total small proteins) are predicted to be solely secreted, and 241 (5%) families, consisting of 43,642 small proteins (9% of the total 467,538 small proteins) are predicted to be both transmembrane and secreted. As expected, 93% (1,207/1,295) of the families that are predicted to be transmembrane are predicted to adopt a helical structure, providing support to our prediction of transmembrane families (Table S3; Data S2).
To pinpoint small proteins that could be specifically important to life within the mammalian gut, we asked which of the predicted transmembrane/secreted families have homologs in other mammalian guts but not in other niches (no other human body sites nor other non-mammalian metagenomes). Our mammalian gut metagenomes include 86 samples originating from diverse mammals, including mouse, rat, multiple non-human primates, panda, and more (Table S1). This narrowed our set from 1,402 to 132 families (transmembrane = 96, secreted = 8, transmembrane and secreted = 28; Table S3) that are found in human as well as other mammalian gut metagenomes.
Family 350024 drew our attention, because it has the highest number of homologs in other non-human mammalian guts. We identified 30 homologs of this small protein in 13 different mammalian gut metagenomic samples. It encodes a 33-amino acid predicted transmembrane and secreted protein with no annotated domain or known function. A homology search of family 350024 against all 1,266 predicted transmembrane families of the ~4,000 small protein families reveals that this small protein is actually even more abundant: there are 22 additional small protein families, ranging in size between 24–40 amino acids (Table S5), that share sequence homology with this family, although they are divergent enough not to be clustered into one big protein family, suggesting rapid evolution (Figure 4A). These predicted transmembrane proteins are often found in mammalian/bird gut samples and are in most cases encoded by diverse Bacteroidetes and Firmicutes species (Figure 4B). A phylogenetic protein tree of homologs of the family, compared to several known housekeeping genes, supports the hypothesis that family 350024 undergoes more rapid evolution than the tested housekeeping or core genes (Figure S5).
Figure 4. Small Proteins that Are Potentially Involved in Cross-Talk.
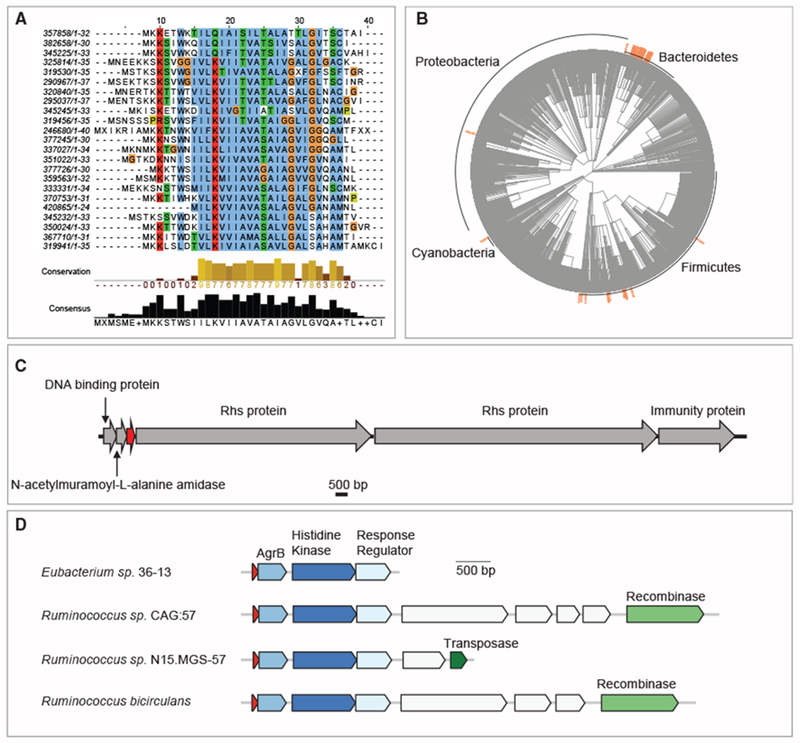
(A–C) Family 350024 is an abundant gut-related predicted transmembrane family potentially involved in bacteria-host or bacteria-bacteria crosstalk. (A) Multiple sequence alignment of representatives of all families that share amino acid sequence homology with family 350024. The length of the protein sequence is indicated after each family ID. (B) Phylogenetic spread of family 350024 and 22 other homologous families. (C) Genomic neighborhood, next to a DNA binding protein and an N-acetylmuramoyl-L-alanine amidase, an enzyme that cleaves the amide bond between N-acetylmuramoyl and L-amino acids in bacterial cell walls. The locus tag of the small predicted transmembrane protein (red) is Ga0104402_10435 (Bacteroides ovatus NLAE-zl-C500).
(D) Putative signaling molecule that is presumably subject to horizontal transfer. Schematic representation of genes encoded on contigs of family 155173. In addition to Agr genes, these contigs typically harbor genes that are associated with horizontal transfer.
The genomic localization of this sORF is also conserved among homologs, adjacent to a DNA binding protein and an N-acetylmuramoyl-L-alanine amidase, an enzyme that cleaves the amide bond between N-acetylmuramoyl and L-amino acids in bacterial cell walls (Figure 4C). Interestingly, the product of an amidase was recently shown to mediate channel formation between bacterial cells that express them (Zheng et al., 2017). In addition, we often observe within close vicinity of these three genes, virulence-related genes as VirE and/or genes encoding for the Rhs protein, a DNase that is delivered to neighboring cells during contact dependent inhibition, as well as the immunity protein that protects the encoding cell from the Rhs’ toxic effect (Koskiniemi et al., 2013). In the proteomic analysis of Bacteroides thetaiotaomicron VPI-5482 described above, we show that a distant homolog (Figure S5) of family 350024, encoded in the intergenic region between an N-acetylmuramoyl-L-alanine amidase (locus tag BT4031) and a DNA binding protein (locus tag BT4032), is expressed. Altogether, we hypothesize that this small protein may be involved in crosstalk with other cells, potentially as part of a novel secretion/inhibition mechanism.
We were intrigued by the genomic neighborhood of family 155173, which was identified in over 40% of gut samples. Homologs of this potentially secreted protein are recurrently found upstream of a transmembrane protein annotated as AgrB, a histidine kinase and a response regulator (Figure 4D). This composition of genes strongly resembles the composition of the quorum sensing Agr operon, which consists of the short signaling peptide (AgrD), a transmembrane protein (AgrB), and a two-component system composed of a histidine kinase (AgrC) and a response regulator (AgrA) (Olson et al., 2014). The small protein identified here was not assigned a domain in our query against CDD domains. However, the genomic localization of this secreted protein in addition to the similarity in size to AgrD, suggest that these four genes encode a quorum sensing system, in which the signaling molecule component is a distant homolog of AgrD. Intriguingly, we also observed that in at least 51/154 homologs of this family, the small gene is encoded in the vicinity of genes that mediate horizontal gene transfer (see below section about horizontal transfer), suggesting that this cluster of genes is subject to horizontal transfer (Figure 4D). The potential of the Agr quorum sensing system to undergo phage-derived horizontal transfer has been suggested before (Hargreaves et al., 2014), and here, we provide additional support to this model.
Small Protein Families with a Potential Role in Bacterial Defense against Phage
Bacteria have evolved a variety of defense systems that protect them from phage attack (Dy et al., 2014; Koonin et al., 2017; Stern and Sorek, 2011) and these tend to cluster in genomic regions denoted “defense islands” (Koonin et al., 2017). This notion has been recently used to identify multiple novel defense systems based on their localization within “defense islands” (Doron et al., 2018). Here, we were interested in identifying small proteins that could be associated with defense against phage. Small defense-related proteins are easily missed in bioinformatic studies, such as the recent systematic study that aimed at identifying CRISPR-Cas-related genes, which applied an inclusion cutoff of 100 amino acids (Shmakov et al., 2018), or studies that rely on domain annotation of protein families (Doron et al., 2018).
To identify small protein families that could be related to bacterial defense against phage, we searched for sORFs that are encoded in the vicinity (within ≤10 genes upstream/downstream) of known defense genes. To identify defense genes, we used a list that was recently compiled that contains 427 different COGs/Pfams of known defense genes (Doron et al., 2018). We were able to identify 869 (869/4,539 = 19%) small protein families in which at least one homolog is encoded in the vicinity of known defense gene/s (Table S3). Of these, 132 families are associated with CRISPR genes.
To increase the confidence that a small protein family is defense-related, we asked whether “defense-relatedness” is conserved among homologs of the same family. For each family, we counted the number of homologs that are encoded within 10 genes of known defense genes and calculated the fraction that are “defense-related” (Table S3). There are 13 families in which at least half of homologs are “defense-related,” of which 5 families are specifically CRISPR-related. Family 395508 is an example of a potential CRISPR-related small protein in which 90% (65/72) of the homologs are encoded within ≤ 10 genes from CRISPR-related genes (Figures 5A and 5B). It encodes a 28-amino acid predicted transmembrane protein (or transmembrane and secreted according to the orthogonal Phobius algorithm). Toxin-antitoxin systems also play role in defense against phage (Rostøl and Marraffini, 2019). In family 588, the small gene is encoded immediately upstream of a known “orphan” toxin that encodes a PIN nuclease in 150/191 contigs. Based on the “guilt by association approach” (Leplae et al., 2011), we hypothesize that family 588 may encode a novel antitoxin protein of a toxin-antitoxin system (Figure 5C).
Figure 5. Small Proteins that Are Potentially Associated with Defense against Phage.
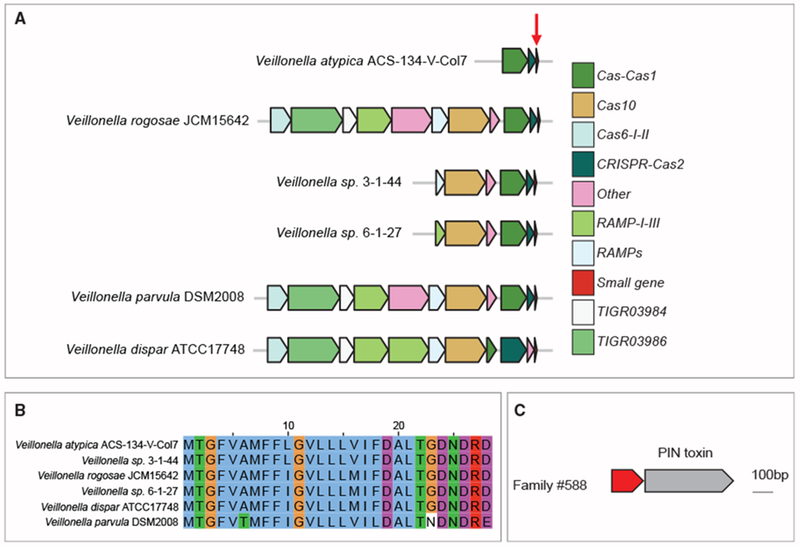
(A and B) Small protein family (395508) possibly associated with a CRISPR anti-phage system. (A) Genomic neighborhood of small protein (red arrow) across 6 different species. Homologs of this small protein are shown in the genomic locus in which they were found among a variety of Veillonella species within HMPI-II data. (B) Multiple sequence alignment of homologs of the family demonstrates a high level of conservation within small protein family 395508.
(C) Small protein of family 588 is encoded upstream of a known toxin.
Small Proteins that Are Part of the “Mobilome” May Play a Role in Bacterial Adaptation
The human gut is presumed to serve as a “melting pot” of horizontal genetic material exchange, which bacteria leverage in evolving to adapt (Liu et al., 2012; Shterzer and Mizrahi, 2015). This phenomenon mediates transfer of antibiotic resistance genes, virulence genes, genes involved in metabolism and stress response, as well as genes involved in defense against phages (Ochman et al., 2000; Soucy et al., 2015; Zaneveld et al., 2008). Phages are among the agents that mediate HGT of advantageous genes between hosts (Colomer-Lluch et al., 2011; Manrique et al., 2017; Virgin, 2014).
Here, we attempted to identify small protein families that could be part of the bacterial “mobilome.” A hallmark of genomic regions that are subject to horizontal gene transfer (HGT) is the presence of genes that mediate horizontal transfer (Oliveira et al., 2017). In addition, because horizontal transfer spreads genes between potentially distant bacterial lineages, genes that are subject to horizontal transfer may display a distribution that is discordant with the organismal tree of life (“patchy distribution”) (Cordero and Hogeweg, 2009). We used these two characteristics to identify families that are potentially subject to HGT.
First, we searched for small protein families whose homologs are recurrently found in the vicinity (within ≤ 10 genes upstream/downstream) of genes that are known to mediate horizontal transfer (STAR Methods). This resulted in a set of 2,646 (58%, 2,646/4,539) small protein families in which at least one homolog is encoded in the vicinity of an HGT-mediating gene (Table S3). To identify families in which homologs are recurrently found in mobile regions, we calculated the fraction of HGT-related homologs from the total number of homologs for each family. Doing so, we identified 329 small protein families that we are highly confident are “HGT-related,” because at least 50% of the homologs of the family are encoded in the vicinity of HGT-mediating gene(s).
Next, we sought to characterize the phylogenetic distribution of these 329 families. Families that display a patchy distribution are more likely to be horizontally transferred. A patchy distribution is associated with families that are identified in a relatively small number of species across multiple clades. However, because a patchy distribution could be a result of sampling biases, our approach is more powered to detect HGT events between higher taxonomic levels, such as between phyla. For a vertically transmitted gene to have a sporadic distribution across phyla, multiple deletion events of the gene across the tree would have occurred, which is less likely. To enrich for small protein families in which the taxonomic classification is more reliable, we filtered out small protein families in which the median percentage of k-mers on the contig of origin that could be classified is <10%. This resulted in 202/329 small protein families (Figures 6A and S6). Firmicutes is the most represented phylum among the 202 small protein families, identified in 68% of the families (Table S3), in line with previous observations that this phylum is a major participant in genetic exchange (Caro-Quintero and Konstantinidis, 2015). Among the 202 small protein families, 57 families were mapped to at least 2 phyla (Figure 6B), representing potential inter-phyla HGT events.
Figure 6. Small Proteins that Are Potentially Subject to HGT between Phyla.
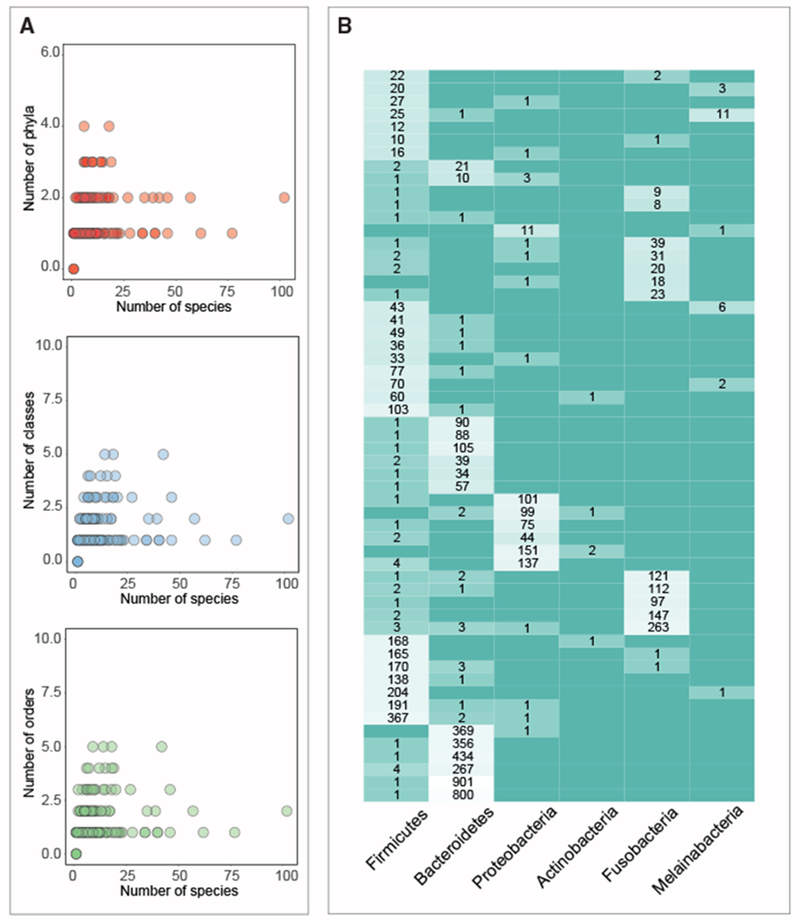
(A) Each dot represents one of 202 families that were identified in the screen of HGT genes in vicinity of small gene and whose median percentage of k-mers that were classified is >10%. Families that are encoded by a small number of species across a larger number of phyla/class/order are more likely to be true positives.
(B) Of the 100 families presented in (A), 57 small protein families that were identified in ≥2 phyla are presented. Only phyla that were identified in at least five different small gene families are shown. Numbers within boxes indicate the total number of individual homologs within the family encoded by the designated phylum. Each row was normalized.
Finally, we sought to identify small proteins that are encoded by phage. By analyzing genomic context and classification of encoding contigs we identified 405 small protein families that either have at least one homolog that was classified as viral or are integrated within a presumable prophage region (Figure S6; STAR Methods).
DISCUSSION
Accumulating evidence suggests that small proteins play important roles in bacterial physiology. However, due to computational and experimental limitations, this class of proteins is consistently overlooked. Here, we focused on small proteins encoded by the human microbiome. We were interested in small proteins within this niche for several reasons. In terms of size, small proteins can represent a “bridge” between the “natural product” world, a rich source of biologically active molecules such as antibiotics, and the larger protein world. As such, they are likely to display a range of activities that would resemble either class and thus operate at microbe-host interface. While natural products have attracted much attention and investigation (Donia et al., 2014; Milshteyn et al., 2018; Trivella and de Felicio, 2018; Wilson et al., 2017), and large proteins are easier to detect and analyze, small proteins in the human microbiome have thus far evaded thorough systematic analysis.
In this study, we applied a combination of computational approaches on 1,773 healthy human metagenomes and identified 4,539 conserved small protein families. We show that most families are not represented in traditional reference genomes and/or do not contain a known protein domain. For a subset of families that could also be detected in an independent metatranscriptomic study, we show that the vast majority are transcribed. By classifying the protein families according to their taxonomic distribution, their prevalence across human body sites and non-human metagenomes, their predicted cellular localization, their genomic neighborhood and more, we assign putative functions to a subset of the families.
Proteins that play housekeeping roles are expected to be unrelated to a specific niche or taxonomic clade. Indeed, among these 14 potential “housekeeping” families, six encode for different ribosomal proteins, a known class of housekeeping proteins. Among the remaining eight families, three were assigned a CDD domain and five were not. We show that the one that contains a domain of unknown function 4295 (DUF4295) likely to be a novel small ribosome-associated protein. We provide evidence that this sORF is indeed transcribed and translated, most probably at high levels. One may wonder how such a protein might escape detection, as ribosomes have been subject of deep investigation spanning several decades of research. We believe that this is due to the focus of prior research on a handful of model organisms (such as E. coli, which lacks this predicted small protein) and the dismissal of small ORFs from bioinformatics analysis pipelines. Many of the genomes that encode this small protein are residents of the human microbiome, whose genomes have mainly been sequenced in the last decade and whose ribosomes have not been studied, in depth. The experimental laboratory strain Bacteroides thetaiotaomicron VPI-5482 encodes this small protein but as is the case for many sORFs, the gene that encodes for this protein remained unannotated.
The continuous arms race between bacteria and bacteriophages has led to the evolution of an arsenal of bacterial anti-phage systems. Some of these systems have important biotechnological applications (i.e., restriction enzymes and CRISPR-Cas), leading to a strong interest in identifying novel systems. However, bioinformatic studies in the field usually fail to detect small proteins, as these do not pass the size inclusion cutoff and are usually devoid of annotation. Using our unbiased approach, we identified 13 small protein families that are presumably found on “defense-islands,” five of which are regions that encode for CRIPSR genes. It is possible that these small proteins are associated with already known or yet unknown defense systems.
The ability of bacteria to rapidly adapt to changing environmental conditions is strongly associated with the acquisition of new genes through horizontal gene transfer. A major clinical challenge is that horizontal gene transfer contributes significantly to the rapid spread of antibiotic resistance. For example, AcrZ, a 49-amino acid membrane protein, can enhance antibiotic resistance through regulation of a multidrug efflux pump (Hobbs et al., 2012). Here, we identify 329 small protein families that are likely horizontally transferred. Of these, 84 families are predicted to be membrane proteins. This list represents an opportunity to search for small proteins that may support adaptation, particularly as regulators of drug pumps.
Because of their short length, small proteins generally consist of one domain and represent a useful model system for protein folding simulations (Imperiali and Ottesen, 1999) and drug design (Martin and Vita, 2000). It has recently been suggested that the number of domains is reaching saturation (Scaiewicz and Levitt, 2018). However, more than 95% of the small protein families identified in our study do not have any known domains. This stresses the possibility that the space of small proteins represents an untapped opportunity for discovery of new building blocks of proteins. Many studies rely on domains assigned to proteins (Doron et al., 2018; Shmakov et al., 2018), emphasizing the benefit of annotating the domains of small proteins, so that they are not overlooked in future studies of this type.
Transmembrane and secreted proteins mediate most of the interactions of a bacterium with its environment, making these classes of proteins relevant targets for medical research. Here, we identified a total of 1,054 families that are predicted to be transmembrane and/or secreted. One of the families (350024) that is presumably very abundant across different mammalian guts encodes for a predicted transmembrane protein that is encoded between a DNA binding protein and an amidase enzyme that cleaves cell wall. A recent paper showed that a similar enzyme is involved in formation of channels for material exchange between cells (Zheng et al., 2017). We suggest that the small protein identified is part of a cluster of genes that could also be involved in channel formation between cells and subsequent DNA translocation.
In light of the increased frequency of resistance to conventional antibiotics, there is an interest in developing antimicrobial peptides as an alternative therapy (Cotter et al., 2013; Lau and Dunn, 2018). While a large fraction of known antimicrobial peptides cause cell death through transmembrane pore formation, a growing number of studies show additional mechanism of action, such as translation inhibition through interaction with the ribosome (Seefeldt et al., 2015). Here, we identify 39 potential novel antimicrobial peptide families that remain to be experimentally validated.
While HGT events within bacteria and archaea are unequivocal (Soucy et al., 2015; Wagner et al., 2017), the frequency and importance of HGT between domains of life is less clear (Husnik and McCutcheon, 2018). Using taxonomic contig classification, we identified multiple families that were mapped to more than one domain of life. While misassembly or misclassification of contigs could possibly account for this, this observation remains intriguing as it suggests either ancient conservation of sORFs or true genetic transfer between evolutionarily distant organisms.
Despite the promise that this approach holds for sORF prediction, it is important to note its limitations. First, our analysis filters out families if they are encoded by <8 different sequences, therefore potentially missing genuine small protein families when these are very rare. Second, small proteins undergoing rapid evolution may fall into separate families in the sequence-based clustering step, which may lead to them being filtered out due to small family size. Third, longer proteins that are undergoing pseudogenization could falsely appear as small proteins. Fourth, since we conduct our analysis on contigs, we are also vulnerable to errors in taxonomic classification that could stem from misassembly and/or misclassification of contigs. Fifth, our prediction of secreted proteins relies on the presence of signal peptides. This is a limited prediction because not all proteins that harbor a signal peptide are secreted outside of the cell (Green and Mecsas, 2016), and because signal peptides contain a hydrophobic region that can be mistaken for a transmembrane region, implying that a subset of the predicted transmembrane proteins could actually be secreted (Krogh et al., 2001). Finally, our analysis of the genomic region of small genes is limited by the number of genes that are encoded on the encoding contig, which is variable in metagenomic data.
In our analysis of published metaproteomics datasets (Zhang et al., 2017, 2018), only a small subset of the predicted small proteins could be validated. While this analysis demonstrates that a small subset of predicted small proteins are indeed transcribed and translated, it also highlights that standard proteomic experimental workflows are limited and there is a need to optimize protocols to enrich for small proteins. In our proteomics analysis of small proteins in Bacteroides thetaiotaomicron, we were able to validate 10% of the its high-confidence small proteins. Our analysis was restricted to one standard growth condition in which we extracted proteins from a saturated culture. Therefore, it is likely that we failed to detect small proteins that are expressed in other conditions or earlier growth stages.
To advance from this study, mechanistic studies will be required. Gene deletion and complementation studies are likely to be highly informative. In light of the relatively low cost of their synthesis, it may be feasible to conduct high-throughput studies in which small genes are synthesized and expressed within cells to study gain of function phenotypes. Finally, interactions of small proteins with human proteins could be studied by applying co-immunoprecipitation protocols.
To facilitate future investigation of these candidate novel small proteins, a comprehensive resource file is presented in this manuscript (Table S3; see also Figure S7). This table provides an exhaustive summary of all attributes associated with each of the 4,539 families and facilitates others to query the database of novel sORFs for families that obey specific attributes of interest. Following such queries, one can extract all DNA/amino acid sequences of homologs from Data S1 and also all underlying contigs according to the guidelines given in the STAR Methods.
Knowledge of small peptides encoded by human associated bacteria is very limited. We hope that the data and computational approach presented here will open a new frontier in the study of the microbiome and enhance our ability to exploit the therapeutic potential of this previously ignored class of macromolecules.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ami S. Bhatt (asbhatt@stanford.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe strains
The bacterial strain used in this study is Bacteroides thetaiotaomicron VPI-5482 (ATCC 29148).
METHODS DETAILS
Identification of sORFs from multiple human associated metagenomes
Contigs from 1,773 HMPI-II human-associated metagenomes from 17 body sites that were shotgun sequenced and contained no less than 5M bp sequenced per sample were downloaded from https://www.hmpdacc.org/hmasm2/. Body sites were collapsed into four groups (Table S1). For each metagenomic sample, all ORFs were predicted using MetaProdigal (Hyatt et al., 2012) with parameters adjusted to include ORFs ≥ 15bp. Small ORFs where filtered to include only those that contain a start and stop codon, resulting in a set of 2,514,099 sORF ≤ 150bp. RBS motifs were extracted from the standard output of MetaProdigal.
Clustering of sORFs into families
Proteins encoded by this set of sORFs were clustered using CD-Hit with the following parameters: -n 2 -p 1 -c 0.5 -d 200 -M 50000 -l 5 -s 0.95 –aL 0.95 –g 1 (the shorter sequences were required to be ≥ 95% length of the representative of the cluster and the alignment must cover ≥ 95% of the longer sequence). This resulted in 444,054 clusters. Each cluster was assigned a ‘cluster representative’ by CD-Hit that was used in subsequent parts of our analysis.
Domain Analysis
The CDD DB was downloaded from ftp://ftp.ncbi.nih.gov/pub/mmdb/cdd/little_endian/Cdd_LE.tar.gz on October 2018. This DB contains models that are in the default CDD database: CD (alignment models curated at NCBI as part of the CDD project), Pfam, Smart, COG, PRK and TIGRFAM. The amino acid sequence of each of the cluster representatives from each one of the 444,054 clusters was searched against this DB, using RPS-blast. A hit was considered significant if the e-value ≤ 0.01 (default CDD e-value threshold) and the small protein aligns to at least 80% of the PSSM’s length. Small protein families were classified according to the PSSM they hit. The same domain may be assigned to multiple families reflecting distant sequence conservation across families (e.g., families 263535, 73615 and 209227 that were all assigned the same PSMM of a small ribosomal protein L36), and families may be assigned multiple domains, reflecting redundancies in CDD (e.g., family 305829, that was assigned two different PSSMs, both of ribosomal protein L34).
Identification of known proteins among the small protein clusters
Small proteins that were studied in depth were divided into two groups: those that have an assigned sequence domain and those that do not. The domains that were assigned to the 444,054 clusters were queried for domains of the first group. The known small proteins that do not have an assigned sequence domain were queried against all 444,054 representative protein sequence, using BLASTp with word-size 2. Hits were considered significant if: e-value ≤ 0.05, the alignment spans ≥ 90% of the protein and the length of the hit was 90%–110% of the length of the small protein.
Analysis of publicly available metatranscriptomics data
Assemblies of 226 human gut metagenomes were downloaded from EBI metagenomics (https://www.ebi.ac.uk/metagenomics/studies/MGYS00003733 and the corresponding metatranscriptomes were downloaded from https://www.ncbi.nlm.nih.gov/bioproject/354235 (Accession PRJNA354235). For each metagenomic sample, all ORFs were predicted using MetaProdigal (Hyatt et al., 2012) with parameters adjusted to include ORFs ≥ 15bp. This set was used to define how many reads are mapped to coding regions. Small ORFs were filtered to include only those that contain a start and stop codon. Representatives of each of the 4,539 small protein families were queried against this set of small proteins, using BLASTp with word-size 2. Hits were considered significant if: e-value ≤ 0.05, the alignment spans ≥ 80% of the protein and the length of the hit was 80%–125% of the length of the small protein. Metatranscriptomic reads (93bp in length) were quality filtered using trim galore version 0.4.0 (Krueger, 2014), a wrapper for cut adapt version 1.8.1 (Martin, 2011) using a default parameters and a quality score cutoff of 30. Reads were mapped to the associated assemblies using bowtie version 1.1.1 (Langmead et al., 2009), using default parameters except allowing for no mismatches. For each predicted ORF, mapped reads were counted using bedtools coverage (Quinlan and Hall, 2010) if ≥ 70% of the read mapped without mismatches to the ORF. To calculate reads per kilobase of transcript, per million mapped reads (RPKM) for each small ORF, the total number of reads mapping to the combined set of small and regular-sized genes was calculated. A small ORF was considered transcribed only if its RPKM ≥ 20.
Analysis of publicly available metaproteomics datasets
Two publicly available metaproteomic datasets were selected. The first is a deep metaproteomics dataset consisting of samples from four children with IBD (Zhang et al., 2017). The second is a random subset of 18 samples from a large pediatric cohort (n = 71) obtained from IBD patients (Zhang et al., 2018). Raw files were searched in MaxQuant version 1.5.2.8(1) (Cox and Mann, 2008), using the search engine Andromeda against the MetaPro-IQ database (374,267 entries published in Zhang et al., 2017) consisting of a gut microbial gene catalog and a human database that were combined with all 72,569 non-redundant sequences composing the ~4k families. The enzyme specificity was set to trypsin and a maximum of 2 missed cleavages were allowed. Search parameters was set to 7-40 amino acids, enzyme specificity: trypsin, max 2 missed cleavages, 4.5 ppm match tolerance for precursor ions and 20 ppm for fragment ions with 1% FDR both at the peptide and protein level. Oxidation on methionine and N-terminal acetylation were set as variable modifications and carbamidomethylation on cysteine as a fixed modification.
Identification of small proteins in Bacteroides thetaiotaomicron VPI-5482 and homologs to ~4k families
Prodigal (Hyatt et al., 2010) with parameters adjusted to include ORFs ≥ 15bp was used to call genes on Bacteroides thetaiotaomicron VPI-5482 GCF_000011065.1, resulting in 5,071 genes. Small ORFs where filtered to include only those that contain a start and stop codon, resulting in a set of 215 sORF ≤ 150bp. Homolog of family 26 is 52aa long in this strain and was added manually after this filtering step. Representatives of families were blasted against the 216 predicted small proteins. Hits were considered significant if: e-value ≤ 0.05, the alignment spans ≥ 80% of the query protein and the length of the hit is 80%–125% of the length of the small protein.
Analysis of Bacteroides thetaiotaomicron VPI-5482 transcriptomics data
Transcriptomics data (Tropini et al., 2018) were downloaded from the Stanford Digital Repository (https://purl.stanford.edu/kw691rt5031). Transcriptomics reads (150 bp in length) were quality filtered using trim galore version 0.4.0 (Krueger, 2014), a wrapper for cutadapt version 1.8.1 (Martin, 2011) using default parameters and a quality score cutoff of 30. Reads were mapped to the Bacteroides thetaiotaomicron VPI-5482 reference genome using bowtie version 1.1.1 (Langmead et al., 2009) using default parameters except allowing for no mismatches. For each predicted ORF, mapped reads were counted using bedtools coverage (Quinlan and Hall, 2010) if ≥ 60% of the read mapped without mismatches to the ORF. If the Bacteroides thetaiotaomicron genome was covered on average above 20-fold in any given condition, then that condition was included in downstream analyses. To calculate reads per kilobase million (RPKM) for each small ORF: 1) Metaprodigal with default parameters was applied on Bacteroides thetaiotaomicron VPI-5482. 2) The set of small ORFs that are homologous to any of the 4,359 small proteins were added. 3) The total number of reads mapping to the combined set was calculated. A small ORF was considered transcribed only if its RPKM ≥ 20.
Ribo-Seq of Bacteroides thetaiotaomicron VPI-5482
Ribosome profiling was performed in duplicates as previously described (Latif et al., 2015) on Bacteroides thetaiotaomicron grown to saturation. Before harvesting, Bacteroides thetaiotaomicron was treated with 0.1 mg of chloramphenicol per mL of culture. After 2 minutes, aliquots of culture were centrifuged in 50 mL tubes at 10,000 x g. Cell pellets were resuspended in Ribo-Seq lysis buffer. As previously described (Latif et al., 2015), the buffer consisted of 25 mM Tris pH 8.0, 25 mM NH4Cl, 10 mM MgOAc, 0.8% Triton X-100, 100 U/mL RNase-free DNase I, 0.3 U/μL Superase-In, 1.55 mM Chloramphenicol, and 17 μM 5′-guanylyl imidodiphosphate (GMPPNP). Lysis was performed using bead beating for 3 minutes in this lysis buffer, using a MiniBeadBeater-16, Model 607 and 1mM zirconia/silica beads. 25 A260 units of RNA were treated with 6000U of MNase using MNase buffer to dilute as necessary. MNase buffer contained 25 mM Tris pH 8.0, 25 mM NH4Cl, 10 mM MgOAc, and 1.55 mM chloramphenicol (Latif et al., 2015). The MNase reaction was incubated at room temperature for 2 hours. All following steps were performed identically to previous literature (Latif et al., 2015), except the tRNA removal steps were excluded. Briefly, 500 mL of polysome binding buffer was used to wash the Sephacryl S400 MicroSpin columns (GE Healthcare Life Sciences) three times - spinning the column for 3 minutes at 4°C at 600 RPM. Polysome binding buffer consisted of 100 μL Igepal CA-630, 500 μL magnesium chloride at 1M, 500 μL EGTA at 0.5 M, 500 μL of NaCl at 5M, 500 μL Tris-HCl pH 8.0. at 1M, and 7.9 mL of RNase-free water. The MNase reaction was applied to the column and centrifuged for 5 minutes at 4°C. The flow through was purified further with miRNAeasy Mini Kit (QIAGEN) using manufacture protocols. Elution was performed at 15 μL volume. rRNA was depleted using RiboZero-rRNA Removal Kit for Bacteria (Illumina) using manufacture protocol, except all reaction volumes and amounts were reduced by 50 percent. This was purified with RNAeasy MinElute Cleanup Kit (QIAGEN), eluting in 20 uL. The reaction, in 18 μL volume, was subjected to T4 PNK Reaction (NEB M0201S) with addition of 1 μL Superase-In (Invitrogen), 2.2 μL 10X T4 PNK Buffer, and 1 μL T4 PNK(10U/μL). This reaction was purified again with RNAeasy MinElute Cleanup (QIAGEN). The concentration was determined with Qubit RNA HS Assay Kit (Illumina). With 100 ng as input, libraries were prepared using NEBNext Small RNA Library Prep Set for Illumina (NEB, E7330), using manufacture protocols. DNA was purified using Minelute PCR Purification Kit (QIAGEN). Libraries were sequenced using a NextSeq 500/550 v2.5 1x75 kit using 50 cycles.
RNA-Seq of Bacteroides thetaiotaomicron VPI-5482
Aliquots of Bacteroides thetaiotaomicron were centrifuged in 50 mL tubes at 10,000 x g. Cell pellets were resuspended in RNA-Seq lysis buffer. The buffer consisted of 25 mM Tris pH 8.0, 25 mM NH4Cl, 10 mM MgOAc, 0.8% Triton X-100, 100 U/mL RNase-free DNase I, and 0.3 U/μL Superase-In. We performed RNA-Seq as follows: 15 μL of proteinase K (Ambion, 20 mg/mL) was added to 600 μL of lysate. After incubation for 10 minutes at room temperature, samples were centrifuged at 21,000 x g for 3 minutes and the supernatant was collected. An equal volume of Phenol/Chloroform/Isoamyl Alcohol 25:24:1 (pH. 5.2) was applied and vortex for three minutes. The mixture was centrifuged at 21,000 x g for three minutes. The aqueous phase was extracted. This was repeated once more. The final aqueous phase was ethanol precipitated. The RNA was further purified using the RNAeasy Mini plus Kit (QIAGEN) according to manufacturer’s protocols. Any remaining DNA was degraded via Baseline-ZERO-DNase (Epicenter), RNA was fragmented for 15 minutes at 70°C using RNA Fragmentation Reagent (Ambion), and the fragmented RNA was purified with miRNAeasy Mini Kit (QIAGEN), all according to the manufacturer’s protocols. Elution was performed at 15 μL. rRNA was depleted using RiboZero-rRNA Removal Kit for Bacteria (Illumina) using half reactions of the manufacturer’s protocols. This was purified with RNAeasy MinElute Cleanup Kit (QIAGEN), eluting in 20 uL. The fragments, in 18 μL volume, were subjected to T4 PNK Reaction (NEB M0201S) with addition of 1 μL Superase-In (Invitrogen), 2.2 μL 10X T4 PNK Buffer, and 1 μL T4 PNK(10U/μL). This reaction was purified again with RNAeasy MinElute Cleanup (QIAGEN). The concentration was determined with Qubit RNA HS Assay Kit (Invitrogen). With 100 ng as input, libraries were prepared using NEBNext Small RNA Library Prep Set for Illumina (NEB, E7330), as per manufacturer’s protocols. DNA was purified using MinElute PCR Purification Kit (QIAGEN). Libraries were sequenced using a NextSeq 500/550 v2.5 1x75 kit using 50 cycles.
Analysis of Bacteroides thetaiotaomicron VPI-5482 RNA-Seq and Ribo-Seq data
Reads (50 bp in length) were quality filtered using trim galore version 0.4.0 (Krueger, 2014), using default parameters and a quality score cutoff of 30. Reads were mapped to the Bacteroides thetaiotaomicron VPI-5482 reference genome using bowtie version 1.1.1 (Langmead et al., 2009) using default parameters except allowing for no mismatches. For each predicted ORF, mapped reads were counted using bedtools coverage (Quinlan and Hall, 2010). To calculate reads per kilobase million (RPKM) for each small ORF: 1) Metaprodigal with default parameters was applied on Bacteroides thetaiotaomicron VPI-5482. 2) The set of small ORFs that are homologous to any of the 4,359 small proteins were added. 3) The total number of reads mapping to the combined set was calculated. A small ORF was considered transcribed only if its RPKM ≥ 20.
Bacteroides thetaiotaomicron VPI-5482 small protein extraction and analysis
Cell Culture: the culture media contained 37 g of brain heart infusion (BHI), sterilized at 121 °C for 20 minutes, and supplemented with freshly prepared, filter-sterilized L-cysteine to a final concentration of 0.1%, hematin solution (0.5 mg/ml in alkaline water) to a final concentration of 5 μg/ml, and NaHCO3 to a final concentration of 0.2%. Culture media were reduced in an anaerobic chamber for a minimum of 6 hours prior to inoculation. The media were inoculated with Bacteroides thetaiotaomicron VPI-5482 and incubated at 37°C under an anaerobic atmosphere for 72 hours until fully saturated. The culture was pelleted by centrifugation at 10,000 rpm for 20 minutes at 4°C, and the pellet was washed with 50 mM Tris-HCl (pH 7.5) to remove residual media and re-pelleted.
Lysis and enrichment for small proteins: cells were lysed on ice for 30 minutes in 80 mL of 50 mM Tris-HCl (pH 7.5), 0.5% SDS, 1 mM EDTA followed by 2 minutes of sonication on ice at output level 8 with a 50% duty cycle. The lysate was clarified by centrifugation at 14,000 rpm at 4°C for 30 minutes. To enrich for small proteins in the lysate, acetic acid was added to the lysate to a final concentration of 0.25% (v/v) followed by incubation on ice for 30 minutes and centrifugation at 14,000 rpm for 20 minutes at 4°C. This addition helps in precipitating larger proteins. A fraction of acid-treated clarified lysate (25 mL) was filtered through a 30-kDa molecular weight cut off (MWCO) filter (Millipore) to further enrich for lower molecular weight proteins in the lysate.
Protein isolation: samples from 3 different preparations were analyzed by LC-MS/MS. First, 10 mL of acid-treated lysate was treated with 40 mL of acetone and incubated at −20°C for 1 hour to precipitate proteins followed by centrifugation at 14,000 rpm at 4°C for 30 minutes. Second, 10 mL of acid-treated 30-kDa MWCO filtrate was treated with 40 mL of acetone and incubated at −20°C for 1 hour to precipitate proteins followed by centrifugation at 14,000 rpm at 4°C for 30 minutes. Third, 1.5 mL of acid-treated 30-kDa MWCO filtrate was dried down in a Vacufuge Concentrator (Eppendorf) at 35°C overnight.
Digestion and Sample Preparation for LC–MS/MS: dried protein pellets were dissolved in 50 mM ammonium bicarbonate in the presence of 0.02% protease max (Promega) and reduced with 10 mM DTT at 55°C for 30 min. Following reduction, proteins were alkylated using 30 mM acrylamide for 30 minutes at room temperature. Digestion was performed with Trypsin/LysC (Promega) in a standard overnight digest at 37°C. After digestion, the reaction was quenched using 1% formic acid and peptides were de-salted on C18 Monospin reversed phase columns (GL Sciences). The de-salted peptides were dried in a speed vac before reconstitution in 20 μL of reconstitution buffer (2% acetonitrile with 0.1% formic acid); 2 μL of this solution was injected on the instrument.
LC–MS/MS Analysis: mass spectrometry experiments were performed using a Q Exactive HF-X Hybrid Quadrupole - Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA) with liquid chromatography using a Nanoacquity UPLC (Waters Corporation, Milford, MA). A flow rate of 450 nL/min was used, where mobile phase A was 0.2% formic acid in water and mobile phase B was 0.2% formic acid in acetonitrile. Analytical columns were prepared in-house with an I.D. of 100 microns packed with Magic 1.8 micron 120Å UChrom C18 stationary phase (nanoLCMS Solutions) to a length of ~25 cm. Peptides were directly injected onto the analytical column using a gradient (2%–45% B, followed by a high-B wash) of 80 minutes. The mass spectrometer was operated in a data dependent fashion using HCD fragmentation for MS/MS spectra generation. For data analysis, the .RAW data files were processed using Byonic v3.2.0 (Protein Metrics, San Carlos, CA) to identify peptides and infer proteins using Bacteroides thetaiotaomicron VPI-5482 database from Uniprot and a custom database of the small proteins predicted in Bacteroides thetaiotaomicron VPI-5482 using Prodigal. Proteolysis was assumed to be semi-specific allowing for N-ragged cleavage with up to two missed cleavage sites. Precursor and fragment mass accuracies were held within 12 ppm. Proteins were held to a false discovery rate of 1%, using standard approaches.
Taxonomic classification of small protein families
1,504,527 contigs encoding the small proteins were classified using the One Codex database 2018 (Minot et al., 2015). Each contig was compared to a database of 83,701 microbial reference genomes. The platform matches all overlapping k-mers in a given contig to the most specific organism possible. Since not all k-mers are unique to a specific operational taxonomic unit (OTU), each k-mer was classified to the lowest common ancestor (LCA). Individual k-mer matches across a given contig were then aggregated to assign the most specific and consistent OTU to the contig. For each contig, the proportion of 31-mers that were classified out of the total 31-mers (rounded to the nearest whole number), was recorded. For each small protein family, the number of different OTUs, phyla, classes, orders, families, genera and species in which it was detected was recorded. Of the 1,504,527 total contigs, 69,974 contigs (4.6%) could not be taxonomically classified. The four families in which all contigs were classified as ‘Homo sapiens’ were excluded from further analysis.
Analysis of small proteins in RefSeq genomes
Protein sequences from 69,681 RefSeq genomes, were download from ftp://ftp.ncbi.nlm.nih.gov/genomes/RefSeq/bacteria/ on July 2017. Representative protein sequences of 4,539 families were blasted against 3,549,250 RefSeq proteins that are ≤ 50 amino acids with word_size 2 and Max number of hits = 500. To call for small genes on these RefSeq genomes, Prodigal (Hyatt et al., 2010) with parameters adjusted to include ORFs ≥ 15bp was run on RefSeq reference genomes. Representatives of families were blasted against 6,931,965 prodigal-predicted proteins that are ≤ 50 amino acids as described above. In both cases, hits were considered significant if: e-value ≤ 0.05, the alignment spans ≥ 90% of the query protein and the length of the hit is 90%–110% of the length of the small protein.
Identification of homologs of small proteins among “long” HMP proteins
MetaProdigal (Hyatt et al., 2012) was used to call for all genes encoded in 1,773 HMPI-II metagenomic samples. The resulting set was filtered to include only those that encode for proteins of at least 100 amino acids and have a start and stop codon, resulting in 82,947,548 proteins. Representatives of the 4,539 small protein families were blasted against this set with word_size 2 and Max number of hits = 500. Hits were considered significant if: e-value <=0.05 and the alignment spans 90-110% of the small protein (no restriction on length of subject sequence).
Analysis of genomic neighborhood of small proteins
MetaProdigal (Hyatt et al., 2012) was used to call for genes on all contigs associated with all 4,539 small protein families. Amino acid sequence of all the genes on all contigs were searched against CDD, using RPS-blast with an e-value threshold of 0.01. A hit was considered significant if the e-value ≤ 0.01 (default CDD e-value threshold) and the protein aligns to at least 80% of the PSSM’s length. Each gene on a contig could have multiple significant domains. All domains of genes identified within 10 genes away from the small gene were recorded. Table S3 lists the associated genes in the longest contig of each family. For example, for family 111917, the small gene on the longest contig associated with the families is the 14th gene on the contig. The list of genes in vicinity includes “10: Phage_holin_4_1, COG4824, holin_tox_secr,” meaning that the 10th gene on the contig was assigned these CDD domains.
To identify small proteins that are in the vicinity of defense genes, domains identified within ≤ 10 genes away of the small protein were queried against the list of COGs and Pfams downloaded from Table S1 in Doron et al. (2018) as well as against the words ‘CRISPR’, cas1 and cas2. To identify HGT-related contigs, the words ‘recombinase’, ‘integrase’, ‘transposon’ and ‘transposase’ were queried. Since for every family of homologs, only part of the underlying contigs are long enough to be considered ‘informative’ (i.e., encode at least 10 genes downstream and upstream of the small gene), for each family, the number of ‘long enough’ contigs were counted (presented in Table S3 under the column ‘Number of contigs in which there are at least 10 genes from each side of the small gene’). To identify contigs that are presumably prophage, the words ‘phage’, ‘terminase’, ‘tail’, ‘caspid’ and ‘portal’ were queried against the CDD ‘short’ domain description. Only families in which the word ‘phage’ as well as one other others in the list were identified, were recorded as ‘phage’. Table S3 lists all domains found on longest contigs of each family.
Identification of homologs of family 350024
Protein sequence of representative of family 350024 was blasted against all 1,295 amino acid sequences of representatives of predicted transmembrane families, using a p-value threshold of 0.05. For each of the homologous families, all contigs associated with the family were annotated using MetaProdigal and proteins on all contigs were subject to RPS-blast against CDD database.
Identification of species that encode for the small protein adjacent to known toxin (family 588)
All contigs associated with family 588 were annotated with MetaProdigal and proteins were queried against CDD to assign domains. Contigs in which the small gene is encoded immediately upstream/downstream of PIN domain/COG3744/COG1848 were classified using the One Codex database, as described above. Number of species in which the small gene and the PIN domain were identified one next to the other, was recorded.
Mapping of small proteins to body parts
For each member in each of the ~4k families, we recorded the human subject and body site from which it originated. A member of a small protein family that was detected more than once in a specific body site of a specific human subject was counted only once (even when identified in multiple different sampling visits). For every family, the total number of appearances in each type of body site was then calculated. The total number of body samples from a specific body site counts multiple samples from the same subject’s body site, as one.
Search against non-human metagenomes
DNA sequences of each of the cluster representatives was blasted against a set of 5,829 non-human metagenomes using blastn with e-value 1e-05, 50% identity and alignment length coverage of 90%. MetaProdigal, adjusted to small gene finding (see above) was applied on the contigs that were hit in the previous step. The proteins that were identified by prodigal were then used as a DB against which protein sequences of all representatives was blasted against. Hits were considered significant if: e-value ≤ 0.05, the alignment spans ≥ 90% of the query protein and the length of the hit is 90%–110% of the length of the small protein.
Cellular Localization
SignalP-5.0 (Almagro Armenteros et al., 2019) was run with default parameters once with ‘gram +’ and once with ‘gram −’ mode on all small proteins encoded by ~4k families. TMHMM (Krogh et al., 2001) was run on the same set of proteins with default parameters. For every family, and for every attribute (transmembrane/signal ‘gram +’/signal ‘gram −‘) the number of transmembrane helices was counted, and whether the protein is predicted to be secreted. The percentage of family members that were predicted to be transmembrane/secreted was calculated and a family was considered transmembrane/secreted if ≥ 80% of the family members were predicted to be such. Phobius (Käll et al., 2004) was also applied (default parameters) on all small proteins encoded by ~4k families. To assess the fraction of transmembrane and secreted protein in ‘regular sized’ proteins, Prodigal was applied (default parameters) on all 1,773 metagenomes. Partial proteins were excluded and complete proteins were analyzed with SignalP-5.0 with ‘gram +’ and ‘gram −‘ mode and TMHMM with default parameters.
Secondary Structure Prediction
PSIPRED (McGuffin et al., 2000), was applied on each of the 4,539 representatives. Fora given protein sequence, PSIPRED first creates a position-specific scoring matrix by identifying homologs of the protein within a given DB. Since typical databases are depleted of small proteins, a DB was created that contains all 467,538 small proteins of 4,539 families. A small protein was categorized as ‘helix’/’coiled’/’beta’ if ≥ 50% of its residues are classified accordingly and as ‘mixed’ if at least 40% of the residues were classified to one category and at least 40% of the residues were classified to another category.
Antimicrobial Peptide prediction
AmPEP (Bhadra et al., 2018) was applied (default parameters) on 4,539 representatives of families.
GUIDELINES FOR EXTRACTION OF ALL CONTIGS ASSOCIATED WITH A SPECIFIC FAMILY OF INTEREST
For the sake of this explanation, family 314163 was chosen. There are 8 homologs in this family. The following four steps can be used to extract all HMPI-II contigs that encode genes from this specific family.
-
Retrieve all amino acid sequences that follow the header “Family: 314163” from the Supplementary file that contains all family sequences (Data S1. In this query, all 8 homologs would appear; for simplicity, only the first homolog entry is listed below.
Family: 314163
> 314163*_*32207*_*Rothia*_*97%*_*GGA/GAG/AGG_rbs_spacer = 5-10bp_Tongue_dorsum_SRS064774_764062976_Female_ 2_prediction_22054_3
MXTHKRLIDVVDPTPKAVDALMRLDLPADVNIEIKL
-
For each sequence, retrieve the field that indicates the sample number and contig number (both in bold in the sequences below) according to the following convention:
> familyID*_*Taxon ID of underlying contig*_*Name of Taxon*_*rbs_motif_rbs_spacer*_*percentage of 31-mers that were classified*_*Body site from which contig originated _SampleID_PatientID_Female/Male_Visit nummer_”prediction”_contig number_locationOfSmallGeneOnContig
Download the relevant sample, according to the Sample ID, from https://www.hmpdacc.org/hmasm2/.
Retrieve the contig of interest from the sample file, according to the contig number, which was given by HMPI-II.
In this example, for the first homolog of family 314163, download the sample SRS06477 and retrieve contig number 22054. This contig encodes the small gene. The small gene is the third gene (‘locationOfSmallGeneOnContig’), if genes are called on this contig with MetaProdigal with parameters modified to include all ORFs as short as 15bp.
QUANTIFICATION AND STATISTICAL ANALYSIS
Assigning p values to small protein families
RNAcode was shown to perform substantially better when the number of input sequences is ≥ 8 (Washietl et al., 2011). Here it was applied on all 11,715 clusters that are composed of ≥ 8 different DNA sequences. Only the 4,539 that were assigned a p-value of ≤ 0.05 were analyzed in subsequent steps of analysis.
DATA AND CODE AVAILABILITY
The raw sequencing reads generated in this study are available at BioProject: PRJNA540869. The published article includes all datasets generated during this study.
Supplementary Material
Figure S1. Basic Statistics of Samples and 4,539 Small Protein Families, Related to Figure 1
A. Protein length distribution of 4,539 families. Proteins of > 50 aa or < 5 aa were filtered out. The smallest protein in our dataset is encoded by family 442207 and is 16aa long. B. Distribution of number of encoding taxa per family. If multiple homologs are mapped to the same taxon, they were counted only once. C. Proportion of samples belonging to different body sites. 128,368,337 contigs were obtained from 1,773 HMPI-II human-associated metagenomes that were shotgun sequenced, spanning 4 different major body sites from 263 healthy individuals. Samples were obtained from individuals in one, two or three subsequent visits. Body sites were collapsed into four groups: anterior nares, buccal mucosa, hard palate, keratinized gingiva, palatine tonsils, saliva, subgingival plaque, supragingival plaque, throat and tongue dorsum to ‘mouth’; mid vagina, posterior fornix and vaginal introitus to ‘vagina’; left retroauricular crease, right retroauricular crease and right antecubital fossa to ‘skin’; stool samples were renamed ‘gut’ here. D. Transmembrane/secreted proteins among the 4,539 families as predicted by TMHMM (Krogh et al., 2001) and signaiP (Almagro Armenteros et al., 2019), respectively. A family is predicted to be transmembrane/secreted if ≥ 80% of the homologs of the family are predicted to be such. In addition to families that passed this threshold, in 180 families, at least one family member, but less than 80% of the family members are predicted to be transmembrane and in 414 families, at least one family member, but less than 80% of family members are predicted to be secreted. An alternative algorithm, Phobius (Käll et al., 2004) was also applied on the 4,539 families. Phobius predicted 1002 families (22%) to be transmembrane. Of these, 965 were also predicted by TMHMM to be transmembrane, providing support to 965/1,295 (75%) of TMHMM predictions. An additional 211 families (60% of the total 348 families that were predicted to be secreted by SignaiP) were also predicted to be secreted by Phobius. Finally, a set of 111 families were predicted to be secreted by Phobius but transmembrane by TMHMM. This could reflect the fact that both secreted and transmembrane proteins contain a hydrophobic region.
Figure S2. Ribosomal Binding Site Presence and Conservation, Related to Figure 1
A. Distribution of the fraction of family members in which an RBS motif was identified. In most families, at least 80% of family members have an RBS. The 40,957 (9%) of all proteins that lack an RBS from were not excluded from further analysis since it has been shown that a non-negligible fraction of bacterial genes lack an RBS (Hockenberry et al., 2017; Omotajo et al., 2015) and that some genes lacking an RBS are still well expressed (Skorski et al., 2006). B. Conservation of RBS motif in families. For each family, the most abundant RBS motif was identified and the number of family members in which the motif was identified out of the total number of members is the ‘conservation score’. In 80% (3,634/4,539) of the families, at least half of the family homologs share the same RBS motif and in 8% (379/4,539) of the families, exactly the same RBS motif is found upstream of all homologs (Table S3) C. Diversity of RBS motifs in families with two different phylogenetic distribution patterns. Family 127388 codes for a 50S ribosomal protein, identified in 10 different phyla and presents a variety of associated RBS motifs. In comparison, family 85442 is Firmicutes-specific and presents substantially less variability in RBS motifs. Only motifs that were identified at least 5 times are presented.
Figure S3. Transcription Profile of Selected Genes from Families that Were Identified in ≥100 Species (Putative “Housekeeping”), Related to Figure 3
(A-F) Small gene indicated with red star.
Figure S4. Core Families, Related to Figure 3.
A. “Core” families across body sites. Venn-diagram representing the distribution of the 458 ‘core’ (identified in ≥ 50% from total samples of specific body site) small protein families across different body sites. Whereas mouth and gut share 9 small protein families that are found in ≥ 50% of the samples in both body sites, other pairs (e.g., skin and gut) do not share core small genes or share a very small number. In line with the fact that 68% of the samples analyzed here are mouth samples, followed by 26% samples that are gut samples, mouth samples ‘contribute’ the largest amount of ‘core’ families, followed by gut, skin and vagina (Figure S1). In most cases, ‘coreness’ of a family is associated with a specific body site but families that are not ‘core’ to a specific body site are not necessarily completely absent from it (Table S3). Only nine families are ‘core’ to both gut and mouth, most of which (8/9) are part of the list of potential housekeeping families identified based on their wide phyletic occurrence (i.e., in ≥ 100 species). The one family that is core in mouth and in gut but has a relatively narrow phylogenetic distribution (identified here in 33 species) is family 125536. This family potentially codes for a 46-amino acid protein predicted to be transmembrane; it is recurrently found downstream of a cluster of genes that includes two transporters and a two-component system (Table S3 and locus tag for example HMPREF1215_00953), suggesting that it could be involved in environmental sensing. This family is potentially subject to horizontal transfer since the small gene is found in the vicinity of genes that are known to participate in mobilization of DNA and the family displays a sporadic distribution across multiple Firmicutes classes. B. Multiple sequence alignment between 3 families that are core to skin and a beta-class phenol-soluble modulin protein. Psm - phenol-soluble modulin. Three of the families that are core to skin and were classified to multiple Staphylococcus species were assigned a beta class phenol-soluble modulin domain, a family of toxins that have multiple roles in staphylococcal pathogenesis (Cheung et al., 2014). Among HMPI-II samples, homologs of this protein family could be detected almost exclusively in skin samples. However, multiple homologs could be detected in environmental microbiomes. In general, families that are core to skin tend to have homologs in environmental samples (11/20 = 55%) more than mouth (56/282 = 20%) or gut (61/158 = 38%) samples (Table S3).
Figure S5. Family 350024 Is Predicted to Undergo Rapid Evolution, Related to Figure 4.
A. Phylogenetic tree of family 350024 suggests that the family undergoes rapid evolution. Proteins of family 350024 are encoded on contigs that were classified into 19 different Bacteroides strains. The protein sequences of 3 housekeeping proteins (rpoB, pyrG and nusA) from the same Bacteroides strains were retrieved from RefSeq whenever available. For each of the 4 sets of proteins, sequences were aligned using T-coffee with default settings and a tree was inferred using maximum likelihood implemented in the PhyML algorithm. Scale bars indicate number of changes per site. The values at the nodes represent bootstrap values from 100 replicates. Phylogenetic analysis was done using the web service Phylogeny.fr. B. Multiple sequence alignment between family 350024, family 291377 and a homolog of family #291377 in Bacteroides thetaiotaomicron VPI-5482. Families 350024 and 291377 are distant homologs. A close homolog of 291377 was identified in the B. theta proteomics analysis. The B. theta homolog is encoded in the intergenic region between BT4031 and BT4032.
Figure S6. Small Proteins Associated with Mobile Elements, Related to Figure 6
A. Scatterplots of 202 small protein families that are found in the vicinity of HGT-mediating genes. Each dot represents the number of species versus number of families/genera that encode for each family. Examples of potential transfer within genus: family 57229 was identified in 19 different Prevotella species; potential transfer within taxonomic family level: family 306379 was identified in 3 different Flavobacteriaceae genera. B. Numbers of small proteins families that were detected in multiple life domains. Disregarding the unclassified homologs, the vast majority of protein families in the 4,539 set are classified as bacteria (4,189/4,539, 92%) (Table S3). There are 8 families that are classified to Eukaryotes and 152 families that are classified to multiple life domains. For example, family 241192 was classified to two life domains (bacteria, eukaryote) as well as to virus. In the contig that was classified to Eukaryotes, ~50% of the k-mers were classified as Candida albicans, an opportunistic pathogenic fungus, common in the human microbiome (Sam et al., 2017). C. Small proteins predicted to be of phage origin. All small-protein encoding contigs were classified against a database that included a set of 19,879 viral genomes. We were able to identify 182 families in which at least one contig was classified as ‘viral’ (green). By far the most common phylogenetic distribution that includes a viral component, observed in 161 (161/182, 88%) of the viral small protein families, is of bacteria-virus (Table S3). Contigs that harbor prophages, bacteriophages integrated into the host’s genome, could theoretically be classified as either ‘bacterial’ or viral, depending on whether the viral reference genome database contains their sequence and on the relative part of sequence that is of viral origin on the contig. To predict prophage regions that were classified as bacterial, we used a common, complementary approach that is based on detection of known viral genes, such as the terminase, capsid, tail and portal proteins (Roux et al., 2015). We screened for a list of ‘phage genes’ encoded on longest contigs of families. Of the 4,539, we detected 223 families in which none of the homologs-encoding contigs was classified as ‘viral’ (in blue) but nevertheless the longest contig encodes for at least one phage gene. Related to Figure 6.
Figure S7. Explanation of Numbers Associated with Each Family as They Appear in Supplementary Tables through Family 221403, Related to Figure 1
Small proteins of this family were identified 14 times across samples (hence, number of members in cluster = 14) originating from 14 metagenomic mouth samples that were sampled from 11 individuals: in 2 individuals the small gene was identified in two subsequent visits (red box) and in 1 individual the small gene was identified in two samples of two different mouth sublocations (Supragingival_plaque and Subgingival_plaque), that were taken from the same individual at the same visit (blue box). To avoid redundancy, we counted each individual only once, hence ‘Number of times found in Mouth’ = 11. Classification of each of the 14 contigs resulted in 2 known OTUs; as all ‘Unclassified’ were counted as a single OTU the total ‘Number of OTUs’ = 3. All small proteins in this family have a predicted transmembrane domain, hence ‘% of family members that are predicted to have a transmembrane domain’ = 1.
KEY RESOURCES TABLE
Highlights.
A genomic approach finds >4,000 conserved small proteins in human microbiomes
The majority of these proteins have no known function or domain
A database provides insights into potential function of these proteins
Over 30% of the small proteins are predicted to be involved in cell-cell communication
ACKNOWLEDGMENTS
We thank the Bhatt laboratory for providing feedback on the study. We also thank Gisela Storz, Noam Livnat, Asaf Levy, Oren Kolodny, Amiyaal Ilany, Ryan Brewster, Matthew Carter, Christopher Severyn, Ramesh Nair, John Hanks (Griznog), Yana Gofman, Andrea Scaiewicz, Ryan Leib, Kratika Singhal (Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University), Michael Bassik, Galen Hess, and Roarke Kamber. This work was supported by the following grants: PhRMA Foundation (H.S.), NIH/NHGRI (T32 HG000044 to H.S.), National Cancer Institute NIH/NCI (K08 CA184420 to A.S.B.), Damon Runyon Clinical Investigator Award (to A.S.B.), NIH (1R01AT01023201 to M.P.S and F.E.), The Foundation Blanceflor Boncompagni Ludovisi, nee Bildt (to F.E.), the US Department of Energy Joint Genome Institute, and a DOE Office of Science User Facility contract (DE-AC02-05CH11231 to G.A.P and N.C.K). G.A.P and N.C.K used resources of the National Energy Research Scientific Computing Center, supported by the Office of Science of the US Department of Energy. This work was supported by the NIH (P30 CA124435) that supports the following Stanford Cancer Institute Shared Resource: the Genetics Bioinformatics Service Center. This work used supercomputing resources provided by the Stanford Genetics Bioninformatics Service Center, supported by NIH S10 Instrumentation Grant (S100D023452). This work was supported in part by NIH (P30 CA124435) utilizing the Stanford Cancer Institute Proteomics/Mass Spectrometry Shared Resource. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.07.016.
DECLARATION OF INTERESTS
N.G. is an employee and shareholder of One Codex. M.P.S. is a cofounder of Personalis, SensOmics, January, Filtricine, Akna, Qbio; he is on the advisory board of the companies he cofounded, along with Genapsys and Jupiter. A.S.B. is on the advisory board of Caribou Biosciences, January, and ArcBio. The authors declare no other competing financial interests.
REFERENCES
- Abu-Ali GS, Mehta RS, Lloyd-Price J, Mallick H, Branck T, Ivey KL, Drew DA, DuLong C, Rimm E, Izard J, et al. (2018). Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nat. Microbiol 3, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan E, Hussain HA, Crawford KR, Miah S, Ascott ZK, Khwaja MH, and Hosie AHF (2007). Genetic variation in comC, the gene encoding competence-stimulating peptide (CSP) in Streptococcus mutans. FEMS Microbiol. Lett 268, 47–51. [DOI] [PubMed] [Google Scholar]
- Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, and Nielsen H (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol 37, 420–23. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra P, Yan J, Li J, Fong S, and Siu SWI (2018). AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep 8, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Quintero A, and Konstantinidis KT (2015). Inter-phylum HGT has shaped the metabolism of many mesophilic and anaerobic bacteria. ISME J. 9, 958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GYC, Joo H-S, Chatterjee SS, and Otto M (2014). Phenol-soluble modulins–critical determinants of staphylococcal virulence. FEMS Microbiol. Rev 38, 698–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukwudi CU, and Good L (2015). The role of the hok/sok locus in bacterial response to stressful growth conditions. Microb. Pathog 79, 70–79. [DOI] [PubMed] [Google Scholar]
- Colomer-Lluch M, Imamovic L, Jofre J, and Muniesa M (2011). Bacteriophages carrying antibiotic resistance genes in fecal waste from cattle, pigs, and poultry. Antimicrob. Agents Chemother 55, 4908–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero OX, and Hogeweg P (2009). The impact of long-distance horizontal gene transfer on prokaryotic genome size. Proc. Natl. Acad. Sci. USA 106, 21748–21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter PD, Ross RP, and Hill C (2013). Bacteriocins - a viable alternative to antibiotics? Nat. Rev. Microbiol 11, 95–105. [DOI] [PubMed] [Google Scholar]
- Couso J-P, and Patraquim P (2017). Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol 18, 575–589. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, and Fischbach MA (2014). A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, and Sorek R (2018). Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval M, and Cossart P (2017). Small bacterial and phagic proteins: an updated view on a rapidly moving field. Curr. Opin. Microbiol 39, 81–88. [DOI] [PubMed] [Google Scholar]
- Dy RL, Richter C, Salmond GPC, and Fineran PC (2014). Remarkable Mechanisms in Microbes to Resist Phage Infections. Annu. Rev. Virol 1, 307–331. [DOI] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, and Li W (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassel M, Möllenkamp T, Puppe W, and Altendorf K (1999). The KdpF subunit is part of the K(+)-translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J. Biol. Chem 274, 37901–37907. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, and Knight R (2018). Current understanding of the human microbiome. Nat. Med 24, 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ER, and Mecsas J (2016). Bacterial Secretion Systems: An Overview. Microbiol. Spectr 4 10.1128/microbiolspec.VMBF-0012-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, and Basu MK (2011). Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol 193, 2745–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DR, and Haft DH (2017). A comprehensive software suite for protein family construction and functional site prediction. PLoS ONE 12, e0171758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves KR, Kropinski AM, and Clokie MRJ (2014). What does the talking?: quorum sensing signalling genes discovered in a bacteriophage genome. PLoS ONE 9, e85131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes CS, Aoki SK, and Low DA (2010). Bacterial contact-dependent delivery systems. Annu. Rev. Genet 44, 71–90. [DOI] [PubMed] [Google Scholar]
- Hobbs EC, Yin X, Paul BJ, Astarita JL, and Storz G (2012). Conserved small protein associates with the multidrug efflux pump AcrB and differentially affects antibiotic resistance. Proc. Natl. Acad. Sci. USA 109, 16696–16701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenberry AJ, Stern AJ, Amaral LAN, and Jewett MC (2017). Diversity of translation initiation mechanisms across bacterial species is driven by environmental conditions and growth demands. Mol. Biol. Evol 35, 582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnik F, and McCutcheon JP (2018). Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol 16, 67–79. [DOI] [PubMed] [Google Scholar]
- Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, and Hauser LJ (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, LoCascio PF, Hauser LJ, and Uberbacher EC (2012). Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230. [DOI] [PubMed] [Google Scholar]
- Imperiali B, and Ottesen JJ (1999). Uniquely folded mini-protein motifs. J. Pept. Res 54, 177–184. [DOI] [PubMed] [Google Scholar]
- Käll L, Krogh A, and Sonnhammer ELL (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol 338, 1027–1036. [DOI] [PubMed] [Google Scholar]
- Karimova G, Davi M, and Ladant D (2012). The β-lactam resistance protein Blr, a small membrane polypeptide, is a component of the Escherichia coli cell division machinery. J. Bacteriol 194, 5576–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Chen HD, Latifi T, and Groisman EA (2012). Reciprocal control between a bacterium’s regulatory system and the modification status of its lipopolysaccharide. Mol. Cell 47, 897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp G, and Cymer F (2014). Small membrane proteins - elucidating the function of the needle in the haystack. Biol. Chem 395, 1365–1377. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Wolf YI (2017). Evolutionary Genomics of Defense Systems in Archaea and Bacteria. Annu. Rev. Microbiol 71,233–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel N, and Balskus EP (2016). Exploring and Understanding the Biochemical Diversity of the Human Microbiota. Cell Chem. Biol 23, 18–30. [DOI] [PubMed] [Google Scholar]
- Koskiniemi S, Lamoureux JG, Nikolakakis KC, t’Kint de Roodenbeke C, Kaplan MD, Low DA, and Hayes CS (2013). Rhs proteins from diverse bacteria mediate intercellular competition. Proc. Natl. Acad. Sci. USA 110, 7032–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, and Sonnhammer ELL (2001). Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J. Mol. Biol 305, 567–580. [DOI] [PubMed] [Google Scholar]
- Krueger F (2014). Trim Galore!. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latif H, Szubin R, Tan J, Brunk E, Lechner A, Zengler K, and Palsson BO (2015). A streamlined ribosome profiling protocol for the characterization of microorganisms. Biotechniques 58, 329–332. [DOI] [PubMed] [Google Scholar]
- Lau JL, and Dunn MK (2018). Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem 26, 2700–2707. [DOI] [PubMed] [Google Scholar]
- Leplae R, Geeraerts D, Hallez R, Guglielmini J, Drèze P, and Van Melderen L (2011). Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res 39, 5513–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen X, Skogerb0 G, Zhang P, Chen R, He S, and Huang D-W (2012). The human microbiome: a hot spot of microbial horizontal gene transfer. Genomics 100, 265–270. [DOI] [PubMed] [Google Scholar]
- Lloyd CR, Park S, Fei J, and Vanderpool CK (2017). The Small Protein SgrT Controls Transport Activity of the Glucose-Specific Phosphotransferase System. J. Bacteriol 199, e00869–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, et al. (2017). Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrique P, Dills M, and Young MJ (2017). The Human Gut Phage Community and Its Implications for Health and Disease. Viruses 9, E141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. (2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39, D225–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45 (D1), D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, and Vita C (2000). Engineering Novel Bioactive Mini-Proteins from Small Size Natural and De Novo Designed Scaffolds. Curr. Protein Pept. Sci 1, 403–430. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal 17, 10–12. [Google Scholar]
- Martin JE, Waters LS, Storz G, and Imlay JA (2015). The Escherichiacoli small protein MntS and exporter MntP optimize the intracellular concentration of manganese. PLoS Genet 11, e1004977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, and Jones DT (2000). The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405. [DOI] [PubMed] [Google Scholar]
- Milshteyn A, Colosimo DA, and Brady SF (2018). Accessing Bioactive Natural Products from the Human Microbiome. Cell Host Microbe 23, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot SS, Krumm N, and Greenfield NB (2015). One Codex: A Sensitive and Accurate Data Platform for Genomic Microbial Identification. bioRxiv. 10.1101/027607. [DOI] [Google Scholar]
- Mitchell AL, Scheremetjew M, Denise H, Potter S, Tarkowska A, Qureshi M, Salazar GA, Pesseat S, Boland MA, Hunter FMI, et al. (2018). EBI Metagenomics in 2017: enriching the analysis of microbial communities, from sequence reads to assemblies. Nucleic Acids Res 46 (D1), D726–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Hopkins AC, and Laub MT (2011). A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev 25, 1328–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Gamez S, Sorg RA, Domenech A, Kjos M, Weissing FJ, van Doorn GS, and Veening J-W (2017). Quorum sensing integrates environmental cues, cell density and cell history to control bacterial competence. Nat. Commun 8, 854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, and Groisman EA (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. [DOI] [PubMed] [Google Scholar]
- Oliveira PH, Touchon M, Cury J, and Rocha EPC (2017). The chromosomal organization of horizontal gene transfer in bacteria. Nat. Commun 8, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Buttner H, Dunman PM, Rohde H, Cech NB, Fey PD, and Horswill AR (2014). Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J. Bacteriol 196, 3482–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omotajo D, Tate T, Cho H, and Choudhary M (2015). Distribution and diversity of ribosome binding sites in prokaryotic genomes. BMC Genomics 16, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaza S, Menschaert G, and Payre F (2017). In search of lost small peptides. Annu. Rev. Cell Dev. Biol 33, 391–416. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, and Maglott DR (2007). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35, D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan R, Rani A, Metwally A, McGee HS, and Perkins DL (2016). Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun 469, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostol JT, and Marraffini L (2019). (Ph)ighting Phages: How Bacteria Resist Their Parasites. Cell Host Microbe 25, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Enault F, Hurwitz BL, and Sullivan MB (2015). VirSorter: mining viral signal from microbial genomic data. Peer J 3, e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam QH, Chang MW, and Chai LYA (2017). The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci 18, E330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaiewicz A, and Levitt M (2018). Unique function words characterize genomic proteins. Proc. Natl. Acad. Sci. USA 115, 6703–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seefeldt AC, Nguyen F, Antunes S, Perebaskine N, Graf M, Arenz S, Inampudi KK, Douat C, Guichard G, Wilson DN, and Innis CA (2015). The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat. Struct. Mol. Biol 22, 470–475. [DOI] [PubMed] [Google Scholar]
- Shmakov SA, Makarova KS, Wolf YI, Severinov KV, and Koonin EV (2018). Systematic prediction of genes functionally linked to CRISPR-Cas systems by gene neighborhood analysis. Proc. Natl. Acad. Sci. USA 115, E5307–E5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shterzer N, and Mizrahi I (2015). The animal gut as a melting pot for horizontal gene transfer. Can. J. Microbiol 61, 603–605. [DOI] [PubMed] [Google Scholar]
- Skorski P, Leroy P, Fayet O, Dreyfus M, and Hermann-Le Denmat S (2006). The highly efficient translation initiation region from the Escherichia coli rpsA gene lacks a shine-dalgarno element. J. Bacteriol 188, 6277–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucy SM, Huang J, and Gogarten JP (2015). Horizontal gene transfer: building the web of life. Nat. Rev. Genet 16, 472–482. [DOI] [PubMed] [Google Scholar]
- Steinbrecher T, Prock S, Reichert J, Wadhwani P, Zimpfer B, Biurck J, Berditsch M, Elstner M, and Ulrich AS (2012). Peptide-lipid interactions of the stress-response peptide TisB that induces bacterial persistence. Biophys.J 103, 1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, and Sorek R (2011). The phage-host arms race: shaping the evolution of microbes. BioEssays 33, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Wolf YI, and Ramamurthi KS (2014). Small proteinscan no longer be ignored. Annu. Rev. Biochem 83, 753–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Ling Y, Yu J, Wu J, and Xiao J (2013). Small proteins: untapped area of potential biological importance. Front. Genet 4, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y-H, de Jong MF, den Hartigh AB, Roux CM, Rolan HG, and Tsolis RM (2012). The small protein CydX is required for function of cytochrome bd oxidase in Brucella abortus. Front. Cell. Infect. Microbiol 2, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivella DBB, and de Felicio R (2018). The Tripod for Bacterial Natural Product Discovery: Genome Mining, Silent Pathway Induction, and Mass Spectrometry-Based Molecular Networking. mSystems 3, e00160–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropini C, Moss EL, Merrill BD, Ng KM, Higginbottom SK, Casavant EP, Gonzalez CG, Fremin B, Bouley DM, Elias JE, et al. (2018). Transient Osmotic Perturbation Causes Long-Term Alteration to the Gut Microbiota. Cell 173, 1742–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW (2014). The virome in mammalian physiology and disease. Cell 157, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, Whitaker RJ, Krause DJ, Heilers J-H, van Wolferen M, van der Does C, and Albers S-V (2017). Mechanisms of gene flow in archaea. Nat. Rev. Microbiol 15, 492–501. [DOI] [PubMed] [Google Scholar]
- Wang H, Yin X, Wu Orr M, Dambach M, Curtis R, and Storz G (2017). Increasing intracellular magnesium levels with the 31-amino acid MgtS protein. Proc. Natl. Acad. Sci. USA 114, 5689–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Findeiss S, Muller SA, Kalkhof S, von Bergen M, Hofacker IL, Stadler PF, and Goldman N (2011). RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA 17, 578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MR, Zha L, and Balskus EP (2017). Natural product discovery from the human microbiome. J. Biol. Chem 292, 8546–8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaneveld JR, Nemergut DR, and Knight R (2008). Are all horizontal gene transfers created equal? Prospects for mechanism-based studies of HGT patterns. Microbiology 154, 1–15. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen W, Ning Z, Mayne J, Mack D, Stintzi A, Tian R, and Figeys D (2017). Deep Metaproteomics Approach for the Study of Human Microbiomes. Anal. Chem 89, 9407–9415. [DOI] [PubMed] [Google Scholar]
- Zhang X, Deeke SA, Ning Z, Starr AE, Butcher J, Li J, Mayne J, Cheng K, Liao B, Li L, et al. (2018). Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun 9, 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Omairi-Nasser A, Li X, Dong C, Lin Y, Haselkorn R, and Zhao J (2017). An amidase is required for proper intercellular communication in the filamentous cyanobacterium Anabaena sp. PCC 7120. Proc. Natl. Acad. Sci. USA 114, E1405–E1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Basic Statistics of Samples and 4,539 Small Protein Families, Related to Figure 1
A. Protein length distribution of 4,539 families. Proteins of > 50 aa or < 5 aa were filtered out. The smallest protein in our dataset is encoded by family 442207 and is 16aa long. B. Distribution of number of encoding taxa per family. If multiple homologs are mapped to the same taxon, they were counted only once. C. Proportion of samples belonging to different body sites. 128,368,337 contigs were obtained from 1,773 HMPI-II human-associated metagenomes that were shotgun sequenced, spanning 4 different major body sites from 263 healthy individuals. Samples were obtained from individuals in one, two or three subsequent visits. Body sites were collapsed into four groups: anterior nares, buccal mucosa, hard palate, keratinized gingiva, palatine tonsils, saliva, subgingival plaque, supragingival plaque, throat and tongue dorsum to ‘mouth’; mid vagina, posterior fornix and vaginal introitus to ‘vagina’; left retroauricular crease, right retroauricular crease and right antecubital fossa to ‘skin’; stool samples were renamed ‘gut’ here. D. Transmembrane/secreted proteins among the 4,539 families as predicted by TMHMM (Krogh et al., 2001) and signaiP (Almagro Armenteros et al., 2019), respectively. A family is predicted to be transmembrane/secreted if ≥ 80% of the homologs of the family are predicted to be such. In addition to families that passed this threshold, in 180 families, at least one family member, but less than 80% of the family members are predicted to be transmembrane and in 414 families, at least one family member, but less than 80% of family members are predicted to be secreted. An alternative algorithm, Phobius (Käll et al., 2004) was also applied on the 4,539 families. Phobius predicted 1002 families (22%) to be transmembrane. Of these, 965 were also predicted by TMHMM to be transmembrane, providing support to 965/1,295 (75%) of TMHMM predictions. An additional 211 families (60% of the total 348 families that were predicted to be secreted by SignaiP) were also predicted to be secreted by Phobius. Finally, a set of 111 families were predicted to be secreted by Phobius but transmembrane by TMHMM. This could reflect the fact that both secreted and transmembrane proteins contain a hydrophobic region.
Figure S2. Ribosomal Binding Site Presence and Conservation, Related to Figure 1
A. Distribution of the fraction of family members in which an RBS motif was identified. In most families, at least 80% of family members have an RBS. The 40,957 (9%) of all proteins that lack an RBS from were not excluded from further analysis since it has been shown that a non-negligible fraction of bacterial genes lack an RBS (Hockenberry et al., 2017; Omotajo et al., 2015) and that some genes lacking an RBS are still well expressed (Skorski et al., 2006). B. Conservation of RBS motif in families. For each family, the most abundant RBS motif was identified and the number of family members in which the motif was identified out of the total number of members is the ‘conservation score’. In 80% (3,634/4,539) of the families, at least half of the family homologs share the same RBS motif and in 8% (379/4,539) of the families, exactly the same RBS motif is found upstream of all homologs (Table S3) C. Diversity of RBS motifs in families with two different phylogenetic distribution patterns. Family 127388 codes for a 50S ribosomal protein, identified in 10 different phyla and presents a variety of associated RBS motifs. In comparison, family 85442 is Firmicutes-specific and presents substantially less variability in RBS motifs. Only motifs that were identified at least 5 times are presented.
Figure S3. Transcription Profile of Selected Genes from Families that Were Identified in ≥100 Species (Putative “Housekeeping”), Related to Figure 3
(A-F) Small gene indicated with red star.
Figure S4. Core Families, Related to Figure 3.
A. “Core” families across body sites. Venn-diagram representing the distribution of the 458 ‘core’ (identified in ≥ 50% from total samples of specific body site) small protein families across different body sites. Whereas mouth and gut share 9 small protein families that are found in ≥ 50% of the samples in both body sites, other pairs (e.g., skin and gut) do not share core small genes or share a very small number. In line with the fact that 68% of the samples analyzed here are mouth samples, followed by 26% samples that are gut samples, mouth samples ‘contribute’ the largest amount of ‘core’ families, followed by gut, skin and vagina (Figure S1). In most cases, ‘coreness’ of a family is associated with a specific body site but families that are not ‘core’ to a specific body site are not necessarily completely absent from it (Table S3). Only nine families are ‘core’ to both gut and mouth, most of which (8/9) are part of the list of potential housekeeping families identified based on their wide phyletic occurrence (i.e., in ≥ 100 species). The one family that is core in mouth and in gut but has a relatively narrow phylogenetic distribution (identified here in 33 species) is family 125536. This family potentially codes for a 46-amino acid protein predicted to be transmembrane; it is recurrently found downstream of a cluster of genes that includes two transporters and a two-component system (Table S3 and locus tag for example HMPREF1215_00953), suggesting that it could be involved in environmental sensing. This family is potentially subject to horizontal transfer since the small gene is found in the vicinity of genes that are known to participate in mobilization of DNA and the family displays a sporadic distribution across multiple Firmicutes classes. B. Multiple sequence alignment between 3 families that are core to skin and a beta-class phenol-soluble modulin protein. Psm - phenol-soluble modulin. Three of the families that are core to skin and were classified to multiple Staphylococcus species were assigned a beta class phenol-soluble modulin domain, a family of toxins that have multiple roles in staphylococcal pathogenesis (Cheung et al., 2014). Among HMPI-II samples, homologs of this protein family could be detected almost exclusively in skin samples. However, multiple homologs could be detected in environmental microbiomes. In general, families that are core to skin tend to have homologs in environmental samples (11/20 = 55%) more than mouth (56/282 = 20%) or gut (61/158 = 38%) samples (Table S3).
Figure S5. Family 350024 Is Predicted to Undergo Rapid Evolution, Related to Figure 4.
A. Phylogenetic tree of family 350024 suggests that the family undergoes rapid evolution. Proteins of family 350024 are encoded on contigs that were classified into 19 different Bacteroides strains. The protein sequences of 3 housekeeping proteins (rpoB, pyrG and nusA) from the same Bacteroides strains were retrieved from RefSeq whenever available. For each of the 4 sets of proteins, sequences were aligned using T-coffee with default settings and a tree was inferred using maximum likelihood implemented in the PhyML algorithm. Scale bars indicate number of changes per site. The values at the nodes represent bootstrap values from 100 replicates. Phylogenetic analysis was done using the web service Phylogeny.fr. B. Multiple sequence alignment between family 350024, family 291377 and a homolog of family #291377 in Bacteroides thetaiotaomicron VPI-5482. Families 350024 and 291377 are distant homologs. A close homolog of 291377 was identified in the B. theta proteomics analysis. The B. theta homolog is encoded in the intergenic region between BT4031 and BT4032.
Figure S6. Small Proteins Associated with Mobile Elements, Related to Figure 6
A. Scatterplots of 202 small protein families that are found in the vicinity of HGT-mediating genes. Each dot represents the number of species versus number of families/genera that encode for each family. Examples of potential transfer within genus: family 57229 was identified in 19 different Prevotella species; potential transfer within taxonomic family level: family 306379 was identified in 3 different Flavobacteriaceae genera. B. Numbers of small proteins families that were detected in multiple life domains. Disregarding the unclassified homologs, the vast majority of protein families in the 4,539 set are classified as bacteria (4,189/4,539, 92%) (Table S3). There are 8 families that are classified to Eukaryotes and 152 families that are classified to multiple life domains. For example, family 241192 was classified to two life domains (bacteria, eukaryote) as well as to virus. In the contig that was classified to Eukaryotes, ~50% of the k-mers were classified as Candida albicans, an opportunistic pathogenic fungus, common in the human microbiome (Sam et al., 2017). C. Small proteins predicted to be of phage origin. All small-protein encoding contigs were classified against a database that included a set of 19,879 viral genomes. We were able to identify 182 families in which at least one contig was classified as ‘viral’ (green). By far the most common phylogenetic distribution that includes a viral component, observed in 161 (161/182, 88%) of the viral small protein families, is of bacteria-virus (Table S3). Contigs that harbor prophages, bacteriophages integrated into the host’s genome, could theoretically be classified as either ‘bacterial’ or viral, depending on whether the viral reference genome database contains their sequence and on the relative part of sequence that is of viral origin on the contig. To predict prophage regions that were classified as bacterial, we used a common, complementary approach that is based on detection of known viral genes, such as the terminase, capsid, tail and portal proteins (Roux et al., 2015). We screened for a list of ‘phage genes’ encoded on longest contigs of families. Of the 4,539, we detected 223 families in which none of the homologs-encoding contigs was classified as ‘viral’ (in blue) but nevertheless the longest contig encodes for at least one phage gene. Related to Figure 6.
Figure S7. Explanation of Numbers Associated with Each Family as They Appear in Supplementary Tables through Family 221403, Related to Figure 1
Small proteins of this family were identified 14 times across samples (hence, number of members in cluster = 14) originating from 14 metagenomic mouth samples that were sampled from 11 individuals: in 2 individuals the small gene was identified in two subsequent visits (red box) and in 1 individual the small gene was identified in two samples of two different mouth sublocations (Supragingival_plaque and Subgingival_plaque), that were taken from the same individual at the same visit (blue box). To avoid redundancy, we counted each individual only once, hence ‘Number of times found in Mouth’ = 11. Classification of each of the 14 contigs resulted in 2 known OTUs; as all ‘Unclassified’ were counted as a single OTU the total ‘Number of OTUs’ = 3. All small proteins in this family have a predicted transmembrane domain, hence ‘% of family members that are predicted to have a transmembrane domain’ = 1.
Data Availability Statement
The raw sequencing reads generated in this study are available at BioProject: PRJNA540869. The published article includes all datasets generated during this study.