

Abstract
The discovery of the RNA-guided DNA nuclease CRISPR-Cas9 has enabled the targeted editing of genomes from diverse organisms, but the permanent and inheritable nature of genome modification also poses immense risks. The potential for accidental exposure, malicious use, or undesirable persistence of Cas9 therapeutics and off-target genome effects highlight the need for detection assays. Here we report a centrifugal microfluidic platform for the measurement of both Cas9 protein levels and nuclease activity. Because Cas9 from many bacterial species have been adapted for biotechnology applications, we developed the capability to detect Cas9 from the widely-used S. pyogenes, as well as S. aureus, N. meningitides, and S. thermophilus using commercially-available antibodies. Further, we show that the phage-derived anti-CRISPR protein AcrIIC1, which binds to Cas9 from several species, can be used as a capture reagent to broaden the species range of detection. As genome modification generally requires Cas9 nuclease activity, a fluorescence-based sedimentation nuclease assay was also incorporated to allow the sensitive and simultaneous measurement of both Cas9 protein and activity in a single biological sample.
Graphical Abstract
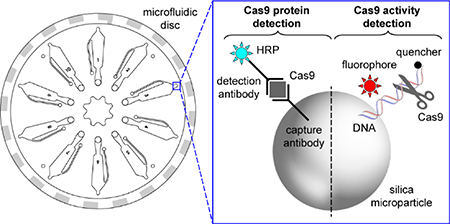
Combined activity- and immunoassays for CRISPR/Cas9 on a portable microfluidic device with integrated sample preparation from clinical sample matrices
Introduction
Although Clustered regularly interspaced palindromic repeats (CRISPR) and CRISPR-associated (Cas) protein-coding genes in prokaryotes and archaea were first described well over a decade ago1, the explosive growth in the field was sparked by the discovery of Cas9 as an RNA-guided nuclease mediating target-specific cleavage of DNA.2 The modular nature of Cas9, with a universal nuclease protein being targeted to cut both strands of a DNA target by the sequence of guide RNA, has made it an incredibly versatile tool for biotechnology. The CRISPR-Cas9 system has been used for targeted cleavage at specific genomic loci in human cell lines3, mice 4, and many other organisms. Repair of the resulting double-stranded DNA breaks by error-prone Non-Homologous End Joining (NHEJ) disrupts the gene coding sequence and results in loss-of-protein.5 When a donor DNA template with homology arms flanking the DNA cut site is introduced along with Cas9 and guide RNA, Homology-Directed Repair (HDR) can be harnessed to perform site-specific integration of any DNA sequence. 6 Additional technologies have been developed based on a dead Cas9 (dCas9) scaffold in which the two nuclease domains have been inactivated by mutation, but the RNA-guided binding of Cas9 to DNA is retained. Fusion of a cytidine deaminase domain to dCas9 has been used for targeted single-base mutations7 and the addition of epigenome effectors to dCas9 can be used modulate transcription8.
Cas9 has already had an enormous impact on basic science research, but the clinical use of CRISPR technologies has immense potential for personalized medicine. Pre-clinical studies have demonstrated excellent results for the treatment of genetic disease in the mouse 9 and human 10 embryo. CRISPR-encoding viral vectors have also shown promise for the post-natal treatment of many genetic diseases in mice11. Safer non-viral delivery vehicles for Cas9 are also actively being developed, including cationic liposomes12 and gold nanoparticles 13. The majority of the reported studies have used Cas9 from S. pyogenes because it was the first to be discovered and characterized, but Cas9 from C. jejuni14, S. aureus15, S. thermophilus16, N. meningitides 17, and G. stearothermophilus 18 also perform genome editing and can be preferable because of their unique DNA sequence specificities, enhanced thermal stability, or smaller size. Because of the known risk of off-target mutations resulting from the persistence of Cas9 beyond the lifetime required for on-target editing19,20, the clinical use of any Cas9 delivery methods will require sensitive assays capable of detecting Cas9 directly in bodily fluids. The potential therapeutic use of multiple Cas9 species, and the use of dCas9 fused to diverse effector domains7,8 makes the ideal clinical assay pan-species-specific and able to detect both Cas9 protein and catalytic nuclease activity.
Experimental
Materials and Reagents
Buffer and bacterial media reagents were from Fisher BioReagents unless otherwise noted. Tris(2-carboxyethyl)phosphine (TCEP), dithiothreitol (DTT), and isopropyl β-D-1-thiogalactopyranoside (IPTG) were from Gold Biotechnology. Antibiotics were from Teknova. Mammalian tissue culture media and fetal bovine serum were from Gibco. The deoxyoligonucleotides were ordered HPLC-purified from Integrated DNA Technologies (IDT). Antibodies were purchased from Abcam, Thermo Fisher, Santa Cruz Biotechnology, and Pierce Biotechnology. Microparticles were purchased from Bangs Laboratories. 3-(Ethyliminomethyleneamino)-N,N-dimethylpropan-1-amine (EDC), N-hydroxysuccinimide (NHS), triethylamine, (sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate) (sulfo-SMCC), and SuperSignal Femto chemiluminescent substrate were purchased from Thermo Fisher. N,N-Dimethylformamide (DMF), dichloromethane (DCM), indole-3-butyric acid were purchased from Sigma-Aldrich. Whole blood, serum, and plasma were purchased from Innovative Research (Novi, MI). These biomaterials were pooled, deidentified, publicly-available specimens from healthy human donors and therefore exempt from IRB/HSB review. All work conducted with risk group 2 biomaterials was reviewed by the IBC (2019–06-05-CK) and universal precautions were observed.
Device components and assembly
Microfluidic discs were made from 0.0625 inch thick clear cast polymethyl methacrylate (PMMA) (McMaster, Part No. 8560K171) and double-sided pressure sensitive adhesive (PSA) film (Fralock, Part No. MYLAR-7–3M-9471LE-2SIDE). The laser cut layers of PMMA and PSA feature alignment holes that allow for easy assembly using a custom jig featuring tapered pins. After manually pressing the layers together, the disc is vacuum sealed and allowed to sit for 24 hours before use.
The rotary control system includes a brushless DC motor with a 12-bit magnetic absolute (Faulhaber, Part No. 2250S024BX4AES-4096), motor controller (Faulhaber, Part No. MCBL3006SAESRS), and reflective object sensor (Optek, Part No. OPB742). The heater is a custom medium-wave twin-tube infrared emitter (Heraeus Noblelight). Optical systems were built using a mix of off-the-shelf optomechanical components (Thorlabs) and custom machined parts. The chemiluminescence detection system was assembled using a photomultiplier tube (PMT) module (Hamamatsu, H10722–01) and set of lenses (Thorlabs, A397TM-A, A230TM-A) for imaging the tip of the chambers. The fluorescence detection system was built using a similar PMT module (Hamamatsu, H10722–20) better optimized for sensitivity in the red spectrum, 4mW 635nm laser diode (Edmund Optics, 57–106), and excitation and emission filters and dichroic beamsplitter (Semrock, FF01–676/29–12.5, FF01–640/14–12.5, Di02-R635–12.5-D). The hardware interfaces with a laptop using a multifunction data acquisition device (National Instruments, USB-6008).
Cas9 cloning and overexpression
S. pyogenes wild-type, D10A, H840A, and dead Cas9 were overexpressed as His-MBP-TEV fusion proteins and purified as described previously. 21 S. aureus and C. jejuni Cas9 were overexpressed and purified as described previously. 21 Plasmids encoding G. stearothermophilus Cas9 (a kind gift of Jennifer Doudna18, Addgene #87703) and N. meningitidis Cas9 (a kind gift of Erik Sontheimer22, Addgene #71474) were expressed as described in their respective papers and purified just as S. aureus and C. jejuni Cas9 from above. All proteins were flash frozen and stored at −80 °C.
AcrIIC1 cloning and overexpression
The AcrIIC1Nme gene23 was ordered as a gene block and cloned into the NdeI/SapI sites of a modified pTXB1 vector (New England Biolabs, Ipswich, MA, USA) containing a His8 tag at the C-terminus of the existing chitin binding domain tag. The insert was verified by Sanger Sequencing. The plasmid was transformed into E. coli BL21(DE3), grown to an OD of 0.5, induced with 0.25 mM IPTG, and grown at 20 °C for 20 h. The pellets were lysed in buffer A [50 mM Tris-HCl, pH 7.5, 0.5 mM TCEP] with 500 mM NaCl and 10% glycerol by an EmulsiFlex-C5 homogenizer (Avestin, Ottawa, ON, Canada). The lysate was clarified and purified by Ni-NTA chromatography with a linear gradient of 10 to 500 mM imidazole in the above buffer and the protein was used directly in conjugation reactions without cleavage of the CBD-His tag.
Microparticle conjugation
Antibodies were oriented onto the microparticles through covalent capture of the nucleotide binding site followed by photocrosslinking using a slightly modified method from literature24. Indole-3-butyric acid (1 mmol) was dissolved in 3 mL of a solution of N,N-dimethylformamide and dichloromethane (3:1, v/v). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (0.9 mmol), N-hydroxysuccinimide (0.9 mmol), and triethylamine (0.072 mmol) were added and the reaction was allowed to proceed for 3h at room temperature to form the succinimidyl ester. Carboxylic acid-functionalized silica microparticles (10 mg, 1 um diameter) were suspended in MES buffer (0.1M 2-(N-morpholino)ethanesulfonic acid, pH 6) and activated with excess EDC/NHS at room temperature for 2h. Particles were spun down and washed three times with 2mL of MES buffer. To the activated particles was then added a solution of Fmoc-NH-PEG(10,000)-NH2 (0.1 mmol, Creative PEGWorks, Chapel Hill, NC, USA) in phosphate buffered saline (PBS; 138 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, pH 7.4). The pH was adjusted to 8.15 with 1M sodium bicarbonate and allowed to react for 4h. The particles were washed in PBS three times and the Fmoc group removed with treatment with 5% aqueous ammonia. The particles are then washed an additional three times with PBS followed by three times in DMF. To the particles was then added a solution of the indole activated ester (0.15 mmol) in 3:1 DMF/DCM with catalytic TEA. The particles were reacted for 5h at room temperature with end-over-end rotation. The particles were then washed three times with PBS and then blocked with 4% (wt/v) BSA for 3h at room temperature. The blocked particles were washed three times with PBS. The antibody was then added to the particles and incubated at room temperature for 1h. The particles were then exposed to UV at 254 nm using a Stratalinker (1.2 J/cm2, Stratagene [now Agilent Technologies], Santa Clara, CA, USA). The particles were then washed three times with PBS and stored in PBS at 4C until used.
AcrIIC1 proteins were immobilized on the microparticles using standard maleimide chemistry to the terminal free cysteine. After functionalization with bisaminoPEG as above, a heterobifunctional crosslinker, sulfo-SMCC, was added to the particles and allowed to react for 2h at room temperature. The particles were washed three times in MES and the AcrIIC1 protein added to the suspension. The reaction proceeded for 2h at room temperature, the particles were washed three times, and blocked with 4% BSA (w/v) for 3h at room temperature. The blocked particles were washed three times in PBS and stored in PBS at 4C until used.
Cas9 protein detection assays
20 uL of density media containing the chemiluminescent substrate were made by dissolving dextran (500 kDa, 30% w/v) and Cas9 reaction buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, pH 6.5) was preloaded into the microfluidic disc and spun for 5s at 4000 rpm. A 5 uL layer of poly(ethylene glycol) (Mn 20,000, 40% w/v in water) was then added to the channels to separate unbound HRP-conjugated antibodies from the chemiluminescent substrate and decrease noise. The disc was then spun again at 4000 rpm for 5s to create an aqueous two-phase system. Capture particles, prepared as above, were suspended in PBS at 20% w/v. 5 uL of sample were added to 10 uL of capture particles and 1 uL of HRP conjugate (1.5 ug/mL, prepared as indicated below) in a microcentrifuge tube and reacted at room temperature for 25m. After incubation, the sample was added to the disc and spun for 1.5m at 6000rpm to allow for signal generation of the HRP while maintaining force on the system to slow diffusion of the layers.
Antibodies were conjugated with HRP using Abcam’s Easy HRP conjugation kit according to the manufacturer’s instructions. Briefly, 30 uL of modifier was added to 300 uL of a 1 mg/mL solution of antibody. The solution was then added to the HRP mix and reacted for 3h at room temperature in the dark. 30 uL of quencher was then added to the solution and reacted at room temperature for 30m. The conjugates were then desalted using Zeba spin purification columns (7 kDa MWCO) to remove low molecular weight byproducts. The conjugates were stored at 4C until use.
Cas9 activity detection assays
A Cy5 dye-labeled, biotinylated oligonucleotide was annealed with a shorter complementary quencher strand. The quenched duplex strand was then incubated with streptavidin-functionalized 5 um silica microparticles for 2h at room temperature and washed three times in Cas9 reaction buffer. The particles were concentrated to 20% w/v from the stock concentration of 1% by centrifugation. The 10 uL of particles were then incubated with 5 uL of sample for 25m. The density medium for this experiment consisted of 2.5M guanidinium hydrochloride to denature the Cas9 proteins bound to the DNA duplex, allowing the quencher strand to dissociate. 75 uL of density medium was added to the disc and spun for 5s at 4000 rpm. The sample suspension was then added to the disc and spun for 1.5m at 6000rpm.
HEK293T Cas9 transfection and cell lysate production
The CRISPR reporter system from PNA Bio (https://www.pnabio.com/products/Reporter.htm), containing a constitutive red fluorescent protein (RFP) and AAVS1 target site flanked by out-of-frame green fluorescent proteins (GFP), was subcloned into a lentiviral expression plasmid with an EF1a promoter. Single-cycle lentivirus was produced via standard protocols and HEK293T cells (ATCC) were transduced, then sorted by flow cytometry to obtain the RFP+ population. These cells were plated at 70% confluence in a 24-well plate and were transfected with pCas-AAVS1 (on-target) or pCas-scramble plasmid (OriGene Technologies) using Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer instructions. An untransfected control was also included. After 48 hours, most of the cells were isolated, washed with PBS, and lysed with Mammalian Protein Extraction Reagent (Thermo Fisher Scientific) containing 1/10th volume murine RNase inhibitor (New England Biolabs). After 15 minutes on ice, the lysate was clarified by spinning at 21,000 g for 15 minutes at 4 °C. The lysate protein concentration for each sample was determined by Bradford Assay using bovine serum albumin as the standard. After 96 hours, the cells that remained in culture were imaged by epifluorescence microscopy to visualize RFP+ and GFP+ cells.
Results and Discussion
Several Cas9 assay technologies have been designed for measuring Cas9 DNA cleavage activity25,26 or binding27,28, but none are suitable for therapeutic protein monitoring or biosurveillance applications. To fill this gap, we have enhanced a centrifugal microfluidic platform29 for the ultrasensitive and simultaneous detection of Cas9 protein and nuclease activity directly in biological sample matrices (Fig. 1). A disposable microfluidic disc (Fig. 1b) is used to confine samples for centrifugation and analysis. The disc is composed of three layers, each fabricated using CO2 laser cutting. The top and bottom are rigid PMMA layers sandwiching a double-sided adhesive layer that joins the assembly and defines the microfluidic architecture. An array of ten chambers are sized for sufficient optical path length (e.g. 600 μm) and volume handling (up to 50 μL) and are spaced to minimize signal crosstalk during the detection phase. The discs are designed to interface with a custom platform (Fig. 1a) consisting of a rotary control system, a temperature control system, and two optical detection systems. Rotary control is accomplished using an encoder-equipped brushless motor, to which the disc is directly mounted, along with an optical switch that senses index markings on the disc for position control purposes. Sample temperature can be controlled via non-contact heating using a medium-wave infrared heater, which is enclosed in a 3D-printed housing that is lowered onto the platform. The two optical systems, one composed of a PMT module and a set of lenses for chemiluminescence detection and the other composed of a 635nm laser diode, PMT module, emission and excitation filter set, dichroic beamsplitter, and focusing lens for laser-induced fluorescence detection, are mounted on the platform with 180° separation, each radially aligned to the detection zone at the tip of the chambers. A LabVIEW interface (Fig 1c) communicates with the motor controller, heater, and optical systems to spin, heat, and scan the microfluidic disc, generating readouts of both chemiluminescence and fluorescence measurements. A detailed list of components and a photo of the platform is included in the Supporting Information (Fig. S1).
Figure 1. Centrifugal microfluidic platform.

(a) Rendering of primary components, including the brushless motor, infrared heater, and optical systems for chemiluminescence and fluorescence-based detections. A bottom view of the heater housing (inset) shows the dual-filament heating element and a top view of the platform (inset) illustrates the alignment of the disc and optical systems. (b) Schematic of the 10-chamber disc. (c) Screen capture of the user interface featuring sample chemiluminescence and fluorescence measurements.
For protein detection, Cas9-containing sample is mixed and incubated with a suspension of capture antibody-functionalized silica microparticles and a horseradish peroxidase-conjugated detection antibody. The resulting suspension is loaded into a disc pre-loaded with chemiluminescent density medium. Upon centrifugation, the microparticles sediment through the density medium and pellet, concentrating the luminescence signal at the bottom of the channel. The entire assay requires only 30 minutes and a 5 μL sample for pg/mL detection of S. pyogenes, S. aureus, S. thermophilus, N. meningitides Cas9 using commercially-available antibodies (Fig. 2a). Because the antibodies impart specificity and the density medium separates the detection signal from any sample contaminants, the limit of detection is nearly identical in buffer, whole blood, serum, and plasma (Fig. 2b). The CRISPR field is rapidly advancing and high affinity antibodies are not yet available for all of the species that are being used for gene editing. To broaden the species of Cas9 that can be detected, we also functionalized silica microparticles with the CRISPR inhibitor AcrIIC1.30 AcrIIC1 binds to the conserved HNH domain from many species of Cas9 and, coupled with specific or even cross-reactive detection antibodies, enabled pg/mL to ng/mL detection of N. mengitides, G. stearothermophilus, and C. jejuni Cas9 (Fig. 2c). Again, the detection of Cas9 using AcrIIC1 as a bait protein is independent of matrix effects, even in whole blood (Fig. S2). These results collectively demonstrate this platform can detect low levels of Cas9 protein from the most commonly used species, directly in the bodily fluids, even if no or only sub-par antibodies are available.
Figure 2. Development of Cas9 protein and activity detection assays.

(a) Detection of Cas9 by capture antibody-functionalized silica microparticles and detection antibodies for S. pyogenes (2 pg/mL), S. aureus (44 pg/mL), S. thermophilus (31 pg/mL), and N. meningitidis (23 pg/mL) using chemiluminescence detection (b) Detection of S. pyogenes Cas9 by capture and detection antibodies have unchanged luminescence profiles regardless of biological matrix. (c) Detection of Cas9 by AcrIIC1-functionalized silica microparticles as a capture reagent. N. meningitidis Cas9 (32 pg/mL) and C. jejuni Cas9 (1293 pg/mL) were detected by species-specific antibodies, while G. stearothermophilus Cas9 (188 pg/mL) could be detected by cross-reactive S. aureus Cas9 antibodies. (d) Microparticle-immobilized fluorescence-based detection of S. pyogenes Cas9 nuclease activity in the presence of the substrate-specific AAVS1 guide RNA. Error bars represent standard deviation, points are average of four replicates, and the limits of detection are indicated in parentheses for each measurement.
Cas9 protein is one component required for genome modification; however, Cas9 nuclease activity and thus bound guide RNA is required to generate a double-stranded DNA break. To sensitively measure Cas9 activity in bodily fluids, we adapted our previous fluorescence Cas9 activity assay21 for detection on this platform. A fluorophore/quencher-labeled DNA duplex (containing the target sequence of interest) is immobilized on silica microparticles. Sample containing Cas9 and guide RNA is incubated with the substrate-functionalized particles, then separated by centrifugation through density medium that denatures the Cas9 protein to release bound DNA. Upon cleavage and denaturation, the quencher-conjugated DNA strand dissociates from the bead-immobilized fluorophore strand to generate an increase in fluorescence from the bead pellet that is proportional to Cas9 RNP concentration (Fig. 2d). When Cas9 activity was measured directly in serum, only a marginal increase in background fluorescence was observed with the Cas9-dependent increase in fluorescence being almost identical to that in buffer (Fig. 2d). As this signal is dependent on Cas9 nuclease activity, and specifically endonuclease activity on a particular target sequence, it is the measurement most directly related to the genome modification potential. Further, we have previously shown that this activity measurement can be readily adapted to other species of Cas9 with different PAM sequences by modulating the DNA sequence and, in some cases, heating the sample. 21 We anticipate that this platform and the general protocols could be easily modified for the detection of protein and activity from nearly any species of Cas9.
To emphasize the importance of simultaneous Cas9 protein and target-specific nuclease activity in a single platform, we analyzed samples consisting of the wt Cas9 protein, the D10A and H840A nickase mutants with one active site disrupted, and the nuclease D10A/H840A dead Cas9. The chemiluminescence signal from all these Cas9 variants was identical, as expected because the capture and detection antibodies are unlikely to contain an epitope overlapping these mutations (Fig. 3a). In contrast, the fluorescence-based Cas9 activity measurements in the presence of guide RNA showed almost no increase in fluorescence with the dCas9 or the nickase variants, but a significant increase in fluorescence with the wt Cas9 (Fig. 3a). These results confirm that our platform can, from a single sample, differentially identify Cas9 protein and activity. To test this capability directly in cells, HEK293T human cells containing an integrated Cas9 reporter were transfected with plasmid encoding Cas9 protein and a scrambled (off-target) or on-target AAVS1 guide. The reporter produces red fluorescent protein (RFP) constitutively and, upon Cas9-mediated editing at the AAVS1 site, green fluorescent protein (GFP) is produced. Four days after transfection, the cells showed the expected presence of GFP in the on-target transfection, but no GFP in the off-target transfection or the untransfected control (Fig. S3). However, just two days after transfection, the cells were lysed and analyzed for Cas9 protein and on-target nuclease activity. Even at this early time, the presence of Cas9 could be clearly identified as chemiluminescence signal in both the on- and off-target guide transfections, but not in the untransfected control (Fig. 3b). Further, from the same lysate Cas9 nuclease activity at the AAVS1 target site could be clearly observed via fluorescence in the on-target transfections, with no Cas9 activity in either the off-target or untransfected controls (Fig. 3b). The reported detection platform is therefore able to measure Cas9 protein and target-specific activity simultaneously, in cell lysates without any sample preparation, before any change in cellular phenotype can be observed.
Figure 3. Simultaneous Cas9 protein and nuclease activity detection.

(a) Detection of chemiluminescent Cas9 protein and fluorescent nuclease activity for wild type (wt), the D10A and H840A nickase mutants with one active site disrupted, and the nuclease D10A/H840A dead Cas9 (dCas9). (b) Detection of Cas9 protein and activity in human HEK293T cells transfected with on-target AAVS1 guide (AAVS1), off-target guide (scrambled), or an untransfected control. Error bars represent standard deviation, points are average of four replicates.
The potential for Cas9-based therapeutics to treat a wide variety of genetic diseases is immense, but many challenges must be overcome. The continued discovery of Cas9 enzymes with favorable properties14 and improvements in Cas9 delivery vehicles31 are significant steps forward, but any clinical use of Cas9 and future biosurveillance applications necessitate the development of robust detection assays. Here we have described significant improvements to a portable microfluidic device for the measurement of orthogonal luminescence and fluorescence signals for monitoring Cas9 protein and target-specific activity. The major advantages of the reported platform are the self-contained and easy-to-use interface, sample temperature control, ability to use small volumes of complex biological samples, the rapid (<30 minute) results, and the ability to detect multiple species of Cas9 that are commonly used in basic research and pre-clinical studies. The simultaneous measurement of both Cas9 protein levels and targeted nuclease activity will be essential to monitoring and understanding the connection between Cas9 persistence and off-target genome modification in clinical models20. The reported device not only meets an urgent unmet need for biodefense but also provides the first-ever clinical detection capabilities for the rapidly-advancing field of CRISPR therapeutics.
Supplementary Material
Acknowledgements
This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, under award number R01AI098853, the Department of Homeland Security Science and Technology Directorate under Agreement HSHQPM-13-X-00222, the DARPA Safe Genes program under contract HR0011-17-2-0043 and the Laboratory Directed Research and Development, Grants 204977 and 200186, at Sandia National Laboratories, a multimission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525.
References
- 1.Mojica FJ, Díez-Villaseñor C, Soria E and Juez G, Mol Microbiol, 2000, 36, 244–246. [DOI] [PubMed] [Google Scholar]
- 2.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA and Charpentier E, Science, 2012, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho SW, Kim S, Kim JM and Kim J-S, Nat Biotechnol, 2013, 31, 230–232. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F and Jaenisch R, Cell, 2013, 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA and Zhang F, Science, 2013, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE and Church GM, Science, 2013, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komor AC, Kim YB, Packer MS, Zuris JA and Liu DR, Nature, 2016, 533, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M and Weissman JS, Cell, 2014, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D and Li J, Cell Stem Cell, 2013, 13, 659–662. [DOI] [PubMed] [Google Scholar]
- 10.Ma H, Marti-Gutierrez N, Park S-W, Wu J, Lee Y, Suzuki K, Koski A, Ji D, Hayama T, Ahmed R, Darby H, Van Dyken C, Li Y, Kang E, Park A-R, Kim D, Kim S-T, Gong J, Gu Y, Xu X, Battaglia D, Krieg SA, Lee DM, Wu DH, Wolf DP, Heitner SB, Belmonte JCI, Amato P, Kim J-S, Kaul S and Mitalipov S, Nature, 2017, 548, 413–419. [DOI] [PubMed] [Google Scholar]
- 11.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D and Gersbach CA, Science, 2016, 351, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang M, Zuris JA, Meng F, Rees H, Sun S, Deng P, Han Y, Gao X, Pouli D, Wu Q, Georgakoudi I, Liu DR and Xu Q, Proc Natl Acad Sci USA, 2016, 113, 2868–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee K, Conboy M, Park HM, Jiang F, Kim HJ, DeWitt MA, Mackley VA, Chang K, Rao A, Skinner C, Shobha T, Mehdipour M, Liu H, Huang W-C, Lan F, Bray NL, Li S, Corn JE, Kataoka K, Doudna JA, Conboy I and Murthy N, Nat Biomed Eng, 2017, 1, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim E, Koo T, Park SW, Kim D, Kim K, Cho H-Y, Song DW, Lee KJ, Jung MH, Kim S, Kim JH, Kim JH and Kim J-S, Nat Commun, 2017, 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA and Zhang F, Nature, 2015, 520, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller M, Lee CM, Gasiunas G, Davis TH, Cradick TJ, Siksnys V, Bao G, Cathomen T and Mussolino C, Molecular Therapy, 2016, 24, 636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou Z, Zhang Y, Propson NE, Howden SE, Chu L-F, Sontheimer EJ and Thomson JA, Proc Natl Acad Sci USA, 2013, 110, 15644–15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrington LB, Paez-Espino D, Staahl BT, Chen JS, Ma E, Kyrpides NC and Doudna JA, Nat Commun, 2017, 8, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senturk S, Shirole NH, Nowak DG, Corbo V, Pal D, Vaughan A, Tuveson DA, Trotman LC, Kinney JB and Sordella R, Nat Commun, 1AD, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu Z, Yang W, Sen Yan A Yin J Gao X Liu Y Zheng J Zheng Z Li S Yang S Li X Guo and Li X-J, Sci Rep, 2017, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seamon KJ, Light YK, Saada EA, Schoeniger JS and Harmon B, Anal. Chem, 2018, 90, 6913–6921. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Rajan R, Seifert HS, Mondragón A and Sontheimer EJ, Mol Cell, 2015, 60, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL and Davidson AR, Cell, 2016, 167, 1829–1838.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alves NJ, Mustafaoglu N and Bilgicer B, Biosens Bioelectron, 2013, 49, 387–393. [DOI] [PubMed] [Google Scholar]
- 25.Liu W, Yu H, Zhou X and Xing D, Anal. Chem, 2016, 88, 8369–8374. [DOI] [PubMed] [Google Scholar]
- 26.Huang M, Zhou X, Wang H and Xing D, Anal. Chem, 2018, 90, 2193–2200. [DOI] [PubMed] [Google Scholar]
- 27.Mekler V, Minakhin L, Semenova E, Kuznedelov K and Severinov K, Nucleic Acids Res, 2016, 44, 2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mekler V, Minakhin L and Severinov K, PNAS, 2017, 114, 5443–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koh C-Y, Schaff UY, Piccini ME, Stanker LH, Cheng LW, Ravichandran E, Singh B-R, Sommer GJ and Singh AK, Anal. Chem, 2015, 87, 922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrington LB, Doxzen KW, Ma E, Liu J-J, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, Kranzusch PJ, Sontheimer EJ, Davidson AR, Maxwell KL and Doudna JA, Cell, 2017, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lino CA, Harper JC, Carney JP and Timlin JA, Drug Deliv, 2018, 25, 1234–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.